

Abstract
Glia and microglia in particular elaborate pro-inflammatory molecules that play key roles in central nervous system (CNS) disorders from neuropathic pain and epilepsy to neurodegenerative diseases. Microglia respond also to pro-inflammatory signals released from other non-neuronal cells, mainly those of immune origin such as mast cells. The latter are found in most tissues, are CNS resident, and traverse the blood–spinal cord and blood–brain barriers when barrier compromise results from CNS pathology. Growing evidence of mast cell–glia communication opens new perspectives for the development of therapies targeting neuroinflammation by differentially modulating activation of non-neuronal cells that normally control neuronal sensitization – both peripherally and centrally. Mast cells and glia possess endogenous homeostatic mechanisms/molecules that can be up-regulated as a result of tissue damage or stimulation of inflammatory responses. Such molecules include the N-acylethanolamine family. One such member, N-palmitoylethanolamine is proposed to have a key role in maintenance of cellular homeostasis in the face of external stressors provoking, for example, inflammation. N-Palmitoylethanolamine has proven efficacious in mast-cell-mediated experimental models of acute and neurogenic inflammation. This review will provide an overview of recent progress relating to the pathobiology of neuroinflammation, the role of microglia, neuroimmune interactions involving mast cells and the possibility that mast cell–microglia cross-talk contributes to the exacerbation of acute symptoms of chronic neurodegenerative disease and accelerates disease progression, as well as promoting pain transmission pathways. We will conclude by considering the therapeutic potential of treating systemic inflammation or blockade of signalling pathways from the periphery to the brain in such settings.
Keywords: mast cells, microglia, neurodegeneration, neuroinflammation, neuroprotection, palmitoylethanolamide
Introduction
Inflammation is fundamentally a protective cellular response aimed at removing injurious stimuli and initiating the healing process. However, when prolonged, inflammation overrides the bounds of physiological control and eventually becomes destructive. Inflammation increasingly surfaces as a key element in the pathobiology of chronic pain, neurodegenerative diseases, stroke, spinal cord injury and perhaps even neuropsychiatric disorders.1–5 A plethora of pro-inflammatory cytokines, eicosanoids and other immune neurotoxins, have been found in cerebrospinal fluid and/or affected brain regions of patients with neurodegenerative disorders.6 Consider also that nuclear factor-κB, a requisite transcription factor for most pro-inflammatory molecules, is activated in the substantia nigra pars compacta of Parkinson's disease patients and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) -intoxicated mice and monkeys, and its selective inhibition protects dopaminergic neurons from MPTP toxicity.7 It is intriguing to note that neuroinflammation may also raise the brain's sensitivity to stress.8 Indeed, a recently published study by Zhang et al.9 reports that inflammation-activated signalling pathways in the brain's hypothalamus control the production of ageing-related hormones. This finding provides a link between inflammation, stress responses and ageing. Inflammation therefore constitutes an important target for neuronal protection in neurodegenerative disorders and neuropathic pain, the latter resulting from damage or disease affecting the somatosensory system.10
Recognition that there is extensive communication between the immune system and the central nervous system (CNS) is, no doubt, one of the more fundamental advances in neuroscience in recent times. Inflammatory cytokines occupy a key niche in this network, regulating host responses to infection, inflammation, stress and trauma. Glial cell activation has been implicated in the pathogenesis of Alzheimer's disease, Parkinson's disease, cerebral ischaemia, multiple sclerosis2,7,11 and motor neuron disease,12 and possibly schizophrenia and depression.4,13 Microglia-mediated neuroinflammatory processes are also proposed to compromise healthy brain aging.14 Found to accumulate at sites of injury or plaques in neurodegenerative CNS diseases,11,15 microglia scavenge dead cells and secrete neuron survival factors; the latter may have beneficial effects in the recovery of injured CNS. However, inappropriate and prolonged activation of glia can cause autoimmune responses leading to brain injury and neuronal cell death.2,11,15 Glia provide a link also between neuroinflammation and neuropathic pain;16 microglia, in particular, show increased activity in multiple pain-processing pathways in response to peripheral injury.17 Systemic inflammation gives rise to signals that communicate with the brain and leads to changes in metabolism and behaviour – including the expression of a pro-inflammatory phenotype by microglia.18 It has been proposed that in multiple chronic disease states, and in ageing, microglia are primed by previous pathology, or by genetic predisposition, to respond more vigorously to subsequent inflammatory stimulation, so transforming an adaptive CNS inflammatory response to systemic inflammation, into one with deleterious consequences.19 It therefore goes without saying that delineating the signalling pathways underlying glial cell activation is crucial in the design of agents capable of antagonizing such signalling steps – which may translate into therapeutic benefit for neurodegenerative disorders and neuropathic pain.
Although it is widely accepted that glial cell activation contributes to neuropathology, one must not forget that microglia and astrocytes also respond to pro-inflammatory signals released from other cells of immune origin. In this view, mast cells represent a potentially important (and underappreciated) peripheral immune signalling link to the brain in an inflammatory setting (Fig. 1). Mast cells share similarities with basophil granulocytes in blood, although the former are likely generated by different precursor cells in the bone marrow.20 In contrast to basophils, mast cells circulate in an immature form until choosing a tissue site to settle – which probably determines their precise characteristics. These effector cells of the innate immune system are present in most tissues in the vicinity of blood vessels, particularly near surfaces exposed to the environment.21 Mast cells participate in innate host defence reactions, occur in peripheral tissues innervated by small calibre sensory nerve fibres and within the endoneurial compartment of peripheral nerves, and in meninges and cerebral blood vessels. During development they enter the brain by way of penetrating blood vessels, with which they remain associated.22 Mast cells can move through the blood–brain barrier of normal brain,23 but may also traverse the blood–spinal cord barrier and blood–brain barrier when compromised by disease. Interestingly, they are capable of phagocytosis and antigen presentation, and can modulate the adaptive immune response (Box 1).
Figure 1.

Electron microscopic image of an isolated tissue mast cell. Note the prominent appearance of numerous cytoplasmic granules.
Mast cells produce a vast array of mediators, which include biogenic amines, cytokines, enzymes, lipid metabolites, ATP, neuropeptides, growth factors and nitric oxide (Table 1).24 Because of their heterogeneity, however, no single mast cell makes all of these. By nature of their immune regulatory role they participate in IgE switching by B cells,25 and the release of chemoattractants that recruit eosinophils26 and monocytes.27 Certain disease states, like those involving autoimmune demyelination are accompanied by an increased absolute number of mast cells within the CNS, as well as those undergoing degranulation.28 Mast cell trypase is elevated in the cerebrospinal fluid of patients with multiple sclerosis.29 At the other end of the equation, activated mast cells can provoke demyelination30 and induce apoptotic oligodendrocyte cell death.31 Brain mast cells might even ‘bridge’ the immune system and anxiety-like behaviour.32 Theoharides et al.33 have suggested that perinatal mast cell activation by infectious, stress-related, environmental or allergic triggers can lead to release of pro-inflammatory and neurotoxic molecules, so contributing to brain inflammation and autism-spectrum disorders pathogenesis, at least in a subgroup of these patients.
Table 1.
Mast cell mediators
| Biogenic amines |
| Biogenic amines [histamine (2–5 pg/cell), serotonin] |
| Cytokines |
| Interleukins 1–6 |
| Leukaemia inhibitory factor |
| Tumour necrosis factor-α |
| Interferon-γ |
| Transforming growth factor-β |
| Granulocyte–macrophage colony-stimulating factor |
| Enzymes |
| Acid hydrolases |
| Chymase |
| Phospholipases |
| Rat mast-cell protease I and II |
| Trypase |
| Lipid metabolites |
| Prostaglandin D2 |
| Leukotriene C4 |
| Platelet-activating factor |
| Other bioactive molecules |
| Neuropeptides (e.g. vasoactive intestinal peptide, substance P) |
| Proteoglycans, mainly heparin (active as an anticoagulant) |
| Nerve growth factor |
| ATP |
| Nitric oxide |
Microglia, mast cells and nervous system disease
Neuropathic pain
Persistent pain represents a substantial and growing unmet medical need, affecting nearly half of people seeking medical care in the USA alone. Among all types of chronic pain, neuropathic pain stands out: this is pain resulting from damage, degeneration or dysfunction of the sensory nervous system, and remains largely untreatable. Central neuropathic pain is found in spinal cord injury, multiple sclerosis and some strokes, whereas the common causes of painful peripheral neuropathies are diabetes and other metabolic conditions. Neuropathic pain is common in cancer as a direct result of cancer on peripheral nerves or as a side effect of chemotherapy. The triggering and maintenance of neuropathic pain states is strongly dependent on Schwann cells, spinal microglia and astrocytes, together with elements of the peripheral immune system.34 Release of interleukin-1β (IL-1β) from spinal microglia as a consequence of inflammation/injury may, by engaging its receptor, induce phosphorylation of the N-methyl-d-aspartate receptor NR1 subunit to strengthen painful signal transmission.35 Under pathological conditions dorsal horn microglia become activated and show up-regulated expression of purinergic receptors,36,37 whose inhibition or deletion strongly attenuates neuropathic pain.36–38
Upon degranulation, mast cells release algogenic (from the Greek ‘algos’ for pain) substances which activate or sensitize nociceptors, thereby contributing directly to neuropathic pain.39 Nerve-resident peripheral nerve mast cells represent the first line of activation at the point of damage and facilitate recruitment of neutrophils and macrophages.40 Mast cell degranulation activates trigemino-cervical and lumbosacral pain pathways and elicits widespread tactile pain hypersensitivity,41 possibly mediated by a sensitizing effect of histamine on nociceptors. Rapid release of nerve growth factor from mast cells also produces sensitization of nociceptors via the latter's high-affinity nerve growth factor-trkA receptors (and indirectly via other peripheral cell types).42 Interestingly, mast cells themselves respond to nerve growth factor, in a paracrine/autocrine fashion.43 These events promote the recruitment of T cells, which reinforce and maintain inflammatory reactions. The released mediators/factors can induce activity in axons and/or undergo retrograde transport to the cell body of dorsal root ganglion neurons, thereby affecting gene expression. Further, mast cells may enhance recruitment of other key immune cell types which, in turn, release pro-nociceptive mediators, such as IL-6.44 Systemic glucocorticoid therapy reduces pain and the number of tumour necrosis factor-α (TNF-α) -positive mast cells in rats with chronic constrictive injury, strengthening a role for mast cells in chronic pain states.45 Mast cells are also important mediators of chronic visceral pain.46
Ischaemia and traumatic brain injury
Stroke and traumatic brain injury are characterized by an inflammatory response in which microglia activation and macrophage/neutrophil infiltration are important elements.47 Left unchecked, this can ultimately lead to secondary injury. Yet, in certain instances attenuation of microglial activation can be beneficial.48 Considerable efforts have been directed also to inhibiting the consequences of blood-borne neutrophil and phagocyte infiltration in ischaemia. In contrast, less emphasis has been placed on brain-resident cell types able to mount an immediate host response in the cerebral parenchyma and meninges, for example, mast cells.49 Analogous to peripheral nerve damage – and contrary to what was long believed50 – mast cell activation is the ‘first responder’ in this injury.51 Although CNS microglia/macrophages and endothelial cells produce TNF-α in response to stimuli, mast cells are ‘primed’ to initiate acute inflammation with their stores of pre-formed TNF-α.52 Treatment with mast cell-stabilizing agents limits the brain damage caused by perinatal hypoxia–ischaemia and transient focal ischaemia.51,53,54 Given their complement of vasoactive and matrix-degrading components like histamine, and proteases able to activate matrix metalloproteinases, mast cells are an early response element in the regulation of acute blood–brain barrier changes after cerebral ischaemia and haemorrhage.55 By regulating acute microvascular gelatinase activation, cerebral mast cells can effect blood–brain barrier disruption following transient cerebral ischaemia.56
Chronic neurodegenerative diseases
Microglia are activated in response to a number of different pathological states within the CNS including neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, multiple sclerosis, amyotrophic lateral sclerosis (ALS), and AIDS dementia complex. There is a vast literature on this facet of neuroinflammation that is beyond the scope of this article. The reader is referred to several excellent recent reviews.57,58 In the case of Parkinson's disease, the oxidative stress response by microglial cells, most notably the activity of the enzyme NADPH oxidase, appears to play a central role in the ensuing pathology.59
The case for mast cells, while somewhat analogous, depends to a greater extent on the specific disease. In patients with multiple sclerosis, especially in chronic active plaques, mast cells have been noted generally clustered around venules and capillaries.60 Mast cell-associated gene transcripts (e.g. tryptase) are also present in multiple sclerosis plaques, whereas mast cell mediators such as tryptase and histamine have been found in cerebrospinal fluid of multiple sclerosis patients, with levels rising during relapses.29,61 In mouse experimental autoimmune encephalomyelitis models of multiple sclerosis, however, there is no clear evidence whether the mast cell effects on experimental autoimmune encephalomyelitis development depend on mouse strain, immunization protocol or type and severity of disease.62 A role for mast cells in Alzheimer's disease pathogenesis remains to be established, although one report claimed that fibrillar amyloid β-peptides trigger CD47-dependent mast cell secretory and phagocytic responses.63 For Parkinson's disease, there is no clear evidence to support a relationship between brain mast cells and Parkinson's disease. As this concerns mast cells and motor neuron disease, this point will be discussed separately below.
Mast cells and ALS: at the crossroads?
Amyotrophic lateral sclerosis, also known as Charcot's or Lou Gehrig's disease, is the most prevalent type of motor neuron disease, affecting around 4–6 per 100 000. ALS is characterized by a progressive dysfunction and degeneration of both upper motor neurons comprising the corticospinal tract, and lower motor neurons arising from the brainstem nuclei and ventral roots of the spinal cord. Neuroinflammation is now established as an important aspect of pathology in ALS.64 There is a marked activation or proliferation of both microglia and astrocytes at specific disease stages in mouse models of ALS65 and in humans in vivo.66 There is also compelling evidence indicating impairment of all neurovascular unit components including the blood–brain and blood–spinal cord barriers in both patients and animal models of ALS.67
As discussed earlier, mast cells participate in innate host defence reactions, and orchestrate neuroinflammatory processes. Meningeal mast cells contain preformed TNF-α (the only cell type to do so) and vasoactive mediators, which are able to regulate blood–brain barrier56 and blood–spinal cord barrier68 function and facilitate the entry of lymphocytes, neutrophils and mast cells themselves69 when the barrier is compromised as a result of CNS pathology, e.g. in ischaemic stroke and ALS, respectively. Indeed, degranulating70 and IL-17-expressing mast cells68 have been observed in the spinal cord of ALS patients, and serum levels of this cytokine are reported to correlate with the duration and severity of the disease.68 An interaction between mast cells and T regulatory lymphocytes can lead to increased IL-6 production by the former,71 in turn promoting the activity of T helper type 17 lymphocytes.72 Moreover, IL-15 and IL-12 are elevated in the serum and cerebrospinal fluid of patients with ALS.73 Interleukin-15 is a mast cell chemoattractant74 (potentially contributing to mast cell recruitment in inflammatory responses), while mast cells are an important source of IL-12.75 Pathogen-associated molecular patterns (PAMPs) are molecules associated with groups of pathogens that are recognized by cells of the innate immune system. One important class of PAMP receptors is the transmembrane Toll-like receptors (TLRs). Interleukin-12 up-regulates expression of mast cell TLR2/TLR476 and proteinase-activated receptor-2,77 emerging targets for neuroinflammation.78
Functional TLR2 and TLR4 expressed by mast cells and microglia respond to molecules called damage-associated molecular patterns, one being the high mobility group Box 1 protein found elevated in the spinal cord of ALS patients.79 Likewise, microglia-released IL-6 and CCL5 could, conceivably affect surface TLR2 and TLR4 expression on mast cells,80 resulting in the up-regulation of numerous chemokines to induce a pro-inflammatory profile in microglia.81 Given that ALS patients frequently experience neuropathic pain, and that a transgenic mouse ALS model develops peripheral nerve inflammation,82 one might ask whether or not mast cells play a role here, as well.
Box 1. Mast cell ID chart
Origin and classification
First described by Paul Ehrlich in 1878 on the basis of their unique staining characteristics and large cytoplasmic granules
Very close to basophil granulocytes in blood; current evidence suggests that mast cells are generated by different precursor cells in the bone marrow
Thought to originate from bone marrow precursors expressing CD34; a distinct subset of mast cells can also be induced upon host responses to inflammation
The haematopoietic lineage development of tissue mast cells is unique compared with other myeloid-derived cells because it is early lineage progenitors, undetectable by histochemistry, that leave the bone marrow to enter the circulation. These immature lineage mast cells immediately undergo transendothelial recruitment into peripheral tissues wherein the appearance of secretory granules with a particular protease phenotype is regulated by the peripheral tissue.
Classified by their species-dependent protease constitution rather than location
Present in most tissues in the vicinity of blood vessels, and are especially prominent near the boundaries between the body's external environment and the internal milieu, such as the skin, mucosa of the lungs and digestive tract, as well as in the mouth, conjunctiva and nose
Mast cells are also found within the nervous system, including meninges, brain parenchyma and nerves.
Physiology
Key role in innate and acquired immunity
Upon activation rapidly release granules into the interstitium
Degranulation is caused by direct injury (e.g. physical or chemical), cross-linking of IgE receptors or by activated complement proteins
Capable of elaborating a vast array of important cytokines and other inflammatory mediators
Express multiple ‘pattern recognition receptors’ thought to be involved in recognizing broad classes of pathogens
Granules carry a variety of bioactive chemicals, proteoglycans, serine proteases, neuropeptides; can be transferred to adjacent cells of the immune system and neurones via transgranulation and their pseudopodia
Role in disease
Allergic reactions
Anaphylactic shock
Neuropathic and inflammatory pain
Acute and chronic neurodegenerative disorders
Microglia and mast cells: the other side of the coin
Activated microglia elaborate a potentially lethal cocktail of compounds capable of damaging neurons, oligodendrocytes or extracellular matrix molecules, and depletion or blockade of microglia and macrophages prevents disease progression.83 Microglia/macrophages, however, may deliver trophic factors,84 and support myelin regeneration by phagocytic removal of obstructive myelin debris85 or through activation and recruitment of endogenous oligodendrocyte precursor cells to the lesion site.86 Microglia are found adjacent to amyloid deposits, and anti-inflammatory drugs that suppress the inflammatory response in microglia attenuate symptoms in a mouse model of Alzheimer's disease.87 Yet, in one study reducing or ablating resident microglia failed to alter amyloid plaque load in transgenic mouse models of Alzheimer's disease.88 Further, deleting the microglial chemokine receptor CCR2 (which mediates the accumulation of mononuclear phagocytes at sites of inflammation) accelerated early disease progression and impaired microglial accumulation in an Alzheimer's disease mouse model.89 Microglia activation via TLR4 signalling appears able to reduce, to some extent, Aβ deposits and preserve cognitive functions from Aβ-mediated neurotoxicity.90 However, prolonged activation of microglia is likely to result in a pro-inflammatory state. This idea is borne out in a newly published study in which the authors used monophosphoryl lipid A (MPL), a lipopolysaccharide-derived TLR4 agonist that exhibits unique immunomodulatory properties at doses that are non-pyrogenic.91 Repeated systemic injections of MPL, but not lipopolysaccharide, significantly improved Alzheimer's disease-related pathology in a transgenic mouse model. Treatment with MPL led to a significant reduction in Aβ load in the brains of these mice, as well as to enhanced cognitive function. MPL induced a potent phagocytic response by microglia while triggering only a moderate inflammatory reaction.114
As discussed earlier, acute CNS injuries are marked by a prolonged inflammatory response involving microglial activation and infiltration of macrophages and neutrophils. However, the fact that microglia accumulation at the lesion site and penumbra hints at a possible neuroprotective role. Indeed, genetic ablation of microglial cells results in a larger infarct after transient middle cerebral artery occlusion,92 while microglia injected into the circulation of Mongolian gerbils home to an ischaemic hippocampal lesion (facilitated by ischaemia-induced blood–brain barrier compromise) and improve neuron survival.93 Further, microglia may protect hippocampal neurons from excitotoxicity.94 Resting (ramified) microglia respond to, and repair, subclinical abnormalities of the brain without a complete activation transformation,48 and are probably also key players in developmental synaptic pruning (discussed in more detail elsewhere95).
Potential beneficial actions of mast cells should not be overlooked. Mast cells of human origin express and store angiogenin (a member of the ribonuclease A superfamily) within their granules, which is released upon stimulation by FcεRI-mediated signals, TLR ligands and nerve growth factor.96 Nerve growth factor-stimulated human mast cells release greater amounts of angiogenin compared with FcεRI cross-linking. Human angiogenin is reported to be neuroprotective and to promote the survival and neuritogenesis of motor neurons,97 and recent studies associating angiogenin gene mutations with ALS98 suggest a possible disease link. Mast cells are a source of serotonin in the hippocampus, which can contribute to behavioural and physiological functions in the hippocampus.99 Outside the nervous system, mast cells may contribute to wound healing.100
A mast cell–glia dialogue
Given their frequent proximity at sites of neuroinflammation, and the potential for peripheral inflammation to influence supraspinal behaviours, it is conceivable that lines of communication exist between these two cell types. There is mounting evidence for this, which will be briefly summarized here (see ref. 95 for a more detailed discussion). There is growing interest in TLR signalling pathways in neurodegenerative disorders and neuropathic pain, and especially TLR2 and TLR4. Engagement of TLR2/TLR4 on mast cells leads to release of cytokines that recruit immune cells to the sites of injury. Likewise, microglial recruitment depends on signalling pathways involving TLR2/TLR4. Mast cell activation will up-regulate chemokine expression, including CCL5/RANTES, which induces a pro-inflammatory profile in microglia. Microglia-released IL-6 and CCL5 could, in turn affect mast cell expression of TLR2/TLR4. ATP, released from damaged cells/tissues, is a potent stimulus for microglia in vitro. ATP may act as an autocrine/paracrine factor for mast cells, and ATP released from one mast cell (e.g. FcεR1 cross-linking, stress) can diffuse across several hundreds of micrometres to elicit a rise in Ca2+ in neighbouring cells.101 ATP binding to P2 receptors may stimulate release of IL-33 from microglia pre-activated (‘primed’) with PAMPs via TLRs.102 Interleukin-33 can induce mast cells to secrete IL-6, IL-13 and CCL2 – in turn modulating microglia activity. Other examples include mast cell tryptase cleavage/activation of proteinase-activated receptor 2 on microglia (resulting in P2X4 receptor up-regulation and brain-derived neurotrophic factor release),103 while microglia-derived IL-6 and TNF-α up-regulate mast cell expression of proteinase-activated receptor 2, resulting in mast cell activation and TNF-α release.104,105 Elements of the complement system are also potential participants in this communication network, as the receptor for the chemoattractant C5a is markedly up-regulated on reactive astrocytes and microglia in inflamed CNS tissue,106 C5a peptide is released in neuroinflammation,107 and there is cross-talk between C5a and TLR4; mast cell C5a receptor is up-regulated upon activation, and C5a receptor is a strong mast cell chemoattractant signal towards C5a peptide. These last findings point to an additional element whereby microglia and mast cells may work in concert to promote neuroinflammation (Table 2).
Table 2.
Potential avenues of mast cell–glia communication
| Biological actions |
|||
|---|---|---|---|
| Effector | Microglia/astrocytes | Mast cells | References |
| TLR2, TLR4 | Microglial release of IL-6 and CCL5 affects surface expression of TLR2/TLR4 on mast cells | Up-regulation of cytokine/chemokine release; CCL5/RANTES induces pro-inflammatory profile in microglia; recruitment of immune cells to site of injury | 108–111,81,112–114 |
| ATP/P2 receptors | ATP stimulates IL-33 release from microglia pre-activated with PAMPs via TLRs | IL-33 binds to its receptor on mast cells and induces secretion of IL-6, IL-13 and monocyte chemoattractant protein 1 which, in turn could modulate microglia activity | 102,115,101 |
| Proteinase-activated receptor 2 (PAR2) | Mast cell tryptase cleaves/activates PAR2 on microglia, resulting in P2X4 receptor up-regulation and BDNF release | IL-6 and TNF-α from microglia can up-regulate mast cell expression of PAR2, which can result in mast cell activation and TNF-α release | 104,105,101,116 |
| CXCR4/CXCL12 | Promotes microglia migration and activation; CXCR4/CXCL12 are both up-regulated in hypoxia/ischaemia | CXCR4 acts as mast cell chemotaxin | 117–120 |
| C5a receptor (C5aR) | C5aR up-regulated upon microglia activation; C5a peptide released in neuroinflammtion; cross-talk between C5a and TLR4 | C5aR up-regulated upon activation; a strong mast cell chemoattractant signal towards C5a peptide; cross-talk between C5a and TLR4 | 106,107,121 |
| CD40/CD40L | Astrocytes display enhanced expression of CD40 – cross-talk with CD40L leads to production of inflammatory cytokines; astrocyte-derived cytokines/chemokines trigger mast cell degranulation | Display enhanced surface expression of CD40L respectively – cross-talk with CD40 leads to production of inflammatory cytokines | 122,123 |
BDNF, brain-derived neurotrophic factor; IL, interleukin; PAMPs, pathogen-associate molecular patterns; PAR, proteinase-activated receptor; TLR, Toll-like receptor; TNF-α, tumour necrosis factor-α. [Modified from Skaper et al.95 (Table 1), Copyright (2012), with permission from the Federation of American Societies for Experimental Biology].
Taping endogenous mechanisms as a therapeutic approach to neuroinflammation
Targeting microglial and mast cell activation is emerging as a promising avenue for neuropathic pain,124 as well as agents that inhibit neurotoxic glial cell activation.125 Given the dangers that neuroinflammation poses to the organism, it would not be surprising if Nature has endowed the body with the capacity for self-defence. Indeed, we now recognize the existence of molecules involved in endogenous protective mechanisms that are activated following different types of tissue damage or stimulation of inflammatory responses and nociceptive fibres. For example, chronic inflammatory processes such as those sustaining neuropathic pain may be counteracted by a programme of resolution that includes the production of lipid mediators able to switch off inflammation.126 Chronic inflammatory conditions may lower the levels or actions of these molecules;127 conceivably, administration of such lipid mediators might provide an avenue ‘to commandeer nature's own anti-inflammatory mechanisms and induce a “dominant” program of resolution’.128 One interesting class of such natural mediators comprises the N-acylethanolamines (NAEs), which are composed of a fatty acid and ethanolamine – the so-called fatty acid ethanolamines. Principal fatty acid ethanolamine family members include the endocannabinoid N-arachidonoylethanolamine (anandamide) and its congeners N-stearoylethanolamine, N-oleoylethanolamine and N-palmitoylethanolamine (PEA, or palmitoylethanolamide).129 PEA and its congeners are formed from N-acylated phosphatidylethanolamine (NAPE) by several enzymatic pathways,130 the principal one involving a membrane-associated NAPE-phospholipase D which generates the respective NAE and phosphatidic acid (Fig. 2).131 This enzyme converts N-palmitoyl-phosphatidyl-ethanolamine into PEA. In the mammalian brain, NAEs are hydrolysed by: (i) fatty acid amide hydrolase in the endoplasmic reticulum, which breaks down NAEs into the corresponding fatty acid and ethanolamine;132 (ii) lysosomal NAE-hydrolysing acid amidase (NAAA) (Fig. 2).133 NAAA is found mainly in macrophages, where it hydrolyses NAEs with less than 18 carbon atoms, i.e. PEA, but not N-oleoylethanolamine and N-stearoylethanolamine. In contrast, fatty acid amide hydrolase hydrolyses all three NAEs.
Figure 2.
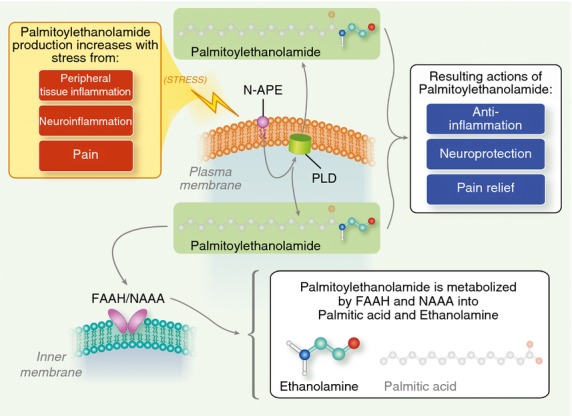
Palmitoylethanolamide synthesis and catabolism. A plasma membrane-associated N-acylated phosphatidylethanolamine-phospholipase D (PLD) converts N-palmitoyl-phosphatidyl-ethanolamine (N-APE) into palmitoylethanolamide and phosphatidic acid. Palmitoy-lethanolamide is metabolized to palmitic acid and ethanolamine by both fatty acid amide hydrolase (FAAH, which also breaks down other fatty acid amides) and the more selective N-acyl ethanolamine-hydrolysing acid amidase (NAAA). Tissue levels of palmitoylethanolamide increase in stressful settings such as peripheral tissue inflammation, neuroinflammation and pain. See text for further details.
Molecules like PEA may, conceivably, function to maintain cellular homeostasis when faced with external stressors provoking, for example, inflammation.134 In fact, PEA is produced/hydrolysed by microglia and mast cells,135,136 it down-modulates mast cell activation,137 and controls microglial cell behaviours.138,139 Tissue levels of PEA are elevated in brain areas involved in nociception and in spinal cord following neuropathic pain induction,140 as well as conditions associated with pain development138,141 and in the interstitium of the trapezius muscle of women with chronic widespread pain and chronic neck–shoulder pain (Table 3).142 The above observations suggest that PEA maintains cellular homeostasis by acting as mediator of resolution of inflammatory processes (Fig. 3). A corollary to this idea is that pathological situations may arise in which endogenous PEA levels are inadequate to deal with the ensuing insult. In these cases, exogenous administration to effectively ‘top up’ the body's own supply may be a therapeutically viable approach. Indeed, a growing number of studies demonstrate the validity of such; these are briefly summarized in Table 4. A more detailed discussion is outside the scope of the review, and will be covered elsewhere (Skaper S.D., unpublished data).
Table 3.
Changes in palmitoylethanolamide levels during neuroinflammation
| Disease; tissue or body fluid | Change | Main finding | References |
|---|---|---|---|
| Chronic relapsing experimental allergic encephalomyelitis; spinal cord | ↑ | ∼Twofold increase | 143 |
| Experimental acute stroke; striatal and cortical infarcted hemisphere | ↑ | ∼25-fold increase compared with controlateral (non-infarcted) areas | 144 |
| Experimental focal cerebral ischaemia; ischaemic cerebral cortex | ↑ | 25-fold increase compared with sham-operated animals, at 24 hr post-focal cerebral ischaemia | 138 |
| Cerebral ischaemia in man; penumbral tissue surrounding the primary ischaemic lesion (microdialysis study) | ↑ | Significantly increased levels within the first day after ischaemia | 145 |
| Chronic migraine or probable chronic migraine and probable analgesic-overuse headache; cerebrospinal fluid | ↑ | Significantly higher levels in the two patient groups (without significant difference between them) compared with control subjects | 146 |
| Chronic widespread pain and chronic neck–shoulder pain in women; microdialysis dialysate of the trapezius | ↑ | Significantly higher levels compared to healthy subjects, and correlation with pain intensity | 142 |
Figure 3.
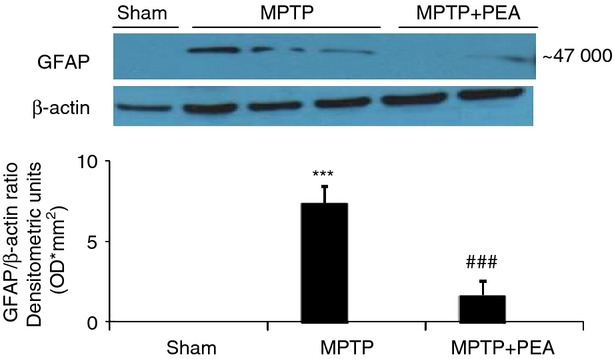
Palmitoylethanolamide (PEA) treatment reduces activation of astrocytes induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) injection. Western blot analysis showed a significant expression of glial fibrillary acidic protein (GFAP) in the substantia nigra. PEA treatment significantly reduced the activation of astrocytes after MPTP injection. The relative expression of the protein band (∼47 000 molecular weight) was standardized for densitometric analysis to β-actin levels, and reported as mean ± SEM from n = 5 or n = 6 brains for each group. ***P < 0·001 versus sham, ###P < 0·001 versus MPTP + vehicle. Modified from Esposito et al.150(Figure 10, © Esposito et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License).
Table 4.
Preclinical studies showing anti-neuroinflammatory and/or neuroprotective effects of palmitoylethanolamide
| Model | Action | References |
|---|---|---|
| Compression model of spinal cord trauma in mice | Reduces spinal inflammation/tissue injury, ameliorates recovery of motor limb function Limits mast cell infiltration and activation; reduces activation of microglia and astrocytes |
147,148 |
| Traumatic brain injury in mice | Reduces edema and infarct size Improves neurobehavioural functions |
149 |
| MPTP mouse model of Parkinson's disease | Protects against MPTP-induced neurotoxicity, microglial and astrocyte activation, and functional deficits | 150 |
| Stroke (middle cerebral artery occlusion in rats) | Reduces oedema and infarct size Improves neurobehavioural functions |
151 |
| β-Amyloid peptide injection in rat brain | Counteracts reactive gliosis Reduces behaviour impairments |
152 |
| Chronic constriction injury in sciatic nerve | Anti-allodynic and anti-hyperalgesic effects Reduces mast cell activation Preservation of nerve structural integrity |
153–155 |
| Acute inflammation (formalin, dextran, carrageenan injection in rat hindpaw) | Reduces mast cell activation, tissue oedema, inflammatory/mechanical hyperalgesia | 156–161 |
| WAG/Rij rat model of absence epilepsy | Anti-epileptic action | 162 |
MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
As NAAA preferentially hydrolyses PEA, inhibition of its degradation represents a complementary therapeutic approach to treat inflammation. A number of selective NAAA inhibitors have been described to date;163–165 these dampen responses induced by inflammatory stimuli in vivo and in vitro, and elevate PEA levels in vitro as well.163 The most recently identified compound, 1-(2-biphenyl-4-yl)ethyl-carbonyl pyrrolidine, is a reversible and competitive NAAA inhibitor: it reduces mRNA expression levels of inducible nitric oxide synthase and IL-6 and increases intracellular PEA levels in mouse macrophages upon lipopolysaccharide-induced inflammation.166
The numerous preclinical studies have encouraged clinical trials of PEA, mainly in the area of chronic and neuropathic pain. PEA reportedly improved myelinated-fibre function in patients with chemotherapy-induced painful neuropathy,167 and reduced neuropathic pain in a patient with multiple sclerosis.168 A recently presented case series described the application and potential efficacy and safety of micronized and ultra-micronized PEA (formulations designed to improve bioavailability) in the treatment of various syndromes associated with chronic pain that is poorly responsive to standard therapies.169 Some 40 or so clinical trials have been conducted to date, with a patient base exceeding 2000.170 There has also been a case study reporting on the effects of ultramicronized PEA in sporadic ALS, in which treatment led to an improved clinical picture, as evidenced by electromyographic analysis and pulmonary function.171 A more detailed description of PEA clinical trials will be discussed elsewhere (Skaper S.D., unpublished data). Importantly, PEA has no adverse effects at pharmacologically relevant doses.
What is the molecular basis underlying the effects of PEA? As suggested in a number of studies, PEA may be a ligand for peroxisome proliferator activated receptor α (PPARα), one of a group of nuclear receptor proteins that function as transcription factors regulating the expression of genes. The α- and γ-isoforms of PPAR in particular are associated with pro-inflammatory events. PEA actions (anti-inflammatory,172 anti-nociceptive/anti-neuropathic155,161,173 and neuroprotective150,154,174) were either absent in PPARα null mice or blocked by PPARα antagonists. An ‘entourage effect’ has also been hypothesized to explain the pharmacological actions of PEA, whereby PEA enhances the anti-inflammatory and anti-nociceptive activity of other endogenous compounds by potentiating their affinity for a receptor or by inhibiting their metabolic degradation.175 Anandamide is a candidate molecule, as it possesses anti-inflammatory and anti-nociceptive effects. Anandamide and its congeners like PEA have in common the transient receptor potential vanilloid type 1 (TRPV1) receptor. The TRPV1 receptor, a non-selective cation channel expressed in small diameter sensory neurons, is activated by noxious heat, low pH and capsaicin. Anandamide itself is a TRPV1 receptor agonist, and PEA enhances anandamide stimulation of the human TRPV1 receptor176 in a cannabinoid CB2 receptor antagonist-sensitive fashion (although PEA shows no appreciable affinity for either CB1 or CB2 receptors) – which could be interpreted as PEA acting indirectly by potentiating anandamide actions.157 Mast cells177 and microglia178 reportedly express TRPV1 receptors.
Concluding remarks
Neuroinflammatory disorders are conditions where immune responses damage components of the nervous system (Karolinska Institute, 2013; http://www.ki.se). Inflammatory effectors derive from the innate and adaptive immune systems, as well as glia within the CNS. Microglia, in particular, act as sensors for disturbed brain tissue homeostasis and accumulate locally in response to neuronal cell injury or entry of foreign material in the brain.179 Few studies until now have been directed to resident brain cell types capable of mounting immediate host responses in the brain and meninges. Mast cells are effector cells of the innate immune system, and represent the ‘first responders’ to injury rather than microglia.51 Mast degranulation/mediator release is very rapid, while longer lasting activation elaborates de novo formed mediators. Mast cell degranulation does not result in cell demise; rather, mast cells are stable, multiple-use cells capable of surviving and delivering several consecutive hits.180
Research to-date has largely focused on detrimental effects of neuroinflammation in association with psychiatric and neurodegenerative diseases, as well as neuropathic pain. Yet, we know little of glial and mast cell changes in human chronic pain – unequivocal demonstration that glial and mast cell activation occurs in hypersensitized patients remains an important gap. We lack also systematic studies that provide a correlation between the magnitude of glial and/or mast cell markers in cerebrospinal fluid or spinal tissue and the intensity of pain in patients.
Today's armamentarium to combat neuropathic pain (antidepressants, anticonvulsants, sodium channel blockers, glutamate receptor antagonists, opioids) treats the symptoms but not the underlying pathophysiology. Further, they provide at best transient relief in only a fraction of neuropathic pain patients and can produce serious CNS side effects. Agents like cromolyn, which stabilize mast cells, suppress development of hyperalgesia but do not affect microglia. Microglial inhibitors (e.g. minocycline) commonly used in pain models rely on their anti-inflammatory properties but may suffer from issues such as non-selectivity in targeting one cell population, as well as the risk of acute or cumulative toxicity. Incomplete understanding of mechanisms underlying the induction and maintenance of neuropathic pain has hindered more effective treatments, including elucidating the role of mast cells and how they might interact with microglia.
Clearly, much remains to be learned about signalling mechanisms that regulate neuroinflammation. One may envision the CNS neuron being subjected to ‘assault’ by a microglia–astrocyte–mast cell network, as a result of inadequate regulation of these non-neuronal cells because of excessive and/or persistent exogenous and/or endogenous stimuli, and/or an inadequate cellular inhibitory capacity (Fig. 4). Targeting endogenous regulators of neuroinflammation may therefore prove to be a viable strategy for affecting a diverse array of nervous system disorders. Within this context, the capacity of PEA to modulate the protective responses of animals during inflammation and pain led to the hypothesis that endogenous PEA may be a component of the complex homeostatic system controlling the basal threshold of both inflammation and pain. The production of PEA during inflammatory conditions supports this role, and emerging data that selective inhibition of PEA degradation is anti-inflammatory provides more direct evidence for the involvement of PEA in the control of pain and inflammation. It also leads one to ponder if we are not missing important therapeutic avenues by studying glia and mast cells in isolation from each other. Future studies should investigate the role of mast cells in inflammatory diseases as a network, which requires a critical examination of specific tissue localization, function and dynamic interaction with endogenous cells.
Figure 4.
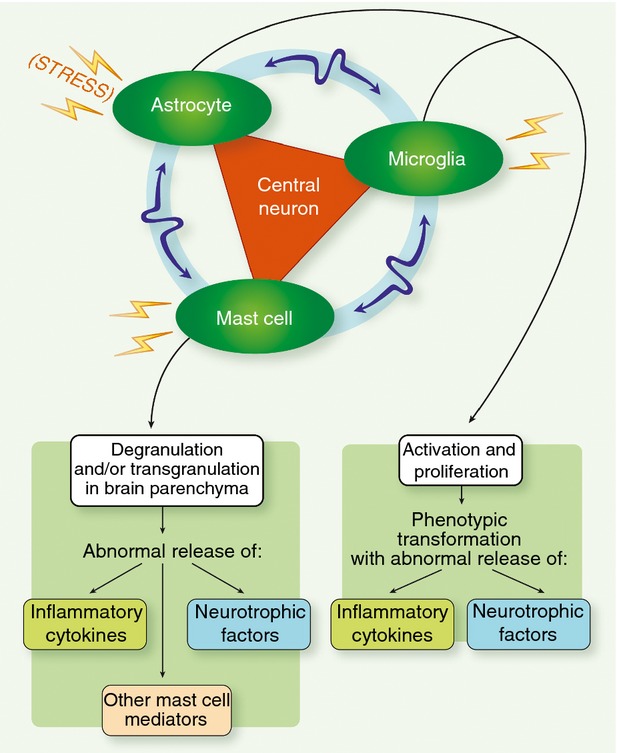
Neuroinflammation may result from central nervous system neurons being impinged upon by a microglia–astrocyte–mast cell network, as a result of inadequate regulation of these non-neuronal cells due to excessive and/or persistent exogenous and/or endogenous stimuli, and/or an inadequate cellular inhibitory capacity. Bidirectional communication may take place between microglia, mast cells and astrocytes, which can act to reinforce the deleterious signals acting on the neuron.
Disclosures
The authors declare no conflicts of interest.
Glossary
- ALS
amyotrophic lateral sclerosis
- CNS
central nervous system
- FAAH
fatty acid amide hydrolase
- IL-1β
interleukin-1β
- MPL
monophosphoryl lipid A
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NAAA
lysosomal NAE-hydrolysing acid amidase
- NAE
N-acylethanolamine
- NAPE
N-acylated phosphatidylethanolamine
- PAMP
pathogen-associated molecular patterns
- PEA
palmitoylethanolamide
- PPAR
peroxisome proliferator activated receptor α
- TLR
Toll-like receptor
- TNF-α
tumour necrosis factor-α
References
- 1.Giovannini MG, Scali C, Prosperi C, Bellucci A, Vannucchi MG, Rosi S, Pepeu G, Casamenti F. β-Amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo. Involvement of the p38MAPK pathway. Neurobiol Dis. 2002;11:257–74. doi: 10.1006/nbdi.2002.0538. [DOI] [PubMed] [Google Scholar]
- 2.Dauer W, Przedborski S. Parkinson disease. Mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 3.Tenorio G, Kulkarni A, Kerr BJ. Resident glial cell activation in response to perispinal inflammation leads to acute changes in nociceptive sensitivity: implications for the generation of neuropathic pain. Pain. 2013;154:71–81. doi: 10.1016/j.pain.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Najjar S, Pearlman DM, Alper K, Najjar A, Devinsky O. Neuroinflammation and psychiatric illness. J Neuroinflammation. 2013;10:43. doi: 10.1186/1742-2094-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagatsu T, Mogi M, Ichinose H, Togari A. Changes in cytokines and neurotrophins in Parkinson disease. J Neural Transm. 2000;60:277–90. doi: 10.1007/978-3-7091-6301-6_19. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh A, Roy A, Liu X, et al. Selective inhibition of NF-κB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson disease. Proc Natl Acad Sci USA. 2007;104:18754–9. doi: 10.1073/pnas.0704908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rivat C, Becker C, Blugeot A, Zeau B, Mauborgne A, Pohl M, Benoliel JJ. Chronic stress induces transient spinal neuroinflammation, triggering sensory hypersensitivity and long-lasting anxiety-induced hyperalgesia. Pain. 2010;150:358–68. doi: 10.1016/j.pain.2010.05.031. [DOI] [PubMed] [Google Scholar]
- 9.Zhang G, Li J, Purkayastha S, et al. Hypothalamic programming of systemic ageing involving IKK-β. NF-κB and GnRH. Nature. 2013;497:211–6. doi: 10.1038/nature12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jensen TS, Baron R, Haanpää M, Kalso E, Loeser JD, Rice AS, Treede RD. A new definition of neuropathic pain. Pain. 2011;152:2204–5. doi: 10.1016/j.pain.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 11.González-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–40. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 12.Appel SH, Zhao W, Beers DR, Henkel JS. The microglial–motoneuron dialogue in ALS. Acta Myol. 2011;30:4–8. [PMC free article] [PubMed] [Google Scholar]
- 13.Hinwood M, Morandini J, Day TA, Walker FR. Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb Cortex. 2012;22:1442–54. doi: 10.1093/cercor/bhr229. [DOI] [PubMed] [Google Scholar]
- 14.Rosano C, Marsland AL, Gianaros PJ. Maintaining brain health by monitoring inflammatory processes: a mechanism to promote successful aging. Aging Dis. 2012;3:16–33. [PMC free article] [PubMed] [Google Scholar]
- 15.Carson MJ. Microglia as liaisons between the immune and central nervous systems. Functional implications for multiple sclerosis. Glia. 2002;40:218–31. doi: 10.1002/glia.10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thacker MA, Clark AK, Marchand F, McMahon SB. Pathophysiology of peripheral neuropathic pain: immune cells and molecules. Anesth Analg. 2007;105:838–47. doi: 10.1213/01.ane.0000275190.42912.37. [DOI] [PubMed] [Google Scholar]
- 17.Gao YJ, Ji RR. Chemokines, neuronal–glial interactions, and central processing of neuropathic pain. Pharmacol Ther. 2010;126:56–68. doi: 10.1016/j.pharmthera.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engler H, Doenlen R, Engler A, et al. Acute amygdaloid response to systemic inflammation. Brain Behav Immun. 2011;25:1384–92. doi: 10.1016/j.bbi.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 19.Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61:71–90. doi: 10.1002/glia.22350. [DOI] [PubMed] [Google Scholar]
- 20.Chen CC, Grimbaldeston MA, Tsai M, Weissman IL, Galli SJ. Identification of mast cell progenitors in adult mice. Proc Natl Acad Sci USA. 2005;102:11408–13. doi: 10.1073/pnas.0504197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilfillan AM, Austin SJ, Metcalfe DD. Mast cell biology: introduction and overview. Adv Exp Med Biol. 2011;716:2–12. doi: 10.1007/978-1-4419-9533-9_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lambracht-Hall M, Dimitriadou V, Theoharides TC. Migration of mast cells in the developing rat brain. Dev Brain Res. 1990;56:151–9. doi: 10.1016/0165-3806(90)90077-c. [DOI] [PubMed] [Google Scholar]
- 23.Silverman AJ, Sutherland AK, Wilhelm M, Silver R. Mast cells migrate from blood to brain. J Neurosci. 2000;20:401–8. doi: 10.1523/JNEUROSCI.20-01-00401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson D, Krenger W. Interactions of mast cells with the nervous system – recent advances. Neurochem Res. 1992;17:939–51. doi: 10.1007/BF00993271. [DOI] [PubMed] [Google Scholar]
- 25.Gauchat JF, Henchoz S, Mazzei G, et al. Induction of human IgE synthesis in B cells by mast cells and basophils. Nature. 1993;365:340–3. doi: 10.1038/365340a0. [DOI] [PubMed] [Google Scholar]
- 26.Wardlaw AJ, Moqbel R, Cromwell O, Kay AB. Platelet-activating factor. A potent chemotactic and chemokinetic factor for human eosinophils. J Clin Invest. 1986;78:1701–6. doi: 10.1172/JCI112765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perry VH, Andersson PB, Gordon G. Macrophages and inflammation in the central nervous system. Trends Neurosci. 1993;16:268–73. doi: 10.1016/0166-2236(93)90180-t. [DOI] [PubMed] [Google Scholar]
- 28.Brenner T, Soffer D, Shalit M, Levi-Schaffer F. Mast cells in experimental allergic encephalomyelitis: characterization, distribution in the CNS and in vitro activation by myelin basic protein and neuropeptides. J Neurol Sci. 1994;122:210–3. doi: 10.1016/0022-510x(94)90300-x. [DOI] [PubMed] [Google Scholar]
- 29.Rozniecki JJ, Hauser SL, Stein M, Lincoln R, Theoharides TC. Elevated mast cell tryptase in cerebrospinal fluid of multiple sclerosis patients. Ann Neurol. 1995;37:63–6. doi: 10.1002/ana.410370112. [DOI] [PubMed] [Google Scholar]
- 30.Theoharides TC, Baloyannis SJ, Manolidis LS. Activated rat peritoneal mast cells can cause syngeneic brain demyelination in vitro. Int J Immunopathol Pharmacol. 1991;4:137–44. [Google Scholar]
- 31.Medic N, Lorenzon P, Vita F, Trevisan E, Marchioli A, Soranzo MR, Fabbretti E, Zabucchi G. Mast cell adhesion induces cytoskeletal modifications and programmed cell death in oligodendrocytes. J Neuroimmunol. 2010;218:57–66. doi: 10.1016/j.jneuroim.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 32.Nautiyal KM, Ribeiro AC, Pfaff DW, Silver R. Brain mast cells link the immune system to anxiety-like behavior. Proc Natl Acad Sci USA. 2008;105:18053–7. doi: 10.1073/pnas.0809479105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Theoharides TC, Angelidou A, Alysandratos KD, Zhang B, Asadi S, Francis K, Toniato E, Kalogeromitros D. Mast cell activation and autism. Biochim Biophys Acta. 2012;1822:34–41. doi: 10.1016/j.bbadis.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 34.DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- 35.Wei F, Dubner R, Ren K. Glial–cytokine–neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–18. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chessell IP, Hatcher JP, Bountra C, et al. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain. 2005;114:386–96. doi: 10.1016/j.pain.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 37.Tsuda M, Kuboyama K, Inoue T, Nagata K, Tozaki-Saitoh H, Inoue K. Behavioral phenotypes of mice lacking purinergic P2X4 receptors in acute and chronic pain assays. Mol Pain. 2009;5:28. doi: 10.1186/1744-8069-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ballini E, Virginio C, Medhurst SJ, et al. Characterization of three diaminopyrimidines as potent and selective antagonists of P2X3 and P2X2/3 receptors with in vivo efficacy in a pain model. Br J Pharmacol. 2011;163:1315–25. doi: 10.1111/j.1476-5381.2011.01322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xanthos DN, Gaderer S, Drdla R, Nuro E, Abramova A, Ellmeier W, Sandkühler J. Central nervous system mast cells in peripheral inflammatory nociception. Mol Pain. 2011;7:42. doi: 10.1186/1744-8069-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zuo Y, Perkins NM, Tracey DJ, Geczy CL. Inflammation and hyperalgesia induced by nerve injury in the rat: a key role of mast cells. Pain. 2003;105:467–79. doi: 10.1016/S0304-3959(03)00261-6. [DOI] [PubMed] [Google Scholar]
- 41.Levy D, Kainz V, Burstein R, Strassman AM. Mast cell degranulation distinctly activates trigemino-cervical and lumbosacral pain pathways and elicits widespread tactile pain hypersensitivity. Brain Behav Immun. 2012;26:311–7. doi: 10.1016/j.bbi.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koda H, Mizumura K. Sensitization to mechanical stimulation by inflammatory mediators and by mild burn in canine visceral nociceptors in vitro. J Neurophysiol. 2002;87:2043–51. doi: 10.1152/jn.00593.2001. [DOI] [PubMed] [Google Scholar]
- 43.Leon A, Buriani A, Dal Toso R, Fabris M, Romanello S, Aloe L, Levi-Montalcini R. Mast cells synthesize, store, and release nerve growth factor. Proc Natl Acad Sci USA. 1994;91:3739–43. doi: 10.1073/pnas.91.9.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leal-Berumen I, Conlon P, Marshall JS. IL-6 production by rat peritoneal mast cells is not necessarily preceded by histamine release and can be induced by bacterial lipopolysaccharide. J Immunol. 1994;152:5468–76. [PubMed] [Google Scholar]
- 45.Hayashi R, Xiao W, Kawamato M, Yuge O, Bennett GJ. Systemic glucocorticoid therapy reduces pain and the number of endoneurial tumor necrosis factor-α (TNF-α)-positive mast cells in rats with a painful peripheral neuropathy. J Pharmacol Sci. 2011;106:559–65. doi: 10.1254/jphs.fp0072181. [DOI] [PubMed] [Google Scholar]
- 46.Wood D. Visceral pain: spinal afferents, enteric mast cells, enteric nervous system and stress. Curr Pharm Des. 2011;17:1573–5. doi: 10.2174/138161211796196918. [DOI] [PubMed] [Google Scholar]
- 47.Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanisch U-K, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–94. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 49.Silver R, Curley JP. Mast cells on the mind: new insights and opportunities. Trends Neurosci. 2013;36:513–21. doi: 10.1016/j.tins.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 50.Chew LJ, Takanohashi A, Bell M. Microglia and inflammation: impact on developmental brain injuries. Ment Retard Dev Disabil Res Rev. 2006;12:105–12. doi: 10.1002/mrdd.20102. [DOI] [PubMed] [Google Scholar]
- 51.Jin Y, Silverman AJ, Vannucci SJ. Mast cells are early responders after hypoxia–ischemia in immature rat brain. Stroke. 2009;40:3107–12. doi: 10.1161/STROKEAHA.109.549691. [DOI] [PubMed] [Google Scholar]
- 52.Gordon JR, Galli SJ. Release of both preformed and newly synthesized tumor necrosis factor α (TNF-α)/cachectin by mouse mast cells stimulated via the Fc εRI. A mechanism for the sustained action of mast cell-derived TNF-α during IgE-dependent biological responses. J Exp Med. 1991;174:103–7. doi: 10.1084/jem.174.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin Y, Silverman AJ, Vannucci SJ. Mast cell stabilization limits hypoxic–ischemic brain damage in the immature rat. Dev Neurosci. 2007;29:373–84. doi: 10.1159/000105478. [DOI] [PubMed] [Google Scholar]
- 54.Strbian D, Karjalainen-Lindsberg ML, Kovanen PT, Tatlisumak T, Lindsberg PJ. Mast cell stabilization reduces hemorrhage formation and mortality after administration of thrombolytics in experimental ischemic stroke. Circulation. 2007;116:411–8. doi: 10.1161/CIRCULATIONAHA.106.655423. [DOI] [PubMed] [Google Scholar]
- 55.Lindsberg PJ, Strbian D, Karjalainen-Lindsberg ML. Mast cells as early responders in the regulation of acute blood–brain barrier changes after cerebral ischemia and hemorrhage. J Cereb Blood Flow Metab. 2010;30:689–702. doi: 10.1038/jcbfm.2009.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mattila OS, Strbian D, Saksi J, Pikkarainen TO, Rantanen V, Tatlisumak T, Lindsberg PJ. Cerebral mast cells mediate blood–brain barrier disruption in acute experimental ischemic stroke through perivascular gelatinase activation. Stroke. 2011;42:3600–5. doi: 10.1161/STROKEAHA.111.632224. [DOI] [PubMed] [Google Scholar]
- 57.Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 2011;87:10–20. doi: 10.1016/j.brainresbull.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010;7:354–65. doi: 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peterson LJ, Flood PM. Oxidative stress and microglial cells in Parkinson's disease. Mediators Inflamm. 2012;2012:401264. doi: 10.1155/2012/401264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Olsson Y. Mast cells in plaques of multiple sclerosis. Acta Neurol Scand. 1974;50:611–8. doi: 10.1111/j.1600-0404.1974.tb02806.x. [DOI] [PubMed] [Google Scholar]
- 61.Lock C, Hermans G, Pedotti R, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–8. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 62.Nelissen S, Lemmens E, Geurts N, Kramer P, Maurer M, Hendriks J, Hendrix S. The role of mast cells in neuroinflammation. Acta Neuropathol. 2013;125:637–50. doi: 10.1007/s00401-013-1092-y. [DOI] [PubMed] [Google Scholar]
- 63.Niederhoffer N, Levy R, Sick E, Andre P, Coupin G, Lombard Y, Gies JP. Amyloid β peptides trigger CD47-dependent mast cell secretory and phagocytic responses. Int J Immunopathol Pharmacol. 2009;22:473–83. doi: 10.1177/039463200902200224. [DOI] [PubMed] [Google Scholar]
- 64.Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 2011;10:253–63. doi: 10.1016/S1474-4422(11)70015-1. [DOI] [PubMed] [Google Scholar]
- 65.Hall ED, Oostveen JA, Gurney ME. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998;23:249–56. doi: 10.1002/(sici)1098-1136(199807)23:3<249::aid-glia7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 66.Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, Leigh PN, Banati R. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15:601–9. doi: 10.1016/j.nbd.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 67.Garbuzova-Davis S, Rodrigues MC, Hernandez-Ontiveros DG, Louis MK, Willing AE, Borlongan CV, Sanberg PR. Amyotrophic lateral sclerosis: a neurovascular disease. Brain Res. 2011;1398:113–25. doi: 10.1016/j.brainres.2011.04.049. [DOI] [PubMed] [Google Scholar]
- 68.Fiala M, Chattopadhay M, La Cava A, et al. IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. J Neuroinflammation. 2010;7:76. doi: 10.1186/1742-2094-7-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sayed BA, Christy AL, Walker ME, Brown MA. Meningeal mast cells affect early T cell central nervous system infiltration and blood–brain barrier integrity through TNF: a role for neutrophil recruitment? J Immunol. 2010;184:6891–900. doi: 10.4049/jimmunol.1000126. [DOI] [PubMed] [Google Scholar]
- 70.Graves MC, Fiala M, Dinglasan LA, Liu NQ, Sayre J, Chiappelli F, van Kooten C, Vinters HV. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5:213–9. doi: 10.1080/14660820410020286. [DOI] [PubMed] [Google Scholar]
- 71.Ganeshan K, Bryce PJ. Regulatory T cells enhance mast cell production of IL-6 via surface-bound TGF-β. J Immunol. 2012;188:594–603. doi: 10.4049/jimmunol.1102389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dudeck A, Suender CA, Kostka SL, von Stebut E, Maurer M. Mast cells promote Th1 and Th17 responses by modulating dendritic cell maturation and function. Eur J Immunol. 2011;41:1883–93. doi: 10.1002/eji.201040994. [DOI] [PubMed] [Google Scholar]
- 73.Rentzos M, Rombos A, Nikolaou C, et al. Interleukin-15 and interleukin-12 are elevated in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Eur Neurol. 2010;63:285–90. doi: 10.1159/000287582. [DOI] [PubMed] [Google Scholar]
- 74.Jackson NE, Wang HW, Tedla N, et al. IL-15 induces mast cell migration via a pertussis toxin-sensitive receptor. Eur J Immunol. 2005;35:2376–85. doi: 10.1002/eji.200526154. [DOI] [PubMed] [Google Scholar]
- 75.Nakano N, Nishiyama C, Kanada S, et al. Involvement of mast cells in IL-12/23 p40 production is essential for survival from polymicrobial infections. Blood. 2007;109:4846–55. doi: 10.1182/blood-2006-09-045641. [DOI] [PubMed] [Google Scholar]
- 76.Yang H, Wei J, Zhang H, Song W, Wei W, Zhang L, Qian K, He S. Upregulation of Toll-like receptor (TLR) expression and release of cytokines from mast cells by IL-12. Cell Physiol Biochem. 2010;26:337–46. doi: 10.1159/000320557. [DOI] [PubMed] [Google Scholar]
- 77.Zhang H, Yang X, Yang H, et al. Modulation of mast cell proteinase-activated receptor expression and IL-4 release by IL-12. Immunol Cell Biol. 2007;85:558–66. doi: 10.1038/sj.icb.7100085. [DOI] [PubMed] [Google Scholar]
- 78.Bushell T. The emergence of proteinase-activated receptor-2 as a novel target for the treatment of inflammation-related CNS disorders. J Physiol. 2007;581(Pt 1):7–16. doi: 10.1113/jphysiol.2007.129577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Casula M, Iyer AM, Spliet WG, Anink JJ, Steentjes K, Sta M, Troost D, Aronica E. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience. 2011;179:233–43. doi: 10.1016/j.neuroscience.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 80.Pietrzak A, Wierzbicki M, Wiktorska M, Brzezińska-Błaszczyk E. Surface TLR2 and TLR4 expression on mature rat mast cells can be affected by some bacterial components and proinflammatory cytokines. Mediators Inflamm. 2011 doi: 10.1155/2011/427473. doi: 10.1155/2011/427473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Skuljec J, Sun H, Pul R, et al. CCL5 induces a pro-inflammatory profile in microglia in vitro. Cell Immunol. 2011;270:164–71. doi: 10.1016/j.cellimm.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 82.Kano O, Beers DR, Henkel JS, Appel SH. Peripheral nerve inflammation in ALS: cause or consequence? Neurology. 2011;78:833–55. doi: 10.1212/WNL.0b013e318249f776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81:302–13. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- 84.Kotter MR, Zhao C, van Rooijen N, Franklin RJM. Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol Dis. 2005;18:166–75. doi: 10.1016/j.nbd.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 85.Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci. 2003;4:703–13. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- 86.Olah M, Amor S, Brouwer N, Vinet J, Eggen B, Biber K, Boddeke HW. Identification of a microglia phenotype supportive of remyelination. Glia. 2012;60:306–21. doi: 10.1002/glia.21266. [DOI] [PubMed] [Google Scholar]
- 87.Fan R, Xu F, Previti ML, Davis J, Grande AM, Robinson JK, Van Nostrand WE. Minocycline reduces microglial activation and improves behavioral deficits in a transgenic model of cerebral microvascular amyloid. J Neurosci. 2007;27:3057–63. doi: 10.1523/JNEUROSCI.4371-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Grathwohl SA, Kälin RE, Bolmont T, et al. Formation and maintenance of Alzheimer's disease β-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1361–3. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–8. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 90.Song M, Jin J, Lim JE, et al. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer's disease. J Neuroinflammation. 2011;8:92. doi: 10.1186/1742-2094-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Michaud JP, Hallé M, Lampron A, et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer's disease-related pathology. Proc Natl Acad Sci USA. 2013;110:1941–6. doi: 10.1073/pnas.1215165110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lalancette-Hébert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Imai F, Suzuki H, Oda J, Ninomiya T, Ono K, Sano H, Sawada M. Neuroprotective effect of exogenous microglia in global brain ischemia. J Cereb Blood Flow Metab. 2007;27:488–500. doi: 10.1038/sj.jcbfm.9600362. [DOI] [PubMed] [Google Scholar]
- 94.Vinet J, van Weering HR, Heinrich A, et al. Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J Neuroinflammation. 2012;9:27. doi: 10.1186/1742-2094-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Skaper SD, Giusti P, Facci F. Microglia and mast cells: two tracks on the road to neuroinflammation. FASEB J. 2012;26:3103–17. doi: 10.1096/fj.11-197194. [DOI] [PubMed] [Google Scholar]
- 96.Kulka M, Fukuishi N, Metcalfe DD. Human mast cells synthesize and release angiogenin, a member of the ribonuclease A (RNase A) superfamily. J Leukoc Biol. 2009;86:1217–26. doi: 10.1189/jlb.0908517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Subramanian V, Crabtree B, Acharya KR. Human angiogenin is a neuroprotective factor and amyotrophic lateral sclerosis associated angiogenin variants affect neurite extension/pathfinding and survival of motor neurons. Hum Mol Genet. 2008;17:130–49. doi: 10.1093/hmg/ddm290. [DOI] [PubMed] [Google Scholar]
- 98.Gellera C, Colombrita C, Ticozzi N, Castellotti B, Bragato C, Ratti A, Taroni F, Silani V. Identification of new ANG gene mutations in a large cohort of Italian patients with amyotrophic lateral sclerosis. Neurogenetics. 2008;9:33–40. doi: 10.1007/s10048-007-0111-3. [DOI] [PubMed] [Google Scholar]
- 99.Nautiyal KM, Dailey CA, Jahn JL, Rodriquez E, Son NH, Sweedler JV, Silver R. Serotonin of mast cell origin contributes to hippocampal function. Eur J Neurosci. 2012;36:2347–59. doi: 10.1111/j.1460-9568.2012.08138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weller K, Foitzik K, Paus R, Syska W, Maurer M. Mast cells are required for normal healing of skin wounds in mice. FASEB J. 2006;20:2366–8. doi: 10.1096/fj.06-5837fje. [DOI] [PubMed] [Google Scholar]
- 101.Osipchuk Y, Cahalan M. Cell-to-cell spread of calcium signals mediated by ATP receptors in mast cells. Nature. 1992;359:241–4. doi: 10.1038/359241a0. [DOI] [PubMed] [Google Scholar]
- 102.Chakraborty S, Kaushik DK, Gupta M, Basu A. Inflammasome signaling at the heart of central nervous system pathology. J Neurosci Res. 2010;88:1615–31. doi: 10.1002/jnr.22343. [DOI] [PubMed] [Google Scholar]
- 103.Trang T, Beggs S, Wan X, Salter MW. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci. 2009;29:3518–28. doi: 10.1523/JNEUROSCI.5714-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang H, Lin L, Yang H, Zhang Z, Yang X, Zhang L, He S. Induction of IL-13 production and upregulation of gene expression of protease activated receptors in P815 cells by IL-6. Cytokine. 2010;50:138–45. doi: 10.1016/j.cyto.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 105.Zhang H, Yang H, He S. TNF increases expression of IL-4 and PARs in mast cells. Cell Physiol Biochem. 2010;26:327–36. doi: 10.1159/000320556. [DOI] [PubMed] [Google Scholar]
- 106.Gasque P, Singhrao SK, Neal JW, Götze O, Morgan BP. Expression of the receptor for complement C5a (CD88) is up-regulated on reactive astrocytes, microglia, and endothelial cells in the inflamed human central nervous system. Am J Pathol. 1997;150:31–41. [PMC free article] [PubMed] [Google Scholar]
- 107.Griffin RS, Costigan M, Brenner GJ, et al. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J Neurosci. 2007;27:8699–708. doi: 10.1523/JNEUROSCI.2018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Orinska Z, Bulanova E, Budagian V, Metz M, Maurer M, Bulfone-Paus S. TLR3-induced activation of mast cells modulates CD8+ T-cell recruitment. Blood. 2005;106:978–87. doi: 10.1182/blood-2004-07-2656. [DOI] [PubMed] [Google Scholar]
- 109.Kim D, Kim MA, Cho IH, et al. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J Biol Chem. 2007;282:14975–83. doi: 10.1074/jbc.M607277200. [DOI] [PubMed] [Google Scholar]
- 110.Ribes S, Adam N, Ebert S, Regen T, Bunkowski S, Hanisch UK, Nau R. The viral TLR3 agonist poly(I:C) stimulates phagocytosis and intracellular killing of Escherichia coli by microglial cells. Neurosci Lett. 2010;482:17–20. doi: 10.1016/j.neulet.2010.06.078. [DOI] [PubMed] [Google Scholar]
- 111.Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci USA. 2005;102:5856–61. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pietrzak A, Wierzbicki M, Wiktorska M, Brzezińska-Błaszczyk E. Surface TLR2 and TLR4 expression on mature rat mast cells can be affected by some bacterial components and proinflammatory cytokines. Mediators Inflamm. 2011 doi: 10.1155/2011/427473. doi: 10.1155/2011/427473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Buchanan MM, Hutchinson M, Watkins LR, Yin H. Toll-like receptor 4 in CNS pathologies. J Neurochem. 2010;114:13–27. doi: 10.1111/j.1471-4159.2010.06736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu S, Liu Y, Hao W, et al. TLR2 is a primary receptor for Alzheimer's amyloid β peptide to trigger neuroinflammatory activation. J Immunol. 2012;188:1098–107. doi: 10.4049/jimmunol.1101121. [DOI] [PubMed] [Google Scholar]
- 115.Burnstock G, Krügel U, Abbracchio MP, Illes P. Purinergic signalling: from normal behaviour to pathological brain function. Prog Neurobiol. 2011;95:229–74. doi: 10.1016/j.pneurobio.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 116.Zhang S, Zeng X, Yang H, Hu G, He S. Mast cell tryptase induces microglia activation via protease-activated receptor 2 signaling. Cell Physiol Biochem. 2012;29:931–40. doi: 10.1159/000171029. [DOI] [PubMed] [Google Scholar]
- 117.Juremalm M, Hjertson M, Olsson N, Harvima I, Nilsson K, Nilsson G. The chemokine receptor CXCR4 is expressed within the mast cell lineage and its ligand stromal cell-derived factor-1α acts as a mast cell chemotaxin. Eur J Immunol. 2000;30:3614–22. doi: 10.1002/1521-4141(200012)30:12<3614::AID-IMMU3614>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 118.Põlajeva J, Sjösten AM, Lager N, et al. Mast cell accumulation in glioblastoma with a potential role for stem cell factor and chemokine CXCL12. PLoS ONE. 2011;6:e25222. doi: 10.1371/journal.pone.0025222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang Y, Huang J, Li Y, Yang GY. Roles of chemokine CXCL12 and its receptors in ischemic stroke. Curr Drug Targets. 2012;13:166–72. doi: 10.2174/138945012799201603. [DOI] [PubMed] [Google Scholar]
- 120.Knerlich-Lukoschus F, von der Ropp-Brenner B, Lucius R, Mehdorn HM, Held-Feindt J. Spatiotemporal CCR1, CCL3(MIP-1α), CXCR4, CXCL12(SDF-1α) expression patterns in a rat spinal cord injury model of posttraumatic neuropathic pain. J Neurosurg Spine. 2011;14:583–97. doi: 10.3171/2010.12.SPINE10480. [DOI] [PubMed] [Google Scholar]
- 121.Soruri A, Grigat J, Kiafard Z, Zwirner J. Mast cell activation is characterized by upregulation of a functional anaphylatoxin C5a receptor. BMC Immunol. 2008;9:29. doi: 10.1186/1471-2172-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kim DY, Hong GU, Ro JY. Signal pathways in astrocytes activated by cross-talk between of astrocytes and mast cells through CD40-CD40L. J Neuroinflammation. 2011;8:25. doi: 10.1186/1742-2094-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–90. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- 124.Gosselin RD, Suter MR, Ji RR, Decosterd I. Glial cells and chronic pain. Neuroscientist. 2010;16:519. doi: 10.1177/1073858409360822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ralay Ranaivo H, Craft JM, Hu W, Guo L, Wing LK, Van Eldik LJ, Watterson DM. Glia as a therapeutic target: selective suppression of human amyloid-β-induced upregulation of brain proinflammatory cytokine production attenuates neurodegeneration. J Neurosci. 2006;26:662–70. doi: 10.1523/JNEUROSCI.4652-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP. The resolution of inflammation. Nat Rev Immunol. 2013;13:59–66. doi: 10.1038/nri3362. [DOI] [PubMed] [Google Scholar]
- 127.Zhu C, Solorzano C, Sahar S, Realini N, Fung E, Sassone-Corsi P, Piomelli D. Proinflammatory stimuli control N-acylphosphatidylethanolamine-specific phospholipase D expression in macrophages. Mol Pharmacol. 2011;79:786–92. doi: 10.1124/mol.110.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339:166–72. doi: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pacher P, Bátkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ueda N, Tsuboi K, Uyama T. Metabolism of endocannabinoids and related N-acylethanolamines: canonical and alternative pathways. FEBS J. 2013;280:1874–94. doi: 10.1111/febs.12152. [DOI] [PubMed] [Google Scholar]
- 131.Leung D, Saghatelian A, Simon GM, Cravatt BF. Inactivation of N-acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry. 2006;45:4720–6. doi: 10.1021/bi060163l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–7. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 133.Tsuboi K, Takezaki N, Ueda N. The N-acylethanolamine-hydrolyzing acid amidase (NAAA) Chem Biodivers. 2007;4:1914–25. doi: 10.1002/cbdv.200790159. [DOI] [PubMed] [Google Scholar]
- 134.Skaper SD, Facci L. Mast cell–glia axis in neuroinflammation and therapeutic potential of the anandamide congener palmitoylethanolamide. Philos Trans R Soc Lond B Biol Sci. 2012;367:3312–25. doi: 10.1098/rstb.2011.0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bisogno T, Maurelli S, Melck D, De Petrocellis L, Di Marzo V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J Biol Chem. 1997;272:3315–23. doi: 10.1074/jbc.272.6.3315. [DOI] [PubMed] [Google Scholar]
- 136.Muccioli GG, Stella N. Microglia produce and hydrolyze palmitoylethanolamide. Neuropharmacology. 2008;54:16–22. doi: 10.1016/j.neuropharm.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Facci L, Dal Toso R, Romanello S, Buriani A, Skaper SD, Leon A. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc Natl Acad Sci USA. 1995;92:3376–80. doi: 10.1073/pnas.92.8.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Franklin A, Parmentier-Batteur S, Walter L, Greenberg DA, Stella N. Palmitoylethanolamide increases after focal cerebral ischemia and potentiates microglial cell motility. J Neurosci. 2003;23:7767–75. doi: 10.1523/JNEUROSCI.23-21-07767.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Luongo L, Guida F, Boccella S, Bellini G, Gatta L, Rossi F, de Novellis V, Maione S. Palmitoylethanolamide reduces formalin-induced neuropathic-like behaviour through spinal glial/microglial phenotypical changes in mice. CNS Neurol Disord Drug Targets. 2013;12:45–54. doi: 10.2174/1871527311312010009. [DOI] [PubMed] [Google Scholar]
- 140.Petrosino S, Palazzo E, de Novellis V, Bisogno T, Rossi F, Maione S, Di Marzo V. Changes in spinal and supraspinal endocannabinoid levels in neuropathic rats. Neuropharmacology. 2007;52:415–22. doi: 10.1016/j.neuropharm.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 141.Jhaveri MD, Richardson D, Robinson I, et al. Inhibition of fatty acid amide hydrolase and cyclooxygenase-2 increases levels of endocannabinoid related molecules and produces analgesia via peroxisome proliferator-activated receptor-α in a model of inflammatory pain. Neuropharmacology. 2008;55:85–93. doi: 10.1016/j.neuropharm.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 142.Ghafouri N, Ghafouri B, Larsson B, Stensson N, Fowler CJ, Gerdle B. Palmitoylethanolamide and stearoylethanolamide levels in the interstitium of the trapezius muscle of women with chronic widespread pain and chronic neck–shoulder pain correlate with pain intensity and sensitivity. Pain. 2013;154:1649–58. doi: 10.1016/j.pain.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 143.Baker D, Pryce G, Croxford JL, et al. Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J. 2001;15:300–2. doi: 10.1096/fj.00-0399fje. [DOI] [PubMed] [Google Scholar]
- 144.Berger C, Schmid PC, Schabitz WR, Wolf M, Schwab S, Schmid HHO. Massive accumulation of N-acylethanolamines after stroke. Cell signalling in acute cerebral ischemia? J Neurochem. 2004;88:1159–67. doi: 10.1046/j.1471-4159.2003.02244.x. [DOI] [PubMed] [Google Scholar]
- 145.Schäbitz WR, Giuffrida A, Berger C, Aschoff A, Schwaninger M, Schwab S, Piomelli D. Release of fatty acid amides in a patient with hemispheric stroke. Stroke. 2002;33:2112–4. doi: 10.1161/01.str.0000023491.63693.18. [DOI] [PubMed] [Google Scholar]
- 146.Sarchielli P, Pini LA, Coppola F, Rossi C, Baldi A, Mancini ML, Calabresi P. Endocannabinoids in chronic migraine: CSF findings suggest a system failure. Neuropsychopharmacology. 2007;32:1384–90. doi: 10.1038/sj.npp.1301246. [DOI] [PubMed] [Google Scholar]
- 147.Genovese T, Esposito E, Mazzon E, et al. Effects of palmitoylethanolamide on signaling pathways implicated in the development of spinal cord injury. J Pharmacol Exp Ther. 2008;326:12–23. doi: 10.1124/jpet.108.136903. [DOI] [PubMed] [Google Scholar]
- 148.Esposito E, Paterniti I, Mazzon E, Genovese T, Di Paola R, Galuppo M, Cuzzocrea S. Effects of palmitoylethanolamide on release of mast cell peptidases and neurotrophic factors after spinal cord injury. Brain Behav Immun. 2011;25:1099–112. doi: 10.1016/j.bbi.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 149.Ahmad A, Crupi R, Impellizzeri D, Campolo M, Marino A, Esposito E, Cuzzocrea S. Administration of palmitoylethanolamide (PEA) protects the neurovascular unit and reduces secondary injury after traumatic brain injury in mice. Brain Behav Immun. 2012;26:1310–21. doi: 10.1016/j.bbi.2012.07.021. [DOI] [PubMed] [Google Scholar]
- 150.Esposito E, Impellizzeri D, Mazzon E, Paterniti I, Cuzzocrea S. Neuroprotective activities of palmitoylethanolamide in an animal model of Parkinson's disease. PLoS ONE. 2012;7:e41880. doi: 10.1371/journal.pone.0041880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ahmad A, Genovese T, Impellizzeri D, Crupi R, Velardi E, Marino A, Esposito E, Cuzzocrea S. Reduction of ischemic brain injury by administration of palmitoylethanolamide after transient middle cerebral artery occlusion in rats. Brain Res. 2012;1477:45–58. doi: 10.1016/j.brainres.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 152.Scuderi C, Esposito G, Blasio A, et al. Palmitoylethanolamide counteracts reactive astrogliosis induced by β-amyloid peptide. J Cell Mol Med. 2011;15:2664–74. doi: 10.1111/j.1582-4934.2011.01267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Costa B, Comelli F, Bettoni I, Colleoni M, Giagnoni G. The endogenous fatty acid amide, palmitoylethanolamide, has anti-allodynic and anti-hyperalgesic effects in a murine model of neuropathic pain: involvement of CB1, TRPV1 and PPARγ receptors and neurotrophic factors. Pain. 2008;139:541–50. doi: 10.1016/j.pain.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 154.De Filippis D, Luongo L, Cipriano M, Palazzo E, Cinelli MP, de Novellis V, Maione S, Iuvone T. Palmitoylethanolamide reduces granuloma-induced hyperalgesia by modulation of mast cell activation in rats. Mol Pain. 2011;10:3. doi: 10.1186/1744-8069-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Di Cesare Mannelli L, D'Agostino G, Pacini A, Russo R, Zanardelli M, Ghelardini C, Calignano A. Palmitoylethanolamide is a disease-modifying agent in peripheral neuropathy: pain relief and neuroprotection share a PPAR-α-mediated mechanism. Mediators Inflamm. 2013;2013:328797. doi: 10.1155/2013/328797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mazzari S, Canella R, Petrelli L, Marcolongo G, Leon L. N-(2-hydroxyethyl)hexadecanamide is orally active in reducing edema formation and inflammatory hyperalgesia by downmodulating mast cell activation. Eur J Pharmacol. 1996;300:227–36. doi: 10.1016/0014-2999(96)00015-5. [DOI] [PubMed] [Google Scholar]
- 157.Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–81. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- 158.Jaggar SI, Hasnie FS, Sellaturay S, Rice AS. The antihyperalgesic actions of the cannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain. 1998;76:189–99. doi: 10.1016/s0304-3959(98)00041-4. [DOI] [PubMed] [Google Scholar]
- 159.Costa B, Conti S, Giagnoni G, Colleoni M. Therapeutic effect of the endogenous fatty acid amide, palmitoylethanolamide, in rat acute inflammation: inhibition of nitric oxide and cyclo-oxygenase systems. Br J Pharmacol. 2002;137:413–20. doi: 10.1038/sj.bjp.0704900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.D'Agostino G, La Rana G, Russo R, et al. Central administration of palmitoylethanolamide reduces hyperalgesia in mice via inhibition of NF-κB nuclear signalling in dorsal root ganglia. Eur J Pharmacol. 2009;613:54–9. doi: 10.1016/j.ejphar.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 161.D'Agostino G, La Rana G, Russo R, et al. Acute intracerebroventricular administration of palmitoylethanolamide, an endogenous peroxisome proliferator-activated receptor-α agonist, modulates carrageenan-induced paw edema in mice. J Pharmacol Exp Ther. 2007;322:1137–43. doi: 10.1124/jpet.107.123265. [DOI] [PubMed] [Google Scholar]
- 162.Citraro R, Russo E, Scicchitano F, et al. Antiepileptic action of N-palmitoylethanolamine through CB1 and PPAR-α receptor activation in a genetic model of absence epilepsy. Neuropharmacology. 2013;69:115–26. doi: 10.1016/j.neuropharm.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 163.Solorzano C, Zhu C, Battista N, et al. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc Natl Acad Sci USA. 2009;106:20966–71. doi: 10.1073/pnas.0907417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Solorzano C, Antonietti F, Duranti A, et al. Synthesis and structure-activity relationships of N-(2-oxo-3-oxetanyl)amides as N-acylethanolamine-hydrolyzing acid amidase inhibitors. J Med Chem. 2010;53:5770–81. doi: 10.1021/jm100582w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yamano Y, Tsuboi K, Hozaki Y, Takahashi K, Jin XH, Ueda N, Wada A. Lipophilic amines as potent inhibitors of N-acylethanolamine-hydrolyzing acid amidase. Bioorg Med Chem. 2012;20:3658–65. doi: 10.1016/j.bmc.2012.03.065. [DOI] [PubMed] [Google Scholar]
- 166.Li Y, Yang L, Chen L, Zhu C, Huang R, Zheng X, Qiu Y, Fu J. Design and synthesis of potent N-acylethanolamine-hydrolyzing acid amidase (NAAA) inhibitor as anti-inflammatory compounds. PLoS ONE. 2012;7:8. doi: 10.1371/journal.pone.0043023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Truini A, Biasiotta A, Di Stefano G, et al. Palmitoylethanolamide restores myelinated-fibre function in patients with chemotherapy-induced painful neuropathy. CNS Neurol Disorders Drug Targets. 2011;10:916–20. doi: 10.2174/187152711799219307. [DOI] [PubMed] [Google Scholar]
- 168.Kopsky DJ, Hesselink JM. Multimodal stepped care approach with acupuncture and PPAR-α agonist palmitoylethanolamide in the treatment of a patient with multiple sclerosis and central neuropathic pain. Acupunct Med. 2012;30:53–5. doi: 10.1136/acupmed-2011-010119. [DOI] [PubMed] [Google Scholar]
- 169.Hesselink JM, Hekker TA. Therapeutic utility of palmitoylethanolamide in the treatment of neuropathic pain associated with various pathological conditions: a case series. J Pain Res. 2012;5:37–42. doi: 10.2147/JPR.S32143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hesselink JMK. New targets in pain, non-neuronal cells, and the role of palmitoylethanolamide. The Open Pain Journal. 2012;5:12–23. [Google Scholar]
- 171.Clemente S. Amytrophic lateral sclerosis treatment with ultramicronized palmitoylethanolamide: a case report. CNS Neurol Disord Drug Targets. 2012;11:933–6. doi: 10.2174/1871527311201070933. [DOI] [PubMed] [Google Scholar]
- 172.Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, Piomelli D. The nuclear receptor peroxisome proliferator-activated receptor-α mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67:15–9. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- 173.de Novellis V, Luongo L, Guida F, et al. Effects of intra-ventrolateral periaqueductal grey palmitoylethanolamide on thermoceptive threshold and rostral ventromedial medulla cell activity. Eur J Pharmacol. 2012;676:41–50. doi: 10.1016/j.ejphar.2011.11.034. [DOI] [PubMed] [Google Scholar]
- 174.Scuderi C, Valenza M, Stecca C, Esposito G, Carratù MR, Steardo L. Palmitoylethanolamide exerts neuroprotective effects in mixed neuroglial cultures and organotypic hippocampal slices via peroxisome proliferator-activated receptor-α. J Neuroinflammation. 2012;9:49. doi: 10.1186/1742-2094-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Smart D, Jonsson KO, Vandevoorde S, Lambert DM, Fowler CJ. ‘Entourage’ effects of N-acyl ethanolamines at human vanilloid receptors. Comparison of effects upon anandamide-induced vanilloid receptor activation and upon anandamide metabolism. Br J Pharmacol. 2002;136:452–8. doi: 10.1038/sj.bjp.0704732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.De Petrocellis L, Davis JB, Di Marzo V. Palmitoylethanolamide enhances anandamide stimulation of human vanilloid VR1 receptors. FEBS Lett. 2001;506:253–6. doi: 10.1016/s0014-5793(01)02934-9. [DOI] [PubMed] [Google Scholar]
- 177.Bíró T, Maurer M, Modarres S, et al. Characterization of functional vanilloid receptors expressed by mast cells. Blood. 1998;91:1332–40. [PubMed] [Google Scholar]
- 178.Kim SR, Kim SU, Oh U, Jin BK. Transient receptor potential vanilloid subtype 1 mediates microglial cell death in vivo and in vitro via Ca2+-mediated mitochondrial damage and cytochrome c release. J Immunol. 2006;177:4322–9. doi: 10.4049/jimmunol.177.7.4322. [DOI] [PubMed] [Google Scholar]
- 179.David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. 2011;12:388–99. doi: 10.1038/nrn3053. [DOI] [PubMed] [Google Scholar]
- 180.Schäfer T, Starkl P, Allard C, Wolf RM, Schweighoffer T. A granular variant of CD63 is a regulator of repeated human mast cell degranulation. Allergy. 2010;65:1242–55. doi: 10.1111/j.1398-9995.2010.02350.x. [DOI] [PubMed] [Google Scholar]