

Abstract
5-methylcytosine (mC) is an epigenetic mark that impacts transcription, development, and genome stability, and aberrant DNA methylation contributes to aging and cancer. Active DNA demethylation involves stepwise oxidation of mC to 5-hydroxymethylcytosine, 5-formylcytosine (fC), and potentially 5-carboxylcytosine (caC), excision of fC or caC by thymine DNA glycosylase (TDG), and restoration of cytosine via follow-on base excision repair. Here, we investigate the mechanism for TDG excision of fC and caC. We find that 5-carboxyl-2′-deoxycytidine ionizes with pKa values of 4.28 (N3) and 2.45 (carboxyl), confirming that caC exists as a monoanion at physiological pH. Calculations do not support the proposal that G·fC and G·caC base pairs adopt a wobble structure that is recognized by TDG. Previous studies show that N-glycosidic bond hydrolysis follows a stepwise (SN1) mechanism, and that TDG activity increases with pyrimidine N1 acidity, i.e., leaving-group quality of the target base. Calculations here show that fC and the neutral tautomers of caC are acidic relative to other TDG substrates, but the caC monoanion exhibits poor acidity and likely resists TDG excision. While fC activity is independent of pH, caC excision is acid catalyzed, and the pH profile indicates that caC ionizes in the enzyme-substrate complex with an apparent pKa of 5.8, likely at N3. Mutational analysis reveals that Asn191 is essential for excision of caC but dispensable for fC activity, indicating that N191 may stabilize N3-protonated forms of caC to facilitate acid catalysis, and suggesting that N191A-TDG could potentially be useful for studying DNA demethylation in cells.
Introduction
The conversion of cytosine (C) to 5-methylcytosine (mC) constitutes the major type of DNA methylation in vertebrates, and this epigenetic signal has a profound influence on biological processes including transcription, development, and genome stability.1 Moreover, aberrant DNA methylation is implicated in aging and in human diseases including cancer.2 The DNA methyltransferases that catalyze DNA methylation are well characterized, but the enzymes responsible for actively reversing the methylation mark, i.e., converting mC back to C, had remained elusive. However, recent biochemical and biological studies have established a pathway for active DNA demethylation involving oxidation of mC and processing of mC derivatives via thymine DNA glycosylase (TDG) and base excision repair (BER), as illustrated in Fig. 1.
Figure 1.

Pathway for active DNA demethylation involving TET enzymes and TDG-initiated BER. Details and abbreviations are provided in the main text.
It was recently discovered that the TET (Ten-eleven translocation) family of dioxygenases can oxidize mC to 5-hydroxymethylcytosine (hmC),3,4 a base that was previously detected in mammalian DNA.5 Subsequent studies revealed that TET enzymes can further oxidize hmC to 5-formylcytosine (fC), and fC to 5-carboxylcytosine (caC), in a stepwise manner.6-8 We showed that TDG can rapidly excise fC from DNA in vitro,9 and this activity was subsequently found in mammalian cells.10-13 TDG also excises caC in vitro,6 albeit substantially slower than fC,9 and this activity is found in mammalian cells.6,11,13 The abasic nucleotide generated by TDG excision fC or caC is replaced by cytosine via down-stream BER, completing the demethylation process.
No mammalian glycosylase can excise hmC, and TDG is the only one that can remove fC or caC.1,9,14,15,16 TDG, and its glycosylase activity in particular, is essential for embryonic development, indicating a critical role for TDG-mediated DNA demethylation in regulating developmental genes.17,18 TDG also functions to maintain genome integrity; it was discovered as an enzyme that selectively excises T from G·T mispairs, an activity needed to protect against mutations caused by mC deamination.19 Here, we investigate the mechanism by which TDG excises fC and caC from DNA, and the chemical properties of these bases that dictate the catalytic requirements for their excision.
Previous kinetic isotope effect (KIE) studies show that 2′-deoxynucleotide hydrolysis reactions, including nonenzymatic and those catalyzed by UNG, MutY, and ricin, follow a stepwise (SN1) mechanism that involves rupture of the N-glycosidic bond to yield a short-lived oxacarbenium ion intermediate and subsequent addition of the nucleophile.20-24 As such, the rate depends on the stability (leaving group ability) of the departing nucleobase, and catalysis can be achieved by activation (protonation) or electrostatic stabilization of the leaving group.22,23,25,26 This chemical precedent indicates that the TDG reaction likely follows a stepwise mechanism,23 rather than a concerted (SN2) mechanism as recently suggested.27 Consistent with this conclusion, we previously showed that TDG activity (kmax) depends on N1 acidity of the target pyrimidine, i.e., activity increases with leaving group ability of the excised base.16 For example, kmax is much greater for 5-chlorouracil (pKaN1 = 8.1) relative to thymine (pKaN1 = 10.2), even though these bases have very similar steric and electrostatic properties.16 This work also revealed that TDG can excise cytosine analogues harboring a C5 substituent (F, Br, OH) that enhances N1 acidity relative to cytosine, which is not excised.16
These previous findings had suggested that TDG could excise fC, given the expected enhancement in N1 acidity afforded by the electronic effect of a formyl group (σm = 0.35 for CHO)28, and this prediction was confirmed.9 We evaluate the chemical properties of fC that are potentially relevant to its enzymatic excision here, including N1 acidity and resonance stabilization of the N1-deprotonated fC anion, and we examine the proposal that fC forms an imino tautomer and a wobble structure with guanine that is recognized by TDG.29,30
The chemical properties of caC that dictate the catalytic requirements for its excision are more complex, due to a number of factors. First, the effect of the carboxyl substituent on N1 acidity depends on its ionization state; acidity is expected to be enhanced by a protonated carboxyl group (σm = 0.37)28 and reduced by a deprotonated carboxyl (σm = -0.10). A previous study reported the calculated N1 acidity for caC with COOH,31 but the carboxyl is expected to be deprotonated (COO-) at physiological pH,32 and N1 acidity for the caC monoanion is unknown. In addition, N3 ionization is expected to dramatically impact N1 acidity, but this has not been previously examined. Here, we report calculated acidities for several different ionization and tautomeric states of caC, and for fC and other relevant pyrimidines. Because N- glycosidic bond hydrolysis can be acid catalyzed, the ionization sites and pKa values are relevant to enzymatic catalysis. A previous study on ionization of the 5-carboxyl-2′-deoxycytidine (5-ca-dC) nucleoside reported a pKa for N3 but not for the carboxyl group (proposed to be <1).32 We reexamined 5-ca-dC ionization here, and provide pKa values for both N3 and the carboxyl group.
We also determined the pH dependence of catalysis for fC and caC, and the results inform the mechanism by which TDG excises these bases. We also determined the role of conserved active-site residues in TDG excision of fC and caC, using site-directed mutagenesis and enzyme kinetics and equilibrium binding experiments. Together, the computational and biochemical results reveal key differences in the chemical properties of fC and caC that are relevant to enzymatic excision, and uncover differences in the mechanism used by TDG to excise these bases, which differ by only a single atom. We also reveal a TDG variant that exhibits normal fC activity but no detectable caC activity, which provides a novel tool to investigate the mechanism of DNA demethylation in cells.
Results and Discussion
Ionization of 5-carboxyl-2′-deoxycytidine
Given that N-glycosidic bond hydrolysis can be acid catalyzed, the pKa values for ionization at N3 and the carboxyl of caC are important for understanding enzymatic excision of this base from DNA. We collected UV absorbance spectra for 5-ca-dC under conditions of pH 0.5 to 8 (Fig. 2a), and the results indicate two ionization events. Observation of isosbestic points for scans at pH ≤ 2.5 (257 nm, 303 nm) and pH >4.5 (267 nm) provide wavelengths that can be used to unambiguously determine each pKa. The pH dependence of A303 reflects a single ionization, yielding a pKa = 4.28 ± 0.02 (Fig. 2b), and the same result is obtained for A257 (pKa = 4.30 ± 0.06, not shown). The pH dependence of A267 gives pKa = 2.49 ± 0.09 (Fig. 2c). Other wavelengths reflect both ionizations. Fitting the pH dependence of A286 gives pKa1 = 4.25 ± 0.04 and pKa2 = 2.41 ± 0.09 (Fig. 2d), in excellent agreement with the values determined at the isosbestic points (Figs. 2b, 2c). For comparison, we find that 2′-deoxycytidine (dC) ionizes at N3 with a pKa = 4.31 ± 0.01 (Supplementary Fig. S1), which is identical to a previous finding.33
Figure 2.

Ionization of N3 and the carboxyl of 5-ca-dC. (a) UV absorbance spectra for 5-ca-dC at pH 0.75 thru 6.0; the absorbance is essentially unchanged for pH 6.0 to 8.0. Isosbestic points are observed for a subset of the scans at 257 nm, 267 nm, and 303 nm; absorbance at these wavelengths depends on only one of the two ionizations. (b) The pH dependence of A303 was fitted to eq. 1, giving pKa = 4.28 ± 0.02. (c) pH dependence of A267 was fitted to eq. 1, giving pKa = 2.49 ± 0.09. (d) pH dependence of A286 was fitted to eq. 2, giving pKa1 = 4.25 ± 0.04 and pKa2 = 2.41 ± 0.09. (e) Ionization of 5-ca-dC as indicated by our findings.
Regarding 5-ca-dC ionization, we assign the pKa of 4.28 to ionization at N3 (Fig. 2e). Given the small electronic effect for a carboxylate substituent (σm = −0.10 for COO−)28, which is probably offset by hydrogen bonding to the vicinal NH2 (Fig. 2e), it is reasonable to find similar N3 pKa values for 5-ca-dC and dC. Assignment of the pKa = 4.25 to N3 is also supported by observation that this ionization exhibits a similar isosbestic point (267 nm) as that observed for N3 of dC (265 nm, Supplementary Fig. S1). Thus, we assign the pKa = 2.45 to the carboxyl of caC, which is consistent with previously determined pKa values for a carboxyl with a vicinal NH2 in aro matic systems.32 We consider the pKa values reported here to be more accurate than previously reported values of pKa = 4 for N3 and pKa <1 for the carboxyl of 5-ca-dC, which were obtained from UV absorbance monitored at a single wave-length (A300).32 Our findings confirm that caC exists as a monoanion at physiological pH, and suggest that acid catalysis of 5-ca-dC hydrolysis is more likely to involve protonation at N3 than at the carboxylate group. Additional studies are needed to determine the pKa values for N3 and the carboxyl of caC in duplex DNA, but they are not expected to be dramatically perturbed from the values reported here.
Amino tautomers of fC and caC likely predominate in DNA
We next consider the tautomeric state of fC and caC that is likely to predominate in DNA under physiological conditions. It was proposed that fC and caC favor an imino tautomeric state (Fig. 3) and thereby adopt a wobble structure when paired with guanine in DNA, similar to the structure of G·T and G·U mispairs, and that a wobble structure is unifying feature of substrate recognition by TDG.29,30 To examine this idea, we calculated the relative stability of the amino and imino tautomers for fC and caC (monoanion). As shown in Fig. 3, the amino tautomers of fC and caC are much more stable than their imino counterparts, in the gas phase and in water. Notably, NMR studies find that the amino tautomer is the predominant form of 5-formyl-2′-deoxycytidine (in DMSO).34 Moreover, DNA melting studies show that the stability of G·fC and G·caC base pairs is equal to or greater than that of G·C pairs.32,35 These findings indicate that G·fC and G·caC pairs adopt the canonical Watson-Crick structure rather than the destabilized wobble structure that would be favored by imino tautomers of fC and caC. Together, these observations do not support the proposal that amino-imino tautomerization of fC and caC can explain how TDG selectively recognizes G·fC and G·caC pairs in DNA.29,30
Figure 3.

Shown are the calculated relative stabilities of the amino and imino tautomers of fC and the caC anion, in the gas phase and water. The values are reported as the difference in free energy (∆∆G, kcal mol-1) with respect to the most stable species (∆∆G = 0).
Calculated acidities of fC, caC, and other pyrimidines
As discussed above, we previously showed that TDG activity depends on pyrimidine N1 acidity, i.e., it increases with leaving group quality of the excised base. 9,16 Although active-site interactions can play a significant role, pyrimidine N1 acidity is an important factor in TDG activity. To understand the catalytic requirements for excision of fC and caC, we calculated the acidities for these bases and other relevant pyrimidines (Fig. 4). The acidities are reported as the free energy required for deprotonation (∆G, kcal mol-1), where a lower value indicates greater acidity. Given the biochemical and structural evidence that the TDG active site is relatively nonpolar,16,36-38 we calculated the acidities in the gas phase as well as in water. Notably, the acidity trends are the same for both media, though the differences are more pronounced in the gas phase. Our results are consistent with previous calculations for limited subsets of these pyrimidines.16,31,39,40 However, N1 acidities have not previously been reported for the caC anion (1) and two of the neutral caC tautomers (2, 4). As shown below, this new information is important for understanding the catalytic requirements for excision of caC from DNA.
Figure 4.

Acidity for N1 and other sites for pyrimidines in the gas phase and bulk solution. The calculated acidities are reported as the free energy (∆G, kcal mol-1) required for deprotonation in the gas phase and in water (parenthetical values).
The calculations indicate that fC is remarkably acidic (N1) compared to other pyrimidines that are excised by TDG, including U, T, and 5-fluorocytosine (5FC). Notably, an intra-molecular hydrogen bond involving the formyl oxygen and the vicinal NH2 of fC33,34 stabilizes one rotamer over the other by about 5 kcal/mol (Supplementary Fig. S2), but it does not substantially alter N1 acidity (compare fC and fC* in Fig. 4). The robust acidity of fC is attributable to the electronic effect of the formyl substituent (σm = 0.35 for CHO)28 and resonance stabilization of the fC anion, via charge delocalization to the formyl oxygen and O2 (Fig. 5) (σp = 0.42, σp- = 1.03; para values given because σm− is not available, to our knowledge).28 The resonance effect likely accounts for the greater acidity of fC relative to 5FC, given the equivalent electronic effects for fluoro and CHO (σm of 0.34 and 0.35, respectively)28. Previous findings that TDG excises fC some 17-fold faster than 5FC9,16 can likely be explained by the much greater N1 acidity of fC relative to 5FC, and perhaps by electrostatic catalysis of fC excision via stabilization of negative charge resonating to the formyl oxygen of the departing fC anion.9,16
Figure 5.
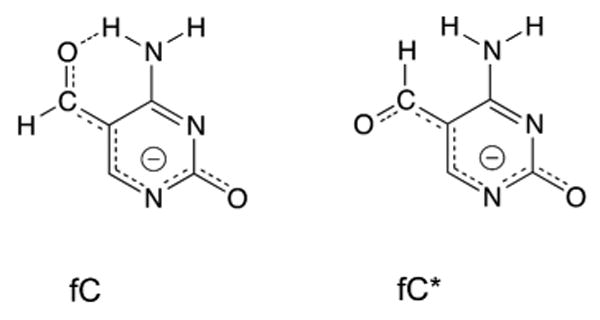
Resonance stabilization of the fC anion.
The poor N1 acidity of hmC (Fig. 4) likely accounts in large part for its resistance to excision by TDG, given previous findings that TDG rapidly excises 5-hydroxymethyluracil (hmU) and can therefore accommodate a hydroxymethyl group at the C5 position of pyrimidines.16
The predominant form of caC expected under physiological conditions, the monoanion (Fig. 2), is likely to resist enzymatic excision, given that the dianion is expected to be unstable and thus a poor leaving group. Consistent with this notion, the caC anion 1 is much less acidic than pyrimidines that are excised by TDG, including fC, U, T, and 5FC (Fig. 4). By contrast, the neutral forms of caC exhibit remarkable acidity, including the zwitterion 2, and the uncharged amino 3 and imino 4 tautomers. Indeed, 2 and 4 are substantially more acidic than U, T, and fC, and 3 exhibits similar acidity to these bases. These relative acidities have implications for catalysis of caC excision, as discussed below. Our findings confirm that ionization of the carboxyl group has a major impact on N1 acidity (Fig. 4, compare 1 and 3), consistent with a large difference in the substituent electronic effects (σm is −0.10 for COO− and 0.37 for COOH).28 The calculations also show that protonation at N3 greatly increases N1 acidity of caC, even if the carboxyl remains deprotonated (compare 1 and 2). As shown above, this N3 ionization occurs with pKa = 4.3 for 5-ca-dC (Fig. 2). Resonance effects likely contribute to acidity of the caC tautomers, as noted above for fC (Fig. 5). Thus, our calculations indicate that protonation of the caC monoanion to give one of the neutral caC tautomers (2, 3, or 4), i.e., acid catalysis, could be an effective strategy for enzymatic excision of caC from DNA.
pH dependence of catalysis for excision of fC and caC
To examine the potential role for acid catalysis of caC excision, we determined the pH dependence of TDG activity for caC and fC. The kinetics experiments were performed, under saturating enzyme conditions, such that the rate constants are not influenced by enzyme-substrate association or events after the chemical step (product release or inhibition).16 As shown in Fig. 6, the fC excision activity of TDG is essentially unchanged for pH 6.0 to 9.0. This is consistent with expectations; while N3-protonation would likely enhance fC excision, it is unlikely to occur given a pKa value of 2.6 for N3 of fC.33 Notably, we previously showed that TDG activity for G·U and G·T substrates is also nearly constant for pH 6 to 9.41 These findings indicate no evidence of acid or base catalysis for G·fC, G·U, or G·T substrates. In stark contrast, caC activity increases with decreasing pH; kobs is 420-fold higher at pH 5.5 than at pH 9.25 (Fig. 6). A previous study found that caC activity is 10-fold higher at pH 5.5 versus pH 8.0 for the catalytic domain of TDG (residues 111-308).29 However, we find that full-length TDG (410 residues) exhibits a much higher 57-fold difference in caC activity for the same pH values (Fig. 6). This discrepancy could indicate that full-length enzyme should be used for examining the pH dependence of catalysis. Previous studies show that the N-terminal region of TDG contributes to binding and base excision, particularly for G·T substrates,42-44 and it could potentially contribute to caC activity.
Figure 6.

pH dependence of TDG activity for G·fC (∆) and G·caC (O) substrates. Fitting the G·caC data to a model for ionization of an essential protonated group (eq. 4; dotted line) gives an apparent pKa = 5.80 ± 0.03 and a limiting kobs of 4.4 ± 0.1 min-1, but fitting is poor for pH > 7. Fitting to a model with an essential protonated group and a second, nonessential group (eq. 5, solid line) gives apparent pKa values of pKa1 = 5.75 ± 0.03 and pKa2 = 8.2 ± 0.7, a limiting kobs of 4.5 ± 0.1 min-1, and a rate enhancement factor (1 + α) of 4.5 (i.e., the fold increase in kobs resulting from deprotonation of the second group).
Because the kinetics experiments were conducted under saturating enzyme conditions, the pH profile for caC excision reflects the ionization of one or more groups in the enzyme-substrate (ES) complex. We fitted the caC data to a standard equation for ionization of a single essential protonated group (Fig. 6, dotted line); this model assumes that no activity remains when the ionizing group is deprotonated (i.e., log linear, slope = -1). While this yields an apparent pKa of 5.80 ± 0.03, the kobs values are displaced increasingly above the fitted curve for pH > 7 (kobs is about 1.7-, 2.8-, and 4.3-fold greater than predicted for pH 8.0, 8.5, and 9.0, respectively). This deviation could reflect ionization of a second, nonessential group that leads to modestly (∼4.3-fold) higher activity upon deprotonation. The fitting is improved for a double-ionization model (Fig 6, solid line), giving apparent pKa values of pKa1 = 5.75 ± 0.03 for the essential protonated group and pKa2 = 8.2 ± 0.7 for the nonessential deprotonated group.
We conclude that the essential protonated group (pKa1 = 5.75) is the caC base, rather than a TDG side chain serving as a general acid (i.e., to protonate caC). Crystal structures reveal only two residues that could potentially serve as an essential general acid, H151 and Y152 (Fig. 7). However, the H151A and Y152F variants exhibit pH profiles for caC excision that are very similar to that of native TDG (Supplementary Fig. S3). Thus, H151 and Y152 do not serve as an essential general acid, nor can they be assigned as the second (nonessential) ionizing group. Together, the data support a model whereby TDG excises a neutral form of the caC base but not the caC anion, and deprotonation of a second group, likely from the enzyme, enhances this activity. Such a model is consistent with the very poor calculated N1 acidity of the caC anion, which suggests that it is unlikely to be excised by TDG.
Figure 7.
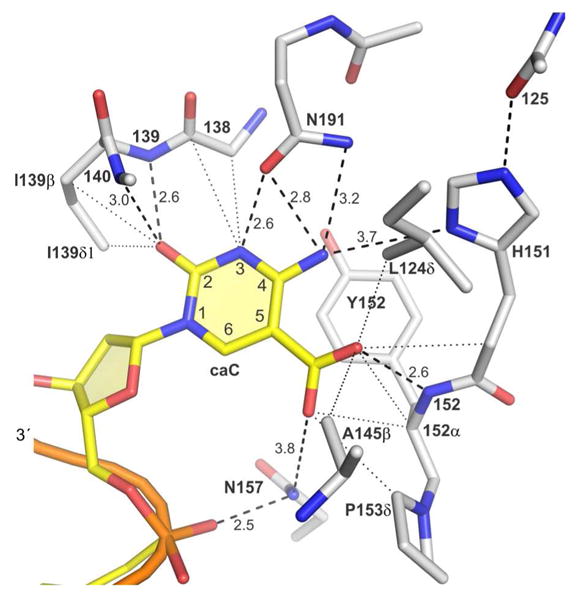
Previously reported structure of TDG (catalytic domain) with a 5-carboxyl-dC analog (non-cleavable) flipped into the active site (PDBID: 3UOB).38 Hydrogen bonds (dashed lines) and van der Waals contacts (dotted lines, d ≤ 3.7 Å) are shown. Similar interactions are observed for a structure of the N140A-TDG variant (catalytic domain) bound to DNA containing an A·caC mismatch (PDBID: 3UO7).38
It seems reasonable that the proton needed for ionization of caC in the ES complex is derived from solvent, given that caC excision is relatively slow (min timescale), and that the TDG active site is relatively permissive and protected from solvent by two loops that move with nucleotide flipping and by residues in the disordered N-terminal region (L124, Fig. 7). Observation that the free 5-ca-dC nucleoside ionizes with pKa values of 4.28 for N3 and 2.45 for the carboxyl (Fig. 2) suggests that protonation of caC in the ES complex (pKa1 = 5.75) occurs at N3 rather than the carboxyl, to give either the zwitterion 2 or the uncharged imino tautomer 4 (Fig. 4). This is supported by findings that activity is much higher for caC versus fC at low pH (Fig. 6) and that N3-protonated forms of caC (2, 4) are more acidic than fC while the carboxyl (COOH) form 3 is less acidic (Fig.4). Additional evidence for protonation of caC at N3 rather than the carboxyl is provided by the mutational studies below. Our finding that caC excision is acid catalyzed with accounts for the initially puzzling observation that caC is excised ∼5-fold slower than fC at pH 7.5,9 even though the N3-protonated forms of caC (2, 4) are more acidic than fC (Fig. 4); the apparent pKa1 = 5.75 indicates a low population of N3-protnated caC and a high population of the excision-resistant anion 1 in the ES complex at pH 7.5.
Mutational analysis of fC and caC excision by TDG
We previously characterized the role of conserved side chains in TDG excision of uracil or thymine (G·U or G·T mispairs),36 but their contribution to fC and caC excision was unknown. Crystal structures of DNA-bound TDG, with either caC (Fig. 7)38 or uracil36 flipped into its active site, reveal potential catalytic interactions between the flipped base and enzyme and side chain and backbone groups. A structure is not available for a TDG-fC complex, but existing structures suggest interactions that could facilitate fC excision. We used mutagenesis and kinetics experiments to examine the role of four conserved side chains in the excision of fC and caC, and the results are given in Table 1. Because the experiments were performed under saturating enzyme conditions, the rate constants (kmax) reflect the maximal rate of product formation without influence from enzyme-substrate association or product release or product inhibition.16,45
Table 1. Glycosylase activity for TDG and variants.
| Substrate | Enzyme | kmax (min-1) | Relative to wild-type TDG |
|---|---|---|---|
| G·fC | TDG | 0.61 ± 0.04 | |
| A145G | 0.22 ± 0.02 | 0.36 | |
| H151A | 0.35 ± 0.06 | 0.57 | |
| Y152F | 0.91 ± 0.09 | 1.5 | |
| N157A | 0.14 ± 0.03 | 0.23 | |
| N191A | 1.00 ± 0.03 | 1.6 | |
| G·caC | TDG | 0.14 ± 0.01 | |
| A145G | 0.60 ± 0.01 | 4.3 | |
| H151A | 0.072 ± .002 | 0.51 | |
| Y152F | 0.29 ± 0.04 | 2.1 | |
| N157A | 0.13 ± 0.001 | 0.89 | |
| N191A | N.D. |
kobs values are the mean and standard deviation for three independent experiments collected at 22 °C under saturating enzyme conditions and analyzed by HPLC. N.D.; activity not detected. Data for wild-type TDG was previously reported.9
Regarding fC excision, we find small (<2-fold) effects on kobs for the H151A, Y152F, and N191A mutations (Table 1), indicating that the H151 and N191 side chains, and the hydroxyl of Y152, contribute minimally to fC excision. The A145G mutation causes a 2.7-fold decrease in kobs. Thus, fC is the only substrate identified to date for which A145 facilitates catalysis; A145 curtails activity for G·T36 and G·caC substrates (Table 1). The crystal structure with flipped caC raises the possibility that the A145 methyl contacts the formyl group of fC (Fig. 7), which could potentially help position the flipped base to optimize catalytic interactions with its formyl or O2 oxygen. The N157A mutation causes a 4-fold loss in kobs for G·fC activity (Table 1). Previous studies found that the same mutation diminishes activity for G·U, G·hmU, and G·T substrates.27,30 This damaging effect for numerous substrates can be reasonably explained by disruption of the contact between the N157 side chain NH2 and the 5′ phosphate of the flipped nucleotide (Fig. 7). Consistent with this idea, the N157D mutation causes a greater loss in G·U and G·hmU activity than the N157A mutation,42,46 as expected for electrostatic repulsion between negatively charged Asp side chain and the 5′-phosphate. Notably, this Asn is conserved in prokaryotic MUG enzymes that are homologous to TDG, and in the more distally related UNG enzymes. Moreover, the Asn-phosphate contact is seen in all DNA-bound structures of TDG, MUG, and UNG.29,36-38,46,47
Our finding that A145, H151, Y152, N157, and N191 contribute minimally or not at all to excision of fC is consistent with its robust N1 acidity (Fig. 4), which renders it inherently amenable to enzymatic excision. Nevertheless, crystal structures suggest that three backbone amide groups of TDG could contact the O2 and formyl oxygens of fC (Fig. 7). These contacts could help retain the flipped fC in the active site and promote C-N bond cleavage by stabilizing the departing fC anion. While these interactions could be sufficient to catalyze fC excision, we cannot rule out a potential role for side chains other than the five examined here.
We next consider the role of the same five active site residues in caC excision by TDG. We find a negligible role for H151 in caC excision (Table 1), which is consistent with findings that the imidazole is distal from the caC base (Fig. 7). Our finding does not support the proposal that the imidazole (protonated) of H151 contributes to recognition of the caC anion.38 The Y152F mutation gives a 2.1-fold increase in activity, demonstrating that the hydroxyl of Y152 is dispensable and suggesting that caC excision is modestly enhanced by the more hydrophobic environment (benzyl versus phenol). Remarkably, the A145G mutation gives a four-fold increase in caC activity (Table 1). Crystal structures indicate that the A145 methyl contacts the caC carboxyl (Fig. 7).38 The nonpolar methyl could potentially disfavor flipping of the caC anion, or impede the formation of optimal catalytic interactions with other active-site groups. This finding is reminiscent of the 13-fold increase in G·T activity caused by the A145G mutation,36 raising the question of why a residue that curtails activity for two biological substrates is strictly conserved in TDG enzymes (vertebrates). Our previous studies indicate that A145 counters aberrant excision of T from A·T pairs36 and we find here that it contributes to fC excision.
The N157A mutation has no significant effect on caC excision (Table 1), even though it does adversely impact activity for G·fC (Table 1), G·U, G·hmU, and G·T substrates.27,30 These findings do not support the proposal that the N157A mutation has no effect on substrate specificity.30 As noted above, N157 contacts the 5′-phosphate of the flipped nucleotide (Fig. 7) and is highly conserved in TDG, MUG, and UNG enzymes. The absence of a damaging effect for N157A on caC activity suggests that the expected loss could be offset by an effect for Ala at position 157 that favors excision of caC but not the other bases (fC, U, hmU, T). One possibility is that the neutral carboxyl (COOH) of caC is stabilized to a greater extent by Ala versus Asn, i.e., Ala could favor neutral forms of caC (3, 4) that are more acidic than the monoanion 1.
Strikingly, the N191A variant has no detectable caC excision activity (Table 1, Fig. 8a), even for extended reaction times (up to 3 h) and enzyme concentrations that greatly exceed the saturating level for nonspecific DNA (5-10 μM ≫ KdNS = 0.2 μM).48 This is not due to a mutational effect on protein stability, because N191A-TDG retains full fC activity (Table 1, Fig. 8a) and substantial activity for G·U mispairs.36 Moreover, an electrophoretic mobility shift assay (EMSA) shows that the N191A mutation does not substantially weaken the binding of TDG to DNA containing a G·caC pair (Fig. 8b). These observations indicate that N191 plays a key role in the chemical step of the reaction for excision of caC, a role that is not needed for fC excision.
Figure 8.

Effect of the N191A mutation on substrate binding and base excision. (a) Electrophoretic glycosylase assay shows that N191A-TDG has normal G·fC activity, markedly reduced G·T activity, and no significant G·caC activity, even for a reaction time of 3 h. Reactions were performed with 0.20 μM enzyme, 0.10 μM substrate, and were quenched after 5 min or 3 h (as indicated). (b) An electrophoretic mobility shift assay (EMSA) shows that N191A-TDG binds G·caC DNA with high affinity, similar to that observed for N140A-TDG. Previous studies show that N140A-TDG binds G·caC and other substrates with essentially the same affinity and/or enzyme-substrate contacts as native TDG,38,45 but it does not excise caC (under the EMSA conditions here). DNA (10 nM) was incubated with enzyme (5-50 nM) at 22 °C for 30 min prior to running the EMSA.
Implications for Catalysis
Together, the calculated acidities, mutational studies, and previous crystal structures suggest that N191 facilitates acid catalysis of caC excision, a function that is not required for fC excision (Fig. 9). Crystal structures indicate that the N191 side chain oxygen (Oδ1) contacts N3 of caC (Fig. 7). This interaction would favor the N3-protonated forms of caC (2, 4) but not the anion or the neutral amino tautomer 3, and could account for findings that the N3 pKa of caC is increased by 1.5 units in the enzyme-substrate complex relative to the 5-ca-dC nucleoside.
Figure 9.

Potential mechanisms for TDG excision of fC and caC as suggested by the results here and previous structural studies.38 (a) Excision of fC does not require a large role for the side chain of any active-site residue examined here, consistent with the robust N1 acidity of fC (Fig. 4). Nevertheless, it seems likely that fC excision involves electrostatic catalysis, where backbone amide groups (139, 140, 152; Fig. 7) stabilize the departing fC anion. (b) In contrast to the results for fC, our findings indicate that excision of the caC anion, the predominant species at neutral pH, requires acid catalysis. Findings that N191 is essential for caC excision but dispensable for fC activity suggest that N191is needed to stabilize an N3-protonated form of caC, which could be the zwitterion 2 or the neutral imino tautomer 4. It seems likely that excision of caC also involves electrostatic catalysis, via the same three backbone amide groups.
While the zwitterion 2 is acidic and should be highly amenable to TDG excision, a nonpolar active site could favor the facile conversion of 2 to the imino tautomer 4 (Fig. 9, Supplementary Fig. S4). Calculations show that 2 is more stable than 4 in water (∆∆Gwater = 5 kcal mol-1), while 4 is much more stable in the gas phase (∆∆Ggas = 9 kcal mol-1; Supplementary Fig. S4). As such, 4 might be favored in the TDG active site, which is relatively nonpolar.16,36-38,62 Indeed, TDG forms many nonpolar contacts, one hydrogen bond, and no ionic interactions with the carboxyl of caC (Fig. 7). Notably, 4 could also be derived directly from the caC anion via coupled abstraction of an amino (NH2) proton by COO− and protonation at N3 (Fig. 9). This would likely be facilitated by N191 and perhaps by the nonpolar contacts with the carboxyl (Fig. 7), which could favor the COOH of 4 over the COO− of 1.
Concluding Remarks
The studies reported here advance our understanding of the chemical properties of fC and caC that dictate the catalytic requirements for their excision from DNA, and the mechanism by which TDG excises these oxidized forms of mC. We show that the 5-ca-dC nucleoside ionizes with pKa values of 4.28 (N3) and 2.45 (carboxyl), confirming that caC exists as a monoanion at physiological pH. The calculated stabilities of the amino and imino tautomers for fC and caC (anion) do not support the proposal that G·fC and G·caC pairs adopt a wobble structure that is recognized by TDG.29,30 The calculated N1 acidities for fC and the neutral forms of caC are comparable to other TDG substrates, but the caC anion exhibits poor acidity, indicating resistance to enzymatic excision. We find that TDG excision of fC is pH independent, while excision of the caC anion is acid catalyzed (Fig. 9). The pH profile indicates that caC ionizes in the enzyme-substrate complex with an apparent pKa of 5.75, likely at N3. Mutational analysis of five conserved active-site side chains reveals that none are critical for excision of fC, in keeping with its robust N1 acidity. Nevertheless, fC excision likely requires electro-static catalysis, and crystal structures suggest this could involve backbone amide groups contacting the formyl and O2 oxygens of fC (Fig. 9). In contrast to the results for fC, N191 is essential for excision of caC. The results suggest N191 facilitates acid catalysis by stabilizing an N3-protonated form of caC (2, 4).
Our finding that N191A-TDG possess normal fC activity but no detectable caC activity suggests that this variant could be useful for examining the role of TDG in active DNA demethylation in mammalian cells. In particular, N191A-TDG could potentially be used to examine the possibility that excision of fC, rather than caC, constitutes the major role of TDG in a Tet-initiated pathway for active DNA demethylation. Observation that the N191A variant also has diminished G·T activity36 should not preclude its potential utility in this re gard, because G·T mispairs do not arise in the Tet-initiated demethylation pathway. A recent study found that N157D-TDG exhibits selective activity for caC over fC at pH 6, but at neutral pH this variant has greatly reduced activity for both caC and fC substrates compared to native TDG.30 Thus, the utility of N157D-TDG for cellular studies is unclear. Nevertheless, a TDG variant that exhibits selective activity for caC over fC under physiological conditions could be useful for studying the function of TDG in active DNA demethylation; our findings could inform the design of such a variant.
Experimental Section
Materials
The duplex DNA substrates consisted of 5′-GGGAGAAGAGGAGGAAxGAAGAGAGCTC, where x = fC, caC, or T, and a complementary strand that places G opposite the target base (x). This 28 bp DNA construct also places the target base (x) in a CpG context, consistent with the specificity of TDG. For reactions analysed by denaturing PAGE, the target strand included a 3′-fluorescein-d
T. The oligodeoxynucleotides were synthesized, purified, and quantified as described.9 Full length human TDG was pressed in E. coli and purified as described.49 The TDG variants A145G, H151A, and N191A were prepared as described.36 Expression plasmids for Y152F-TDG and N157A-TDG were generated using site-directed mutagenesis45 (primers provided in Supplementary Table S1) and the mutation was confirmed by DNA sequencing. The Y152F-TDG and N157A-TDG variants was expressed and purified using the protocol for native TDG.36,49 5-carboxyl-2′-deoxycytidine was from Berry & Associates.
pKa determinations
Ionization of 5-ca-dC (100 μM) was monitored by UV absorbance in buffers of varying pH (0.5 to 8.0) consisting of 25 mM sodium phosphate, 25 mM sodium acetate, and 50 mM NaCl. The pH dependence of absorbance a given wavelength was fitted by non-linear regression using equations for a single ionization (eq. 1) or double ionization (eq. 2) with Grafit 6.0.50
| (1) |
| (2) |
Glycosylase activity
The kinetics experiments used to determine the glycosylase activity of TDG and the TDG variants were performed essentially as described,36 using a saturating concentration of enzyme (5 or 10 μM) and DNA substrate concentrations of 0.5 μM or 1.0 μM, in HEMN.1 buffer (0.02 M HEPES pH 7.5, 0.1 M NaCl, 0.2 mM EDTA, 2.5 mM MgCl2) at 22 °C. The reactions were monitored by electrophoresis9 or anion-exchange HPLC under denaturing conditions.16 The HPLC data were fitted by non-linear regression to a single exponential equation (eq. 3).
| (3) |
where A is the amplitude, kobs is the observed rate constant, and t is reaction time. Because the experiments were performed under saturating enzyme conditions ([E] > [S] ≫ Kd), the rate constants (kobs) reflect the maximal rate of product formation (i.e., kobs ≈ kmax) and are not influenced by product release or product inhibition. The attainment of saturating enzyme conditions was confirmed by observation that using a two-fold higher enzyme concentration yielded the same kobs value (within error). Notably, previous findings from kinetics and equilibrium binding experiments show that TDG binds very tightly to GT, G·fC, and G·caC substrates, and tightly to nonspecific DNA (Kd = 0.1-0.3 μM).16,36,45,48,49,51
pH dependence of activity
The dependence of TDG activity (kobs) on pH was monitored for G·fC and G·caC substrates 22 °C using a buffer consisting of 0.01 M NaMES, 0.01 M Na-HEPES, 0.01 M Tris, 0.01 M NaCHES, 0.1 M NaCl, 2.5 mM MgCl2, and 0.2 mM EDTA. The caC activity of TDG could not be determined for pH < 5.5 or pH > 9.25, due likely to loss of TDG structural integrity, consistent with our previous observations for pH dependence of activity for G·U and G·T substrates and pH effects on enzyme stability.41 As noted above, kinetics experiments were performed under saturating enzyme conditions, as confirmed by observation of equivalent kobs values for multiple TDG concentrations (ranging from 2.5 μM to 15 μM). The pH profile for caC activity was fitted by non-linear regression to equations involving ionization of a single group (eq. 4) or two groups (eq. 5) in the enzyme-substrate complex:
| (4) |
| (5) |
For eqs. 4 and 5, kobs is the observed rate constant and k1 is the limiting rate constant.
Equilibrium binding experiments
The equilibrium binding of enzyme to DNA was monitored using an electrophoretic mobility shift assay (EMSA), essentially as previously described.48 The binding reactions (30 ul) were performed by incubating G·caC DNA (10 nM) with varying concentrations of enzyme (5-50 nM) at room temperature for 30 min in binding buffer (25 mM HEPES pH 7.5, 0.1 M NaCl, 1 mM EDTA, 1 mM DTT, 0.1 mg/ml BSA, 5% glycerol). Samples were loaded to a precast 6% native polyacrylamide gel (Invitrogen), and electrophoresis was performed for 60 min at 100 V, 5 °C. The gels were analyzed using a Typhoon 9400 imager (GE Healthcare) in the fluorescence mode to detect the 3′-fluorescein labeled DNA.
Computational studies
The gas phase calculations were conducted at the B3LYP/6-31+G(d) level of theory using Gaussian 03 and Gaussian 09.52-56 Structures were fully optimized in the gas phase, and frequencies calculated (no imaginary frequencies were found). Gas-phase acidity, and relative stability values are reported as ∆G in kcal mol-1. The only exception is the caC zwitterion 2 (Fig. 4), which is not a stable minimum in the gas phase and therefore is partially optimized. For the caC zwitterion 2, ∆G values are estimated from ∆E values. Dielectric medium calculations were done using the conductor-like polarizable continuum solvent model (CPCM, fully optimized at B3LYP/6-31+G(d) with UFF cavity) as implemented in Gaussian 03.57-59 The one exception is the N3-H acidity of 4 (Fig. 4); the deprotonated structure is not stable in solution so the gas-phase optimized structure was used. The ″total free energy in solution″ (∆G) values are reported, and these account for the free energy of solvation of a proton (-265.9 kcal mol-1).60,61
Supplementary Material
Acknowledgments
This work was supported by the U.S. National Institutes of Health (GM72711 to ACD) and the U.S. National Science Foundation (to AZM and JKL).
Abbreviations
- 5-ca-dC
5-carboxyl-2′-deoxycytidine
- BER
base excision repair
- caC
5-carboxylcytosine
- fC
5-formylcytosine
- hmC
5-hydroxymethylcytosine
- mC
5-methylcytosine
- TDG
thymine DNA glycosylase
- TET
Ten-eleven translocation
- UNG
uracil DNA glycosylase
Footnotes
Supporting Information. Supplementary Figures S1-S4, Supplementary Table S1, and the Cartesian coordinates and energies (in Hartree) for the calculated species. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
References
- 1.Nabel CS, Manning SA, Kohli RM. ACS Chem Biol. 2012;7:20. doi: 10.1021/cb2002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones PA, Baylin SB. Nat Rev Genet. 2002;3:415. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 3.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Science. 2009;324:930. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Nature. 2010;466:1129. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Penn NW, Suwalski R, O'Riley C, Bojanowski K, Yura R. Biochem J. 1972;126:781. doi: 10.1042/bj1260781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Science. 2011;333:1303. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Science. 2011;333:1300. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfaffeneder T, Hackner B, Truss M, Munzel M, Muller M, Deiml CA, Hagemeier C, Carell T. Angew Chem Int Ed Engl. 2011;50:7008. doi: 10.1002/anie.201103899. [DOI] [PubMed] [Google Scholar]
- 9.Maiti A, Drohat AC. J Biol Chem. 2011;286:35334. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raiber EA, Beraldi D, Ficz G, Burgess H, Branco MR, Murat P, Oxley D, Booth MJ, Reik W, Balasubramanian S. Genome Biol. 2012;13:R69. doi: 10.1186/gb-2012-13-8-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nabel CS, Jia H, Ye Y, Shen L, Goldschmidt HL, Stivers JT, Zhang Y, Kohli RM. Nat Chem Biol. 2012;8:751. doi: 10.1038/nchembio.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song CX, Szulwach KE, Dai Q, Fu Y, Mao SQ, Lin L, Street C, Li Y, Poidevin M, Wu H, Gao J, Liu P, Li L, Xu GL, Jin P, He C. Cell. 2013;153:678. doi: 10.1016/j.cell.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen L, Wu H, Diep D, Yamaguchi S, D'Alessio AC, Fung HL, Zhang K, Zhang Y. Cell. 2013;153:692. doi: 10.1016/j.cell.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masaoka A, Matsubara M, Hasegawa R, Tanaka T, Kurisu S, Terato H, Ohyama Y, Karino N, Matsuda A, Ide Hl. Biochemistry. 2003;42:5003. doi: 10.1021/bi0273213. [DOI] [PubMed] [Google Scholar]
- 15.Morera S, Grin I, Vigouroux A, Couve S, Henriot V, Saparbaev M, Ishchenko AA. Nucleic Acids Res. 2012:40, 9917. doi: 10.1093/nar/gks714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennett MT, Rodgers MT, Hebert AS, Ruslander LE, Eisele L, Drohat AC. J Am Chem Soc. 2006;128:12510. doi: 10.1021/ja0634829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cortazar D, Kunz C, Selfridge J, Lettieri T, Saito Y, Macdougall E, Wirz A, Schuermann D, Jacobs AL, Siegrist F, Steinacher R, Jiricny J, Bird A, Schar P. Nature. 2011;470:419. doi: 10.1038/nature09672. [DOI] [PubMed] [Google Scholar]
- 18.Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, Abramowitz LK, Bartolomei MS, Rambow F, Bassi MR, Bruno T, Fanciulli M, Renner C, Klein-Szanto AJ, Matsumoto Y, Kobi D, Davidson I, Alberti C, Larue L, Bellacosa A, et al. Cell. 2011;146:67. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiebauer K, Jiricny J. Proc Natl Acad Sci U S A. 1990;87:5842. doi: 10.1073/pnas.87.15.5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Werner RM, Stivers JT. Biochemistry. 2000;39:14054. doi: 10.1021/bi0018178. [DOI] [PubMed] [Google Scholar]
- 21.Dinner AR, Blackburn GM, Karplus M. Nature. 2001;413:752. doi: 10.1038/35099587. [DOI] [PubMed] [Google Scholar]
- 22.McCann JAB, Berti PJ. J Am Chem Soc. 2008;130:5789. doi: 10.1021/ja711363s. [DOI] [PubMed] [Google Scholar]
- 23.Berti PJ, McCann JA. Chem Rev. 2006;106:506. doi: 10.1021/cr040461t. [DOI] [PubMed] [Google Scholar]
- 24.Chen XY, Berti PJ, Schramm VL. J Am Chem Soc. 2000;122:1609. [Google Scholar]
- 25.Drohat AC, Stivers JT. J Am Chem Soc. 2000;122:1840. [Google Scholar]
- 26.Drohat AC, Jagadeesh J, Ferguson E, Stivers JT. Biochemistry. 1999;38:11866. doi: 10.1021/bi9910878. [DOI] [PubMed] [Google Scholar]
- 27.Hashimoto H, Zhang X, Cheng X. DNA Repair (Amst) 2013;12:535. doi: 10.1016/j.dnarep.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansch C, Leo A, Taft RW. Chemical Reviews. 1991;91:165. [Google Scholar]
- 29.Hashimoto H, Hong S, Bhagwat AS, Zhang X, Cheng X. Nucleic Acids Res. 2012;40:10203. doi: 10.1093/nar/gks845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hashimoto H, Zhang X, Cheng X. J Mol Biol. 2013;425:971. doi: 10.1016/j.jmb.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams RT, Wang Y. Biochemistry. 2012;51:6458. doi: 10.1021/bi300797q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sumino M, Ohkubo A, Taguchi H, Seio K, Sekine M. Bioorg Med Chem Lett. 2008;18:274. doi: 10.1016/j.bmcl.2007.10.081. [DOI] [PubMed] [Google Scholar]
- 33.La Francois CJ, Jang YH, Cagin T, Goddard WA, Sowers LC. Chem Res Toxicol. 2000;13:462. doi: 10.1021/tx990209u. [DOI] [PubMed] [Google Scholar]
- 34.LaFrancois CJ, Fujimoto J, Sowers LC. Chemical Research in Toxicology. 1998;11:75. doi: 10.1021/tx970186o. [DOI] [PubMed] [Google Scholar]
- 35.Munzel M, Lischke U, Stathis D, Pfaffeneder T, Gnerlich FA, Deiml CA, Koch SC, Karaghiosoff K, Carell T. Chemistry. 2011;17:13782. doi: 10.1002/chem.201102782. [DOI] [PubMed] [Google Scholar]
- 36.Maiti A, Noon MS, Mackerell AD, Jr, Pozharski E, Drohat AC. Proc Natl Acad Sci U S A. 2012;109:8091. doi: 10.1073/pnas.1201010109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maiti A, Morgan MT, Pozharski E, Drohat AC. Proc Natl Acad Sci USA. 2008;105:8890. doi: 10.1073/pnas.0711061105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L, Lu X, Lu J, Liang H, Dai Q, Xu GL, Luo C, Jiang H, He C. Nat Chem Biol. 2012;8:328. doi: 10.1038/nchembio.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhachkina A, Lee JK. J Am Chem Soc. 2009;131:18376. doi: 10.1021/ja906814d. [DOI] [PubMed] [Google Scholar]
- 40.Liu M, Li TT, Amegayibor FS, Cardoso DS, Fu YL, Lee JK. J Org Chem. 2008;73:9283. doi: 10.1021/jo801822s. [DOI] [PubMed] [Google Scholar]
- 41.Maiti A, Drohat AC. DNA Repair. 2011;10:545. doi: 10.1016/j.dnarep.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallinari P, Jiricny J. Nature. 1996;383:735. doi: 10.1038/383735a0. [DOI] [PubMed] [Google Scholar]
- 43.Steinacher R, Schar P. Curr Biol. 2005;15:616. doi: 10.1016/j.cub.2005.02.054. [DOI] [PubMed] [Google Scholar]
- 44.Guan X, Madabushi A, Chang DY, Fitzgerald M, Shi G, Drohat AC, Lu AL. Nucleic Acids Res. 2007;35:6207. doi: 10.1093/nar/gkm678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maiti A, Morgan MT, Drohat AC. J Biol Chem. 2009;284:36680. doi: 10.1074/jbc.M109.062356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barrett TE, Scharer OD, Savva R, Brown T, Jiricny J, Verdine GL, Pearl LH. EMBO J. 1999;18:6599. doi: 10.1093/emboj/18.23.6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, Tainer JA. Proc Natl Acad Sci USA. 2000;97:5083. doi: 10.1073/pnas.97.10.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morgan MT, Maiti A, Fitzgerald ME, Drohat AC. Nucleic Acids Res. 2011;39:2319. doi: 10.1093/nar/gkq1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morgan MT, Bennett MT, Drohat AC. J Biol Chem. 2007;282:27578. doi: 10.1074/jbc.M704253200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leatherbarrow RJ. GraFit 5.0. Erithacus Software Ltd.; Staines, U.K.: 1998. [Google Scholar]
- 51.Fitzgerald ME, Drohat AC. J Biol Chem. 2008;283:32680. doi: 10.1074/jbc.M805504200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]
- 53.Lee CT, Yang WT, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 54.Kohn W, Becke AD, Parr RG. J Phys Chem. 1996;100:12974. [Google Scholar]
- 55.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JJA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick KK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. GAUSSIAN 03. Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]
- 56.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JJA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick KK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. GAUSSIAN 09. Gaussian, Inc.; Wallingford CT: 2009. [Google Scholar]
- 57.Barone V, Cossi M. J Phys Chem A. 1998;102:1995. [Google Scholar]
- 58.Cossi M, Rega N, Scalmani G, Barone V. J Comput Chem. 2003;24:669. doi: 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- 59.Takano Y, Houk KN. J Chem Theory Comput. 2005;1:70. doi: 10.1021/ct049977a. [DOI] [PubMed] [Google Scholar]
- 60.Camaioni DM, Schwerdtfeger CA. J Phys Chem A. 2005;109:10795. doi: 10.1021/jp054088k. [DOI] [PubMed] [Google Scholar]
- 61.Kelly CP, Cramer CJ, Truhlar DG. J Phys Chem B. 2007;111:408. doi: 10.1021/jp065403l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kurinovich MA, Lee JK. J Am Chem Soc. 2000;122:6258. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.