

Abstract
Increasing evidence has demonstrated that Toll-like receptor 4 (TLR4) -mediated systemic inflammatory response syndrome accompanied by multiple organ failure, is one of the most common causes of death in patients with severe acute pancreatitis. Recent reports have revealed that heparan sulphate (HS) proteoglycan, a component of extracellular matrices, potentiates the activation of intracellular pro-inflammatory responses via TLR4, contributing to the aggravation of acute pancreatitis. However, little is known about the participants in the HS/TLR4-mediated inflammatory cascades. Our previous work provided a clue that a membrane potassium channel (MaxiK) is responsible for HS-induced production of inflammatory cytokines. Therefore, in this report we attempted to reveal the roles of MaxiK in the activation of macrophages stimulated by HS. Our results showed that incubation of RAW264.7 cells with HS up-regulated MaxiK and TLR4 expression levels. HS could also activate MaxiK channels to promote the efflux of potassium ions from cells, as measured by the elevated activity of caspase-1, whereas this was significantly abolished by treatment with paxilline, a specific blocker of the MaxiK channel. Moreover, it was found that paxilline substantially inhibited HS-induced activation of several different transcription factors in macrophages, including nuclear factor-κB, p38 and interferon regulatory factor-3, followed by decreased production of tumour necrosis factor-α and interferon-β. Taken together, our investigation provides evidence that the HS/TLR4-mediated intracellular inflammatory cascade depends on the activation of MaxiK, which may offer an important opportunity for a new approach in therapeutic strategies of severe acute pancreatitis.
Keywords: heparan sulphate, MaxiK, paxilline, Toll-like receptor 4
Introduction
Toll like receptors (TLRs), the evolutionarily conserved transmembrane proteins of the interleukin-1 receptor superfamily, have recently emerged as the key components of the innate immune system that recognize conserved products of microbial metabolisms and trigger host defence responses.1–3 To date, a total of 13 TLRs have been found in mammals, and among them, TLR4 is the most heavily studied.4 Initially, this receptor was identified as the specific receptor of lipopolysaccharide (LPS), the principal component of the outer membrane of Gram-negative bacteria. Reports in recent years have discovered that TLR4 also plays an essential role in triggering the innate immune response to endogenous mediators released during tissue damage and inflammation independent of infectious state (known as damage-associated molecular patterns).5,6
Generally, TLR4 acts as a sensor of bacterial invasion or sterile tissue injury and mediates release of the cytokines necessary for the development of effective immunity. However, sometimes TLR4 can be excessively activated by its ligands and contribute to strong inflammatory cascade responses, resulting in severe complications. Heparan sulphate (HS), a negatively charged glycosaminoglycan in extracellular matrices, is reported to be capable of inducing TLR4-mediated systemic inflammatory response syndrome during acute pancreatitis.7 The activated pancreatic enzymes, especially elastase, might cleave extracellular matrices and subsequently release soluble HS, which can be recognized by TLR4, consequently provoking considerable production of inflammatory cytokines such as tumour necrosis factor-α (TNF-α) and thereby contributing to the resultant aggravation of acute pancreatitis.5,7,8
Although there is much evidence for the critical role of HS in the TLR4-mediated inflammatory cascade, very little is known about the molecules involved in the HS–TLR4 interaction and consequent signalling process. The finding from Blunck et al.' work that activation of a large-conductance potassium channel (MaxiK) is an early step in the transmembrane signal transduction of LPS in macrophages will shed new light on our understanding of the mechanism underlying the HS–TLR4 inflammatory cascade.9 In particular, other researchers have stated that MaxiK blockade inhibited the LPS-induced nuclear factor-κB (NF-κB) signalling pathway in macrophages.10 These results imply the important role of MaxiK channel in TLR4 activation after recognizing LPS. It will, accordingly, trigger our interest in revealing the function of MaxiK in the HS-induced TLR4 signalling cascade, because HS is also a known agonist of TLR4. In our preliminary experiment, TNF-α production in response to HS by murine peritoneal macrophages has been shown to be dependent on the activation of MaxiK (unpublished data). Hence, this study extended our original analysis to evaluate the role of MaxiK in the HS-induced intracellular inflammatory signal cascade.
Materials and methods
Reagents
Antibodies against interferon regulatory factor-3 (IRF-3), phosphorylated-IRF-3 (p-IRF-3), p38, p-p38, extracellular signal-regulated kinase 1/2 (ERK1/2), p-ERK1/2, c-jun-N-terminal kinase (JNK), p-JNK, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and horseradish peroxidase (HRP) -conjugated goat anti-rabbit antibody were obtained from Cell Signaling Technology (Beverly, MA). Rat anti-mouse TLR4 monoclonal antibody (MTS510) and its control IgG were from Santa Cruz Biotechnology (Santa Cruz, CA). HS, LPS (from Escherichia coli O111:B4), tetraethylammonium (TEA) and paxilline were purchased from Sigma (St Louis, MO). To exclude the evoked responses due to possible LPS contamination, which is an important consideration in the current study, HS stock solution (1 mg/ml) was quantified by using the Limulus amoebocyte lysate assay. The analysis showed a concentration of < 0·1 EU/ml LPS in the HS stock solution, suggesting that the amount of contaminated LPS in HS working solutions (1000 times diluted) could be ignored.11 Paxilline was suspended in 0·05% DMSO and diluted in PBS when used.
Expression of MaxiK and TLR4 in macrophages
Mouse RAW264.7 cells (from the American Type Culture Collection, Manassas, VA) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum at 37° with 5% CO2. Cells were incubated for 6 hr at 37° with HS (1 μg/ml) or LPS (10 ng/ml) in the absence and presence of paxilline (2 μg/ml), respectively. Subsequently total cellular RNA was isolated from the cells using the RNeasy Mini kit (Qiagen, Valencia, CA) and reverse transcribed with oligo(dT) primers. Then PCR was performed with 1% of Taq polymerase and the primers used for the α-subunit of MaxiK were 5′-ATGCAGTTTGATGACAGCATCG-3′ and 5′-CAGATCACCATAACAACCACCA-3′. The primers for TLR4 were 5′-CAGAGTTGCTTTCAATGGCATC-3′ and 5′-AGACT GTAATCAAGAACCTGGAGG-3′. Primers used for the amplification of GAPDH mRNA were 5′-GTCGCTGTTGAAGTCAGAGG-3′ and 5′-GAAACTGTGGCGTGATGG-3′. PCR conditions were 30 cycles of denaturation at 95° for 30 seconds, annealing at 62° for 40 seconds and extension at 72° for 60 seconds. The PCR products were separated by electrophoresis in 1% agarose gels containing 0·1 μg/ml ethidium bromide and visualized under ultraviolet light.
To determine the protein expression of MaxiK and TLR4, whole cell lysate was separated on SDS–polyacrylamide gels followed by transfer to a PVDF membrane (Millipore, Billerica, MA) after the protein concentration was quantified by the BCA protein determination kit (Pierce, Rockford, IL). The membrane was incubated with blocking solution (10 mm Tris–HCl, pH 7·4, 150 mm NaCl, 1% Triton X-100, and 0·25% BSA) containing 5% skimmed milk for more than 1 hr at room temperature and subsequently incubated with anti-MaxiK or TLR4 antibody (Santa Cruz Biotechnology, Santa Cruz, CA; 1 : 1000 dilution) overnight at 4°, respectively. After 1 hr of incubation with HRP-conjugated secondary antibody (1 : 2000 dilutions) at room temperature, the protein bands were visualized by the chemiluminescent detection system (Amersham Biosciences, Piscataway, NJ).
Caspase-1 enzymatic activity assay
As caspase-1 activation is sensitive to the efflux of cellular K+, we determined the enzymatic activity of caspase-1 in RAW264.7 cells to evaluate the HS-induced activation of the MaxiK channel using the colorimetric assay. Briefly, RAW264.7 cells were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum at 37° with 5% CO2. Then cells were incubated for 4 hr with HS (1 μg/ml) or LPS (10 ng/ml) in the absence and presence of paxilline (2 μg/ml), respectively. Cells treated with only PBS served as the control. The inhibitory effects of TEA (1 mg/ml), an unselective blocker of potassium channels, and the anti-TLR4 monoclonal antibody (MTS510, 20 μg/ml) on HS-caused caspase-1 activation were also evaluated. Then cells (5 × 106 in 500 μl) were lysed in a lysis buffer (20 mm HEPES, pH 7·5, 1·5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 0·1 mm PMSF and protease inhibitor cocktail) for 15 min on ice. After centrifugation at 10 000 g for 4 min, the supernatants were collected and incubated with 40 μm YVAD-pNA (Santa Cruz Biotechnology), the substrate of caspase-1, followed by incubation at 37° for 2 hr. The caspase-1 activity was measured at 405 nm using a microplate reader.
NF-κB activation assay
Electrophoretic mobility shift assay was performed to determine the activity of NF-κB in the nuclear extract. Briefly, RAW264.7 cells were incubated for 1 hr at 37° with HS (1 μg/ml) or LPS (10 ng/ml) in the absence and presence of paxilline (2 μg/ml), respectively. Cells treated with only PBS served as negative control. The nuclear proteins were extracted from macrophages as described by Schreiber et al.12 and the protein content was determined using an assay kit (BCA protein assay reagent; Pierce). The NF-κB activity was determined using an electrophoretic mobility shift assay kit according to the manufacturer's instructions (Promega, Madison, WI). Briefly, nuclear extract (4 μg) was incubated with 1 μl of 32P-labelled oligonucleotide (5′-AGT TGA GGG GAC TTT CCC AGG-3′) for 20 min. Binding reactions were then performed in 1 × binding buffer (0·5 mm EDTA, 1 mm MgCl2, 50 mm NaCI, 10 mm Tris–HCl, 4% glycerol, 0·5 mm dithiothreitol) and 0·05 mg/ml poly(dI-dC) in a total volume of 10 μl. Then 1 μl of loading buffer (50% glycerol, 0·1% bromophenol blue, 0·1% xylene cyanol) was added and the reactions were loaded on 4% non-denaturing polyacrylamide gels. After electrophoresis, the gels were dried for autoradiography.
Western blotting assay for IRF-3 and mitogen-activated protein kinases
RAW264.7 cells were grouped and treated as described above. Cells treated with only PBS served as the negative control. After incubation for 30 min at 37°, whole cell lysate was prepared to determine the phosphorylated levels of IRF-3, p38, ERK1/2 and JNK, respectively. The BCA protein assay kit (Pierce) was employed to determine protein concentration. Lysate was separated by SDS–polyacrylamide gels and followed by transferring to a PVDF membrane (Millipore). The membrane was incubated with blocking solution containing 5% skimmed milk for more than 1 hr at room temperature and probed by 1 : 1000 dilutions of rabbit polyclonal antibodies against IRF-3, p-IRF-3, p38, p-p38, JNK, p-JNK, ERK1/2 and p-ERK1/2, respectively. After washing, membrane was incubated with secondary goat anti-rabbit antibody conjugated to HRP for 1 hr at room temperature. Immunoreactive proteins were visualized by chemiluminescence using the Western blotting detection system (Amersham Biosciences).
Cytokines release from RAW264.7 cells
RAW264.7 cells were incubated with HS (1 μg/ml) or LPS (10 ng/ml) at 37° in the absence and presence of paxilline (2 μg/ml), respectively. Cells treated with only PBS served as negative control. The concentrations of TNF-α (4 hr later) or interferon-β (IFN-β, 12 hr later) in the supernatants were analysed using the ELISA kits (R&D Systems, Minneapolis, MN) as instructed by the manufacturer.
Statistical and presentation of data
All experimental values are expressed as mean ± SE. Statistical comparisons were examined by the two-tailed Student's t-test. Differences in values were considered significant if P-values were < 0·05.
Results
HS stimulated expression of TLR4 and MaxiK in macrophages
We first detected the mRNA levels of TLR4 and MaxiK in mouse RAW264.7 macrophages under the treatment of the stimulators by PCR amplification of reverse-transcribed RNA using gene-specific primers for TLR4 and the pore-forming α-subunit of MaxiK, respectively. As shown in Fig. 1, treatment of HS or LPS significantly up-regulated mRNA and protein expression of MaxiK in macrophages, which were significantly suppressed by the addition of paxilline. However, paxilline was unable to exert similar inhibitory effects on substantially elevated mRNA and protein expression of TLR4 in response to HS or LPS stimulation.
Figure 1.
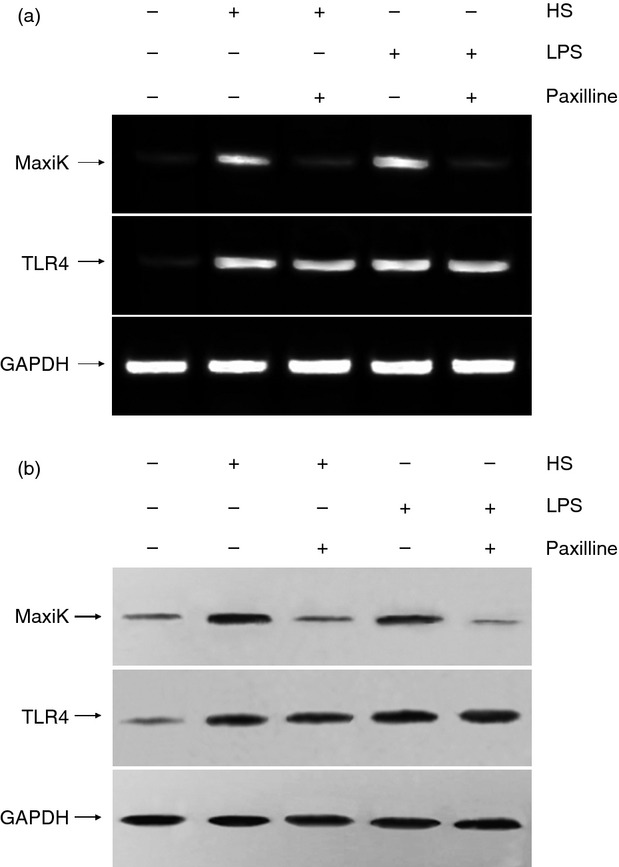
The mRNA and protein expression of MaxiK and Toll-like receptor 4 (TLR4) in RAW264.7 cells. RAW264.7 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum at 37° with 5% CO2. Cells were incubated for 6 hr at 37° with heparan sulphate (HS; 1 μg/ml) or lipopolysaccharide (LPS; 10 ng/ml) in the absence and presence of paxilline (2 μg/ml), respectively. (a) To determine the mRNA expression levels of MaxiK and TLR4, total cellular RNA was isolated from the cells and reverse transcribed with primers. After amplification by PCR, the products were separated by electrophoresis in 1% agarose gels containing 0·1 μg/ml ethidium bromide and visualized under ultraviolet light. (b) To detect the protein expression of MaxiK and TLR4, whole cell lysate was separated on SDS–polyacrylamide gels followed by transfer to a PVDF membrane. The membrane was incubated with blocking solution for more than 1 hr at room temperature and subsequently incubated with anti-MaxiK or TLR4 antibody (1 : 1000 dilutions) overnight at 4°, respectively. After 1-hr incubation at room temperature with horseradish peroxidase-conjugated secondary antibody (1 : 2000 dilution), the protein bands were visualized by the chemiluminescent detection system. Results of one out two experiments with similar results are shown.
HS-stimulated activation of caspase-1 in macrophages was inhibited by paxilline
Activation of the MaxiK channel is accompanied by an efflux of cellular potassium ions, which results in the elevated activity of caspase-1. Therefore, we accordingly measured the enzymatic activity of caspase-1 in HS- or LPS-challenged mouse macrophages. Compared with the unstimulated RAW264.7 cells, remarkable activation of caspase-1 was found when cells were co-incubated with HS. In contrast, TEA, as well as the TLR4 monoclonal antibody, almost completely abolished HS-induced caspase-1 activity. Although the MaxiK channel blocker paxilline could also effectively suppress HS-caused activation of caspase-1, the inhibitory effect was relatively weak compared with TEA or TLR4 monoclonal antibody (Fig. 2a). Moreover, a similar result was found in LPS-treated cells, as shown in Fig. 2(b).
Figure 2.
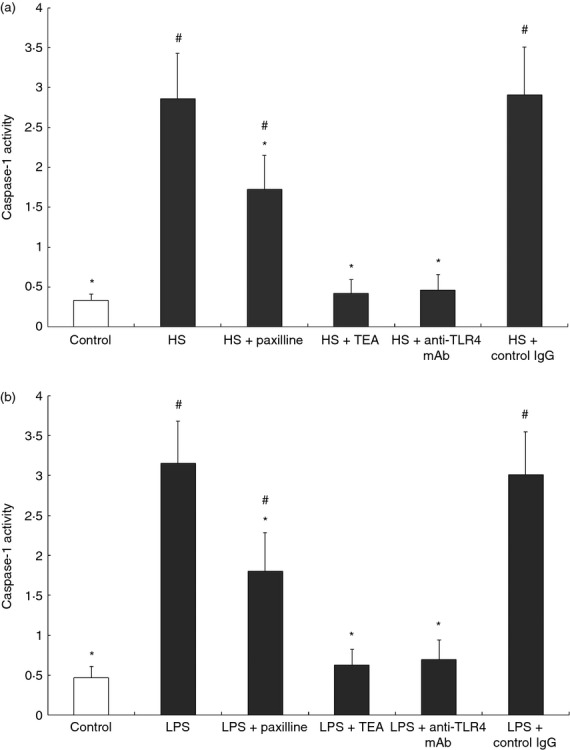
Heparan sulphate (HS) or lipopolysaccharide (LPS) -induced activation of caspase-1 in RAW264.7 cells was suppressed by paxilline. RAW264.7 cells were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum at 37° with 5% CO2. (a) Cells were incubated for 4 hr with HS (1 μg/ml) alone or in the presence of paxilline (2 μg/ml) or tetraethylammonium (TEA; 1 mg/ml), or anti-Toll-like receptor 4 (TLR4) monoclonal antibody (20 μg/ml), respectively. (b) Cells were also incubated for 4 hr with LPS (10 ng/ml) alone or in the presence of paxilline (2 μg/ml) or TEA (1 mg/ml), or anti-TLR4 monoclonal antibody (20 μg/ml), respectively. Cells treated with only PBS served as control. After cells (5 × 106 in 500 μl) were lysed and centrifuged at 10 000 g for 4 min, the supernatants were collected and incubated with 40 μm YVAD-pNA, followed by incubation at 37° for 2 hr. The caspase-1 activity was measured at 405 nm using a microplate reader. *P < 0·05, compared with HS or LPS; #P < 0·05, compared with control. The data (mean ± SE) of one out three experiments run in triplicates with similar results are shown.
HS-induced NF-κB, IRF-3, and p38 mitogen-activated protein kinase activation was inhibited by paxilline
It is known that NF-κB is a ubiquitous transcription factor that controls pro-inflammatory gene expression in response to LPS. In the present study, the effect of HS on NF-κB DNA binding in nuclear extracts prepared from RAW264.7 cells was evaluated. Furthermore, we asked whether the increased NF-κB DNA binding activity induced by HS was MaxiK dependent. Our data illustrated that HS also acted as a potent agonist of NF-κB and the MaxiK blocker paxilline substantially abolished either HS- or LPS-induced NF-κB activation (Fig. 3). Similarly, treatment of RAW264.7 cells with HS or LPS provoked markedly augmented phosphorylation of IRF-3, which was attenuated in the presence of paxilline. In addition, the exposure to HS or LPS increased the phosphorylated level of p38, JNK and ERK1/2. Blocking MaxiK channel by paxilline only reduced the phosphorylation of p38, but had no influence on JNK and ERK1/2 (Fig. 4).
Figure 3.
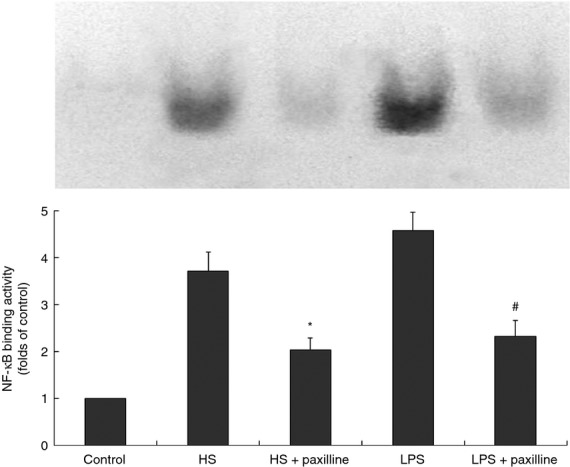
Heparan sulphate (HS) -induced nuclear factor-κB (NF-κB) activation in RAW264.7 cells was inhibited by paxilline. RAW264.7 cells were incubated for 1 hr at 37° with HS (1 μg/ml) in the absence and presence of paxilline (2 μg/ml), respectively. The nuclear proteins were extracted from macrophages and incubated with 1 μl of 32P-labelled oligonucleotide for 20 min. Binding reactions were then performed in 1 × binding buffer and 0·05 mg/ml poly(dI-dC) in a total volume of 10 μl. Then, 1 μl of loading buffer was added and the reactions were loaded on a 4% non-denaturing polyacrylamide gel. After electrophoresis the gels were dried for autoradiography. Cells treated with lipopolysaccharide (LPS; 10 ng/ml) in the absence and presence of paxilline, respectively, served as control. *P < 0·05, HS + paxilline versus HS; #P < 0·05, LPS + paxilline versus LPS. Results of one out two experiments with similar results are shown.
Figure 4.
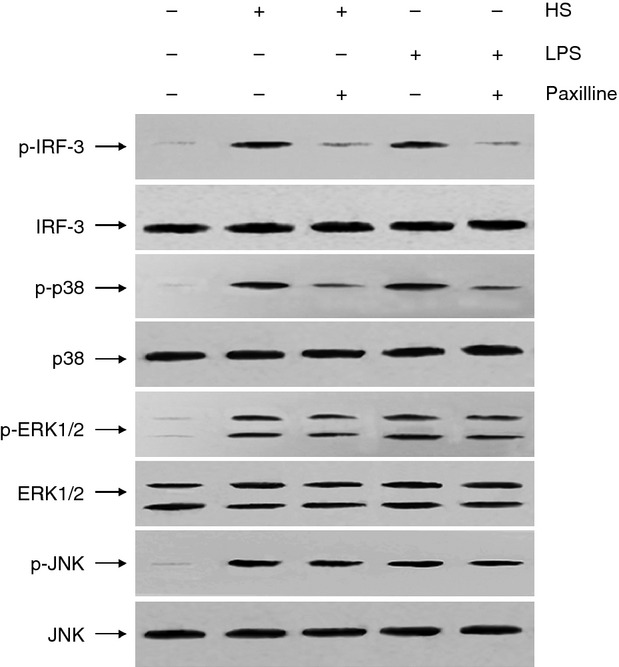
Heparan sulphate (HS) -induced interferon regulatory factor-3 (IRF-3) and p38 activation in RAW264.7 cells was suppressed by paxilline. The mouse RAW264.7 macrophages were incubated for 30 min at 37° with HS (1 μg/ml) in the absence and presence of paxilline (2 μg/ml), respectively. Subsequently whole cell lysate was prepared and separated by SDS-polyacrylamide gels, followed by transferring to a PVDF membrane. The membrane was incubated with blocking solution for more than 1 hr at room temperature and probed by 1 : 1000 dilutions of rabbit polyclonal antibodies against IRF-3, p-IRF-3, p38, p-p38, c-jun N-terminal kinase (JNK), p-JNK, extracellular signal-regulated kinase 1/2 (ERK1/2) and p-ERK1/2, respectively. After washing, membrane was incubated with secondary goat anti-rabbit antibody conjugated to horseradish peroxidase (HRP) for 1 hr at room temperature. Immunoreactive proteins were visualized by chemiluminescence using the Western blotting detection system. Cells treated with LPS (10 ng/ml) in the absence and presence of paxilline, respectively served as control. The results shown are from one out two experiments.
HS-caused cytokines release in macrophages was suppressed by paxilline
Tumour necrosis factor-α plays a pivotal role in the pro-inflammatory cytokine cascade through NF-κB and mitogen-activated protein kinase pathways.13 In contrast, the production of IFN-β is mediated by the IRF-3 signalling pathway.14 To further verify the inhibitory effect of MaxiK blockade on HS-induced inflammatory responses, we measured the amount of TNF-α and IFN-β produced by HS-treated RAW264.7 cells in the presence of paxilline. In agreement with the results above, the augmented production of TNF-α and IFN-β by macrophages in response to HS, as well as LPS, was efficiently diminished when the MaxiK channel was selectively blocked by paxilline (Fig. 5).
Figure 5.
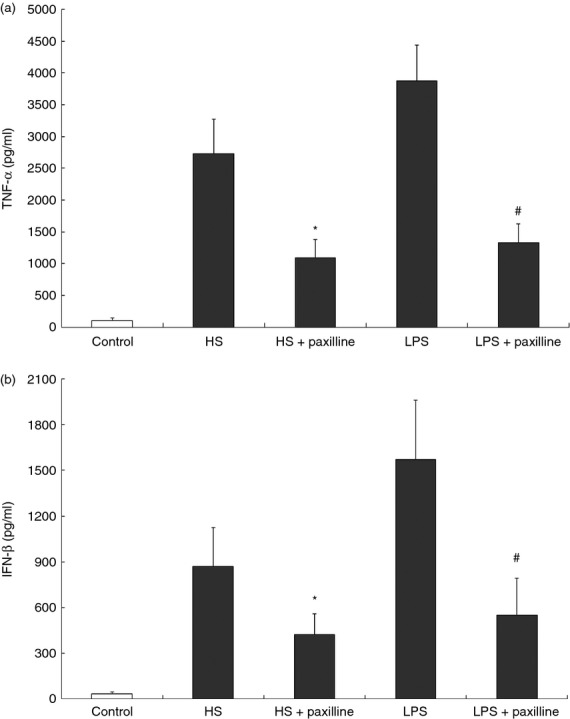
Heparan sulphate (HS) -stimulated production of cytokines in RAW264.7 cells was inhibited by paxilline. (a) RAW264.7 cells were incubated for 4 hr at 37° with HS (1 μg/ml) or lipopolysaccharide (LPS; 10 ng/ml) in the absence and presence of paxilline (2 μg/ml), respectively. After centrifugation, the concentrations of tumour necrosis factor-α (TNF-α) in the supernatants were analysed using an ELISA kit. (b) RAW264.7 cells received the same treatment as mentioned above. After 12 hr, the concentrations of interferon-β (IFN-β) were determined by ELISA. Cells treated with only PBS served as negative control. *P < 0·05, HS + paxilline versus HS; #P < 0·05, LPS + paxilline versus LPS. Data are representative of one out of three experiments.
Discussion
As an important receptor of the innate immune system, TLR4 can be activated by stimulation of cells with distinct agonists to evoke the inflammatory cascade reactions, finally leading to different complications. Activation of TLR4 signalling pathways by bacterial LPS is a critical upstream event in the pathogenesis of Gram-negative sepsis.15,16 Likewise, the aggravation of acute pancreatitis is largely attributed to the HS-induced systemic inflammatory responses through TLR4,5,7,8 suggesting that inhibition on inappropriate activation of TLR4 may be of potential therapeutic benefit in those complications. Previous research highlighted the association between MaxiK and TLR4-mediated transmembrane signal transduction, making this ion channel an attractive target for anti-TLR4 therapy.9,10 MaxiK channel blockage conferred significant inhibition against macrophage activation induced by LPS, the well-known exogenous ligand of TLR4, indicating the probability that MaxiK plays similar roles in HS-induced inflammatory cascade responses during acute pancreatitis. For this reason, the beneficial effects of MaxiK blockade by its inhibitor on HS-stimulated intracellular signal activation in a murine macrophage line were evaluated. Here, we found that HS unregulated the expression of MaxiK, and meanwhile provoked activation of the transcription factors, followed by increased production of cytokines, which could be effectively suppressed by paxilline, the blocker of the MaxiK channel. These results linked the MaxiK channel and intracellular inflammatory responses when macrophages were exposed to HS, and provided the likely evidence that MaxiK is required for the HS-mediated inflammatory cascades.
Caspase-1 is a cysteine protease, which is regarded as a key mediator involved in inflammation and apoptosis.17 In the present observation, the determined enzymatic activity of caspase-1 served as a marker of MaxiK activation based on the previous finding that opening of the MaxiK channel is accompanied by an efflux of potassium ions from the cell, resulting in caspase-1 activation.18 Paxilline, the specific MaxiK channel blocker, showed a significant, but not complete, inhibitory effect on HS-caused increase of cellular caspase-1 activity. Compared with paxilline, TEA, an unselected inhibitor of potassium channel, almost completely abolished the HS-caused caspase-1 activation, providing a clue that other potassium channels probably participate in HS-induced efflux of potassium ions and further activation of macrophages, which is in agreement with the findings from previous reports.19,20 Furthermore, the observation that TLR4 blockade by its antibody resulted in significant attenuation of the activity of caspase-1 just revealed the functional association of TLR4 and potassium ion channels in macrophages when they are activated by the ligands of TLR4.
The herein described ability of HS to activate the inflammatory signalling process via the MaxiK channel was similar to LPS, an exogenous ligand of TLR4. This finding suggested that a common mechanism of TLR4 initiation via MaxiK is probably shared by both molecules, in spite of the different molecular structures. Given this background, we further hypothesized that activation of the MaxiK channel by various stimulators is probably non-specific. From these findings and considerations, there are some interesting questions that need to be addressed by future research. For example, besides LPS and HS, are there any other molecules possessing similar potency to activate MaxiK? Is the modulation of MaxiK by these molecules involved in normal physiological functions? Actually, it is known that the MaxiK channel is distributed ubiquitously in a wide variety of mammalian tissues including smooth muscles, skeletal muscles, neurons, kidney and immune cells except in heart myocytes, function in modulating neurotransmitter release21,22 and endocrine secretion,23 tuning hair cells firing frequencies in the auditory system,24,25 and regulating vascular,26–28 urinary bladder29 and respiratory tone.30,31 Therefore, the adverse effects caused by MaxiK blockade should not be negligible, though blocking it will bring potential beneficial effects in controlling HS- or LPS-induced systemic inflammatory responses. In this case, one of our further goals is to develop specific inhibitors of MaxiK, which could antagonize the TLR4-mediated inflammatory cascade meanwhile without affecting the other normal physiological roles of this channel.
Taken together, the findings from our study have thrown some light on the elucidation of mechanisms underlying the HS-induced intracellular signalling cascade and, for the first time, have shown the possibility that blockade of the MaxiK channel might confer therapeutic benefits on the HS/TLR4-mediated activation of intracellular inflammatory cascades and even the resultant aggravation of acute pancreatitis. Further studies to elucidate the relationship between MaxiK and the function of other signal molecules in inflammatory signal transduction are warranted.
Acknowledgments
This work was supported by grants from the funds of the National Nature Science Foundation of China (81000185 to Jian-Dong Ren) and the Military Medical and Health Research Foundation of China (08z012 to Fu-Zhou Tian). Jian-Dong Ren and Kai-Hua Fan designed this study, for which Jian-Dong Ren and Fu-Zhou Tian secured funding. Bo-Tao Yu and Wei-Hua Jin performed experiments and analysed data. Jian-Dong Ren wrote the manuscript, which was revised by Kai-Hua Fan. Li Fan provided experimental support. Yong-Hong Tan and Long Cheng contributed methodological advice.
Glossary
- ERK
extracellular signal-regulated kinase
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HRP
horseradish peroxidase
- HS
heparan sulphate
- IFN-β
interferon-β
- IRF-3
interferon regulatory factor-3
- JNK
c-jun N-terminal kinase
- LPS
lipopolysaccharide
- NF-κB
nuclear factor- κB
- TEA
tetraethylammonium
- TLR4
Toll-like receptors 4
- TNF-α
tumour necrosis factor α
Disclosures
The authors declare no conflict of interests.
References
- 1.Beutler BA. TLRs and innate immunity. Blood. 2009;113:1399–407. doi: 10.1182/blood-2008-07-019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–80. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 3.Brightbill HD, Modlin RL. Toll-like receptors: molecular mechanisms of the mammalian immune response. Immunology. 2000;101:1–10. doi: 10.1046/j.1365-2567.2000.00093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vogel SN, Fitzgerald KA, Fenton MJ. TLRs: differential adapter utilization by Toll-like receptors mediates TLR-specific patterns of gene expression. Mol Interv. 2003;3:466–77. doi: 10.1124/mi.3.8.466. [DOI] [PubMed] [Google Scholar]
- 5.Johnson GB, Brunn GJ, Platt JL. Cutting edge: an endogenous pathway to systemic inflammatory response syndrome (SIRS)-like reactions through Toll-like receptor-4. J Immunol. 2004;172:20–4. doi: 10.4049/jimmunol.172.1.20. [DOI] [PubMed] [Google Scholar]
- 6.Lee KM, Seong SY. Partial role of TLR4 as a receptor responding to damage associated molecular pattern. Immunol Lett. 2009;125:31–9. doi: 10.1016/j.imlet.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 7.Liu H, Li Y, Wang L, Chen H, Guan J, Zhou Z. Aggravation of acute pancreatitis by heparan sulfate in mice. Scand J Gastroenterol. 2009;44:626–32. doi: 10.1080/00365520902745047. [DOI] [PubMed] [Google Scholar]
- 8.Axelsson J, Norrman G, Malmström A, Weström B, Andersson R. Initiation of acute pancreatitis by heparan sulphate in the rat. Scand J Gastroenterol. 2008;43:480–9. doi: 10.1080/00365520701733814. [DOI] [PubMed] [Google Scholar]
- 9.Blunck R, Scheel O, Müller M, Brandenburg K, Seitzer U, Seydel U. New insights into endotoxin-induced activation of macrophages: involvement of a K+ channel in transmembrane signaling. J Immunol. 2001;166:1009–15. doi: 10.4049/jimmunol.166.2.1009. [DOI] [PubMed] [Google Scholar]
- 10.Papavlassopoulos M, Stamme C, Thon L, Adam D, Hillemann D, Seydel U, Schromm AB. MaxiK blockade selectively inhibits the lipopolysaccharide-induced I κB-α/NF-κB signaling pathway in macrophages. J Immunol. 2006;177:4086–93. doi: 10.4049/jimmunol.177.6.4086. [DOI] [PubMed] [Google Scholar]
- 11.Gorbet MB, Sefton MV. Endotoxin: the uninvited guest. Biomaterials. 2005;26:6811–7. doi: 10.1016/j.biomaterials.2005.04.063. [DOI] [PubMed] [Google Scholar]
- 12.Schreiber E, Matthias P, Müller MM, Schaffner W. Rapid detection of octamer binding proteins with “miniextracts”, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malleo G, Mazzon E, Siriwardena AK, Cuzzocrea S. Role of tumor necrosis factor-α in acute pancreatitis: from biological basis to clinical evidence. Shock. 2007;28:130–40. doi: 10.1097/shk.0b013e3180487ba1. [DOI] [PubMed] [Google Scholar]
- 14.Sato M, Suemori H, Hata N. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity. 2000;13:539–48. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 15.Roger T, Froidevaux C, Le Roy D, et al. Protection from lethal Gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc Natl Acad Sci USA. 2009;106:2348–52. doi: 10.1073/pnas.0808146106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leon CG, Tory R, Jia J, Sivak O, Wasan KM. Discovery and development of toll-like receptor 4 (TLR4) antagonists: a new paradigm for treating sepsis and other diseases. Pharm Res. 2008;25:1751–61. doi: 10.1007/s11095-008-9571-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol. 2003;15:725–31. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 18.Kahlenberg JM, Dubyak GR. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am J Physiol Cell Physiol. 2004;286:C1100–8. doi: 10.1152/ajpcell.00494.2003. [DOI] [PubMed] [Google Scholar]
- 19.Jo HY, Kim SY, Lee S, Jeong S, Kim SJ, Kang TM, Lee KY. Kir3.1 channel is functionally involved in TLR4-mediated signaling. Biochem Biophys Res Commun. 2011;407:687–91. doi: 10.1016/j.bbrc.2011.03.076. [DOI] [PubMed] [Google Scholar]
- 20.Qiu MR, Campbell TJ, Breit SN. A potassium ion channel is involved in cytokine production by activated human macrophages. Clin Exp Immunol. 2002;130:67–74. doi: 10.1046/j.1365-2249.2002.01965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu H, Shao LR, Chavoshy S, et al. Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J Neurosci. 2001;21:9585–97. doi: 10.1523/JNEUROSCI.21-24-09585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robitaille R, Charlton MP. Presynaptic calcium signals and transmitter release are modulated by calcium-activated potassium channels. J Neurosci. 1992;12:297–305. doi: 10.1523/JNEUROSCI.12-01-00297.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Findlay I, Dunne MJ, Petersen OH. High-conductance K+ channel in pancreatic islet cells can be activated and inactivated by internal calcium. J Membr Biol. 1985;83:169–75. doi: 10.1007/BF01868748. [DOI] [PubMed] [Google Scholar]
- 24.Art JJ, Wu YC, Fettiplace R. The calcium-activated potassium channels of turtle hair cells. J Gen Physiol. 1995;105:49–72. doi: 10.1085/jgp.105.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu YC, Art JJ, Goodman MB, Fettiplace RP. A kinetic description of the calcium-activated potassium channel and its application to electrical tuning of hair cells. Prog Biophys Mol Biol. 1995;63:131–58. doi: 10.1016/0079-6107(95)00002-5. [DOI] [PubMed] [Google Scholar]
- 26.Brenner R, Peréz GJ, Bonev AD, et al. Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–6. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- 27.Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–5. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- 28.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–7. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 29.Herrera GM, Etherton B, Nausch B, Nelson MT. Negative feedback regulation of nerve-mediated contractions by KCa channels in mouse urinary bladder smooth muscle. Am J Physiol Regul Integr Comp Physiol. 2005;289:R402–9. doi: 10.1152/ajpregu.00488.2004. [DOI] [PubMed] [Google Scholar]
- 30.Kume H, Takai A, Tokuno H, Tomita T. Regulation of Ca2+-dependent K+-channel activity in tracheal myocytes by phosphorylation. Nature. 1989;341:152–4. doi: 10.1038/341152a0. [DOI] [PubMed] [Google Scholar]
- 31.Kotlikoff MI. Potassium channels in airway smooth muscle: a tale of two channels. Pharmacol Ther. 1993;58:1–12. doi: 10.1016/0163-7258(93)90064-k. [DOI] [PubMed] [Google Scholar]