

Abstract
Neuroinflammatory changes are a characteristic of several, if not all, neurodegenerative diseases including Alzheimer's disease and are typified by increased microglial activation. Microglia express several receptors making them highly reactive and plastic cells, and, at least in vitro, they adopt different phenotypes in a manner analogous to their peripheral counterparts, macrophages. Microglia also express numerous cell surface proteins enabling them to interact with cells and the evidence indicates that maintenance of microglia in a quiescent state relies, at least to some extent, on an interaction with neurons by means of specific ligand–receptor pairs, for example CD200–CD200R. It is clear that microglia also interact with T cells and recent evidence indicates that co-incubation of microglia with T helper type 1 cells markedly increases their activation. Under normal conditions, small numbers of activated T cells gain entry to the brain and are involved in immune surveillance but infiltration of significant numbers of T cells occurs in disease and following injury. The consequences of T cell infiltration appear to depend on the conditions, with descriptions of both neurodestructive and neuroprotective effects in animal models of different diseases. This review will discuss the modulatory effect of T cells on microglia and the impact of infiltration of T cells into the brain with a focus on Alzheimer's disease, and will propose that infiltration of interferon-γ-producing cells may be an important factor in triggering inflammation that is pathogenic and destructive.
Keywords: Alzheimer's disease, inflammation, macrophages/monocytes, neuroimmunology
Introduction
Although the pathological hallmarks of Alzheimer's disease (AD) were described over a decade ago, the pathogenesis of the late-onset form of the disease remains poorly understood. One theory, the amyloid hypothesis, suggests that the accumulation of β-amyloid (Aβ), because of a decrease in Aβ clearance, perhaps accompanied by increased processing of amyloid precursor protein (APP), triggers a cascade of damaging reactions. Another theory proposes that tau hyperphosphorylation is key to the demise of neurons. However, the trigger leading to the initial changes has not been identified, and the likelihood is that there is not a single factor but a collection of coincident events that superimpose upon already existing age-related changes. For some time now it has been recognized that inflammatory changes may be a factor in accelerating disease onset. Indeed many of the risk factors for the disease, for example atherosclerosis, neurotrauma, type 1 diabetes and infection, are associated with inflammatory changes.
Neuroinflammation and AD
A great deal of evidence indicates that inflammatory changes are a feature of AD and it is agreed that a key change is an increase in microglial activation, which was originally described by Alzheimer over 100 years ago. Several reports have confirmed this observation and have shown that activated cells cluster around Aβ-containing plaques.1,2 An increase in expression of inflammatory cytokines, interleukin-1β (IL-1β), IL-6 and tumour necrosis factor-α (TNF-α) has been observed in the AD brain and IL-1-positive microglia co-locate with Aβ-containing plaques, as do activated astrocytes. These changes are associated with other indicators of inflammatory changes including increased expression of several chemokines and elements of the complement activation pathway.3–5 However, it is still debated whether these changes are a consequence of the disease or contribute to its pathogenesis; this is a difficult issue to dissect because the evidence from model systems indicates the existence of a cycle in which inflammatory changes drive Aβ accumulation while Aβ stimulates glia to produce inflammatory mediators. A possible resolution to this question was presented when early epidemiological evidence indicated that the incidence or severity of AD was reduced in individuals who were treated with non-steroidal anti-inflammatory drugs (NSAID).6–8 These observations suggested that anti-inflammatory agents might be a useful therapeutic option for the treatment of the disease, but several clinical trials have failed to identify a beneficial effect of anti-inflammatory agents.9–11 It has been argued that, because inflammatory changes occur early in the disease, only very early intervention with anti-inflammatory treatments is likely to be valuable,12 and indeed in a study where participants were aged ≥ 65 years on enrolment (older than participants in other studies), the use of NSAID was detrimental with adjusted hazard ratio of risk for AD of 1·57.13 Interestingly, post-mortem examination of brain tissue from non-demented elderly individuals indicated a correlative decrease in microglial activation and senile plaque number in those treated with NSAID but not steroids,14 although other data have not shown this.15
Microglia and macrophages share certain properties
Microglia are the primary immune cells in the brain and they are often referred to as macrophages of the brain, although we now know that microglia are an ontogenically distinct population of cells derived from primitive haematopoietic cells in the yolk sac.16 Therefore microglia, being confined to the brain, are protected by the blood–brain barrier (BBB) from exposure to circulating high molecular weight molecules as well as the vast array of stimuli encountered by macrophages. Morphologically, microglia and macrophages are somewhat distinct. Under resting conditions microglia, unlike macrophages, are multi-processed cells and the processes are constantly motile, enabling the cells to sample their microenvironment and react, when necessary, to noxious stimuli. Activation of microglia results in retraction of processes and the cells adopt an amoeboid morphology. However, the cells share many functions and both are phagocytic and function as antigen-presenting cells (APC).
Microglia are highly reactive cells and, like macrophages, express a multitude of cell surface receptors17 including pathogen recognition receptors, complement receptor, Fc receptors, chemokine and cytokine receptors, and receptors for numerous neurotransmitters.17,18 Macrophages are regularly exposed to pathogen-associated molecular patterns, whereas brain infections are relatively rare so activation of pathogen recognition receptors on microglia is more likely to be induced by damage-associated molecular patterns; these endogenously generated molecules include ATP and high mobility group box-1, that are released from damaged/dying cells.19 However, an association between AD and certain pathogens has been identified20 and infections can accelerate cognitive decline in AD patients.21,22 Evidence has indicated that a significant proportion of AD brains were positive for Chlamydia pneumoniae23 whereas intranasal inoculation of mice with C. pneumoniae induced deposition of fibrillar Aβ associated with reactive glia.24 Peripheral challenge with the Toll-like receptor agonists, lipopolysaccharide (LPS) or polyriboinosinic-polyribocytidilic acid (PolyI:C), induced amyloid pathology in some,25,26 but not all,27 animal models of AD. Herpes simplex virus DNA has also been found in the cortex of a high proportion of individuals with AD28 and the risk of developing AD is exacerbated by the virus in individuals with the type 4 allele of the apolipoprotein E.29
Several factors act to down-regulate microglial activation
Under resting conditions, microglia are maintained in a relatively quiescent state and, although the evidence suggests that cell–cell interactions are primarily responsible for this, it is also known that some neurotransmitters, particularly noradrenaline and acetylcholine, also modulate the activation of microglia.30 Other factors that assist in maintaining the ‘resting’ state of microglia are the low numbers of T cells in the brain and therefore the minimal expression of potent activators like interferon-γ (IFN-γ), the presence of electrically active neurons that suppress glial expression of MHC and co-stimulatory molecules31 and the production by neurons and astrocytes of transforming growth factor-β (TGF-β) and IL-10.32,33
Microglia physically interact with other cells and, in the case of microglia–neuron interactions, several ligand–receptor pairs have been shown to support this interaction. Perhaps the two best studied pairs are CD200–CD200R and CX3CL1–CX3CR1. CD200 is expressed on many cell types including neurons whereas CD200 receptor expression is confined to cells of the myeloid lineage;34,35 consequently co-culturing microglia with neurons decreases LPS-induced or Aβ-induced microglial activation in a CD200-dependent manner.36,37 CD200 deficiency increases inflammatory changes in models of multiple sclerosis,38 Parkinson's disease39 and experimental autoimmune uveoretinitis40 and these mice also exhibit exaggerated responses to inflammatory challenges.37,41 Perhaps predictably, a fusion protein CD200Fc, which acts on CD200R to induce its signalling, attenuates the inflammatory changes associated with age and Aβ treatment42,43 and decreases the symptoms and inflammatory changes in experimental autoimmune encephalomyelitis44 and collagen-induced arthritis.45,46 Interestingly CD200 is decreased in tissue from individuals with AD47 and in tissue adjacent to the plaques in multiple sclerosis,48,49 and CD200R signalling is dysfunctional in macrophages from individuals with Parkinson's disease.50
The largely complementary expression of CX3CL1 on neurons and its receptor on microglia suggests an interaction similar to that described for CD200–CD200R, and evidence to support this has been reported.51 Several studies have reported that disruption in CX3CL1–CX3CR1 interactions is associated with microglial activation and increased secretion of inflammatory cytokines.52 However, in the APP/PS1 transgenic mouse model of AD, knockdown of CX3CR1 was associated with increased clearance of Aβ,53 indicating that the impact of CX3CR1 activation is complex with respect to microglial activation. Interestingly, expression of both ligand and receptor is decreased in hippocampal and cortical tissue in AD.54
Other ligand–receptor pairs include CD45 and signal regulatory protein 1α, which are expressed predominantly on microglia and interact with neuronally expressed CD22 and CD47, respectively.55–59 These interactions function as ‘off’ signals, whose role is to keep microglia in a resting state; other ‘off’ signals include secreted CD22 and fractalkine, secreted neurotrophins and anti-inflammatory cytokines such as IL-10 and IL-455,60 and several of these factors attenuate IFN-γ-induced, LPS-induced and Aβ-induced microglial activation.60,61
Astrocytes, like neurons, express CD200 and they also have the ability to interact with microglia to down-regulate their activation. Indeed incubation of microglia with CD200-bearing astrocytic membrane preparations attenuates the LPS-induced increase in mRNA expression of IL-1β, TNF-α and IL-6 and the LPS-induced increase in release of TNF-α and IL-6.62 However, soluble factors secreted by astrocytes also modulate microglial activation63 and it has been shown that conditioned medium obtained from astrocytes decreased hydrogen peroxide-induced reactive oxygen species production, increased expression and activity of the antioxidant enzyme, haemoxygenase-1, and decreased IFN-γ-induced inducible nitrous oxide synthase (iNOS) expression in microglia.64 The modulatory effect of astrocytes on microglial activation perhaps derives from the fact that astrocytes are GABA-ergic cells, which impact on GABA receptors expressed by microglia.65 However, it is clear that astrocytes can also release factors that trigger microglial activation and therefore contribute to the changes that occur as a consequence of chronic inflammation.66,67
T cells can also interact with microglia to modulate their function (Table 1). Co-culture experiments showed that T helper type 1 (Th1) cells up-regulated expression of markers that are typical of APC in microglia, including MHCII and CD40 induced, whereas Th2 cells were unable to do so.68 Conversely, microglia induced Th1 cells to release IFN-γ but were unable to trigger release of IL-4 from Th2 cells.68 In addition, conditioned medium obtained from Th1 cells, but not Th2 cells, increased expression of CD80, CD86, CD40 and the adhesion molecule CD54 on microglia; these changes were mimicked by IFN-γ and inhibited by anti-IFN-γ antibody, although only partially, prompting the authors to conclude that the effect of Th1 cells cannot be attributed exclusively to IFN-γ release.69 We have also reported that T cells, specifically Th1 and Th17 cells, interact with microglia and induce their activation in vitro.70,71 Aβ-specific Th1 cells increased production of inflammatory cytokines IL-1β, IL-6 and TNF-α and increased expression of MHCII and CD86 on microglia.70 Th17 cells exerted similar effects, whereas Th2 cells exerted little effect. However, Th2 cells attenuated the effect of Th17 cells on microglia but did not inhibit the effect of Th1 cells. Myelin oligodendrocyte glycoprotein-specific Th1 cells and Th1/Th17 cells also increased microglial production of inflammatory cytokines and expression of MHCII, CD80 and CD86.71 Consistent with these findings, it has also been shown that co-culture of organotypic slices with ovalbumin-specific or myelin basic protein-specific Th1 cells increased microglial activation whereas Th2 cells exerted no effect.72 These in vitro data all suggest that the APC function of microglia is increased when cells interact with Th1 and Th17 cells and, interestingly, up-regulation of APC function induced by Th1 cells appears to be associated with a switch in microglial phenotype away from one that is efficient at phagocytosis.73 The evidence from our recent studies suggests that the modulatory effect of T cells on microglial function observed in vitro also occurs in vivo. Focusing on analysis of the effects in a model of amyloid over-expression for AD, we found that intravenous administration of Aβ-specific Th1 cells into APP/PS1 mice, which tracked to the brain, increased microglial activation and enhanced Aβ pathology and this effect was attenuated in APP/PS1 mice treated with an anti-IFN-γ antibody.74 The effect of Th17 cell administration was less profound whereas injection of Th2 cells was essentially without effect in these mice. The lack of effect of Th2 cells is at variance with an earlier report, which suggested a beneficial effect of transfer of Aβ-specific Th2 cells on behavioural deficits and pathology in APP/PS1 mice.75 This group also reported that Th2 cells reduced the numbers of activated microglia surrounding Aβ-containing plaques.
Table 1.
Effect of T cells on microglial activation
| Main finding | Preparation | Reference |
|---|---|---|
| Ovalbumin (OVA) -specific T helper type 1 (Th1) cells increased expression of MHCII, CD40 and CD54 on microglia | Microglia from BALB/c mice. | 68 |
| Th2 cells had little effect | T cells prepared from transgenic mice carrying a T-cell receptor (TCR) DO11.10 for cOVA323–339 peptide mouse | |
| Conditioned medium from Th1 cells increased expression of CD80, CD86, CD40 and CD54 and release of tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6) and CXCL10 | Microglia from human tissue obtained following surgery | 69 |
| Medium from Th2 cells had no significant effect | T cells from healthy volunteers | |
| Amyloid β (Aβ) -specific Th1 cells increased expression of MHCII and CD86 and release of IL-1β, TNF-α and IL-6 | Mixed glia and T cells from C57BL/6 mouse | 70 |
| Aβ-specific Th17 cells exerted a similar effect | ||
| Th2 cells had little effect | ||
| Myelin oligodendrocyte glycoprotein (MOG) -specific Th1 cells increased expression of MHCII, CD80, CD86 and CD40 and release of IL-1β, TNF-α | Mixed glia and T cells from C57BL/6 mouse | 71 |
| MOG-specific Th1/Th17 cells exerted a similar effect | ||
| Myelin basic protein-specific and OVA-specific Th1 cells increased CD80 expression (with or without cell contact) but decreased CD86 expression (slices already expressed CD86) | Organotypic entorhinal–hippocampal slice cultures prepared from 11-day-old mice | 72 |
| Th1 and Th2 cell lines from transgenic mice carrying a TCR for myelin basic protein peptide Ac1-11 or transgenic mice carrying a TCR DO11.10 for cOVA323–339 peptide | ||
| Adoptive transfer of Aβ-specific Th1 cells increased CD11b immunoreactivity and Aβ accumulation in APP/PS1 transgenic mice | T cells from C57BL/6 mice adoptively transferred into APP/PS1 mice | 74 |
| Th2 cells had little effect | ||
| Adoptive transfer of Aβ-specific Th2 cells decreased plaque-associated microglial numbers | Th2 cells from congenic mice adoptively transferred into APP/PS1 mice | 75 |
| T cell infusion reduced area covered by microglia surrounding Aβ deposits | T cell-enriched infusate prepared from Tg2576 mice | 96 |
| Th1 cell-enriched infusion was less effective | Adoptively transferred into 8·5-month-old Tg2576 mice |
Do microglia adopt the M1 and M2 phenotypes observed in macrophages?
It has been known for a few decades that macrophages adopt different activation states, identified by up-regulation of specific markers, in response to different signals.76 These are broadly described as M1, an inflammatory phenotype and M2, an anti-inflammatory phenotype. The Th1 cell-derived cytokine IFN-γ induces classical activation (M1), and this phenotype is identified by up-regulation of TNF-α and iNOS. The term alternative activation (M2a phenotype) was first used to describe a macrophage that adopted a phenotype distinct from that induced by IFN-γ and LPS.77 These cells were not capable of producing NO and so were not cytotoxic and, although MHCII expression was increased, the cells were not efficient APC and prevented proliferation of T cells.78 This phenotype is induced by the Th2 cell-derived cytokines, IL-4, IL-5 and IL-13 and is identified by up-regulation of mRNA expression of arginase I, mannose receptor, chitinase 3-like 3 (YM1) and found in inflammatory zone-1. Because the cells were shown to inhibit inflammatory cytokine production, they were recognized as regulatory macrophages.79 Acquired deactivation (M2c phenotype) is induced by the immunosuppressive cytokines, IL-10 and TGF-β, which are derived from regulatory T cells, and this phenotype is associated with up-regulation of anti-inflammatory cytokines like IL-10 and TGF-β and down-regulation of factors that contribute to APC function like MHCII.76 Like macrophages, microglia respond to IL-10 and administration of IL-10 to aged rats decreases microglial activation and neuroinflammatory changes,80 suggesting that microglia can also adopt the acquired deactivated state. It has been shown that this state, which is characterized by an increase in receptors like scavenger receptors, which support their phagocytic function, can also be induced by phagocytosis of apoptotic cells, or TGF-β though the data are largely derived from in vitro studies.81
In the past 5 years or so, significant interest has developed in determining whether microglia react to the different stimuli in the same way as macrophages. Although there are data that support this, it remains to be established whether the characteristics of these activation states, and their patterns of stimulation, faithfully translate from the in vitro into the in vivo situation. It is also not entirely clear that the phenotypic markers that apply to macrophages are relevant in the context of microglia. A further issue relates to the nature of the stimuli that trigger classical and alternative activation states because resident cells in the brain produce limited IFN-γ and IL-4. It must therefore be considered that infiltrating cells are responsible for production of these cytokines and consequently for triggering polarization of microglia into classically and alternatively activated phenotypes, or that other polarizing stimuli substitute in the brain. Despite these caveats, it is known that IFN-γ potently activates microglia and among the changes observed is increased expression of TNF-α and iNOS as well as up-regulation of other inflammatory cytokines and markers typical of an APC in vivo82,83 as well as in vitro.84 A recent comprehensive study in microglia also identified that cyclo-oxygenase 2 mRNA was markedly increased in IFN-γ-treated cells whereas mannose receptor and arginase-1 mRNA were up-regulated by IL-4.85 Alternatively activated macrophages have a key role in tissue repair and restructuring of the extracellular matrix;81 whereas alternatively activated microglia may also play a role in tissue repair, a key role is likely to be re-establishing homeostasis following an inflammatory stimulus.
Identifying factors that enable switching of microglia from an inflammatory to an anti-inflammatory phenotype is an important goal because it is likely to point towards strategies that might prevent chronic inflammation. One potential molecular switch is activation of peroxisome proliferator-activated receptor γ;86 its activation by pioglitazone increased expression of several markers of alternative activation in 12-month-old APP/PS1 mice. This was associated with decreased Aβ pathology, suggesting that alternatively activated microglia are more phagocytic.87 Inhibition of NADPH oxidase or functional deletion of p47phox have also been identified as factors that potentially control the polarization of microglia from the classically to the alternatively activated state in the brain.88 Similarly, knockdown of NOD-like receptor family, pyrin domain containing 3 (NLRP3) in APP/PS1 mice switches microglia from the classical to the alternative activation state.89 The tetrapeptide, (threonine-lysine-proline-arginine), tuftsin, also induces an anti-inflammatory phenotype in vitro and in vivo90 whereas CD45, which inhibits the interaction between CD40 ligand and receptor, appears to switch off the inflammatory M1 state in vivo.91 In contrast, age induces microglia to adopt an inflammatory phenotype.92 Recent evidence has revealed that specific microRNAs may be key factors in polarization of microglia, and it has been suggested that miR-155 skews microglia towards an M1 phenotype, at least in cultured cells.93 However, changes in up-regulation of other microRNAs including miR-101 and miR-125b, together with down-regulation of miR-92, also appear to be characteristic of the M1 phenotype.94
What microglial phenotype is observed in AD and is this linked with Aβ accumulation?
With the focus on the amyloid hypothesis of AD, the question of which microglial phenotype is associated with Aβ accumulation and with greater phagocytic capability has been the subject of intense interest. In the context of AD in particular, it is important to unravel the factors that contribute to the apparent inability of microglia to phagocytose Aβ despite the proximity of activated cells to the Aβ-containing plaques. The consensus suggests that an inflammatory milieu inhibits the phagocytic function of microglia. Hence, it has been reported that Th1 cytokines inhibit microglial phagocytosis of Aβ, whereas IL-4 and IL-10 have the opposite effect.73 Jimenez et al. demonstrated that the microglial phenotype changed with age in APP/PS1 mice, shifting from an alternative altivation state, which was associated with phagocytic capability, to a classically activated phenotype, which was associated with pro-inflammatory cytokine production. Aβ-immunoreactivity was observed in tomato lectin-stained microglia that surrounded plaques in 6-month-old APP/PS1 mice and the evidence indicated that these cells were alternatively activated microglia because they stained positively for YM-1. By 18 months of age, microglia had adopted a classically activated phenotype and these cells appeared not to be phagocytic.92 It was also shown that knocking out IFN-γR1 in APP mice was associated with reduced Aβ deposition in cortex and hippocampus and this correlated with decreased gliosis.95 Additionally, alternative activation of microglia in APP/PS1 mice, which was induced by the peroxisome proliferator-activated receptor γ activator pioglitazone87 or NLRP3 knockdown89 was associated with an increase in Aβ clearance. We have found that adoptive transfer of Aβ-specific Th1 cells markedly increased microglial activation, inflammatory changes and Aβ pathology but Th2 cells were without effect.74 This contrasts with the findings of others, which suggested that injection of Th2 cells decreased plaque burden.75 This finding followed up a previous report from this group, which demonstrated that injection of a mixed population of Aβ-specific T cells decreased Aβ pathology in 8·5-month-old APP/PS1 mice, although this effect was not evident if an enriched Th1 preparation was injected.96 Our evidence has indicated that, even in 6- to 7-month-old APP/PS1 mice, there was evidence of an inflammatory phenotype,97,98 and in a recent study we were unable to detect any evidence of alternatively activated microglia in 12-month-old APP/PS1 mice (A. Minogue et al., unpublished data). This may vary between models of AD because increased mRNA expression of TNF-α (although not iNOS), as well as mannose receptor and arginase-1, was observed in cortical tissue prepared from 60-week-old Tg2576 mice, suggesting a heterogeneity in microglial phenotypes in at least this model of AD.79 This heterogeneity was also observed in transgenic mice that over-expressed APP only but was more marked in APP mice with a deficiency in NOS2.99 However, this group have suggested that amyloid deposition is greater in circumstances in which there is up-regulation of markers of alternatively activated microglia, which is at variance with most of the literature.
It has been suggested that macrophages rather than microglia are the primary phagocytes in the brain100 and that these infiltrating cells are key to reparative processes.101 However, the evidence suggests that alternatively activated macrophages, like microglia, have enhanced phagocytic capability102 and the possibility exists that the inflammatory microenvironment in the AD brain, which inhibits efficient phagocytosis,103 prevents the macrophages from adopting the alternatively activated state. This may explain the finding that the macrophages that infiltrate the brain in AD patients fail to phagocytose Aβ efficiently.104 Therefore, a therapeutic value of anti-inflammatory therapies given at the appropriate time in the disease, may be a reduction in Aβ pathology.
Does infiltration of peripheral cells contribute to the onset or progression of AD?
In AD and amyloid-based transgenic models of AD, microglia adopt an inflammatory phenotype,97,98 and increased expression TNF-α and iNOS, which are markers of classical activation, have been reported in the brain of mouse models of AD and in post-mortem tissue from AD patients.89,99,105 A link between microglial activation and tau hyperphosphorylation has also been reported,106 though accumulating tau in a mouse model in which tau is over-expressed leads to an LPS-induced up-regulation of markers of alternative activation.107 If it is accepted that IFN-γ is required for inducing classical activation of microglia, then it follows that there must be infiltration of IFN-γ-producing cells, for example Th1 cells, since IFN-γ is generally not produced to any significant degree by resident cells in the brain. However, it must be considered that factors other than IFN-γ can induce changes that mimic classical activation and, in this context it is known that several of the changes induced by IFN-γR activation, including up-regulation of TNF-α and iNOS, as well as cell surface markers of activation like MHCII and CD40, are also triggered by Aβ and Toll-like receptor agonists.42,108
It is widely accepted that T cell entry into the central nervous system under normal circumstances is very limited and the evidence suggests that their role is immunosurveillance;109,110 it has been proposed that cells gain entry at the choroid plexus under these conditions when there is no evidence of neuroinflammation.111 However, significant infiltration of immune cells occurs in neuroinflammatory conditions.111 The role of these cells in the pathogenesis of multiple sclerosis is well rehearsed,112,113 but a recent study revealed that T cell infiltration also occurs in Parkinson's disease114 and the evidence from animal studies suggests that the presence in the brain of CD4+ cells significantly contributes to the demise of dopaminergic cells.
The first evidence that T cells were present in the brain of AD patients was presented 25 years ago115,116 and similar findings have been sporadically reported since.12,117–122 These cells were found to be in close apposition with plaques and activated glia.119 CD8+ cells have been found in the post-mortem brain of individuals with mild to moderate AD but also in non-demented controls, though some evidence suggests that their numbers were decreased in the AD brains.12,121 We have recently shown that there is significant infiltration of T cells, particularly IFN-γ-positive and IL-17-positive T cells, into the brains of 6- to 8-month-old APP/PS1 mice74 and this infiltration increases with age so that infiltration in 12-month-old animals was significantly greater123. T cell infiltration has also been shown in 18-month-old, but not 6-month-old, APP/PS1 mice.92
Infiltration of immune cells may result from the creation of a chemotactic gradient as a consequence of increased expression of chemokines in brain; CCL3, CXCL10 and CCL5 have established lymphocyte chemotactic properties124–126 and increased expression of these chemokines has been reported in AD.127–129 Interestingly, expression of CCR5 and CXCR3 on T cells obtained from AD patients has been reported.130–132 A loss of BBB integrity, which will also enable cell infiltration, has been described in AD; increased fibrinogen immunoreactivity, altered immunohistochemical staining for von Willebrand's factor and dystrophic vessels have been reported.133 This was accompanied by marked glial activation; activated microglia were co-located with fibrinogen immunoreactivity, suggesting that fibrinogen induces cell activation.134 Interestingly, intrahippocampal injection of Aβ induced BBB permeability in the rat hippocampus, as revealed by fibrinogen immunoreactivity, whereas we have recently found that the age-related accumulation of endogenous Aβ in APP/PS1 mice was accompanied by increased BBB permeability, infiltration of CD4+ IFNγ+ cells and increased expression of markers of classical activation of microglia (Minogue et al., unpublished data).
Conclusions
A proposed sequence of events presented in Fig. 1 suggests that BBB permeability, which is increased in AD, together with the creation of a chemotactic gradient, leads to infiltration of IFN-γ-producing T cells. Inflammatory T cells together with IFN-γ, produced by these cells, induces classical activation of microglia, which leads to inflammatory cytokine and chemokine production. These changes act to increase APP processing and Aβ accumulation and also induce BBB permeability with further infiltration of cells, and so a cycle of damaging events ensues. It is proposed that interupting this cycle is key to limiting disease progression in AD. Validation (or otherwise) of this hypothesis requires significant investigation. For example, it predicts that preventing BBB permeability or infiltration of IFN-γ-producing T cells will reduce inflammation and pathology. It also predicts that IFN-γ-induced changes in microglia, and up-regulation of chemokines, induce further BBB permeability whereas the effects of microglia, which have an anti-inflammatory phenotype, do not. It remains to be established whether these changes impact in the predicted way on neuronal and cognitive function.
Figure 1.
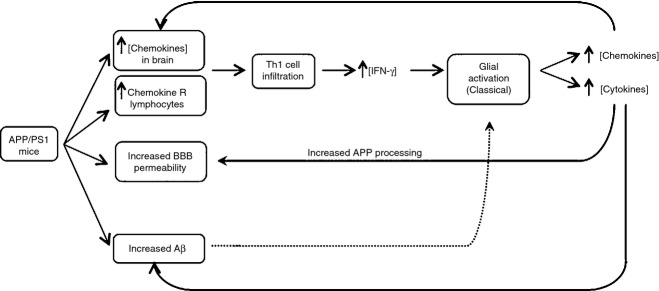
Proposed sequence of events leading to amyloid pathology and microglial activation in AD.
Disclosures
The author has no financial disclosures.
References
- 1.McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79:195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- 2.Guillot-Sestier MV, Town T. Innate immunity in Alzheimer's disease: a complex affair. CNS Neurol Disord Drug Targets. 2013;20:593–607. doi: 10.2174/1871527311312050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer's disease, role of cytokines. Scientific World Journal. 2012;2012:756357. doi: 10.1100/2012/756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solito E, Sastre M. Microglia function in Alzheimer's disease. Front Pharmacol. 2012;3:14. doi: 10.3389/fphar.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Launer L. Nonsteroidal anti-inflammatory drug use and the risk for Alzheimer's disease: dissecting the epidemiological evidence. Drugs. 2003;63:731–9. doi: 10.2165/00003495-200363080-00001. [DOI] [PubMed] [Google Scholar]
- 7.Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J. Nonsteroidal anti-inflammatory drugs in Alzheimer's disease. Neurology. 1995;45:51–5. doi: 10.1212/wnl.45.1.51. [DOI] [PubMed] [Google Scholar]
- 8.Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997;48:626–32. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- 9.Aisen PS, Schafer KA, Grundman M, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289:2819–26. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- 10.Reines SA, Block GA, Morris JC, et al. Rofecoxib: no effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62:66–71. doi: 10.1212/wnl.62.1.66. [DOI] [PubMed] [Google Scholar]
- 11.Imbimbo BP, Solfrizzi V, Panza F. Are NSAIDs useful to treat Alzheimer's disease or mild cognitive impairment? Front Aging Neurosci. 2010;2:19. doi: 10.3389/fnagi.2010.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parachikova A, Agadjanyan MG, Cribbs DH, Blurton-Jones M, Perreau V, Rogers J, et al. Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol Aging. 2007;28:1821–33. doi: 10.1016/j.neurobiolaging.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Breitner JC, Haneuse SJ, Walker R, Dublin S, Crane PK, Gray SL, Larson EB. Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology. 2009;72:1899–905. doi: 10.1212/WNL.0b013e3181a18691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mackenzie IR. Postmortem studies of the effect of anti-inflammatory drugs on Alzheimer-type pathology and associated inflammation. Neurobiol Aging. 2001;22:819–22. doi: 10.1016/s0197-4580(01)00304-9. [DOI] [PubMed] [Google Scholar]
- 15.Halliday GM, Shepherd CE, McCann H, Reid WG, Grayson DA, Broe GA, Kril JJ. Effect of anti-inflammatory medications on neuropathological findings in Alzheimer disease. Arch Neurol. 2000;57:831–6. doi: 10.1001/archneur.57.6.831. [DOI] [PubMed] [Google Scholar]
- 16.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–5. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64:110–22. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30:527–35. doi: 10.1016/j.tins.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Savage CD, Lopez-Castejon G, Denes A, Brough D. NLRP3-inflammasome activating DAMPs stimulate an inflammatory response in glia in the absence of priming which contributes to brain inflammation after injury. Front Immunol. 2012;3:288. doi: 10.3389/fimmu.2012.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Honjo K, van Reekum R, Verhoeff NP. Alzheimer's disease and infection: do infectious agents contribute to progression of Alzheimer's disease? Alzheimers Dement. 2009;5:348–60. doi: 10.1016/j.jalz.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Holmes C, El-Okl M, Williams AL, Cunningham C, Wilcockson D, Perry VH. Systemic infection, interleukin 1β, and cognitive decline in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2003;74:788–9. doi: 10.1136/jnnp.74.6.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, Culliford D, Perry VH. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73:768–74. doi: 10.1212/WNL.0b013e3181b6bb95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammond CJ, Hallock LR, Howanski RJ, Appelt DM, Little CS, Balin BJ. Immunohistological detection of Chlamydia pneumoniae in the Alzheimer's disease brain. BMC Neurosci. 2010;11:121. doi: 10.1186/1471-2202-11-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Little CS, Hammond CJ, MacIntyre A, Balin BJ, Appelt DM. Chlamydia pneumoniae induces Alzheimer-like amyloid plaques in brains of BALB/c mice. Neurobiol Aging. 2004;25:419–29. doi: 10.1016/S0197-4580(03)00127-1. [DOI] [PubMed] [Google Scholar]
- 25.Krstic D, Madhusudan A, Doehner J, et al. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J Neuroinflammation. 2012;9:151. doi: 10.1186/1742-2094-9-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid β peptide in APPswe transgenic mice. Neurobiol Dis. 2003;14:133–45. doi: 10.1016/s0969-9961(03)00069-x. [DOI] [PubMed] [Google Scholar]
- 27.Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer's disease. J Neurosci. 2005;25:8843–53. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wozniak MA, Mee AP, Itzhaki RF. Herpes simplex virus type 1 DNA is located within Alzheimer's disease amyloid plaques. J Pathol. 2009;217:131–8. doi: 10.1002/path.2449. [DOI] [PubMed] [Google Scholar]
- 29.Itzhaki RF, Lin WR, Shang D, Wilcock GK, Faragher B, Jamieson GA. Herpes simplex virus type 1 in brain and risk of Alzheimer's disease. Lancet. 1997;349:241–4. doi: 10.1016/S0140-6736(96)10149-5. [DOI] [PubMed] [Google Scholar]
- 30.Carnevale D, De Simone R, Minghetti L. Microglia–neuron interaction in inflammatory and degenerative diseases: role of cholinergic and noradrenergic systems. CNS Neurol Disord Drug Targets. 2007;6:388–97. doi: 10.2174/187152707783399193. [DOI] [PubMed] [Google Scholar]
- 31.Neumann H. Control of glial immune function by neurons. Glia. 2001;36:191–9. doi: 10.1002/glia.1108. [DOI] [PubMed] [Google Scholar]
- 32.Lacmann A, Hess D, Gohla G, Roussa E, Krieglstein K. Activity-dependent release of transforming growth factor-β in a neuronal network in vitro. Neuroscience. 2007;150:647–57. doi: 10.1016/j.neuroscience.2007.09.046. [DOI] [PubMed] [Google Scholar]
- 33.Fernandez AM, Fernandez S, Carrero P, Garcia-Garcia M, Torres-Aleman I. Calcineurin in reactive astrocytes plays a key role in the interplay between proinflammatory and anti-inflammatory signals. J Neurosci. 2007;27:8745–56. doi: 10.1523/JNEUROSCI.1002-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wright GJ, Cherwinski H, Foster-Cuevas M, et al. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J Immunol. 2003;171:3034–46. doi: 10.4049/jimmunol.171.6.3034. [DOI] [PubMed] [Google Scholar]
- 35.Wright GJ, Puklavec MJ, Willis AC, Hoek RM, Sedgwick JD, Brown MH, Barclay AN. Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity. 2000;13:233–42. doi: 10.1016/s1074-7613(00)00023-6. [DOI] [PubMed] [Google Scholar]
- 36.Lyons A, McQuillan K, Deighan BF, et al. Decreased neuronal CD200 expression in IL-4-deficient mice results in increased neuroinflammation in response to lipopolysaccharide. Brain Behav Immun. 2009;23:1020–7. doi: 10.1016/j.bbi.2009.05.060. [DOI] [PubMed] [Google Scholar]
- 37.Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KH, Lynch MA. CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J Neurosci. 2007;27:8309–13. doi: 10.1523/JNEUROSCI.1781-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoek RM, Ruuls SR, Murphy CA, et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290:1768–71. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- 39.Wang XJ, Ye M, Zhang YH, Chen SD. CD200–CD200R regulation of microglia activation in the pathogenesis of Parkinson's disease. J Neuroimmune Pharmacol. 2007;2:259–64. doi: 10.1007/s11481-007-9075-1. [DOI] [PubMed] [Google Scholar]
- 40.Broderick C, Hoek RM, Forrester JV, Liversidge J, Sedgwick JD, Dick AD. Constitutive retinal CD200 expression regulates resident microglia and activation state of inflammatory cells during experimental autoimmune uveoretinitis. Am J Pathol. 2002;161:1669–77. doi: 10.1016/S0002-9440(10)64444-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lyons A, McQuillan K, Deighan BF, et al. Decreased neuronal CD200 expression in IL-4-deficient mice results in increased neuroinflammation in response to lipopolysaccharide. Brain Behav Immun. 2009;23:1020–7. doi: 10.1016/j.bbi.2009.05.060. [DOI] [PubMed] [Google Scholar]
- 42.Lyons A, Downer EJ, Costello DA, Murphy N, Lynch MA. Dok2 mediates the CD200Fc attenuation of Aβ-induced changes in glia. J Neuroinflammation. 2012;9:107. doi: 10.1186/1742-2094-9-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cox FF, Carney D, Miller AM, Lynch MA. CD200 fusion protein decreases microglial activation in the hippocampus of aged rats. Brain Behav Immun. 2012;26:789–96. doi: 10.1016/j.bbi.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y, Bando Y, Vargas-Lowy D, Elyaman W, Khoury SJ, Huang T, Reif K, Chitnis T. CD200R1 agonist attenuates mechanisms of chronic disease in a murine model of multiple sclerosis. J Neurosci. 2010;30:2025–38. doi: 10.1523/JNEUROSCI.4272-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gorczynski RM, Chen Z, Lee L, Yu K, Hu J. Anti-CD200R ameliorates collagen-induced arthritis in mice. Clin Immunol. 2002;104:256–64. doi: 10.1006/clim.2002.5232. [DOI] [PubMed] [Google Scholar]
- 46.Gorczynski RM, Chen Z, Yu K, Hu J. CD200 immunoadhesin suppresses collagen-induced arthritis in mice. Clin Immunol. 2001;101:328–34. doi: 10.1006/clim.2001.5117. [DOI] [PubMed] [Google Scholar]
- 47.Walker DG, Dalsing-Hernandez JE, Campbell NA, Lue LF. Decreased expression of CD200 and CD200 receptor in Alzheimer's disease: a potential mechanism leading to chronic inflammation. Exp Neurol. 2009;215:5–19. doi: 10.1016/j.expneurol.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koning N, Bo L, Hoek RM, Huitinga I. Downregulation of macrophage inhibitory molecules in multiple sclerosis lesions. Ann Neurol. 2007;62:504–14. doi: 10.1002/ana.21220. [DOI] [PubMed] [Google Scholar]
- 49.Koning N, Swaab DF, Hoek RM, Huitinga I. Distribution of the immune inhibitory molecules CD200 and CD200R in the normal central nervous system and multiple sclerosis lesions suggests neuron–glia and glia–glia interactions. J Neuropathol Exp Neurol. 2009;68:159–67. doi: 10.1097/NEN.0b013e3181964113. [DOI] [PubMed] [Google Scholar]
- 50.Luo XG, Zhang JJ, Zhang CD, Liu R, Zheng L, Wang XJ, Chen SD, Ding JQ. Altered regulation of CD200 receptor in monocyte-derived macrophages from individuals with Parkinson's disease. Neurochem Res. 2010;35:540–7. doi: 10.1007/s11064-009-0094-6. [DOI] [PubMed] [Google Scholar]
- 51.Lyons A, Lynch AM, Downer EJ, Hanley R, O'Sullivan JB, Smith A, Lynch MA. Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J Neurochem. 2009;110:1547–56. doi: 10.1111/j.1471-4159.2009.06253.x. [DOI] [PubMed] [Google Scholar]
- 52.Wolf Y, Yona S, Kim KW, Jung S. Microglia, seen from the CX3CR1 angle. Front Cell Neurosci. 2013;7:26. doi: 10.3389/fncel.2013.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces β-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. 2010;177:2549–62. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cho SH, Sun B, Zhou Y, Kauppinen TM, Halabisky B, Wes P, Ransohoff RM, Gan L. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem. 2011;286:32713–22. doi: 10.1074/jbc.M111.254268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 56.Adams S, van der Laan LJ, Vernon-Wilson E, Renardel de Lavalette C, Dopp EA, Dijkstra CD, Simmons DL, van den Berg TK. Signal-regulatory protein is selectively expressed by myeloid and neuronal cells. J Immunol. 1998;161:1853–9. [PubMed] [Google Scholar]
- 57.Reinhold MI, Lindberg FP, Plas D, Reynolds S, Peters MG, Brown EJ. In vivo expression of alternatively spliced forms of integrin-associated protein (CD47) J Cell Sci. 1995;108(Pt 11):3419–25. doi: 10.1242/jcs.108.11.3419. [DOI] [PubMed] [Google Scholar]
- 58.van Beek EM, Cochrane F, Barclay AN, van den Berg TK. Signal regulatory proteins in the immune system. J Immunol. 2005;175:7781–7. doi: 10.4049/jimmunol.175.12.7781. [DOI] [PubMed] [Google Scholar]
- 59.Mott RT, Ait-Ghezala G, Town T, et al. Neuronal expression of CD22: novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia. 2004;46:369–79. doi: 10.1002/glia.20009. [DOI] [PubMed] [Google Scholar]
- 60.Lyons A, Griffin RJ, Costelloe CE, Clarke RM, Lynch MA. IL-4 attenuates the neuroinflammation induced by amyloid-β in vivo and in vitro. J Neurochem. 2007;101:771–81. doi: 10.1111/j.1471-4159.2006.04370.x. [DOI] [PubMed] [Google Scholar]
- 61.Clarke RM, Lyons A, O'Connell F, Deighan BF, Barry CE, Anyakoha NG, Nicolaou A, Lynch MA. A pivotal role for interleukin-4 in atorvastatin-associated neuroprotection in rat brain. J Biol Chem. 2008;283:1808–17. doi: 10.1074/jbc.M707442200. [DOI] [PubMed] [Google Scholar]
- 62.Cox FF, Berezin V, Bock E, Lynch MA. The neural cell adhesion molecule-derived peptide, FGL, attenuates lipopolysaccharide-induced changes in glia in a CD200-dependent manner. Neuroscience. 2013;235:141–8. doi: 10.1016/j.neuroscience.2012.12.030. [DOI] [PubMed] [Google Scholar]
- 63.Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP – thirty-one years (1969–2000) Neurochem Res. 2000;25:1439–51. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- 64.Min KJ, Yang MS, Kim SU, Jou I, Joe EH. Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci. 2006;26:1880–7. doi: 10.1523/JNEUROSCI.3696-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee M, Schwab C, McGeer PL. Astrocytes are GABAergic cells that modulate microglial activity. Glia. 2011;59:152–65. doi: 10.1002/glia.21087. [DOI] [PubMed] [Google Scholar]
- 66.Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A. 2012;109:E197–205. doi: 10.1073/pnas.1111098109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.von Bernhardi R, Ramirez G. Microglia–astrocyte interaction in Alzheimer's disease: friends or foes for the nervous system? Biol Res. 2001;34:123–8. doi: 10.4067/s0716-97602001000200017. [DOI] [PubMed] [Google Scholar]
- 68.Aloisi F, De Simone R, Columba-Cabezas S, Penna G, Adorini L. Functional maturation of adult mouse resting microglia into an APC is promoted by granulocyte–macrophage colony-stimulating factor and interaction with Th1 cells. J Immunol. 2000;164:1705–12. doi: 10.4049/jimmunol.164.4.1705. [DOI] [PubMed] [Google Scholar]
- 69.Seguin R, Biernacki K, Prat A, et al. Differential effects of Th1 and Th2 lymphocyte supernatants on human microglia. Glia. 2003;42:36–45. doi: 10.1002/glia.10201. [DOI] [PubMed] [Google Scholar]
- 70.McQuillan K, Lynch MA, Mills KH. Activation of mixed glia by Aβ-specific Th1 and Th17 cells and its regulation by Th2 cells. Brain Behav Immun. 2010;24:598–607. doi: 10.1016/j.bbi.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 71.Murphy AC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24:641–51. doi: 10.1016/j.bbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 72.Wolf SA, Gimsa U, Bechmann I, Nitsch R. Differential expression of costimulatory molecules B7-1 and B7-2 on microglial cells induced by Th1 and Th2 cells in organotypic brain tissue. Glia. 2001;36:414–20. doi: 10.1002/glia.1127. [DOI] [PubMed] [Google Scholar]
- 73.Townsend KP, Town T, Mori T, et al. CD40 signaling regulates innate and adaptive activation of microglia in response to amyloid β-peptide. Eur J Immunol. 2005;35:901–10. doi: 10.1002/eji.200425585. [DOI] [PubMed] [Google Scholar]
- 74.Browne TC, McQuillan K, McManus RM, O'Reilly JA, Mills KH, Lynch MA. IFN-γ production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer's disease. J Immunol. 2013;190:2241–51. doi: 10.4049/jimmunol.1200947. [DOI] [PubMed] [Google Scholar]
- 75.Cao C, Arendash GW, Dickson A, Mamcarz MB, Lin X, Ethell DW. Aβ-specific Th2 cells provide cognitive and pathological benefits to Alzheimer's mice without infiltrating the CNS. Neurobiol Dis. 2009;34:63–70. doi: 10.1016/j.nbd.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 77.Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287–92. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–12. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 79.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moore ME, Piazza A, McCartney Y, Lynch MA. Evidence that vitamin D3 reverses age-related inflammatory changes in the rat hippocampus. Biochem Soc Trans. 2005;33:573–7. doi: 10.1042/BST0330573. [DOI] [PubMed] [Google Scholar]
- 81.Colton CA, Wilcock DM. Assessing activation states in microglia. CNS Neurol Disord Drug Targets. 2010;9:174–91. doi: 10.2174/187152710791012053. [DOI] [PubMed] [Google Scholar]
- 82.Maher FO, Clarke RM, Kelly A, Nally RE, Lynch MA. Interaction between interferon γ and insulin-like growth factor-1 in hippocampus impacts on the ability of rats to sustain long-term potentiation. J Neurochem. 2006;96:1560–71. doi: 10.1111/j.1471-4159.2006.03664.x. [DOI] [PubMed] [Google Scholar]
- 83.Downer EJ, Cowley TR, Cox F, Maher FO, Berezin V, Bock E, Lynch MA. A synthetic NCAM-derived mimetic peptide, FGL, exerts anti-inflammatory properties via IGF-1 and interferon-γ modulation. J Neurochem. 2009;109:1516–25. doi: 10.1111/j.1471-4159.2009.06076.x. [DOI] [PubMed] [Google Scholar]
- 84.Kim HS, Whang SY, Woo MS, Park JS, Kim WK, Han IO. Sodium butyrate suppresses interferon-γ-, but not lipopolysaccharide-mediated induction of nitric oxide and tumor necrosis factor-α in microglia. J Neuroimmunol. 2004;151:85–93. doi: 10.1016/j.jneuroim.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 85.Chhor V, Le CT, Lebon S, et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav Immun. 2013;32:70–85. doi: 10.1016/j.bbi.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chawla A. Control of macrophage activation and function by PPARs. Circ Res. 2010;106:1559–69. doi: 10.1161/CIRCRESAHA.110.216523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease. J Neurosci. 2012;32:10117–28. doi: 10.1523/JNEUROSCI.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Choi SH, Aid S, Kim HW, Jackson SH, Bosetti F. Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J Neurochem. 2012;120:292–301. doi: 10.1111/j.1471-4159.2011.07572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–8. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wu M, Nissen JC, Chen EI, Tsirka SE. Tuftsin promotes an anti-inflammatory switch and attenuates symptoms in experimental autoimmune encephalomyelitis. PLoS ONE. 2012;7:e34933. doi: 10.1371/journal.pone.0034933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Salemi J, Obregon DF, Cobb A, Reed S, Sadic E, Jin J, et al. Flipping the switches: CD40 and CD45 modulation of microglial activation states in HIV associated dementia (HAD) Mol Neurodegener. 2011;6:3. doi: 10.1186/1750-1326-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jimenez S, Baglietto-Vargas D, Caballero C, et al. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci. 2008;28:11650–61. doi: 10.1523/JNEUROSCI.3024-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cardoso AL, Guedes JR, Pereira de Almeida L, Pedroso de Lima MC. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology. 2012;135:73–88. doi: 10.1111/j.1365-2567.2011.03514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guedes J, Cardoso AL, Pedroso de Lima MC. Involvement of MicroRNA in microglia-mediated immune response. Clin Dev Immunol. 2013;2013:186872. doi: 10.1155/2013/186872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. Interferon-γ and tumor necrosis factor-α regulate amyloid-β plaque deposition and β-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–92. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ethell DW, Shippy D, Cao C, Cracchiolo JR, Runfeldt M, Blake B, Arendash GW. Aβ-specific T-cells reverse cognitive decline and synaptic loss in Alzheimer's mice. Neurobiol Dis. 2006;23:351–61. doi: 10.1016/j.nbd.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 97.Gallagher JJ, Finnegan ME, Grehan B, Dobson J, Collingwood JF, Lynch MA. Modest amyloid deposition is associated with iron dysregulation, microglial activation, and oxidative stress. J Alzheimers Dis. 2012;28:147–61. doi: 10.3233/JAD-2011-110614. [DOI] [PubMed] [Google Scholar]
- 98.Gallagher JJ, Minogue AM, Lynch MA. Impaired performance of female APP/PS1 Mice in the Morris water maze is coupled with increased Aβ accumulation and microglial activation. Neurodegener Dis. 2013;11:33–41. doi: 10.1159/000337458. [DOI] [PubMed] [Google Scholar]
- 99.Wilcock DM, Zhao Q, Morgan D, Gordon MN, Everhart A, Wilson JG, Lee JE, Colton CA. Diverse inflammatory responses in transgenic mouse models of Alzheimer's disease and the effect of immunotherapy on these responses. ASN Neuro. 2011;3:249–58. doi: 10.1042/AN20110018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 101.Shechter R, Schwartz M. Harnessing monocyte-derived macrophages to control central nervous system pathologies: no longer ‘if’ but ‘how’. J Pathol. 2013;229:332–46. doi: 10.1002/path.4106. [DOI] [PubMed] [Google Scholar]
- 102.Chinetti-Gbaguidi G, Baron M, Bouhlel MA, et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARγ and LXRα pathways. Circ Res. 2011;108:985–95. doi: 10.1161/CIRCRESAHA.110.233775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar β-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25:8240–9. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fiala M, Lin J, Ringman J, et al. Ineffective phagocytosis of amyloid-β by macrophages of Alzheimer's disease patients. J Alzheimers Dis. 2005;7:221–32. doi: 10.3233/jad-2005-7304. discussion 55-62. [DOI] [PubMed] [Google Scholar]
- 105.Rao JS, Rapoport SI, Kim HW. Altered neuroinflammatory, arachidonic acid cascade and synaptic markers in postmortem Alzheimer's disease brain. Transl Psychiatry. 2011;1:e31. doi: 10.1038/tp.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 106.Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68:19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee DC, Rizer J, Selenica ML, et al. LPS-induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation. 2010;7:56. doi: 10.1186/1742-2094-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Downer EJ, Johnston DG, Lynch MA. Differential role of Dok1 and Dok2 in TLR2-induced inflammatory signaling in glia. Mol Cell Neurosci. 2013;56C:148–58. doi: 10.1016/j.mcn.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 109.Hickey WF. Migration of hematogenous cells through the blood–brain barrier and the initiation of CNS inflammation. Brain Pathol. 1991;1:97–105. doi: 10.1111/j.1750-3639.1991.tb00646.x. [DOI] [PubMed] [Google Scholar]
- 110.Hickey WF. Basic principles of immunological surveillance of the normal central nervous system. Glia. 2001;36:118–24. doi: 10.1002/glia.1101. [DOI] [PubMed] [Google Scholar]
- 111.Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood–brain barriers. Trends Immunol. 2012;33:579–89. doi: 10.1016/j.it.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 112.Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162:1–11. doi: 10.1111/j.1365-2249.2010.04143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weissert R. The immune pathogenesis of multiple sclerosis. J Neuroimmune Pharmacol. 2013 doi: 10.1007/s11481-013-9467-3. doi: 10.1007/s11481-013-9467-3. [DOI] [PubMed] [Google Scholar]
- 114.Brochard V, Combadiere B, Prigent A, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119:182–92. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rogers J, Luber-Narod J, Styren SD, Civin WH. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer's disease. Neurobiol Aging. 1988;9:339–49. doi: 10.1016/s0197-4580(88)80079-4. [DOI] [PubMed] [Google Scholar]
- 116.Itagaki S, McGeer PL, Akiyama H. Presence of T-cytotoxic suppressor and leucocyte common antigen positive cells in Alzheimer's disease brain tissue. Neurosci Lett. 1988;91:259–64. doi: 10.1016/0304-3940(88)90690-8. [DOI] [PubMed] [Google Scholar]
- 117.McGeer PL, Akiyama H, Itagaki S, McGeer EG. Immune system response in Alzheimer's disease. Can J Neurol Sci. 1989;16:516–27. doi: 10.1017/s0317167100029863. [DOI] [PubMed] [Google Scholar]
- 118.Hartwig M. Immune ageing and Alzheimer's disease. NeuroReport. 1995;6:1274–6. doi: 10.1097/00001756-199506090-00011. [DOI] [PubMed] [Google Scholar]
- 119.Togo T, Akiyama H, Iseki E, et al. Occurrence of T cells in the brain of Alzheimer's disease and other neurological diseases. J Neuroimmunol. 2002;124:83–92. doi: 10.1016/s0165-5728(01)00496-9. [DOI] [PubMed] [Google Scholar]
- 120.Town T, Tan J, Flavell RA, Mullan M. T-cells in Alzheimer's disease. Neuromolecular Med. 2005;7:255–64. doi: 10.1385/NMM:7:3:255. [DOI] [PubMed] [Google Scholar]
- 121.Pirttila T, Mattinen S, Frey H. The decrease of CD8-positive lymphocytes in Alzheimer's disease. J Neurol Sci. 1992;107:160–5. doi: 10.1016/0022-510x(92)90284-r. [DOI] [PubMed] [Google Scholar]
- 122.Monsonego A, Zota V, Karni A, et al. Increased T cell reactivity to amyloid β protein in older humans and patients with Alzheimer disease. J Clin Invest. 2003;112:415–22. doi: 10.1172/JCI18104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.McManus R, Higgins S, Mills KHG, Lynch MA. Respiratory infection promotes T cell infiltration and amyloid-β deposition in APP/PS1 mice. Neurobiol Aging. 2014;35:109–21. doi: 10.1016/j.neurobiolaging.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 124.Murooka TT, Rahbar R, Platanias LC, Fish EN. CCL5-mediated T-cell chemotaxis involves the initiation of mRNA translation through mTOR/4E-BP1. Blood. 2008;111:4892–901. doi: 10.1182/blood-2007-11-125039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Agostini C, Facco M, Siviero M, et al. CXC chemokines IP-10 and mig expression and direct migration of pulmonary CD8+/CXCR3+ T cells in the lungs of patients with HIV infection and T-cell alveolitis. Am J Respir Crit Care Med. 2000;162:1466–73. doi: 10.1164/ajrccm.162.4.2003130. [DOI] [PubMed] [Google Scholar]
- 126.Schall TJ, Bacon K, Camp RD, Kaspari JW, Goeddel DV. Human macrophage inflammatory protein α (MIP-1α) and MIP-1β chemokines attract distinct populations of lymphocytes. J Exp Med. 1993;177:1821–6. doi: 10.1084/jem.177.6.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tripathy D, Thirumangalakudi L, Grammas P. Expression of macrophage inflammatory protein 1α is elevated in Alzheimer's vessels and is regulated by oxidative stress. J Alzheimers Dis. 2007;11:447–55. doi: 10.3233/jad-2007-11405. [DOI] [PubMed] [Google Scholar]
- 128.Tripathy D, Thirumangalakudi L, Grammas P. RANTES upregulation in the Alzheimer's disease brain: a possible neuroprotective role. Neurobiol Aging. 2010;31:8–16. doi: 10.1016/j.neurobiolaging.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT. Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer's disease. J Neuroimmunol. 2000;108:227–35. doi: 10.1016/s0165-5728(00)00285-x. [DOI] [PubMed] [Google Scholar]
- 130.Man SM, Ma YR, Shang DS, et al. Peripheral T cells overexpress MIP-1α to enhance its transendothelial migration in Alzheimer's disease. Neurobiol Aging. 2007;28:485–96. doi: 10.1016/j.neurobiolaging.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 131.Liu YJ, Guo DW, Tian L, et al. Peripheral T cells derived from Alzheimer's disease patients overexpress CXCR2 contributing to its transendothelial migration, which is microglial TNF-α-dependent. Neurobiol Aging. 2010;31:175–88. doi: 10.1016/j.neurobiolaging.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 132.Reale M, Iarlori C, Feliciani C, Gambi D. Peripheral chemokine receptors, their ligands, cytokines and Alzheimer's disease. J Alzheimers Dis. 2008;14:147–59. doi: 10.3233/jad-2008-14203. [DOI] [PubMed] [Google Scholar]
- 133.Ryu JK, McLarnon JG. A leaky blood–brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer's disease brain. J Cell Mol Med. 2009;13:2911–25. doi: 10.1111/j.1582-4934.2008.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H, Degen JL, Akassoglou K. The fibrin-derived γ377–395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med. 2007;204:571–82. doi: 10.1084/jem.20061931. [DOI] [PMC free article] [PubMed] [Google Scholar]