

Abstract
Ectopia lentis (EL) is a condition that can either herald underlying systemic conditions, or be isolated. The recent expansion in the genetics of these conditions has furthered the understanding of the underlying molecular aetiology. It is becoming apparent that novel genes, and in particular the ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) family, are important in ocular development. The common link in these genes seems to be EL. The clinical management of EL is challenging. In particular, the options for addressing surgically induced aphakia in the context of an ectopic capsule are varied. Little evidence exists to direct management of these issues. This review summarises the molecular pathogenesis of EL and conditions associated with it, using the genetic aetiology as a framework. Furthermore, it summarises some of the issues involved in its clinical management.
Keywords: ectopia lentis, vitreolensectomy, Marfan syndrome
The Austrian ophthalmologist Karl Stellwag1 is credited with coining the term ectopia lentis (EL) in 1856. It describes abnormal movement of the crystalline lens from within its natural position (Figure 1). Although trauma is a common cause, a genetic predisposition was first proposed by Williams,2 in 1875, by describing a family with EL in two generations. The genetic aetiology of EL is an ever growing topic.
Figure 1.

Ectopia lentis—note attenuated zonules offering very limited capsular support.
Anatomy
The critical structures for maintaining the crystalline lens in its natural position are the zonular filaments (ZF). These form a circular structure between the equatorial lens and the ciliary body through a triangular structure with the apex of the triangle on the equatorial margin of the lens covering an area of up to 55 nm23 that is rich in fibrillin fibres.4 They were first described as being part of the family of microfibrils in 19715 and the most important macromolecular component of ZF are fibrillins.
The three distinct fibrillins are fibrillin-1, -2, and -3, which are encoded by the genes FBN1 (OMIM 134797), FBN2 (OMIM 612570), and FBN3 (OMIM 608529), respectively. FBN1 is a 237-kb gene consisting of 65 exons located at 15q21.1.6 It encodes fibrillin-1, the most abundant macromolecule in ZF. This protein consists of 47 epidermal growth factor domains, 43 of which are calcium binding (cbEGF) and two cysteine rich domains (transforming growth factor-binding protein-like domain).7 The latter of these are unique to this family of proteins, and their specific function is unclear, although they may have a role in integrin binding.8 cbEGF domains, in particular, form intradomain disulphide bonds and also contain a calcium-binding consensus sequence. When calcium binds to these bonds, the molecule strengthens and is more resistant to degradation. It is these structures that are crucial to the extracellular matrix (ECM) function of these proteins. It is proposed that fibrillin-1 provides force-bearing structural support, whereas fibrillin-2 acts mostly in the early process of fibre assembly.9
The structure of the fibrillin microfibril is of a ‘beads on a string' of 10–15 nm diameter with beads 50 nm apart.10 The bead-like structures are mostly encoded for by exon 24 of FBN1.11 Mutations in this exon lead to severe Marfan syndrome (MFS). Other constituents of ZF include elastin, proteoglycans, and GAGs. The most important associated glycoprotein is MAGP-1, which probably has a role in cross linking the microfibrils.10
FBN1 and Marfan syndrome
Heterozygous mutations in FBN1 lead to haploinsufficiency of fibrillin-1. This results in disrupted microfibrillar architecture in the ECM.12 Mutations in this gene result in classical MFS, neonatal MFS, autosomal dominant ascending aortic aneurysms, familial arachnodactyly, Shprintzen–Goldberg syndrome and severe progressive kyphoscoliosis, the ‘MASS' phenotype (myopia, mitral valve prolapse, borderline aortic root enlargement, skin and skeletal findings), mitral valve prolapse syndrome, and autosomal dominant EL. There may be significant overlap between these conditions.
As fibrillin is a significant element of the ciliary zonules, it is unsurprising that EL manifest in up to 60% of MFS cases.13 MFS is an autosomal dominant disorder encompassing characteristic features of the cardiovascular, skeletal, and ocular systems.14 The ocular features include EL, flat corneal curvature, glaucoma, and axial myopia.15 The former of these (EL), with the genetic and cardiovascular features form the major components of the diagnostic Ghent criteria.14
The Ghent criteria for MFS were updated in 2010,14 emphasising the importance of the aortic root diameter in this syndrome. Diagnostic dilatation of the aortic root in the presence of either a diagnostic FBN1 mutation, or EL or seven points of skeletal features results in the diagnosis of MFS. The update in 2010 to the Ghent criteria had important implications for ophthalmologists and geneticists. Specifically, this was in relation to patients with EL and a FBN1 mutation with no aortic root dilatation. If the particular mutation had been previously described in classical MFS, these patients' diagnosis is now MFS. A summary of this is shown in Figure 2.
Figure 2.
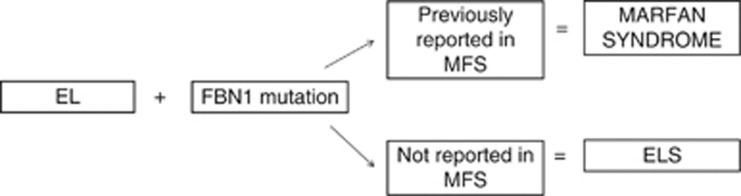
Summary of the importance of the Ghent criteria 2010 in those without diagnostic cardiovascular features of MFS. ELS, ectopia lentis syndrome; FBN1, fibrillin-1 gene.
Over 800 pathogenic mutations in FBN1 have been described.16 It is suggested that a significantly higher proportion of missense mutations involving cysteine residues (responsible for the structurally critical disulphide bonds) and mutations at the 5′ end are causative in EL,16, 17 in particular within the first 15 exons. These exons encode the N-terminus of fibrillin-1. This portion of the protein is thought to be integral to homodimer formation of the fibrillin-1 molecules, which eventually leads to polymers of fibrillin-1 and thus microfibrils.18
The mutations in FBN1 result in abnormal distribution and structure of microfibrillary bundles in the capsule of MFS patients, particularly at the site of zonule attachment,4, 19 conjunctiva,20 and zonules themselves.21
Alternative genetic causes of EL
It became apparent in the intervening years since MFS was first described that numerous other genetic conditions are associated with EL. Although no clear epidemiological data is available, it has been suggested that congenital EL may have a prevalence of six per 100 000.22 It is probable that the second most common cause of EL is a condition termed isolated ectopia lentis (IEL).22, 23 As the term suggests, this condition does not manifest features of other conditions, ocular or systemic; in particular MFS. It is thought to be inherited in an autosomal dominant (OMIM 129600) or autosomal recessive (OMIM 225100) manner.
Autosomal dominant EL
There are many reports of pedigrees in which the classical features of MFS are not reported, but EL segregates in an autosomal dominant fashion.17, 24, 25 Heterozygous mutations in FBN1 cause this condition, and it has thus occasionally been termed autosomal dominant isolated ectopia lentis (OMIM 129600). However, these patients are likely to represent part of a phenotypic spectrum of MFS,26 and the exclusion of MFS in these patients must be undertaken carefully and in view of the most recent Ghent criteria for MFS,14 as described above. In particular, detailed understanding of the history of any FBN1 mutation found must be gained before MFS can be excluded.
Examples of the importance of this recent alteration include a report by Edwards and colleagues,27 who presented a family with autosomal dominant EL with some mild skeletal features but no cardiological features of MFS, hence diagnosing IEL. However, the mutation they found in FBN1 (R240C) was subsequently described in a family with classical MFS.28 Because of the report of this mutation in MFS, the diagnosis would alter in the family to MFS. Further examples of such misdiagnoses continue,29, 30 suggesting that the updated Ghent criteria should be highlighted to those involved in an EL research. Furthermore, it has been demonstrated that patients with EL secondary to FBN1 mutations may progress to develop cardiovascular features of MFS.31, 32 It is thus recommended that these patients have long-term cardiology follow-up.14 The authors of the 2010 Ghent criteria14 therefore suggested that those with EL and a previously unreported FBN1 mutation should be termed ectopia lentis syndrome, to illustrate the potential for cardiovascular complications. This may be somewhat misleading, as these patients may never develop extraocular features of fibrillin-1 dysfunction. We would therefore simply recommend the term ‘isolated' not be used in the context of FBN1 mutations. We suggest simply ‘dominant ectopia lentis' in this situation.
Autosomal recessive IEL
This has been established for over 70 years.33, 34 al-Salem,35 in 1990, was the first to describe detailed ocular phenotypes of two consanguineous families from Iraq and Jordan with recessive IEL. Ahram and colleagues36 19 years later described a homozygous nonsense mutation in ADAMTSL4 on 1q21.2 in the Jordanian family. Further mutations in this gene have been described causing autosomal recessive IEL37, 38, 39, 40 and ectopia lentis et pupillae (EL&P).40, 41, 42 The genotype/phenotype similarity of IEL and EL&P most likely represents a spectrum of anterior segment dysgenesis.
The role of this protein and gene are unclear, and it has been suggested to interact with fibrillin-1 in microfibril biosynthesis.43 However, recessive mutations in ADAMTSL4 result in a more severe ocular phenotype with earlier onset of EL and greater axial length, than dominant mutations in FBN1.40 In addition, the gene and protein are found throughout the human eye.44 It is therefore probable that this gene may have a role in ocular development, independent of an interaction with fibrillin-1.
Other autosomal recessive mutations resulting in EL
Weill-Marchesani syndrome (WMS: OMIM 277600) is a very rare condition characterised by short stature, brachydactyly, and joint stiffness.45 The characteristic ocular features include myopia and EL (commonly manifesting as microspherophakia). Although causative heterozygous mutations in FBN1 has been described,46 it is more commonly inherited in an autosomal recessive manner, caused by mutations in ADAMTS1047, 48 on 19p13.2. EL is described in the majority of both dominant and recessive cases.49
In 2009, Morales and colleagues47 described homozygous mutations in ADAMTS17 on 15q24 in a consanguineous family causing EL and short stature. This family did not fulfil the diagnosis of WMS. The authors therefore coined this condition as ‘Weill-Marchesani-like syndrome' (OMIM 613195) (WML). Distinguishing this from WMS is challenging. The only further report to date of a mutation in this gene causing this condition was later published by the same group in 2012.50 Finally, mutations in LTBP2 on 14q24.3 have been described to cause EL, both in the context of WMS51 and isolated with other ocular features.52 Unlike other members of its protein family, LTBP2 does not bind to latent transforming growth factor, and instead its C-terminus has high affinity for the N-terminus of fibrillin-1. An ocular phenotype including EL caused by mutations in this gene may therefore not be surprising.
Homocystinuria (OMIM 236200) is a rare metabolic disorder of sulphur metabolism, owing to recessive mutations in cystathionine beta-synthase (CBS) on 21q22. Systemic manifestations include mental retardation, hypopigmentation of skin and hair, thromboembolic events, and marfanoid habitus. Ocular manifestations of untreated homocystinuria include myopia and EL.53, 54
Although congenital vitreoretinopathies commonly cause lenticular changes55 Knobloch syndrome (OMIM 267750, 608454) (KNO) is the only one in which EL is commonly a feature. This is an autosomal recessive condition first described in 1971,56 with the cardinal features consisting of high myopia, vitreoretinopathy, and occipital defects. Mutations in COL18A1 on 21q22.3 are causative of this condition.57 The encoded protein COL18A1 predominates in basement membranes, including the inner limiting membrane of the neural retina.58, 59 Mutations in this gene therefore understandably contribute towards the preponderance for rhegmatogenous retinal detachment in KNO. A similar disruption of the basement membrane of the lens capsule, particularly at the insertion of the zonules, may contribute to EL being the most common further ophthalmic feature of this condition. Very recently, a proband was described with features similar to KNO including EL, smooth irides, high myopia, and cone-rod dystrophy.60 This patient was found to have a homozygous mutation in VSX2, a gene on 14q24.3. Although this protein has a role in retinal development,61 its potential role in EL is a yet unexplained.
It must be remembered that mutations in PAX6, causing aniridia, can also manifest with EL.62 In addition, it may be expected that pseudoexfoliation and high myopia, both of which are known to manifest weakening of the ZF, may result in EL. However, spontaneous EL has rarely been reported in these conditions. A summary of the genetic aetiology of EL is presented in Table 1.
Table 1. Genes confirmed to cause ectopia lentis.
| Gene | Inheritance | Condition | Reference |
|---|---|---|---|
| FBN1 | Autosomal dominant | Marfan syndrome | Dietz et al (1991)73 |
| Dominant ectopia lentis | Edwards et al (1994)27 | ||
| Dominant Weill Marchesani Syndrome | Faivre et al (2003)46 | ||
| ADAMTSL4 | Autosomal recessive | Isolated ectopia lentis | Ahram et al (2009)36 |
| Ectopia lentis et pupillae | Christensen et al (2010)41 | ||
| Ectopia lentis and craniosynostosis | Chandra et al (2012)40 | ||
| CBS | Autosomal recessive | Homocystinuria | Kraus (1994)74 |
| ADAMTS10 | Autosomal recessive | Weill Marchesani Syndrome | Daganeau et al (2004)48 |
| ADAMTS17 | Autosomal recessive | Weill Marchesani Like | Morales et al (2009)47 |
| COL18A1 | Autosomal recessive | Knobloch | Sertie et al (2000)57 |
| PAX6 | Autosomal recessive | Aniridia | Jin et al (2012)62 |
| LTBP2 | Autosomal recessive | Weill Marchesani Syndrome | Haji-Seyed-Javadi et al (2012)51 |
| Megalocornea, spherophakia | Desir et al (2010)75 | ||
| VSX2 | Autosomal recessive | High myopia, EL, cone-rod dystrophy | Khan et al (2013)60 |
The continual discovery of genes causing EL has led to the recent interest in the apparent role of the ADAMTS family in ocular disease and development. This is a group of 19 proteases which have roles in ECM degradation, connective tissue structure, cell migration, and angiogenesis.63 Included in this family are the seven ADAMTS-like proteins that lack the catalytic domain of the ADAMTS family and are thus thought to have a regulatory role of the ADAMTS enzymes.64 To date four members of this superfamily of genes have been described to cause ocular phenotypes (Table 2), with EL as a common feature. The specific role of the ADAMTS family in ocular development remains to be clarified.
Table 2. Ocular manifestations of recessive mutations in the ADAMTS genes.
| Gene | Ocular phenotype | Reference |
|---|---|---|
| ADAMTSL4 | Isolated ectopia lentis (OMIM 225100) | Ahram et al (2009)36 Greene et al (2010)37 Aragon Martin et al (2010)38 Neuhann et al (2010)39 Chandra et al (2012)40 |
| Ectopia lentis et papillae (OMIM 225200) | Christensen et al (2010)41 Chandra et al (2012)40 Sharifi et al (2013)42 | |
| Ectopia lentis and craniosynostosis (OMIM 603595) | Chandra et al (2012)40 | |
| ADAMTS10 | Weill-Marchesani (OMIM 277600) | Dagoneau et al (2004)48 Kutz et al (2008)76 Morales et al (2009)47 |
| ADAMTS17 | Weill-Marcahasni-like (microsherophakia and short stature) (OMIM 613915) | Morales et al (2009)47 Khan et al (2012)50 |
| ADAMTS18 | Microcornea, myopic chorioretinal atrophy and telecanthus (MMCAT) | Aldahmesh et al (2013)77 |
| Early-onset retinal dystrophy | Peluso et al (2013)78 |
Specific ophthalmic features of the different conditions associated with EL have not been defined. Although superior movement of the crystalline lens has been suggested in MFS13 and anterior dislocation in homocystinuria,53 these have not been convincingly replicated. A recently devised clinical grading system65 may help characterise these features to clarify such suggestions in the future.
Clinical management
Many cases of lens subluxation can be managed without surgical intervention. Appropriate spectacle or contact lens correction will for most individuals provide adequate stable visual acuity.66 Patients should be counselled that after childhood, lens displacement is very unlikely to progress. Surgery may be indicated where there is progressive lens subluxation, cataract formation, lens instability or less commonly pupil block glaucoma, or retinal detachment. Lens instability can easily be overlooked as patients are frequently unaware that this is the cause of their visual problems.
Subluxed lenses with limited zonular support may in some cases be managed with phacoemulsification surgery, however long-term stability following phacoemulsification has not been consistently reported. Pars plana lensectomy together with vitrectomy has therefore often been utilised to provide stable long-term results. We have previously reported successful outcome from pars plana surgery for subluxed lenses in MFS, initially leaving patients aphakic (many were previous contact lens wearers).67
Various options exist for lens replacement following pars lensectomy. Sutured posterior chamber lenses can provide excellent visual results.68, 69 We have documented that late suture breakage (using 10/0 proline) is an important problem using this technique70 and have therefore moved away from suturing lenses as a technique of choice. Modern designs of anterior chamber lens implant generally have low levels of complications71 and are suitable for this patient population. Similarly newer designs of iris-supported IOLs72 and intra-scleral fixation of IOL haptics have been reported with good results recently, although long-term follow-up for these techniques in aphakic eyes has not yet been reported. Currently, there is no consensus on which type of IOL is most suitable for these patients,71 therefore decisions must be made on individual cases.
Summary
Inherited EL is a condition that may herald numerous syndromes or be isolated. Differentiating these conditions is critical, and this review has summarised some of the important genetic aetiologies involved. Understanding these would help involved ophthalmologists and eye scientists in the crucial role they would have in this process.
The authors declare no conflict of interest.
References
- Karl Stellwag von Carion. BMJ. 1904;2 (2293:1615–1616. [Google Scholar]
- Williams E. Rare cases, with practical remarks. Trans Am Ophthalmol Soc. 1875;2:291–301. [PMC free article] [PubMed] [Google Scholar]
- Streeten BW. The zonular insertion: a scanning electron microscopic study. Invest Ophthalmol Vis Sci. 1977;16 (4:364–375. [PubMed] [Google Scholar]
- Traboulsi EI, Whittum-Hudson JA, Mir SH, Maumenee IH. Microfibril abnormalities of the lens capsule in patients with Marfan syndrome and ectopia lentis. Ophthalmic Genet. 2000;21 (1:9–15. [PubMed] [Google Scholar]
- Robert B, Szigeti M, Derouette JC, Robert L. Studies on the nature of the ‘microfibrillar' component of elastic fibers. Eur J Biochem. 1971;21 (4:507–516. doi: 10.1111/j.1432-1033.1971.tb01496.x. [DOI] [PubMed] [Google Scholar]
- Biery NJ, Eldadah ZA, Moore CS, Stetten G, Spencer F, Dietz HC. Revised genomic organization of FBN1 and significance for regulated gene expression. Genomics. 1999;56 (1:70–77. doi: 10.1006/geno.1998.5697. [DOI] [PubMed] [Google Scholar]
- Robinson PN, Godfrey M. Marfan Syndrome: A Primer for Clinicians and Scientists. Landes Bioscience/Eurekah.com: Georgetown, TX, USA; 2004. [Google Scholar]
- Yuan X, Downing AK, Knott V, Handford PA. Solution structure of the transforming growth factor beta-binding protein-like module, a domain associated with matrix fibrils. Embo J. 1997;16 (22:6659–6666. doi: 10.1093/emboj/16.22.6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Hu W, Ramirez F. Developmental expression of fibrillin genes suggests heterogeneity of extracellular microfibrils. J Cell Biol. 1995;129 (4:1165–1176. doi: 10.1083/jcb.129.4.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis EC, Roth RA, Heuser JE, Mecham RP. Ultrastructural properties of ciliary zonule microfibrils. J Struct Biol. 2002;139 (2:65–75. doi: 10.1016/s1047-8477(02)00559-2. [DOI] [PubMed] [Google Scholar]
- Werneck CC, Trask BC, Broekelmann TJ, Trask TM, Ritty TM, Segade F, et al. Identification of a major microfibril-associated glycoprotein-1-binding domain in fibrillin-2. J Biol Chem. 2004;279 (22:23045–23051. doi: 10.1074/jbc.M402656200. [DOI] [PubMed] [Google Scholar]
- Eldadah ZA, Brenn T, Furthmayr H, Dietz HC. Expression of a mutant human fibrillin allele upon a normal human or murine genetic background recapitulates a Marfan cellular phenotype. J Clin Invest. 1995;95 (2:874–880. doi: 10.1172/JCI117737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maumenee IH. The eye in the Marfan syndrome. Trans Am Ophthalmol Soc. 1981;79:684–733. [PMC free article] [PubMed] [Google Scholar]
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47 (7:476–485. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- Nemet AY, Assia EI, Apple DJ, Barequet IS. Current concepts of ocular manifestations in Marfan syndrome. Surv Ophthalmol. 2006;51 (6:561–575. doi: 10.1016/j.survophthal.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81 (3:454–466. doi: 10.1086/520125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre L, Collod-Beroud G, Callewaert B, Child A, Loeys BL, Binquet C, et al. Pathogenic FBN1 mutations in 146 adults not meeting clinical diagnostic criteria for Marfan syndrome: further delineation of type 1 fibrillinopathies and focus on patients with an isolated major criterion. Am J Med Genet A. 2009;149A (5:854–860. doi: 10.1002/ajmg.a.32809. [DOI] [PubMed] [Google Scholar]
- Trask TM, Ritty TM, Broekelmann T, Tisdale C, Mecham RP. N-terminal domains of fibrillin 1 and fibrillin 2 direct the formation of homodimers: a possible first step in microfibril assembly. Biochem J. 1999;340 (Pt 3:693–701. [PMC free article] [PubMed] [Google Scholar]
- Mir S, Wheatley HM, Hussels IE, Whittum-Hudson JA, Traboulsi EI. A comparative histologic study of the fibrillin microfibrillar system in the lens capsule of normal subjects and subjects with Marfan syndrome. Invest Ophthalmol Vis Sci. 1998;39 (1:84–93. [PubMed] [Google Scholar]
- Ganesh A, Smith C, Chan W, Unger S, Quercia N, Godfrey M, et al. Immunohistochemical evaluation of conjunctival fibrillin-1 in Marfan syndrome. Arch Ophthalmol. 2006;124 (2:205–209. doi: 10.1001/archopht.124.2.205. [DOI] [PubMed] [Google Scholar]
- Kielty CM, Davies SJ, Phillips JE, Jones CJ, Shuttleworth CA, Charles SJ. Marfan syndrome: fibrillin expression and microfibrillar abnormalities in a family with predominant ocular defects. J Med Genet. 1995;32 (1:1–6. doi: 10.1136/jmg.32.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs J, Rosenberg T. Congenital ectopia lentis. A Danish national survey. Acta Ophthalmol Scand. 1998;76 (1:20–26. doi: 10.1034/j.1600-0420.1998.760105.x. [DOI] [PubMed] [Google Scholar]
- Chandra A.Isolated Ectopia Lentis Orphanet EncyclopediaAvailable at http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=1885 .
- Vanita V, Singh JR, Singh D, Varon R, Robinson PN, Sperling K. A recurrent FBN1 mutation in an autosomal dominant ectopia lentis family of Indian origin. Mol Vis. 2007;13:2035–2040. [PubMed] [Google Scholar]
- Zhao JH, Jin TB, Liu QB, Chen C, Hu HT. Ophthalmic findings in a family with early-onset isolated ectopia lentis and the p.Arg62Cys mutation of the fibrillin-1 gene (FBN1) Ophthalmic Genet. 2012;34:21–26. doi: 10.3109/13816810.2012.718029. [DOI] [PubMed] [Google Scholar]
- Ades LC, Holman KJ, Brett MS, Edwards MJ, Bennetts B. Ectopia lentis phenotypes and the FBN1 gene. Am J Med Genet A. 2004;126A (3:284–289. doi: 10.1002/ajmg.a.20605. [DOI] [PubMed] [Google Scholar]
- Edwards MJ, Challinor CJ, Colley PW, Roberts J, Partington MW, Hollway GE, et al. Clinical and linkage study of a large family with simple ectopia lentis linked to FBN1. Am J Med Genet. 1994;53 (1:65–71. doi: 10.1002/ajmg.1320530114. [DOI] [PubMed] [Google Scholar]
- Loeys B, Nuytinck L, Delvaux I, De Bie S, De Paepe A. Genotype and phenotype analysis of 171 patients referred for molecular study of the fibrillin-1 gene FBN1 because of suspected Marfan syndrome. Arch Intern Med. 2001;161 (20:2447–2454. doi: 10.1001/archinte.161.20.2447. [DOI] [PubMed] [Google Scholar]
- Yang G, Chu M, Zhai X, Zhao J. A novel FBN1 mutation in a Chinese family with isolated ectopia lentis. Mol Vis. 2012;18:945–950. [PMC free article] [PubMed] [Google Scholar]
- Li H, Qu W, Meng B, Zhang S, Yang T, Huang S, et al. Identification and study of a FBN1 gene mutation in a Chinese family with ectopia lentis. Mol Vis. 2012;18:504–511. [PMC free article] [PubMed] [Google Scholar]
- Pepe G, Lapini I, Evangelisti L, Attanasio M, Giusti B, Lucarini L, et al. Is ectopia lentis in some cases a mild phenotypic expression of Marfan syndrome? Need for a long-term follow-up. Mol Vis. 2007;13:2242–2247. [PubMed] [Google Scholar]
- Zadeh N, Bernstein JA, Niemi AK, Dugan S, Kwan A, Liang D, et al. Ectopia lentis as the presenting and primary feature in Marfan syndrome. Am J Med Genet A. 2011;155A (11:2661–2668. doi: 10.1002/ajmg.a.34245. [DOI] [PubMed] [Google Scholar]
- Falls HF, Cotterman CW. Genetic studies on ectopia lentis: a pedigree of simple ectopia of the lens. Arch Ophthalmol. 1943;30:610–620. [Google Scholar]
- Ruiz C, Rivas F, Villar-Calvo VM, Serrano-Lucas JI, Cantu JM. Familial simple ectopia lentis. A probable autosomal recessive form. Ophthalmic Paediatr Genet. 1986;7 (2:81–84. doi: 10.3109/13816818609076113. [DOI] [PubMed] [Google Scholar]
- al-Salem M. Autosomal recessive ectopia lentis in two Arab family pedigrees. Ophthalmic Paediatr Genet. 1990;11 (2:123–127. doi: 10.3109/13816819009012957. [DOI] [PubMed] [Google Scholar]
- Ahram D, Sato TS, Kohilan A, Tayeh M, Chen S, Leal S, et al. A homozygous mutation in ADAMTSL4 causes autosomal-recessive isolated ectopia lentis. Am J Hum Genet. 2009;84 (2:274–278. doi: 10.1016/j.ajhg.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene VB, Stoetzel C, Pelletier V, Perdomo-Trujillo Y, Liebermann L, Marion V, et al. Confirmation of ADAMTSL4 mutations for autosomal recessive isolated bilateral ectopia lentis. Ophthalmic Genet. 2010;31 (1:47–51. doi: 10.3109/13816810903567604. [DOI] [PubMed] [Google Scholar]
- Aragon-Martin JA, Ahnood D, Charteris DG, Saggar A, Nischal KK, Comeglio P, et al. Role of ADAMTSL4 mutations in FBN1 mutation-negative ectopia lentis patients. Hum Mutat. 2010;31 (8:E1622–E1631. doi: 10.1002/humu.21305. [DOI] [PubMed] [Google Scholar]
- Neuhann TM, Artelt J, Neuhann TF, Tinschert S, Rump A. A homozygous microdeletion within ADAMTSL4 in patients with isolated ectopia lentis: evidence of a founder mutation. Invest Ophthalmol Vis Sci. 2011;52 (2:695–700. doi: 10.1167/iovs.10-5740. [DOI] [PubMed] [Google Scholar]
- Chandra A, Aragon-Martin JA, Hughes K, Gati S, Reddy MA, Deshpande C, et al. A genotype-phenotype comparison of ADAMTSL4 and FBN1 in isolated ectopia lentis. Invest Ophthalmol Vis Sci. 2012;53 (8:4889–4896. doi: 10.1167/iovs.12-9874. [DOI] [PubMed] [Google Scholar]
- Christensen AE, Fiskerstrand T, Knappskog PM, Boman H, Rodahl E. A novel ADAMTSL4 mutation in autosomal recessive ectopia lentis et pupillae. Invest Ophthalmol Vis Sci. 2010;51 (12:6369–6373. doi: 10.1167/iovs.10-5597. [DOI] [PubMed] [Google Scholar]
- Sharifi Y, Tjon-Fo-Sang MJ, Cruysberg JR, Maat-Kievit AJ. Ectopia lentis et pupillae in four generations caused by novel mutations in the ADAMTSL4 gene. Br J Ophthalmol. 2013;97:583–587. doi: 10.1136/bjophthalmol-2012-302367. [DOI] [PubMed] [Google Scholar]
- Gabriel LA, Wang LW, Bader H, Ho JC, Majors AK, Hollyfield JG, et al. ADAMTSL4, a secreted glycoprotein widely distributed in the eye, binds fibrillin-1 microfibrils and accelerates microfibril biogenesis. Invest Ophthalmol Vis Sci. 2012;53 (1:461–469. doi: 10.1167/iovs.10-5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra A, Jones M, Cottrill P, Eastlake K, Limb GA, Charteris DG. Gene expression and protein distribution of ADAMTSL-4 in human iris, choroid and retina. Br J Ophthalmol. 2013;97:1208–1212. doi: 10.1136/bjophthalmol-2013-303353. [DOI] [PubMed] [Google Scholar]
- Faivre L, Dollfus H, Lyonnet S, Alembik Y, Megarbane A, Samples J, et al. Clinical homogeneity and genetic heterogeneity in Weill-Marchesani syndrome. Am J Med Genet A. 2003;123A (2:204–207. doi: 10.1002/ajmg.a.20289. [DOI] [PubMed] [Google Scholar]
- Faivre L, Gorlin RJ, Wirtz MK, Godfrey M, Dagoneau N, Samples JR, et al. In frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndrome. J Med Genet. 2003;40 (1:34–36. doi: 10.1136/jmg.40.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales J, Al-Sharif L, Khalil DS, Shinwari JM, Bavi P, Al-Mahrouqi RA, et al. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am J Hum Genet. 2009;85 (5:558–568. doi: 10.1016/j.ajhg.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagoneau N, Benoist-Lasselin C, Huber C, Faivre L, Megarbane A, Alswaid A, et al. ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am J Hum Genet. 2004;75 (5:801–806. doi: 10.1086/425231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsilou E, MacDonald IM.Weill-Marchesani SyndromeIn: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP (eds) GeneReviews University of Washington: Seattle, WA, USA; 1993 [PubMed] [Google Scholar]
- Khan AO, Aldahmesh MA, Al-Ghadeer H, Mohamed JY, Alkuraya FS. Familial spherophakia with short stature caused by a novel homozygous ADAMTS17 mutation. Ophthalmic Genet. 2012;33 (4:235–239. doi: 10.3109/13816810.2012.666708. [DOI] [PubMed] [Google Scholar]
- Haji-Seyed-Javadi R, Jelodari-Mamaghani S, Paylakhi SH, Yazdani S, Nilforushan N, Fan JB, et al. LTBP2 mutations cause Weill-Marchesani and Weill-Marchesani-like syndrome and affect disruptions in the extracellular matrix. Hum Mutat. 2012;33 (8:1182–1187. doi: 10.1002/humu.22105. [DOI] [PubMed] [Google Scholar]
- Khan AO, Aldahmesh MA, Alkuraya FS. Congenital megalocornea with zonular weakness and childhood lens-related secondary glaucoma - a distinct phenotype caused by recessive LTBP2 mutations. Mol Vis. 2011;17:2570–2579. [PMC free article] [PubMed] [Google Scholar]
- Harrison DA, Mullaney PB, Mesfer SA, Awad AH, Dhindsa H. Management of ophthalmic complications of homocystinuria. Ophthalmology. 1998;105 (10:1886–1890. doi: 10.1016/S0161-6420(98)91035-1. [DOI] [PubMed] [Google Scholar]
- Burke JP, O'Keefe M, Bowell R, Naughten ER. Ocular complications in homocystinuria—early and late treated. Br J Ophthalmol. 1989;73 (6:427–431. doi: 10.1136/bjo.73.6.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AO. Clinical features of the congenital vitreoretinopathies. Eye (Lond) 2008;22 (10:1233–1242. doi: 10.1038/eye.2008.38. [DOI] [PubMed] [Google Scholar]
- Knobloch WH, Layer JM. Retinal detachment and ecephalocele. J Pediat Ophthal. 1971;8:181–184. [Google Scholar]
- Sertie AL, Sossi V, Camargo AA, Zatz M, Brahe C, Passos-Bueno MR. Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome) Hum Mol Genet. 2000;9 (13:2051–2058. doi: 10.1093/hmg/9.13.2051. [DOI] [PubMed] [Google Scholar]
- Ponsioen TL, van Luyn MJ, van der Worp RJ, van Meurs JC, Hooymans JM, Los LI. Collagen distribution in the human vitreoretinal interface. Invest Ophthalmol Vis Sci. 2008;49 (9:4089–4095. doi: 10.1167/iovs.07-1456. [DOI] [PubMed] [Google Scholar]
- Fukai N, Eklund L, Marneros AG, Oh SP, Keene DR, Tamarkin L, et al. Lack of collagen XVIII/endostatin results in eye abnormalities. Embo J. 2002;21 (7:1535–1544. doi: 10.1093/emboj/21.7.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AO, Aldahmesh MA, Noor J, Salem A, Alkuraya FS. Lens subluxation and retinal dysfunction in a girl with homozygous VSX2 mutation. Ophthalmic Genet. 2013. [DOI] [PubMed]
- Liang L, Sandell JH. Focus on molecules: homeobox protein Chx10. Exp Eye Res. 2008;86 (4:541–542. doi: 10.1016/j.exer.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Jin C, Wang Q, Li J, Zhu Y, Shentu X, Yao K. A recurrent PAX6 mutation is associated with aniridia and congenital progressive cataract in a Chinese family. Mol Vis. 2012;18:465–470. [PMC free article] [PubMed] [Google Scholar]
- Le Goff C, Cormier-Daire V. The ADAMTS(L) family and human genetic disorders. Hum Mol Genet. 2011;20 (R2:R163–R167. doi: 10.1093/hmg/ddr361. [DOI] [PubMed] [Google Scholar]
- Apte SS. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem. 2009;284 (46:31493–31497. doi: 10.1074/jbc.R109.052340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra A, Banerjee PJ, Charteris DG. Grading in ectopia lentis (GEL): a novel classification system. Br J Ophthalmol. 2013;97:942–943. doi: 10.1136/bjophthalmol-2012-302921. [DOI] [PubMed] [Google Scholar]
- Neely DE, Plager DA. Management of ectopia lentis in children. Ophthalmol Clin North Am. 2001;14 (3:493–499. doi: 10.1016/s0896-1549(05)70247-9. [DOI] [PubMed] [Google Scholar]
- Hubbard AD, Charteris DG, Cooling RJ. Vitreolensectomy in Marfan's syndrome. Eye (Lond) 1998;12 (Pt 3a:412–416. doi: 10.1038/eye.1998.97. [DOI] [PubMed] [Google Scholar]
- Johnston RL, Charteris DG, Horgan SE, Cooling RJ. Combined pars plana vitrectomy and sutured posterior chamber implant. Arch Ophthalmol. 2000;118 (7:905–910. [PubMed] [Google Scholar]
- Yang YF, Bunce C, Dart JK, Johnston RL, Charteris DG. Scleral-fixated posterior chamber intraocular lenses in non-vitrectomized eyes. Eye (Lond) 2006;20 (1:64–70. doi: 10.1038/sj.eye.6701804. [DOI] [PubMed] [Google Scholar]
- Vote BJ, Tranos P, Bunce C, Charteris DG, Da Cruz L. Long-term outcome of combined pars plana vitrectomy and scleral fixated sutured posterior chamber intraocular lens implantation. Am J Ophthalmol. 2006;141 (2:308–312. doi: 10.1016/j.ajo.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Wagoner MD, Cox TA, Ariyasu RG, Jacobs DS, Karp CL. Intraocular lens implantation in the absence of capsular support: a report by the American Academy of Ophthalmology. Ophthalmology. 2003;110 (4:840–859. doi: 10.1016/s0161-6420(02)02000-6. [DOI] [PubMed] [Google Scholar]
- Cleary C, Lanigan B, O'Keeffe M. Artisan iris-claw lenses for the correction of aphakia in children following lensectomy for ectopia lentis. Br J Ophthalmol. 2012;96 (3:419–421. doi: 10.1136/bjophthalmol-2011-300579. [DOI] [PubMed] [Google Scholar]
- Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352 (6333:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- Kraus JP. Komrower lecture. molecular basis of phenotype expression in homocystinuria. J Inherit Metab Dis. 1994;17 (4:383–390. doi: 10.1007/BF00711354. [DOI] [PubMed] [Google Scholar]
- Desir J, Sznajer Y, Depasse F, Roulez F, Schrooyen M, Meire F, et al. LTBP2 null mutations in an autosomal recessive ocular syndrome with megalocornea, spherophakia, and secondary glaucoma. Eur J Hum Genet. 2010;18 (7:761–767. doi: 10.1038/ejhg.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutz WE, Wang LW, Dagoneau N, Odrcic KJ, Cormier-Daire V, Traboulsi EI, et al. Functional analysis of an ADAMTS10 signal peptide mutation in Weill-Marchesani syndrome demonstrates a long-range effect on secretion of the full-length enzyme. Hum Mutat. 2008;29 (12:1425–1434. doi: 10.1002/humu.20797. [DOI] [PubMed] [Google Scholar]
- Aldahmesh MA, Alshammari MJ, Khan AO, Mohamed JY, Alhabib FA, Alkuraya FS. The syndrome of microcornea, myopic chorioretinal atrophy, and telecanthus (MMCAT) is caused by mutations in ADAMTS18. Hum Mutat. 2013;34 (9:1195–1199. doi: 10.1002/humu.22374. [DOI] [PubMed] [Google Scholar]
- Peluso I, Conte I, Testa F, Dharmalingam G, Pizzo M, Collin RW, et al. The ADAMTS18 gene is responsible for autosomal recessive early onset severe retinal dystrophy. Orphanet J Rare Dis. 2013;8 (1:16. doi: 10.1186/1750-1172-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]