

Abstract
The binding of NKG2D to its ligands strengthens the cross-talk between natural killer (NK) cells and dendritic cells, particularly at early stages, before the initiation of the adaptive immune response. We found that retinoic acid early transcript-1ε (RAE-1ε), one of the ligands of NKG2D, was persistently expressed on antigen-presenting cells in a transgenic mouse model (pCD86-RAE-1ε). By contrast, NKG2D expression on NK cells, NKG2D-dependent cytotoxicity and tumour rejection, and dextran sodium sulphate-induced colitis were all down-regulated in this mouse model. The down-regulation of NKG2D on NK cells was reversed by stimulation with poly (I:C). The ectopic expression of RAE-1ε on dendritic cells maintained NKG2D expression levels and stimulated the activity of NK cells ex vivo, but the higher frequency of CD4+ NKG2D+ T cells in transgenic mice led to the down-regulation of NKG2D on NK cells in vivo. Hence, high levels of RAE-1ε expression on antigen-presenting cells would be expected to induce the down-regulation of NK cell activation by a regulatory T-cell subset.
Keywords: natural killer, NKG2D, regulation, retinoic acid early transcript-1, transgenic mouse
Introduction
Innate and adaptive immunity are coordinated in protective immune responses. The cross-talk between dendritic cells (DCs) and natural killer (NK) cells has an impact not only on the innate immune response, but also on the initiation, development and outcome of the acquired immune response.1,2 The DCs activate resting NK cells by direct cell-to-cell contact (e.g. NKG2D ligand–NKG2D, CD70–CD28, CD48–2B43,4), and the secretion of cytokines [e.g. interferon-α (IFN-α), tumour necrosis factor-α (TNF-α), interleukin-12 (IL-12), IL-15, IL-18], and they increase anti-tumour immunity by triggering NK cell functions.5,6 Natural killer cells can promote DC maturation by producing IFN-γ or TNF-α, and they can kill immature DCs through signals mediated by NKp30 or TRAIL.7,8
MHC class I-related chain A or B (MICA/B) and human cytomegalovirus UL16-binding proteins (ULBP1–6) are ligands of human NKG2D, which transduces activating signals in NK cells.9,10 Mouse NKG2D ligands include retinoic acid early induced transcript-1 (RAE-1α, -β, -γ, -δ, -ε), H60 and murine ULBP-like transcript 1 (MULT-1).11–13 MICA/B antigens are expressed predominantly on the gastrointestinal epithelium, tumour cells, some virus-infected cells and other stressed cells. Monocytes and immature DCs display low-level MICA expression, but some infections, such as Mycobacterium tuberculosis14 and influenza virus infections,15 and some cytokines (e.g. TNF-α, IL-15) and lipopolysaccharide up-regulate MICA expression in DCs and macrophages.16,17
The ectopic expression of MICA on cancer cells promotes NK cell activation ex vivo, but sustained MICA expression on either cancer cells or normal cells down-regulates NKG2D expression on NK cells and CD8+ T cells, as demonstrated in both H2-Kb : MICA18 and β-actin : Rae-1ε transgenic mice.19 In addition, soluble MICA shedding from cancer cells also induces NKG2D down-regulation.20,21 The persistent binding of NKG2D to its ligands inhibits NKG2D binding to DAP10, but also decreases the activation of downstream signal molecules.22 We generated a mouse transgenic for the RAE-1ε gene under control of the CD86 promoter, to investigate the effects on NKD2D expression and NK cell function of the persistent expression of high levels of RAE-1 on DCs. RAE-1ε was strongly expressed on DCs, B cells and macrophages in these transgenic mice. NKG2D expression on NK cells was down-regulated to a moderate level and CD4+ NKG2D+ T cells were induced to mediate regulatory functions through the production of transforming growth factor-β (TGF-β) in CD86-RAE-1ε mice.
Materials and methods
DNA construct and the generation of CD86-RAE-1 transgenic mice
The promoter of the mouse CD86 gene (1·3 kb) was amplified with primers (forward: 5′-CCGACTAGTTAGAAGCTAGAGGAGTCAAGGAT-3′; reverse: 5′-CGCAAGCTTGTCTGG TTGTTCAAGTCCGT-3′) based on the CD86 promoter sequences in GenBank (GI: 56406302) flanked with SpeI and HindIII sites. The promoter region of the pcDNA3.1 (+) vector was replaced with CD86 promoter gene sequences. RAE-1ε cDNA (supplied by Lewis L Lanier; University of California, San Francisco, CA) was inserted into CD86-pcDNA3.1 (+), between the KpnI and NotI sites.
The transgene vector was digested with SpeI and DraIII, and the resulting 2·6-kb fragment including the CD86-RAE-1 transgene and the bovine growth hormone polyadenylation signal was isolated, for microinjection into the pronuclei of one-cell embryos from C57BL/6 mice. Neonatal mice were bred and maintained under specific pathogen-free conditions in the animal model research centre of Nanjing University.
For the screening of founder mice, tail DNA was isolated by the SDS–proteinase K method. Founders were genotyped by PCR, with specific primers spanning the CD86-MICA transgene. The oligonucleotide 5′-TAGCAAGTCAATTTCCGCATAC-3′ was used as a forward primer, and 5′-TTCCCAGGTGGCACTAGG-3′ was used as a reverse primer. Founder mice were crossed with normal C57BL/6 mice; littermates in the progeny were then crossed to generate homozygous mice. We finally obtained three homozygous lines, lines 48, 1 and 17, which we used for subsequent studies.
Analysis of RAE-1ε expression by Western blotting
About 2 mg of liver tissues were chopped and ground in tissue lysis buffer (Dakewei, Shenzhen, China). The resulting whole-cell lysates were centrifuged and separated by electrophoresis in 1-mm-thick 5–12% Tris–glycine gels. The bands obtained were then transferred to PVDF membranes. The PVDF membranes were blocked by incubation with 5% (weight/volume) non-fat dry milk powder in Tris-buffered saline (TBS). Anti-RAE-1 monoclonal antibody (mAb) (AF1136; R&D Systems, Minneapolis, MN) was diluted 1/500 in TBST (TBS-Tween) buffer supplemented with 2 mg/ml BSA and incubated with the membrane for 2 hr at 37°. The PVDF membranes were then incubated with horseradish peroxidase-conjugated rabbit anti-goat IgG or goat anti-rabbit IgG (1/3000 in TBST; Invitrogen, Carlsbad, CA) for 1 hr. Membranes were thoroughly washed in TBST, and immunoreactive bands were visualized by incubation with a chemiluminescence reagent (Dakewei).
RT-PCR analysis
Natural killer cells were isolated, by sorting CD3− NK1.1+ cells from the spleen of transgenic or wild-type mice by flow cytometry. RNA was extracted from NK cells and transcribed as previously described (TaKaRa, Otsu, Shiga, Japan). The primer sequences used for NKG2D-L and NKG2D-S amplification were 5′-CAGGAAGCAGAGGCAGATTATCTC-3′ (5′NKG2D-L) and 5′-TCCCTTCTCTGCTCAGAG-3′ (5′NKG2D-S). The common 3′ NKG2D primer was 5′-TTACACCGCCCTTTTCATGCAGATG-3′. The other primers used were: DAP10 forward (5′-ATGGACCCCCCAGGCTACCTC-3′) and DAP10 reverse (5′-TCAGCCTCTGCCAGGCATGTT-3′); DAP12 forward (5′-ACTTTCCCAAGATGCGAC-3′) and DAP12 reverse (5′-GTACCCTGTGGATCTGTA-3′); β-actin forward (5′-TACCACTGGCATCGTGATGGACT-3′) and β-actin reverse (5′-TCCTTCTGCATCCTGCGGCAAT-3′).
Splenocyte isolation and stimulation
The spleen was isolated and homogenized. Splenocytes were then obtained by passing the homogenate through a filter with 40-μm pores (BD Biosciences, San Jose, CA). For the sorting of B, CD4+ T and CD11c+ cells, we used magnetic beads coated with antibodies against CD19, CD4 and CD11c, respectively (Miltenyi Biotec, Bergisch Gladbach, Germany). CD4+ NKG2D+ T cells, CD4+ NKG2D− T cells and NK1.1+ CD3− cells were obtained by flow cytometry sorting (BD FACSAria). Spleen DCs were isolated on the basis of their binding to magnetic beads bearing the anti-CD11c antibody. They were cultured with NK cells in a 1 : 2 ratio. CD4+ NKG2D+ T cells and CD4+ NKG2D− T cells were also mixed with NK cells at a ratio of 1 : 2 and incubated overnight.
Antibodies and flow cytometry
The antibodies for flow cytometry were obtained from Biolegend (San Diego, CA) or eBioscience (San Diego, CA). We used antibodies against CD3 (17A2), CD8 (53.67), CD4 (GK1.5), NK1.1 (PK136), CD49b (DX5), CD19 (6D5), Gr-1 (RB6-8C5), CD11c (N418), CD11b (M1/70), CD69 (LG.3A10), NKG2D (CX5), CD107a (1D4B), IFN-γ (XMG1.2), 2B4 (458.1), NKp46 (29A1.4), NKG2A (16A11), TGF-β (TW7-16B4), Ly49D (4E5), Ly49H (3D10) and KLRG1 (2F1). Neutralized TGF-β antibody (1D11), RAE-1ε mAb (205001) and recombinant NKG2D-immunoglobulin were obtained from R&D Systems. Alexa 488-conjugated anti-human IgG was obtained from Invitrogen. All cells were blocked by incubation with mouse serum and then incubated with antibodies at 4° for 30 min. They were then washed and analysed by flow cytometry with cellquest (BD FACSCalibur) or FACS diva (BD FACSAria software).
Immunofluorescence staining
Mouse tissues were embedded in optimal cutting temperature compound and snap-frozen in liquid nitrogen for the cutting of cryostat sections. The sections were fixed in 4% paraformaldehyde or cold acetone, blocked by incubation with donkey serum and stained by incubation with goat anti-mouse RAE-1ε antibody (AF1136; R&D Systems) and rat anti-mouse I-A/I-E antibody (M5/114.15.2) at 4° overnight. The sections were then washed in PBS and incubated with Alexa 546-labelled donkey anti-goat secondary antibody and Alexa 488-labelled donkey anti-rat antibody at room temperature for 60 min. The sections were thoroughly washed in PBS and covered with 30 μl of mounting medium containing DAPI (Vector Laboratories, Burlingame, CA). Fluorescence was detected with an Eclipse E600 microscope (Nikon).
NK cell degranulation and cytotoxicity
For the detection of NK cell degranulation, we added fluorochrome-conjugated anti-CD107a mAb or isotype control antibody when the effectors were mixed with target cells. After 2 hr of co-incubation, we added monensin (GolgiStop; BD Biosciences) at a dilution of 1 : 100 and incubated the mixture for another 2 hr. Surface staining was performed by incubating cells with anti-CD3 and anti-NK1.1 antibodies for 30 min on ice, and splenocytes were analysed by flow cytometry. NK cells were co-cultured with YAC-1 cells for 4 hr and cytotoxicity was estimated in the lactate dehydrogenase release assay, according to the manufacturer's protocol.
Intracellular staining
Interferon-γ production was assessed with an intracellular staining kit (eBioscience). In brief, splenocytes were cultured with PMA/ionomycin, Ba/F3 cells or Ba/F3-RAE cells, in the presence of brefeldin A, for 4 hr at 37°. The NK cells were then stained by incubation with phycoerythrin-conjugated anti-CD3, Alexa-647-conjugated anti-NK1.1 antibodies, for 30 min at 4°. Cells were then fixed, permeabilized, stained with FITC-labelled anti-IFN-γ mAb or isotype antibody and analysed by flow cytometry.
In vivo cytotoxicity
In vivo cytotoxicity assays were carried out as follows. For target cell preparations, erythrocytes were removed from spleen suspensions by osmotic lysis, and the remaining cells were then washed and labelled with CFSE (5 μm; Invitrogen) for cells from transgenic mice, or with PKH26 (2 μm; Sigma, St Louis, MO) for cells from control mice. For intravenous injection, we mixed approximately equal numbers of cells from each population and inoculated each mouse with 1 × 107 cells in 100 ml of PBS. Twelve hours after the injection, we analysed spleen cell suspensions by flow cytometry, to determine the ratio of cells displaying CFSE and PKH26 fluorescence. For specific lysis, we used the following formulae: ratio = (percentage PKH26/percentage CFSE); and percentage specific lysis = [1 − (ratio of injected mix/recovery ratio)] × 100. For NK cell activation in vivo, we injected 100 μg poly (I:C) (Sigma) intraperitoneally into the mice 12 hr before the experiments.
Enzyme-linked immunosorbent assays
Serum soluble RAE-1 concentration was determined with a sandwich ELISA kit from R&D Systems. In brief, we added 50 μl of serum to the plates, which we had previously coated with RAE-1 antibody by incubation for 1 hr at 37°. We then washed the plates with PBS and added another horseradish peroxidase-labelled antibody. The plates were thoroughly washed with PBS, and substrate and stop solution were then added separately, in that order. We then determined the absorbance at 450 nm (A450) and determined the concentration of RAE-1 in the serum from a standard curve. Serum anti-RAE-1 antibody was measured by an indirect ELISA method. In brief, we coated plastic wells with recombinant mouse RAE-1ε (2 μg; R&D Systems) by incubation at 4° overnight. We then added a serum sample to each well. The plates were washed with PBS and horseradish peroxidase-labelled goat anti-mouse IgG was added. The plates were then thoroughly washed and substrate was added. We determined the A450 value after adding stop solution to the wells.
Mouse models
B16BL6 or B16BL6-RAE cells (n = 6, 2 × 106 cells) were implanted subcutaneously into the back of both CD86-RAE transgenic mice and control mice. Tumour area was determined by measuring the dimensions of the tumour with digital calipers and then calculating the area from the following formula: A = length × width. Colitis was induced by the administration of dextran sodium sulphate (DSS; 2·5% weight/volume; molecular weight 36 000–50 000; MP Biomedicals, Santa Ana, CA) in the drinking water for 7 days (n = 5). Body weight was recorded daily.
Statistical analysis
Differences between groups were analysed with Student's t-test. Values of P < 0·05 were considered significant (GraphPad, GraphPad Software, Inc., La Jolla, CA).
Results
Generation and identification of CD86-RAE-1ε transgenic mice
A transgene fragment containing the coding sequence of RAE-1ε and the CD86 promoter (Fig. 1a) was introduced into the germline of C57BL/6 mice. Offspring displaying high levels of RAE-1 expression in liver tissues were selected (Fig. 1b), and three transgenic lines (lines 48, 1, 17) were established by repeated backcrossing with C57BL/6 mice. All CD86-RAE-1ε mice appeared healthy, with no overt signs of autoimmunity or infertility. No significant differences were found between CD86-RAE-1ε mice and wild-type mice, in the frequencies of CD4+ T cells, CD8+ T cells and NK lymphocytes in the spleen (Fig. 2a and see Supplementary material, Fig. S1).
Figure 1.

Generation and identification of CD86-RAE transgenic mice. The CD86-RAE-1ε transgene encompassing the CD86 promoter, the retinoic acid early transcript-1ε (RAE-1ε) coding sequence, and the bovine growth hormone polyadenylation signal (2·6-kb) (a). Founder mice identified by PCR were further screened by Western blotting. Top, RAE-1ε expression was detected in liver tissues F0-1, -12, -17, -48, -40 mice. Bottom, α-tubulin blot (b). RAE-1ε expression on spleen lymphocytes, as identified by flow cytometry. Grey area: isotype control; dotted line: control mice; solid line: CD86-RAEε mice (c). Immunofluorescence histology of the thymus, intestine, liver, lung and kidney stained with anti-I-A/I-E antibody (green) and anti-RAE-1 antibody (red) and counterstained with DAPI. Similar patterns were obtained for the mice of lines 48, 1 and 17. The photographs shown are from a line 48 mouse, and were taken at a magnification of × 200 (d). Changes in RAE-1ε expression on myeloid cells and lymphocytes in response to overnight stimulation with poly (I:C) ex vivo (50 ng/ml) (e) and in vivo (100 μg) (f). Grey area: before stimulation; red line: after stimulation. The figures in black and red show the results before and after stimulation, respectively.
Figure 2.

Weak NKG2D-dependent function of natural killer (NK) cells in CD86-RAE-1ε mice. NKG2D expression on freshly isolated NK cells, gated on CD3− NK1.1+ cells (a). Transcript levels for NKG2D-L, NKG2D-S, DAP10, and DAP12 in NK cells from wild-type and transgenic mice (b). Killing activities were assessed by both CD107a staining and the lactate dehydrogenase release assay (c). After the incubation of NK cells with Ba/F3-RAE or Ba/F3 cells, CD107a expression on NK cells was assessed by flow cytometry (d). Splenocytes from transgenic mice or wild-type mice were stained with CFSE and PKH-26, respectively, mixed in equal proportions and injected intravenously into recipient mice. After 12 hr, fluorescence ratios were determined for the cells recovered from spleens, by flow cytometry (e). Interferon-γ (IFN-γ) production by NK cells from transgenic mice and wild-type mice stimulated with PMA/ionomycin, Ba/F3 or Ba/F3-RAE cells. Results are expressed as means ± SD. Asterisks indicate P < 0·05 (f).
Most CD11c+, CD11b+, Gr+ and CD19+ cells from CD86-RAE-1ε mice strongly expressed RAE-1ε, whereas CD3+ and NK1.1+ cells did not express RAE-1ε (Fig. 1c). I-A/I-E+ cells in the thymus, intestine and liver also displayed high levels of RAE-1 expression in transgenic mice. As expected, RAE-1 was expressed on epithelial cells in the intestine, but not on the epithelial cells of the liver, lung and kidney of wild-type or transgenic mice. I-A/I-E+ cells in the lung and kidney of transgenic mice did not express RAE-1 either, indicating that CD86 promoter activity was weak in the DCs and macrophages of these organs in physiological conditions (Fig. 1d).
We checked that RAE-1ε expression was regulated by the CD86 promoter, by analysing the effect of inflammatory conditions on RAE-1 expression. RAE-1 expression was evaluated on spleen-derived myeloid cells and lymphocytes before and after poly (I:C) stimulation. Myeloid cells (CD11c+, CD11b+, Gr-1+ cells) from both transgenic and wild-type mice displayed a sharp increase in RAE-1ε expression following ex vivo poly (I:C) stimulation. Lymphocytes (CD19+, CD3+, NK1.1+ cells) from transgenic mice displayed a small increase in RAE-1ε expression, whereas B and NK cells from control mice showed no significant change in RAE-1ε expression. RAE-1ε expression on the T cells of control mice also increased slightly, as CD86 could be induced on T cells (Fig. 1e). When transgenic mice and control mice were stimulated with poly (I:C) in vivo the lymphocytes of transgenic mice displayed a slight increase in RAE-1ε expression, whereas no significant change in expression level was observed on the lymphocytes of control mice. RAE-1ε expression on the myeloid cells of control mice increased to moderate levels following poly (I:C) stimulation in vivo, whereas no significant change was observed for the myeloid cells of transgenic mice (Fig. 1f).
Down-regulation of NKG2D on the NK cells in CD86-RAE-1ε mice
We analysed NKG2D expression on NK cells and CD8+ T cells in more detail. The proportion of NK cells expressing NKG2D was about 50% in CD86-RAE-1ε mice, whereas that in wild-type mice was close to 100% (Fig. 2a). In contrast, the proportion of CD8+ T cells expressing NKG2D did not differ significantly between wild-type and transgenic mice (see Supplementary material, Fig. S2). We also analysed the levels of transcripts for NKG2D-L, NKG2D-S, DAP10 and DAP12 in NK cells from transgenic and control mice. Transcript levels for NKG2DL, NKG2DS, DAP10 and DAP12 were all moderately lower in transgenic mice than in control mice (Fig. 2b).
The NK cell cytotoxicity to YAC-1 cells was also lower in transgenic mice than in control mice (Fig. 2c). YAC-1 cells express multiple ligands for NK cell activation. We therefore evaluated NK cell degranulation in response to stimulation with Ba/F3 cells negative for the NKG2D ligand and with stably transfected Ba/F3-RAE cells.23 We found that fewer NK cells from transgenic mice than from control mice entered a state of degranulation in response to stimulation with Ba/F3-RAE cells, whereas no significant difference in degranulation was observed between wild-type and control mice following stimulation with Ba/F3 cells (Fig. 2d). Hence, the NK cells of transgenic mice had lower levels of NKG2D expression and NKG2D-dependent cytotoxicity.
We also analysed NK cell cytotoxicity to RAE-1+ target cells in vivo. Splenocytes from transgenic mice or wild-type mice were stained with CFSE and PKH-26 respectively, and a mixture of equivalent numbers of the two types of cells was injected intravenously into mice. Twelve hours later we recovered spleen cells and calculated fluorescence ratios, to determine cytotoxicity. Before injection, splenocytes from transgenic mice were shown to be more susceptible to attack by IL-2-activated NK cells than those from wild-type mice, and this killing was blocked by NKG2D-immunoglobulin protein (see Supplementary material, Fig. S3). RAE-1-targeted natural cytotoxicity in CD86-RAE-1ε mice was significantly lower than that in wild-type mice (Fig. 2e). The IFN-γ production of NK cells from CD86-RAE-1ε mice was not significantly affected by stimulation with PMA/ionomycin, but decreased in response to Ba/F3-RAE cell stimulation (Fig. 2f).
Impaired NKG2D-dependent NK cell function in CD86-RAE-1ε mice
We then investigated whether the down-regulation of NKG2D on NK cells in CD86-RAE-1ε mice was a result of their particular microenvironment. Splenocytes from wild-type mice were stained with CFSE and used for adoptive transfer into transgenic or wild-type mice. NK cells transferred into CD86-RAE-1ε mice displayed a decrease in NKG2D expression to levels similar to those on the recipient cells, whereas NK cells transferred into wild-type mice retained their original level of NKG2D expression (Fig. 3a). In addition, CD86-RAE-1ε mice displayed lower levels of immune surveillance against the tumour formed by B16-RAE cells, whereas no significant difference was observed for the surveillance of growth for B16 cells (Fig. 3b,c).
Figure 3.
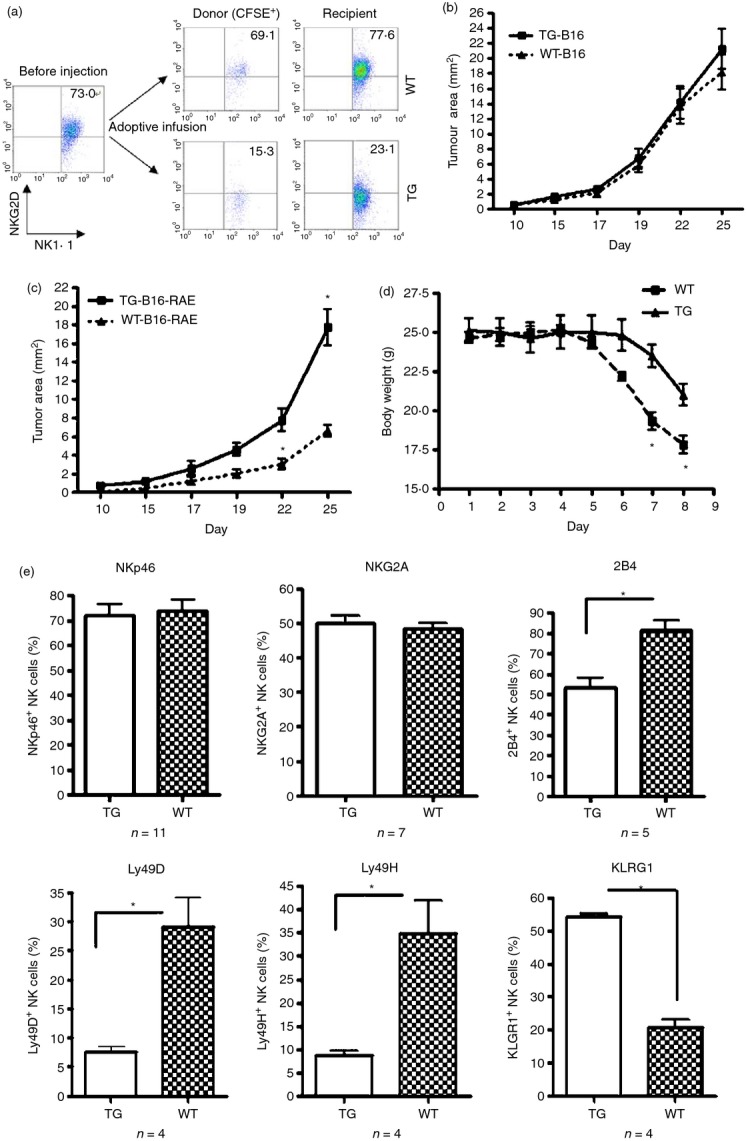
The adoptive transfer of normal natural killer (NK) cells into CD86-RAE-1ε mice decreased NKG2D expression. Splenocytes from wild-type mice were stained with CFSE and intravenously injected into transgenic mice or wild-type mice. Fluorescent cells were recovered after 12 hr. Frequencies of CD3− NK1.1+ NKG2D+ cells in donor mice were analysed before and after injection, and the frequencies of spleen CD3− NK1.1+ NKG2D+ cells in recipient mice were also determined (a). B16BL6-RAE (b) or B16BL6 cells (c) (2 × 106) were injected subcutaneously into the back of the animal (n = 6). Tumour area was determined daily. CD86-RAE-1ε mice and wild-type mice (n = 5) were supplied with drinking water containing 2·5% dextran sodium sulphate (DSS). Body weight was measured daily (d). Expression levels of NKp46, NKG2A, 2B4, Ly49D, Ly49H and KLRG1 on NK cells, identified by gating on CD3− NK1.1+ cells (e). Results are expressed as means ± SD. Asterisks indicate P < 0·05. Each experiment was carried out three times.
Dendritic cells in the intestines of transgenic mice strongly expressed RAE-1. We therefore investigated the local immunological contribution of this expression. The experimental disease-inducing DSS-colitis protocol was performed in both transgenic and wild-type mice.24 The onset of colitis occurred much later in DSS-treated CD86-RAE-1ε mice than in DSS-treated wild-type C57BL/6 mice (Fig. 3d). We also analysed the expression of certain receptors, such as NKp46, NKG2A, 2B4, Ly49D, Ly49H and KLRG1, on NK cells. Transgenic mice had lower frequencies of 2B4+, Ly49D+ and Ly49H+ NK cells than wild-type mice, but higher frequencies of KLRG1+ NK cells, whereas no difference in the frequencies of NKp46+ NK cells or NKG2A+ NK cells was found between transgenic and wild-type mice (Fig. 3e). The phenotypes of NK cells in transgenic mice indicated that NK cell function was down-regulated by both NKG2D-dependent and NKG2D-independent mechanisms.
NKG2D down-regulation in CD86-RAE-1ε mice can be reversed by poly (I:C) stimulation
We investigated the reversibility of NKG2D down-regulation on NK cells in CD86-RAE-1ε mice, by assessing phenotypic and functional changes to NK cells following stimulation with poly (I:C) ex vivo and in vivo. The cytotoxicity of NK cells to YAC-1 cells in transgenic mice was restored to a level similar to that of NK cells in wild-type mice, by poly (I:C) stimulation ex vivo (Fig. 4a). Meanwhile, no significant difference was found between transgenic and wild-type mice in terms of the increase in the killing of Ba/F3-RAE or Ba/F3 cells by NK cells following poly (I:C) treatment (Fig. 4b). With poly (I:C) stimulation, NKG2D and 2B4 expression on the NK cells of transgenic mice increase to wild-type levels (Fig. 4c). If CD86-RAE-1ε and wild-type mice received peritoneal injections of poly (I:C) 12 hr before the experiment, NK cell cytotoxicity to RAE-1+ targets was similar in CD86-RAE-1ε mice and wild-type mice in vivo (Fig. 4d). Hence, NKG2D down-regulation and the resulting cytotoxicity in transgenic mice were reversed by poly (I:C) stimulation.
Figure 4.
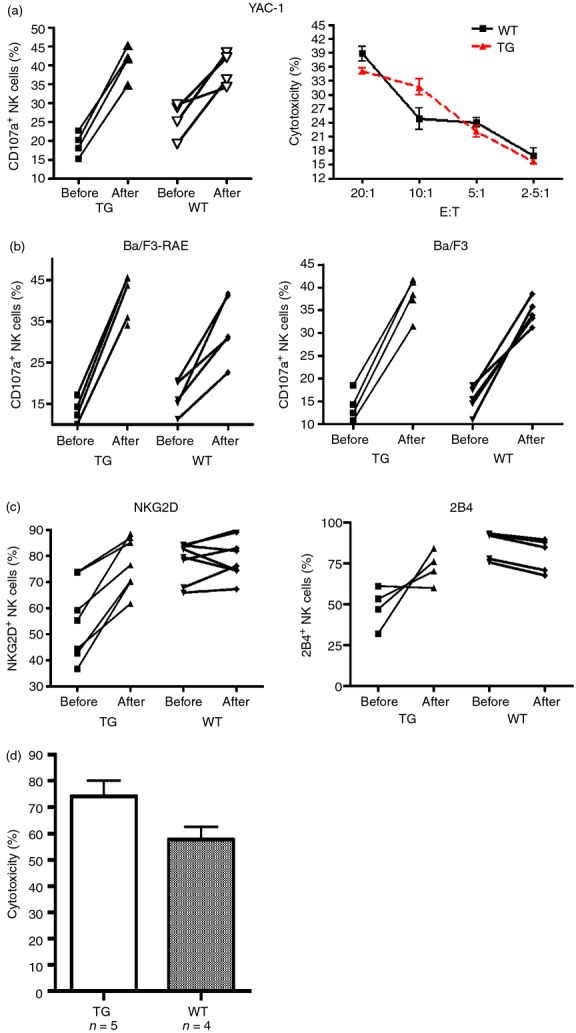
Poly (I:C) stimulation restored NKG2D expression and NKG2D-dependent functions. Natural killer (NK) cell degranulation on incubation with YAC-1 cells was detected before and after poly (I:C) stimulation (Left). The killing of YAC-1 cells was assessed by the lactate dehydrogenase release method (Right) (a). NK cells co-cultured with Ba/F3-RAE (left) or Ba/F3 cells (right), at a ratio of 3 : 1, for 4 hr, then stained with CD107a antibody, and analysed by flow cytometry (b). NKG2D (left) and 2B4 (right) expression on NK cells before and after poly (I:C) stimulation (c). Both groups of mice had received intraperitoneal injections of poly (I:C) (100 μg) 12 hr before the experiment. We then injected equal numbers of splenocytes (CFSE) from transgenic mice and splenocytes (PKH-26) from wild-type mice into these mice. After 12 hr, fluorescent cells were recovered from the spleen and fluorescence ratios were determined by flow cytometry to calculate cytotoxicity. Results are expressed as means ± SD. Asterisks indicate P < 0·05 (d).
Sustained RAE-1ε expression on DCs activates NK cells ex vivo
We investigated whether the down-regulation of NKG2D expression on NK cells in transgenic mice was a direct result of the high level of RAE-1ε expression on DCs. Spleen DCs were isolated from transgenic mice or wild-type mice, with magnetic beads coated with an antibody against CD11c, and co-cultured with normal NK cells at a ratio of 1 : 2 ex vivo. Both DCs from wild-type mice and DCs-RAE from transgenic mice promoted NK cell activation after an incubation period of 12 hr. Stimulation with DCs-RAE resulted in slightly stronger activation than stimulation with DCs. However, the incubation of DCs-RAE with NK cells for 5 days did not lead to a decrease in NK cell function of a magnitude similar to that observed for CD107a+ NK cells co-cultured with YAC-1, Ba/F3 or Ba/F3-RAE cells. In addition, IFN-γ production changed in a similar manner following stimulation with DCs and DCs-RAE (Fig. 5a).
Figure 5.
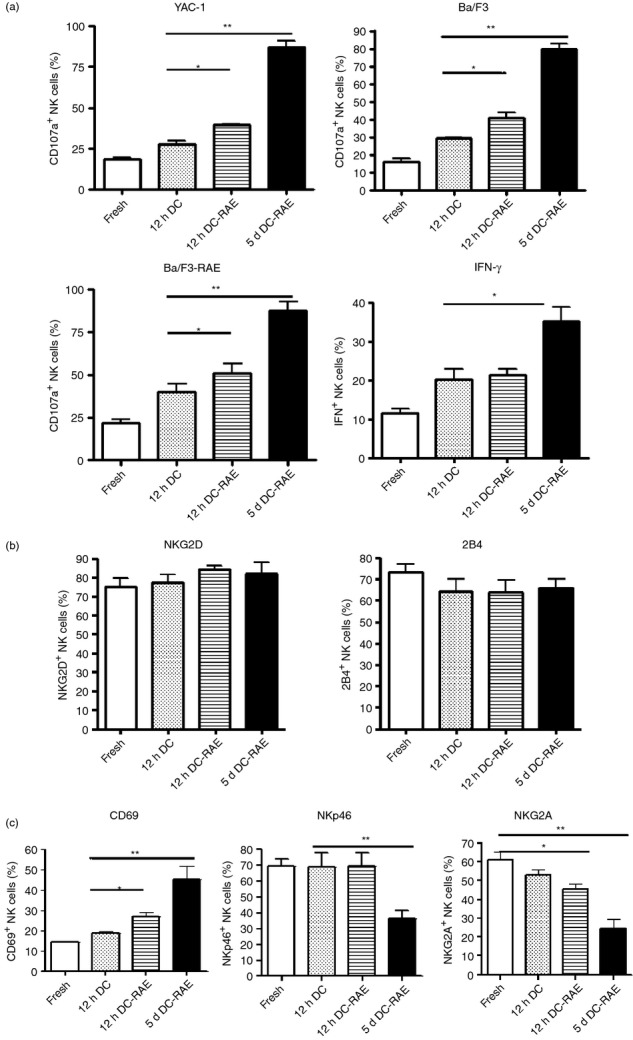
Sustained retinoic acid early transcript (RAE) expression on dendritic cell-activated natural killer (NK) cells ex vivo. CD107a expression and interferon-γ (IFN-γ) production were assessed for freshly isolated NK cells and after stimulation with dendritic cells from a wild-type mouse or a CD86-RAE-1ε mouse, at a ratio of 1 : 2, for 12 hr or 5 days (a). NKG2D, 2B4 (b), CD69, NKp46, NKG2A (c) expression on NK cells stimulated with dendritic cells at a ratio of 1 : 3 for 12 hr, as measured by flow cytometry, with gating on CD3− NK1.1+ cells.
We also investigated the changes in NKG2D, 2B4, CD69, NKp46 and NKG2A expression on NK cells in response to stimulation with DCs or DCs-RAE. Like the cytotoxicity of NK cells, the expression of NKG2D and 2B4 remained at a high level following DCs-RAE stimulation for 5 days (Fig. 5b), or even 10 days (see Supplementary material, Fig. S4). CD69 expression on NK cells was maximal after co-culture with DCs for 5 days. However, NKp46 expression on NK cells remained at a high level after stimulation with DCs or DCs-RAE for 12 hr, but decreased significantly after 5 days of co-culture. NKG2A expression decreased significantly after as little as 12 hr of co-culture (Fig. 5c). As IL-15 is a key cytokine for the maintenance of NK cell phenotype and function, the down-regulation of NKp46 and NKG2A may be associated with a shortage of IL-15 in the culture system.
Serum from CD86-RAE-1ε mice does not induce NKG2D down-regulation
We investigated the reasons for NKG2D down-regulation on NK cells in CD86-RAE-ε transgenic mice, by checking the concentration in the serum of soluble RAE-1, which has been reported to down-regulate NKG2D expression on NK cells. Soluble RAE-1 concentrations in sera were similar for CD86-RAE-1ε transgenic mice and wild-type mice (Fig. 6a), confirming that the GPI-anchored RAE-1 molecule was not cleaved by proteolysis.25 We found no differences in the levels of serum antibodies against RAE-1 between the two groups of mice (Fig. 6b).26 The incubation of NK cells with sera from transgenic mice or wild-type mice did not induce NKG2D down-regulation (Fig. 6c). We therefore conclude that the NKG2D down-regulation on NK cells could not be attributed to serum from the CD86-RAE-1ε mouse.
Figure 6.
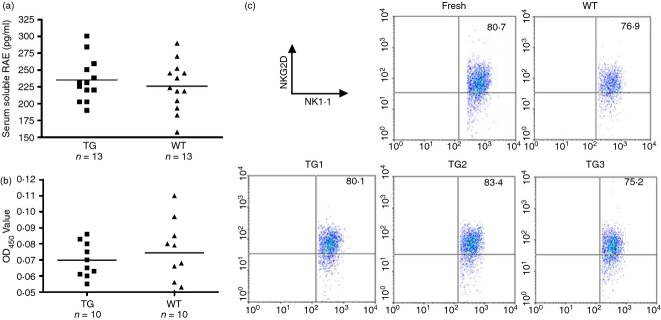
Serum from CD86-RAE-1ε mice did not induce NKG2D down-regulation. The concentration of soluble retinoic acid early transcript (RAE) in the serum was determined with a sandwich ELISA kit (a). Serum levels of antibodies against RAE-1 were determined in an indirect ELISA assay (b). NKG2D expression was assessed on freshly isolated natural killer (NK) cells, and NK cells incubated with serum from a wild-type or transgenic mouse. NK cells were isolated by gating on CD3− NK1.1+ cells (c). The experiment was carried out twice.
CD4+ NKG2D+ T cells contribute to NKG2D down-regulation on NK cells
It has been reported that a subset of CD4+ T cells expresses NKG2D and down-regulates the activity of bystander cells, such as CD4+ NKG2D− T cells and CD8+ T cells, via soluble and membrane FasL, and through the secretion of IL-10 and TGF-β.27,28 Furthermore, TGF-β has been shown to play an important role in down-regulating NKG2D expression on NK cells.29 We determined the frequency of spleen CD4+ NKG2D+ T cells in both groups of mice. The spleens of CD86-RAE-1ε mice contained about twice as many CD4+ NKG2D+ T cells as the spleens of wild-type mice (Fig. 7a). Higher levels of TGF-β production were observed in CD4+ NKG2D+ T cells, but not in CD4+ NKG2D− T cells (Fig. 7b). We then co-cultured normal NK cells with CD4+ NKG2D− T cells or CD4+ NKG2D+ T cells from transgenic mice, or with CD4+ NKG2D+ T cells in the presence of anti-TGF-β antibody. NKG2D expression was found to be down-regulated when NK cells were co-cultured with CD4+ NKG2D+ T cells. Anti-TGF-β antibody blocked the down-regulation of NKG2D on NK cells (Fig. 7c). Finally, the CD4+ NKG2D+ T cells did not express Foxp3 (Fig. 7d), indicating a difference between these regulatory T-cell subsets and conventional CD4+ CD25+ Foxp3+ T cells.
Figure 7.

CD4+ NKG2D+ T cells contributed to the down-regulation of NKG2D on natural killer (NK) cells. Frequencies and absolute numbers of CD4+ NKG2D+ T cells in the spleen were compared between CD86-RAE-1ε mice and wild-type mice (a). Transforming growth factor-β (TGF-β) was detected by flow cytometry, with intracellular staining and gating on CD4+ NKG2D− T cells and CD4+ NKG2D+ T cells (b). NKG2D expression on CD3− NK1.1+ cells was determined following the co-culture of normal NK cells, in a 1 : 1 ratio, with CD4+ NKG2D− T cells, CD4+ NKG2D+ T cells, or CD4+ NKG2D+ T cells in the presence of anti-TGF-β antibody, for 24 hr (c). CD4+ NKG2D+ T cells did not express Foxp3, as shown by flow cytometry (d). Asterisks indicate that P < 0·05. The experiment was carried out three times.
Discussion
In this study, we generated a transgenic mouse in which RAE-1ε was constitutively expressed on antigen-presenting cells, to investigate changes in NK cell function in vivo. The RAE-1 α1/α2 domain displays sequence similarity to MICA/B ectodomains. Hence, the changes induced by RAE-1 binding to mouse NKG2D may be functionally equivalent to those induced by MICA/B binding to human NKG2D.29 The persistent expression of RAE-1ε on DCs, macrophages and B cells in CD86-RAE-1ε transgenic mice resulted in levels of NKG2D expression that were slightly lower than those in control mice, with a correspondingly lower level of NKG2D-mediated cytotoxicity to NKG2D ligand-positive tumour cells. The tumour rejection mediated by NK cells was also weaker in the transgenic mice, and DSS-induced colitis was delayed. With poly (I:C) stimulation, it was possible to restore NKG2D expression on NK cells to normal levels. The high level of expression of RAE-1 on DCs kept NKG2D expression levels high and stimulated NK cell activity ex vivo, but the high frequency of CD4+ NKG2D+ T cells in transgenic mice led to a down-regulation of NKG2D expression on NK cells in vivo. Hence, high levels of RAE-1 expression on DCs or other antigen-presenting cells in vivo induce the generation of a new subset of T cells, to down-regulate NKG2D-dependent NK cell function.
The mechanisms of NKG2D down-regulation on NK cells involve persistent ligand-binding or effects of TGF-β. Sustained NKG2D ligand expression on normal or tumour cells, or soluble NKG2D ligands,21 may induce the down-regulation of NKG2D expression on NK cells. Such down-regulation has been demonstrated in H-2Kb-MICA transgenic mice18 and β-actin-RAE-1ε transgenic mice, and the inducible expression of RAE-1ε on normal epithelium has been reported for involucrin : Rae-1ε mice.19 No down-regulation of NKG2D expression was observed in an RAE-1ε transgenic mouse in which the transgene was under the control of the rat insulin promoter, in which RAE-1ε expression was restricted to the β-islet cells of the pancreas, possibly as the result of low levels of contact between spleen NK cells and the islets.30 Moreover, significant NKG2D down-regulation was also observed when NK cells were exposed to RMA-H60 for 72 hr ex vivo.20,31
The ligand-induced down-regulation of NKG2D is associated with post-translational mechanisms, whereas TGF-β, which has also been shown to down-regulate NKG2D, is associated with transcriptional mechanisms.29,32 NOD mice33 and H-2Kb-MICA transgenic mice display low levels of NKG2D expression in NK cells and weak NKG2D-dependent function, but with no impairment of transcription for NKG2D and DAP10. Sustained exposure of NK cells to RMA-60 cells in vitro20,31 has no effect on the transcription of NKG2DL, NKG2DS, DAP10 or DAP12, but results in lower levels of the corresponding proteins. In addition, in β-actin-RAE-1ε transgenic mice and in conditions of sustained ex vivo exposure of NK cells to RMA-60, the expression of other receptors, such as Ly49D and Ly49H is maintained, with no impairment of the antiviral functions of these receptors.25,34 We conclude that NKG2D down-regulation is mediated principally by TGF-β in CD86-RAE-1ε transgenic mice. First, the levels of transcripts for NKG2DL, NKG2DS, DAP10 and DAP12 in NK cells were moderately lower than those in control mice (Fig. 2b). Second, the Ly49D and Ly49H activating receptors were down-regulated, whereas KLRG1, which down-regulates NK cell cytotoxicity, was up-regulated. Third, TGF-β not only down-regulates NKG2D expression, but also decreases 2B4 expression on NK cells.35
NKG2D expression can be up-regulated by IL-2 or poly (I:C) stimulation.36,37 The stimulation of NK cells from CD86-RAE-1ε transgenic mice with poly (I:C) ex vivo led to an increase in NKG2D expression and NKG2D-dependent cytotoxicity to levels similar to those in control mice. However, NKG2D expression on NK cells did not reach wild-type levels in H-2Kb-MICA transgenic mice or β-actin-RAE-1ε transgenic mice. The magnitude of NKG2D down-regulation in CD86-RAE-1ε transgenic mice was smaller than that in H-2Kb-MICA transgenic mice or β-actin-RAE-1ε transgenic mice.
Serum from CD86-RAE-1ε transgenic mice did not induce NKG2D down-regulation. We can therefore rule out an effect of high levels of TGF-β in the serum of transgenic mice. The high frequency of CD4+ NKG2D+ T cells with regulatory function in CD86-RAE-1ε transgenic mice suggested that the induction of CD4+ NKG2D+ T cells was associated with RAE-1 on DCs, B cells and macrophages. A high frequency of human CD4+ NKG2D+ T cells has been found in patients bearing MICA-positive tumours28 and in patients with juvenile-onset lupus.27 These CD4+ NKG2D+ T cells can be induced by co-stimulation with CD3 antibody and soluble MICA protein or with autologous B cells from lupus patients ex vivo. In CD86-RAE-1ε transgenic mice, there are two key elements for CD4+ NKG2D+ T-cell induction: MHC II molecules and RAE-1ε. The co-culture of CD4+ NKG2D− T cells from normal mice with B cells from transgenic mice led to the induction of CD4+ NKG2D− T cells expressing NKG2D (data not shown). Human CD4+ NKG2D+ T cells down-regulate the activities of conventional CD4+ NKG2D− T cells and CD8+ T cells through membrane and soluble FasL, IL-10 and TGF-β. We are currently carrying out detailed analyses of the phenotype and activity of murine CD4+ NKG2D+ T cells.
Some CD4+ NKG2D+ T cells mediate inflammatory function by secreting IFN-γ and TNF-α in rheumatoid arthritis38 and cytomegalovirus infection,39 or by secreting IL-17 in Crohn's disease.40,41 These inflammatory T-cell subsets are induced by IL-15 or TNF-α. NKG2D antibody may have therapeutic effects in colitis induced by the adoptive transfer of CD4+ CD25− T cells.42 We now need to identify other cell markers or typical transcriptional factors for distinguishing between regulatory or inflammatory CD4+ NKG2D+ T cells. We also suggest that regulatory CD4+ NKG2D+ T cells and CD4+ CD25+ Foxp3+ Treg cells43,44 are different regulatory T-cell subsets, because the CD4+ NKG2D+ T cells studied here were Foxp3-negative (Fig. 7d).
The CD86-RAE-1ε transgenic mouse can provide insight into at least one pathogenic mechanism. As DCs express NKG2D ligand in some pathological conditions, CD4+ NKG2D+ T cells may be induced to mediate immune evasion. Dendritic cells displayed an increase in NKG2D ligand expression in response to stimulation with poly (I:C) (Fig. 1e,f); a similar increase has also been observed in response to stimulation with IL-15 or TNF-α and in some viral or bacterial infections.14–17 MICA was constitutively expressed on monocyte-derived DCs in healthy individuals and colorectal cancer patients (data not shown). The induction of a regulatory T-cell subset may be necessary in some inflammatory situations in the body, because high levels of RAE-1 expression on DCs are likely to activate NK cells. The acute up-regulation of RAE-1 in the epidermis has also been shown to activate tissue-resident T-cell receptor-γδ+ intraepithelial T cells, mediating tissue immune surveillance of cancer45 and atopy.46 In this case, inflammation-associated disease would occur if the activation got out of control.
In conclusion, we confirm here that high levels of RAE-1ε expression on DCs activate NK cells ex vivo, but show that sustained RAE-1ε expression on antigen-presenting cells in vivo may down-regulate NKG2D expression on NK cells by inducing regulatory CD4+ NKG2D+ T cells. The sustained expression of MICA acts as a regulatory element via several pathways in vivo, such as the down-regulation of NKG2D expression on NK cells,18,19 the inhibition of NKG2D ligation with DAP10 or DAP12,22 and the induction of regulatory CD4+ NKG2D+ T lymphocytes28 or T-cell receptor-αβ CD4 CD8αα double-positive intraepithelial lymphocytes.47
Acknowledgments
This work was supported by the National Natural Science Foundation (No. 81172785, No. 81273214, No. 30671917), the Natural Science Fund of Jiangsu Province (BK2011449, BK2008215), and the Science and Technology Innovation fund for undergraduates of the Educational Committee of Jiangsu Province. We thank the Animal Model Research Centre of Nanjing University for generating the transgenic mice.
Disclosures
The authors declare that there are no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. CD4+ T and CD8+ T-cell frequencies do not differ significantly between CD86-RAE-1 transgenic and wild-type mice.
Figure S2. Frequencies of CD8+ NKG2D+ T cells in CD86-RAE-1ε transgenic mice and control mice, as determined by flow cytometry (Top).
Figure S3. The splenocytes of CD86-RAE-1ε mice were more sensitive to killing by interleukin-2-activated natural killer cells, which was blocked by incubation with NKG2D-Ig.
Figure S4. No significant down-regulation of NKG2D was observed on natural killer cells co-cultured with dendritic cells from RAE-1ε transgenic mice for 10 days.
References
- 1.Jacobs B, Ullrich E. The interaction of NK cells and dendritic cells in the tumor environment: how to enforce NK cell and DC action under immunosuppressive conditions? Curr Med Chem. 2012;19:1771–9. doi: 10.2174/092986712800099857. [DOI] [PubMed] [Google Scholar]
- 2.Marcenaro E, Carlomagno S, Pesce S, Moretta A, Sivori S. NK/DC crosstalk in anti-viral response. Adv Exp Med Biol. 2012;946:295–308. doi: 10.1007/978-1-4614-0106-3_17. [DOI] [PubMed] [Google Scholar]
- 3.Morandi B, Costa R, Falco M, et al. Distinctive lack of CD48 expression in subsets of human dendritic cells tunes NK cell activation. J Immunol. 2005;175:3690–7. doi: 10.4049/jimmunol.175.6.3690. [DOI] [PubMed] [Google Scholar]
- 4.Srivastava RM, Varalakshmi C, Khar A. The ischemia-responsive protein 94 (Irp94) activates dendritic cells through NK cell receptor protein-2/NK group 2 member D (NKR-P2/NKG2D) leading to their maturation. J Immunol. 2008;180:1117–30. doi: 10.4049/jimmunol.180.2.1117. [DOI] [PubMed] [Google Scholar]
- 5.Newman KC, Riley EM. Whatever turns you on: accessory-cell-dependent activation of NK cells by pathogens. Nat Rev Immunol. 2007;7:279–91. doi: 10.1038/nri2057. [DOI] [PubMed] [Google Scholar]
- 6.Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity. 2007;26:503–17. doi: 10.1016/j.immuni.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vitale M, Della-Chiesa M, Carlomagno S, Pende D, Aricò M, Moretta L, Moretta A. NK-dependent DC maturation is mediated by TNF-α and IFN-γ released upon engagement of the NKp30 triggering receptor. Blood. 2005;106:566–71. doi: 10.1182/blood-2004-10-4035. [DOI] [PubMed] [Google Scholar]
- 8.Castriconi R, Cantoni C, Della-Chiesa M, et al. Transforming growth factor β1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci USA. 2003;100:4120–5. doi: 10.1073/pnas.0730640100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayakawa Y. Targeting NKG2D in tumor surveillance. Expert Opin Ther Targets. 2012;16:587–99. doi: 10.1517/14728222.2012.681378. [DOI] [PubMed] [Google Scholar]
- 10.Champsaur M, Lanier LL. Effect of NKG2D ligand expression on host immune responses. Immunol Rev. 2010;235:267–85. doi: 10.1111/j.0105-2896.2010.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress inducible MICA. Science. 1999;285:727–9. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 12.Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci USA. 2001;98:11521–6. doi: 10.1073/pnas.201238598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takada A, Yoshida S, Kajikawa M, et al. Two novel NKG2D ligands of the mouse H60 family with differential expression patterns and binding affinities to NKG2D. J Immunol. 2008;180:1678–85. doi: 10.4049/jimmunol.180.3.1678. [DOI] [PubMed] [Google Scholar]
- 14.Krmpotic A, Hasan M, Loewendorf A, et al. NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J Exp Med. 2005;201:211–20. doi: 10.1084/jem.20041617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Groh V, Rhinehart R, Randolph-Habecker J, Topp MS, Riddell SR, Spies T. Costimulation of CD8+ T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat Immunol. 2001;2:255–60. doi: 10.1038/85321. [DOI] [PubMed] [Google Scholar]
- 16.Spies T. Induction of T cell alertness by bacterial colonization of intestinal epithelium. Proc Natl Acad Sci USA. 2002;99:2584–6. doi: 10.1073/pnas.062058399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jinushi M, Takehara T, Tatsumi T, et al. Autocrine/paracrine IL-15 that is required for type I IFN-mediated dendritic cell expression of MHC class I-related chain A and B is impaired in hepatitis C virus infection. J Immunol. 2003;171:5423–9. doi: 10.4049/jimmunol.171.10.5423. [DOI] [PubMed] [Google Scholar]
- 18.Wiemann K, Mittrücker HW, Feger U, Welte SA, Yokoyama WM, Spies T, Rammensee HG, Steinle A. Systemic NKG2D down-regulation impairs NK and CD8 T cell responses in vivo. J Immunol. 2005;175:720–9. doi: 10.4049/jimmunol.175.2.720. [DOI] [PubMed] [Google Scholar]
- 19.Oppenheim DE, Roberts SJ, Clarke SL, Filler R, Lewis JM, Tigelaar RE, Girardi M, Hayday AC. Sustained localized expression of ligand for the activating NKG2D receptor impairs natural cytotoxicity in vivo and reduces tumor immunosurveillance. Nat Immunol. 2005;6:928–37. doi: 10.1038/ni1239. [DOI] [PubMed] [Google Scholar]
- 20.Kaiser BK, Yim D, Chow IT, et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature. 2007;447:482–6. doi: 10.1038/nature05768. [DOI] [PubMed] [Google Scholar]
- 21.Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419:734–8. doi: 10.1038/nature01112. [DOI] [PubMed] [Google Scholar]
- 22.Coudert JD, Scarpellino L, Gros F, Vivier E, Held W. Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood. 2008;111:3571–8. doi: 10.1182/blood-2007-07-100057. [DOI] [PubMed] [Google Scholar]
- 23.Qian L, Ji MC, Pan XY, Gong WJ, Tian F, Duan QF. Construction of a plasmid for co-expression of mouse membrane-bound form of IL-15 and RAE-1ε and its biological activity. Plasmid. 2011;65:239–45. doi: 10.1016/j.plasmid.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 24.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemical induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–6. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 25.Cerwenka A, Bakker AB, McClanahan T, Wagner J, Wu J, Phillips JH, Lanier LL. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 2000;12:721–7. doi: 10.1016/s1074-7613(00)80222-8. [DOI] [PubMed] [Google Scholar]
- 26.Jinushi M, Vanneman M, Munshi NC, et al. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci USA. 2008;105:1285–90. doi: 10.1073/pnas.0711293105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai Z, Turtle CJ, Booth GC, Riddell SR, Gooley TA, Stevens AM, Spies T, Groh V. Normally occurring NKG2D+ CD4+ T cells are immunosuppressive and inversely correlated with disease activity in juvenile-onset lupus. J Exp Med. 2009;206:793–805. doi: 10.1084/jem.20081648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Groh V, Smythe K, Dai Z, Spies T. Fas-ligand-mediated paracrine T cell regulation by the receptor NKG2D in tumor immunity. Nat Immunol. 2006;7:755–62. doi: 10.1038/ni1350. [DOI] [PubMed] [Google Scholar]
- 29.Park YP, Choi SC, Kiesler P, Gil-Krzewska A, Borrego F, Weck J, Krzewski K, Coligan JE. Complex regulation of human NKG2D-DAP10 cell surface expression: opposing roles of the γc cytokines and TGF-β1. Blood. 2011;118:3019–27. doi: 10.1182/blood-2011-04-346825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Markiewicz MA, Wise EL, Buchwald ZS, Pinto AK, Zafirova B, Polic B, Shaw AS. RAE1ε ligand expressed on pancreatic islets recruits NKG2D receptor-expressing cytotoxic T cells independent of T cell receptor recognition. Immunity. 2012;36:132–41. doi: 10.1016/j.immuni.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coudert JD, Zimmer J, Tomasello E, Cebecauer M, Colonna M, Vivier E, Held W. Altered NKG2D function in NK cells induced by chronic exposure to NKG2D ligand-expressing tumor cells. Blood. 2005;106:1711–7. doi: 10.1182/blood-2005-03-0918. [DOI] [PubMed] [Google Scholar]
- 32.Dasgupta S, Bhattacharya-Chatterjee M, O'Malley BW, Jr, Chatterjee SK. Inhibition of NK cell activity through TGF-β1 by down-regulation of NKG2D in a murine model of head and neck cancer. J Immunol. 2005;175:5541–50. doi: 10.4049/jimmunol.175.8.5541. [DOI] [PubMed] [Google Scholar]
- 33.Ogasawara K, Hamerman JA, Hsin H, et al. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity. 2003;18:41–51. doi: 10.1016/s1074-7613(02)00505-8. [DOI] [PubMed] [Google Scholar]
- 34.Champsaur M, Beilke JN, Ogasawara K, Koszinowski UH, Jonjic S, Lanier LL. Intact NKG2D-independent function of NK cells chronically stimulated with the NKG2D ligand Rae-1. J Immunol. 2010;185:157–65. doi: 10.4049/jimmunol.1000397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun C, Fu B, Gao Y, Liao X, Sun R, Tian Z, Wei H. TGF-β1 down-regulation of NKG2D/DAP10 and 2B4/SAP expression on human NK cells contributes to HBV persistence. PLoS Pathog. 2012;8:e1002594. doi: 10.1371/journal.ppat.1002594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diefenbach A, Tomasello E, Lucas M, Jamieson AM, Hsia JK, Vivier E, Raulet DH. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat Immunol. 2002;3:1142–9. doi: 10.1038/ni858. [DOI] [PubMed] [Google Scholar]
- 37.Thaxton JE, Nevers T, Lippe EO, Blois SM, Saito S, Sharma S. NKG2D blockade inhibits poly(I:C)-triggered fetal loss in wild type but not in IL-10−/− mice. J Immunol. 2013;190:3639–47. doi: 10.4049/jimmunol.1203488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci USA. 2003;100:9452–7. doi: 10.1073/pnas.1632807100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sáez-Borderías A, Gumá M, Angulo A, Bellosillo B, Pende D, López-Botet M. Expression and function of NKG2D in CD4+ T cells specific for human cytomegalovirus. Eur J Immunol. 2006;36:3198–206. doi: 10.1002/eji.200636682. [DOI] [PubMed] [Google Scholar]
- 40.Pariente B, Mocan I, Camus M, et al. Activation of the receptor NKG2D leads to production of Th17 cytokines in CD4+ T cells of patients with Crohn's disease. Gastroenterology. 2011;141:217–26. doi: 10.1053/j.gastro.2011.03.061. [DOI] [PubMed] [Google Scholar]
- 41.Allez M, Tieng V, Nakazawa A, et al. CD4+ NKG2D+ T cells in Crohn's disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology. 2007;132:2346–58. doi: 10.1053/j.gastro.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 42.Kjellev S, Haase C, Lundsgaard D, Urso B, Tornehave D, Markholst H. Inhibition of NKG2D receptor function by antibody therapy attenuates transfer-induced colitis in SCID mice. Eur J Immunol. 2007;37:1397–406. doi: 10.1002/eji.200636473. [DOI] [PubMed] [Google Scholar]
- 43.Goodman WA, Cooper KD, McCormick TS. Regulation generation: the suppressive functions of human regulatory T cells. Crit Rev Immunol. 2012;32:65–79. doi: 10.1615/critrevimmunol.v32.i1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee HM, Bautista JL, Hsieh CS. Thymic and peripheral differentiation of regulatory T cells. Adv Immunol. 2011;112:25–71. doi: 10.1016/B978-0-12-387827-4.00002-4. [DOI] [PubMed] [Google Scholar]
- 45.Strid J, Roberts SJ, Filler RB, et al. Acute upregulation of an NKG2D ligand promotes rapid reorganization of a local immune compartment with pleiotropic effects on carcinogenesis. Nat Immunol. 2008;9:146–54. doi: 10.1038/ni1556. [DOI] [PubMed] [Google Scholar]
- 46.Strid J, Sobolev O, Zafirova B, Polic B, Hayday A. The intraepithelial T cell response to NKG2D-ligands links lymphoid stress surveillance to atopy. Science. 2011;334:1293–7. doi: 10.1126/science.1211250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park EJ, Takahashi I, Ikeda J, et al. Clonal expansion of double-positive intraepithelial lymphocytes by MHC class I-related chain A expressed in mouse small intestinal epithelium. J Immunol. 2003;171:4131–9. doi: 10.4049/jimmunol.171.8.4131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. CD4+ T and CD8+ T-cell frequencies do not differ significantly between CD86-RAE-1 transgenic and wild-type mice.
Figure S2. Frequencies of CD8+ NKG2D+ T cells in CD86-RAE-1ε transgenic mice and control mice, as determined by flow cytometry (Top).
Figure S3. The splenocytes of CD86-RAE-1ε mice were more sensitive to killing by interleukin-2-activated natural killer cells, which was blocked by incubation with NKG2D-Ig.
Figure S4. No significant down-regulation of NKG2D was observed on natural killer cells co-cultured with dendritic cells from RAE-1ε transgenic mice for 10 days.