

Abstract
Loss of muscle and bone mass with age are significant contributors to falls and fractures among the elderly. Myostatin deficiency is associated with increased muscle mass in mice, dogs, cows, sheep and humans, and mice lacking myostatin have been observed to show increased bone density in the limb, spine, and jaw. Transgenic overexpression of myostatin propeptide, which binds to and inhibits the active myostatin ligand, also increases muscle mass and bone density in mice. We therefore sought to test the hypothesis that in vivo inhibition of myostatin using an injectable myostatin propeptide (GDF8 propeptide-Fc) would increase both muscle mass and bone density in aged (24 mo) mice. Mice were injected weekly (20 mg/kg body weight) with recombinant myostatin propeptide-Fc (PRO) or vehicle (VEH; saline) for four weeks. There was no difference in body weight between the two groups at the end of the treatment period, but PRO treatment significantly increased mass of the tibialis anterior muscle (+7%) and increased muscle fiber diameter of the extensor digitorum longus (+16%) and soleus (+6%) muscles compared to VEH treatment. Bone volume relative to total volume (BV/TV) of the femur calculated by microCT did not differ significantly between PRO- and VEH-treated mice, and ultimate force (Fu), stiffness (S), toughness (U) measured from three-point bending tests also did not differ significantly between groups. Histomorphometric assays also revealed no differences in bone formation or resorption in response to PRO treatment. These data suggest that while developmental perturbation of myostatin signaling through either gene knockout or transgenic inhibition may alter both muscle and bone mass in mice, pharmacological inhibition of myostatin in aged mice has a more pronounced effect on skeletal muscle than on bone.
Keywords: sarcopenia, osteoporosis, fractures, anabolic therapy
1. Introduction
Globally, the size of the aging population is increasing rapidly, and as a corollary the prevalence of age-related musculoskeletal disorders such as osteoarthritis, sarcopenia, and osteoporosis is also increasing (Sanchez-Riera et al., 2010). A primary contributor to the morbidity and mortality associated with aging is an increased frequency of falls, and falls are often accompanied by bone fractures. Indeed, falls are the primary factor in more than 90% of bone fractures (Jarvinen et al., 2008). In many cases bone fractures limit subsequent capacity for normal daily activities, ambulation, and independence, ultimately leading to assisted living situations which can be financially burdensome. The disability following a fall and fracture also contributes directly to an increase in comoribities such as respiratory infections, which in turn contribute to greater overall age-related mortality (Bertram et al., 2011).
The growth, development, and aging of muscle and bone are closely linked. Pediatric gains in bone mass are normally preceded by gains in muscle mass, and loss of muscle mass with age typically precedes peak rates of loss in bone density (Hamrick et al., 2010a). The close coupling of muscle and bone across the lifespan therefore suggests that changes in one tissue may be mechanistically linked with changes in another. Indeed, there are multiple mechanisms linking the two tissues such as mechanical loading, muscle-derived trophic factors (myokines), as well as systemic factors such as sex steroids and growth factors that have anabolic effects on both muscle and bone. The functional and perhaps molecular integration between the two tissues therefore suggests that therapeutic strategies targeting one particular tissue may have positive effects on the other, or that certain pharmacologic approaches (e.g., androgen-receptor modulators, vitamin D receptor agonists) could positively impact both tissues at once (Hamrick 2010, 2011, 2012).
Given the very close linkages between muscle and bone referenced above, we sought to test the hypothesis that pharmacologic inhibition of myostatin (GDF-8) could increase both muscle and bone mass in an aged animal model. Our rationale for pursuing this hypothesis is based on several key observations. The first is that mice lacking myostatin show increased muscle mass as well as increased bone density in the spine, limb, and jaw (Elkasrawy et al., 2010). The second is that recent studies have demonstrated that a myostatin antibody (LeBrasseur et al., 2009; Murphy et al., 2010) and a decoy myostatin receptor (Chiu et al., 2013) can increase muscle mass in aged mice. The decoy receptor (ActR-IIB) was also found to increase bone formation and bone density (Chiu et al., 2013). These findings suggest that therapeutic modulation of myostatin in vivo may be an effective strategy for preserving muscle and bone mass with age, and so we employed a mouse model to evaluate this hypothesis. Specifically, we have previously shown that C57BL6 mice lose significant muscle mass, bone density, and bone strength with age, such that mice 24 months of age show a marked decrease in these measures compared to mice at 12 months of age (Hamrick et al., 2006a). This study utilizes myostatin propeptide treatment in aged (24 months) C57BL6 mice as an in vivo model for assessing the potential effects of myostatin inhibition on age associated muscle and bone loss. The propeptide fragment is utilized here because it has previously been shown to enhance muscle and bone repair in vivo, and binds the active myostatin ligand with high affinity (Bogdanovich et al., 2005; Hamrick et al, 2010b).
2. Materials & Methods
2.1 Production and validation of myostatin propeptide
The myostatin propeptide-Fc fusion protein was produced in CHO cells as described previously (Jiang et al., 2004). To measure the activity of myostatin and the efficacy of the myostatin propeptide a luciferase reporter gene assay was developed where the vector pGL3(CAGA)12 – neo was stably transfected into A204 (human rhabdomyosarcoma) cells. Addition of myostatin to the A204 CAGA cells, and the binding of myostatin to its receptors, initiates the Smad signal transduction pathway and activates the luciferase reporter gene. The level of activation is proportional to the luciferase activity and the linear portion of the activity curve is in the ng/ml range (Fig. 1), which is what is expected for a protein in the TGFβ superfamily. The addition of the myostatin propeptide prevents the binding of myostatin to its receptors, and the IC50 for the propeptide is approximately 2.0 nM (Fig. 1).
Figure 1.

Myostatin-induced luciferase activity declines significantly with increasing concentration of myostatin propeptide. The pGL3(CAGA)12 – neo reporter vector contains 12 CAGA boxes previously reported to be TGF-β-responsive elements (Dennler et al. (1998) EMBO J. 17:3091–3100), a neo resistance gene, and the basic luciferase reporter plasmid pGL3 (Promega Corporation).
2.2 Animals and treatment for aging study
C57BL6 mice were purchased from the aged rodent colony at the National Institute on Aging, National Institutes of Health (USA) at 22 months of age and delivered to Georgia Regents University, Augusta GA. Animals were allowed to acclimate for one week and were maintained at the Laboratory Animal Service Facility of Georgia Regents University. An earlier dose-response study was used to evaluate the efficacy of a myostatin propeptide in vivo (Hamrick et al., 2010b). Adult mice (5–6 mo.) were treated with the propeptide at 0, 10, 20, or 50 mg/kg at day 0, 5, and 10 and then sacrificed one week after the last treatment. Those data showed that propeptide treatment increased fore- and hindlimb muscle mass by 10% at the 10 mg/kg dose and increased muscle mass by more than 15% at the 20 mg/kg dose, but the 50 mg/kg dose did not increase muscle mass beyond the increase observed in the 20 mg/kg group (Hamrick et al., 2010b). The 20 mg/kg dose was therefore used in this study. Mice were divided into two treatment groups: a vehicle group (VEH; n=14) and a myostatin propeptide group (PRO; n=15). Mice received i.p. injections every five days for 25 days with a dosage of 20 mg/kg body weight at a volume of 0.2 ml. Myostatin propeptide [4.48mg/ml] was obtained from Pfizer Inc (Cambridge, MA, USA). Mice were given calcein i.p. injections to label actively mineralizing bone surfaces four days and 24 hours prior to sacrifice.
2.3 Tissue collection
Animals were euthanized by CO2 overdose and thoracotomy following procedures approved by the Institutional Animal Care and Use Committee of Georgia Regents University. The extensor digitorum longus (EDL) and soleus (SOL) muscles from one limb were dissected out, cut in half and embedded in OCT for cryostat sectioning and muscle fiber size measurements. The tibialis anterior from one limb was dissected out, weighed, snap frozen and homogenized for RT-PCR of the following myogenic markers: myostatin, Mafbx, Murf 1, MHC, and IGF-1. The right tibias were disarticulated and fixed in 70% ethanol for μCT and plastic sectioning while the left tibias were stored damp at minus 20°C for biomechanical testing followed by RT-PCR for bone formation and osteoblast differentiation markers Runx2, Osx and BMP-2. The femora were fixed in 4% paraformaldehyde, decalcified and embedded in paraffin for sectioning and stained for osteoclasts (TRAP kit from Sigma 386A-1KT) and osteoblasts (Celestine blue/van Geison’s).
2.4 Bone Histomorphometry
Osteoblast and osteoclast counts were performed as previously described (Wenger et al., 2010) on 4–5 μm sections after the specimens were decalcified in 4% EDTA for 1 week, dehydrated, cleared in xylene, then embedded in paraffin. Osteoblasts were counted on sections stained using von Giessen, and osteoclasts counted on sections stained for tartrate-resistant acid phosphatase (TRAP) activity. Standardized peripheral locations from the metaphysis were measured in a fixed region of interest. Mineralizing surfaces were measured from calcein-labeled, undecalcified bone sections. Tibias fixed in 70% ethanol were dehydrated and embedded in methyl methacrylate and sectioned in the horizontal (transverse) plane. Sections were viewed using a Zeiss Axioplan2 fluorescent microscope and captured using a SPOT® digital camera to image labeled bone surfaces. Forming surface was calculated as the percentage of non-eroded, single-labeled surface/total surface × 100 (MS/BS).
2.5 Biomechanical testing and micro-computed tomography (uCT)
Left tibias were stored damp at −20°C before being allowed to thaw at room temperature in PBS for 1 hr. Specimens were tested in three-point bending using a Vitrodyne V1000 Material Testing system as described previously (Hamrick et al., 2006b, 2008). Tibias were mounted antero-posteriorly on stainless steel fixtures 5mm apart, approximately 2.5 mm either side of center. Testing was linear displacement control with a displacement speed of 10 μm/sec using a Transducer Techniques 5 kg load cell. Structural, or extrinsic, properties including ultimate force (Fu; height of curve) and stiffness (S; slope of curve) were calculated from load-displacement curves. MicroCT images of the right tibia were scanned using Bruker Skyscan1174 compact micro-CT (Belgium), software version 1.5 (build 9) using NRecon version 1.6.4.8 for reconstructed images.
2.6 PCR and Western blotting
Total RNA was extracted using Trizol and cDNA was synthesized using Quantitect reverse transcription kit (catalog no. 205310; Qiagen). Expression was analyzed quantitatively by means of the Quantitect SYBR Green PCR kit (catalog no. 204143, Qiagen), and QuantiTect Primer Assays. We used specific primers provided by QuantiTect Primer Assays for Myostatin, Murf1, MaFbx, MHC, IGF-1, Runx2, Osx, BMP-2 and 18S, GAPDH and β-actin (internal controls; Table 1). Half of each extensor digitorum longus muscle was snap-frozen in liquid nitrogen for Western blotting. Each muscle was placed in phosphate buffered saline (PBS) and subjected to homogenization using Fisherbrand Tissuemiser® rotary homogenizer until large pieces of muscle were no longer visible. Samples were subjected to two freeze–thaw cycles to disrupt the plasma membrane then centrifuged briefly. Protein concentrations were measured using a commercial BCA reagent (Pierce, Rockford, IL) to ensure equal loading. 30 μg of proteins from whole tissue lysates were mixed 1:1 with 2× sample buffer and then applied to 4–20 % polyacrylamide gels. Samples were electrophoretically separated and transferred to nitrocellulose membrane (Invitrogen, Carlsbad, CA). The membranes were incubated with specific primary antibodies MURF1 (Abcam cat. 77577) or MAFbx (Santa Cruz Biotech cat. 166806) and then incubated with anti-mouse or anti-rabbit peroxidase-conjugated secondary antibodies (Santa Cruz, CA). After the incubation, the membranes were washed three times for 15 min each with 1× TTBS solution and then incubated with 1 ml of chemiluminescence reagent (Invitrogen). The protein bands were visualized using X-ray films (Fisher Scientific, Rochester, NY).
Table 1.
Nucleotide sequences of mouse primers used for RT-PCR
| Gene | Primer | Reference/Accession Number |
|---|---|---|
| GAPDH | CAT GGC CTC CAA GGA GTA AGA GAG GGA GAT GCT CAG TGT TGG |
M32599 |
| 18S | AGT GCG GGT CAT AAG CTT GC GGG CCT CAC TAA AC CAT CCA |
V00851 |
| β-actin | GTT TGA GAC CTT CAA CAC CCC GTG GCC ATC TCC TGC TCG AAG TC |
Meredith et al 2011* |
| Mstn | ACT GGA CCT CTC GAT AGA ACA CTC ACT TAG TGC TGT GTG TGT GGA GAT |
NM_010834.2 |
| IGF-1 | CAG ACA GGA GCC CAG GAA AG AAG TGC CGT ATC CCA GAG GA |
NM_184052 |
| MHC | ACA GTC AGA GGT GTG ACTC AGC CG CCG ACT TGC GGA GGA AAG GTG C |
NM_001099635 |
| Murf1 | GGAGCAGCTGGAAAAGTCCACC AGCTGCTTGGCACTTGAGAGGA |
NM_001039048.2 |
| Mafbx | CAGCTTCGTGAGCGACCTC GGCAGTCGAGAAGTCCAGTC |
NM_026346 |
| BMP-2 | TGT TTG GCC TGA AGC AGA GA TGA GTG CCT GCG GTA CAG AT |
NM_007553.2 |
| RUNX-2 | GGA AAG GCA CTG ACT GAC CTA ACA AAT TCT AAG CTT GGG AGG A |
NM_009820 |
| Osx | ACT ACC CAC CCT TCC CTC AC ACT AGG CAG GCA GTC AGA CG |
AY803733 |
Meredith ME, Harrison FE, May JM. Differential regulation of the ascorbic acid transporter SVCT2 during development and in response to ascorbic acid depletion. Biochem Biophys Res Commun. 2011 Nov 4;414(4):737–42.
2.7 Statistical analysis
Single-factor ANOVA with treatment group as the factor was used to for pairwise comparisons of morphometric and histomorphometric parameters. For analysis of gene expression data, the control genes of 18S and Actin were averaged to obtain an average control gene for muscle tissue while GAPDH was used as the control gene for bone. Difference in control gene Ct expression between GDF-8 and vehicle was assessed using a two-sample t-test. Delta Ct values for each treatment group were calculated as ΔCt = CtTarget gene − CtControl gene. The difference in ΔCt expression between GDF-8 and vehicle was assessed using a two-sample t-test. The magnitude of the difference between the groups was estimated using deltadelta Ct values for each target gene and these were calculated as ΔΔCt = ΔCtGDF-8 − ΔCtvehicle and fold change was calculated as 2 to the power −ΔΔCt. SAS® version 9.3 (SAS Institute, Inc., Cary, NC) was used for all analyses and alpha=0.05 was used to determine statistical significance.
3. Results
3.1 Myostatin propeptide increases muscle mass and fiber size in aged mice
Body weight of the vehicle- and propeptide-treated animals was similar at the end of the study (Fig 2A). Each treatment group did, however, lose some weight over the treatment period but this was less dramatic for the treated animals, such that their decrease in body weight from day 0 to day 25 was significantly less than that of the vehicle-treated mice (Fig. 2B). Muscle mass of the tibialis anterior was significantly increased in the treated mice, both absolutely (Fig. 3A) and relative to body weight (Fig. 3B). Fiber size of the predominantly fast-twitch extensor digitorum longus (EDL) muscle was also significantly increased by more than 15% in the treated mice (Fig. 3C,D), whereas the increase in muscle fiber size in the predominantly slow-twitch soleus (SOL) muscle was also increased significantly (Fig. 3E) but by a lesser magnitude (~5%). Propeptide treatment produced a slight but non-significant increase in the expression of myostatin itself, as well as expression of myosin heavy chain and IGF-1 (Fig. 4A). Surprisingly, expression of the ubiquitin ligases Murf1 and Mafbx was significantly increased with propeptide treatment (Fig. 4A), and the PCR data were further validated by Western blot (Fig. 4B).
Figure 2.

Body mass (A) and change in body weight (B) for animals treated with either saline (VEH) or myostatin propeptide (PRO) weekly for over four weeks.
Figure 3.



Muscle parameters for mice treated with saline (VEH) or myostatin propeptide (PRO) weekly for a period of four weeks. (A) Tibialis anterior mass, (B) tibialis anterior mass relative to body weight, (C) extensor digitorum longus fiber diameter (EDL), (D) alpha-laminin stained cryostat sections of the EDL, (E) soleus muscle fiber diameter.
Figure 4.

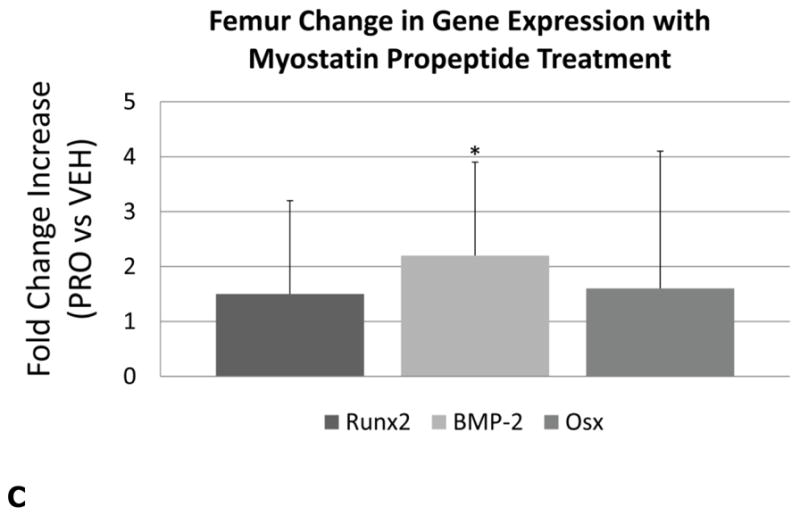
Real-time PCR data for mice treated with saline (VEH) or myostatin propeptide (PRO) weekly for a period of four weeks showing increased expression of Murf1 and Mafbx in PRO-treated mice (A), Western blot showing similar increases in Murf1 and Mafbx with PRO-treatmend (B), and increased expression of BMP-2 in mice treated with propeptide (C). *P<.05.
3.2 Myostatin inhibitor does not alter bone formation or bone strength in aged mice
MicroCT data from the tibia show that bone mineral density is actually slightly higher (3%) in the tibias of vehicle-treated mice (Table 2), but other parameters such as bone volume relative to total volume, trabecular number, and trabecular thickness are similar between the two groups (Table 2). Likewise, three-point bending tests of tibias show that ultimate force, stiffness, and toughness (energy to fracture) are also similar between the vehicle- and propeptide-treated mice (Table 2). Bone histomorphometry data reveal that osteoblast and osteoclast numbers do not differ between the experimental groups (Table 3). Fluorochrome labeling showed double-labels in only three mice from each group, and so single-labeled surfaces were compared. Actively mineralizing surfaces were also similar between the two groups of mice (Table 3). Gene expression data show no significant differences in the expression of osteogenic genes Osx or Runx2 with propeptide treatment, however the expression of BMP-2 is increased in animals receiving the propeptide (Fig. 4C).
Table 2.
microCT and biomechanical testing of the proximal tibia for mice treated with saline (VEH) or myostatin propeptide (PRO; 20 mg/kg).
| Parameter | VEH (n=14) | PRO (n=15) | p value |
|---|---|---|---|
| BMD | 1.43±0.06 | 1.38±0.05 | .01 |
| BV/TV | 6.67±2.37 | 6.14±2.16 | .24 |
| Tb. Th | 0.11±0.02 | 0.11±0.01 | .47 |
| Tb. N | 0.59±0.14 | 0.54±0.16 | .23 |
| Fu (kg) | 2.21±.40 | 2.18±.34 | .39 |
| U (kg/um2) | 740.6±417.5 | 670.3±309 | .31 |
| S (g/um) | 4.6±2.0 | 4.7±2.0 | .44 |
BMD=bone mineral density, BV/TV=bone volume relative to total volume, Tb.Th=trabecular thickness, Tb.N=trabecular number, Fu=ultimate force, U=energy-to-fracture, S=stiffness.
Table 3.
Bone histomorphometry data for the distal femur of mice treated with saline (VEH) or myostatin propeptide (PRO; 20 mg/kg).
| Parameter | VEH (n=15) | PRO (n=14) | p value |
|---|---|---|---|
| N.Ob/BS | 27.26±17.49 | 25.09±9.31 | .14 |
| MS/BS | 0.41±0.17 | 0.43±0.13 | .34 |
| N.Oc/BS | 6.33±2.61 | 6.18±3.82 | .38 |
N.Ob/BS=osteoblast number per bone surface, MS/BS=mineralizing surface (single-label) relative to bone surface, N.Oc/BS=osteoclast number per bone surface.
4. Discussion
Pharmacological inhibition of myostatin has, to date, been pursued using a variety of in vivo approaches. These include utilization of myostatin-specific antibodies (Bogdanovich et al., 2002; Wagner et al., 2008; LeBrasseur et al., 2009; Murphy et al., 2010), a decoy myostatin receptor (ActRIIB-Fc; Lee et al., 2005; Bialek et al. 2008; Borgstein et al., 2009; Chiu et al., 2013), and myostatin propeptide (Bogdanovich et al., 2005; Hamrick et al., 2010b). Published data now exist in which each of these therapies has been evaluated in aged rodents, so that some comparison of the different approaches can be undertaken. Our data using a myostatin propeptide are consistent with data from studies using myostatin antibodies, where these myostatin inhibitors were found to have significant, positive effects on muscle mass, fiber size, and muscle force production. Specifically, LeBrasseur et al. (2009) used a slightly higher dose (25 mg/kg) than we used in our study (20 mg/kg), but also used weekly injections of a myostatin inhibitor (PF-354 antibody) over a period of 4 weeks in mice 24 months of age. They too found a moderate (<10%) in muscle mass and a significant increase in muscle mass relative to body weight (+12–17%), as we did for the tibialis anterior muscle relative to body weight (+~15%). Murphy et al. (2010), like LeBrasseur et al. (2009), used weekly doses of the PF-354 antibody but used a lower dose (10 mg/kg) for a longer treatment period—14 weeks of treatment starting in mice aged 18 months. These authors found increases in overall muscle mass (<10%) in the gastrocnemius and quadriceps of the aged mice following 14 weeks of treatment, and a significant increase in (+12%) in muscle fiber cross-sectional area of the tibialis anterior muscle, that were similar in magnitude to the changes we observed with propeptide treatment. Together, these studies using myostatin antibodies and our study using the myostatin propeptide show similar increases in muscle fiber size and muscle mass using either a low dose (10 mg/kg) over a longer (14 week) treatment period, or a higher dose (20–25 mg/kg) over a shorter treatment period (4 weeks).
Data from in vivo studies using either the myostatin antibody or the propeptide differ in two important ways from those utilizing the decoy myostatin receptor (ActRIIB-Fc). First, a 10 mg/kg dose of ActRIIB-Fc administered twice weekly for four weeks increased tibialis anterior mass by 30% and quadriceps mass by 25% (Chiu et al., 2013). These increases in muscle mass are much greater than those observed with either the myostatin antibody or propeptide, which as noted above generated increases in total muscle mass of <10%. It is possible that these differences could be due to the more frequent administration of the ActRIIB-Fc, but the ActRIIB-Fc dose is much lower than that used in either our study or the study by LeBrasseur et al. (2009), suggesting that the ActRIIB-Fc is a more potent molecule for increasing muscle mass in aged mice. The reason for the greater potency of the ActRIIB-Fc for increasing muscle mass is likely because this molecule can bind several ligands in addition to myostatin, including activin, BMP-3, BMP-7, BMP-9, BMP-10, and GDF-11 (Souza et al., 2008). Some of these molecules, such as activin, are also likely to play a role in regulating muscle mass, which is further indicated by the fact that ActRIIB-Fc treatment can increase muscle mass in mice that lack myostatin altogether (Lee et al., 2005). The second way in which our data differ from those using ActRIIB-Fc is related to the effects on bone. ActRIIB-Fc treatment was previously documented to increase bone formation and bone mass in young, growing mice (Bialek et al., 2008; Yan et al., 2008), and the data from Chiu et al. (2013) are consistent with this earlier report in showing that ActRIIB-Fc increases bone density and serum markers of bone formation in aged mice after just 30 days of treatment. In contrast, our data revealed no bone effects with myostatin propeptide treatment. These data may indicate that, as proposed by Chiu et al. (2013), the anabolic effects of ActRIIB-Fc on bone are due to antagonizing effects on ligands other than myostatin, such as various BMPs or activin.
Previous work in our lab showed that myostatin can inhibit the proliferation of aged bone marrow stromal cells (Bowser et al., 2013), that bone marrow stromal cells from mice lacking myostatin show increased proliferation (Elkasrawy et al., 2011), and that myostatin can inhibit chondrogenesis in vivo and in vitro (Elksrawy et al., 2012). These data may at least in part explain the increased fracture callus size following osteotomy in mice lacking myostatin (Kellum et al., 2009), and increased fracture callus bone volume in mice treated with myostatin propeptide following osteotomy (Hamrick et al., 2010). That is, myostatin seems to play a key role in musculoskeletal injury repair, one in which myostatin secretion from muscle is elevated following muscle damage, and then mediates the repair response in adjacent bone by modulating progenitor cell proliferation (Elkawrawy et al., 2012). On the other hand, myostatin appears to have a more limited role in mature, intact bone. This is indicated by the fact that myostatin itself is not expressed at a significant level by osteoblasts (Digirolamo et al., 2011), and that myostatin inhibition via propeptide treatment in adult mice does not significantly alter osteoblast number, mineralizing surfaces, or bone strength (Tables 1 and 2). Thus, therapeutic targeting of myostatin specifically via antibody or propeptide treatment may have clinical application in the context of improving muscle mass alone, or improving the healing of muscle and bone following injury, but is not likely to have a significant impact on bone formation in the intact, aged animal. In contrast, the decoy myostatin receptor (ActRIIB-Fc) appears capable of increasing muscle mass, bone formation, and bone strength in aged rodents, suggesting that this molecule may have potential clinical use for age-associated loss of both muscle and bone in the form of sarcopenia and osteoporosis.
Muscle and bone are closely associated spatially and in terms of structure and function during growth, development, and aging. Muscle in particular has been considered a driving force for bone modeling and remodeling, in that muscle is the primary source of mechanical stimuli for bone and bone tissue is thought to adapt its gross structure in response to muscle-derived stimuli. Thus, targeting muscle therapeutically is thought to be one approach for improving bone health, simply by enhancing the mechanical relationship between muscle and bone. On the other hand, a large portion of osteoporotic fractures do not occur in individuals with low bone density as measured by two-dimensional densitometry, and so fall prevention alone may be another strategy for reducing falls and fall-associated morbidity and mortality in the elderly (Jarvinen et al., 2008). Behavioral interventions such as resistance exercise or nutritional interventions such as vitamin D supplementation (Girgis et al., 2013) may improve muscle strength and/or neuromuscular control and proprioception, perhaps reducing fall risk. The extent to which myostatin inhibition may augment such strategies remains relatively unexplored. Mice are relatively small in body weight and their bones are capable of withstanding loads many times their own body mass--for example it takes more than 2 kg of force to fracture the tibia of a 32 g mouse (Fig. 1, Table 2). Thus, increases in muscle mass in these small mammals may not significantly alter the mechanical environment of their bones. Additional studies in patient populations are needed to determine the extent to which therapeutic targeting of muscle alone via a myostatin antibody or propeptide, perhaps in conjunction with an exercise regimen, could reduce the incidence of bone fractures versus a molecule such as ActRIIB-Fc, that may potentially increase the mass and strength of both muscle and bone.
5. Conclusions
We tested the hypothesis that in vivo inhibition of myostatin using an injectable myostatin propeptide (GDF8 propeptide-Fc) would increase both muscle mass and bone density in aged (24 mo) mice. Our goal was to evaluate this potential therapeutic for its capacity to increase both muscle and bone mass in the setting of age-associated sarcopenia and osteoporosis. Mice were injected weekly (20 mg/kg body weight) with recombinant myostatin propeptide-Fc (PRO) or vehicle (VEH; saline) for four weeks. The data show that PRO treatment significantly increases muscle fiber size and muscle mass, both absolutely and relative to body weight. In contrast bone volume, bone strength, and histomorphometric parameters of bone formation and bone resorption were unchanged with PRO treatment. Our findings are consistent with previous studies utilizing a myostatin antibody in aged mice showing that targeting myostatin increases muscle fiber size and mass; however, our data differ from work utilizing a decoy myostatin receptor (ActRIIB-Fc) to inhibit myostatin function in that ActRIIB-Fc appears particularly effective at increasing bone density and bone formation whereas the propeptide does not. The anabolic effects of ActRIIB-Fc on aged bone are likely due to the ability of this molecule to antagonize other ligands besides myostatin, such as activin or bone morphogenetic proteins. Clinical trials evaluating the potential of these molecules to prevent falls and fractures are needed to determine the optimal approaches for reducing musculoskeletal diseases and complications in the elderly.
Acknowledgments
Funding for this research was provided by the Congressionally Directed Medical Research Programs, Department of the Army (CDMRP093619) and the National Institute on Aging (P01 AG036675).
References
- Bertram M, Norman R, Kemp L, Vos T. Review of the long-term disability associated with hip fractures. Inj Prev. 2011;17:365–70. doi: 10.1136/ip.2010.029579. [DOI] [PubMed] [Google Scholar]
- Bialek P, Parkington J, Warner L, St Andre M, Jian L, Gavin D, Wallace C, Zhang J, Yan G, Root A, Seeherman H, Yaworsky P. Mice treated with a myostatin/GDF-8 decoy receptor, ActRIIB-Fc, exhibit a tremendous increase in bone mass. Bone. 2008;42:S46. [Google Scholar]
- Bogdanovich S, Krag T, Barton ER, Morris LD, Whittemore LA, Ahima RS, Khurana T. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–21. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- Bogdanovich S, Perkins K, Krag T, Whittemore L, Khurana T. Myostatin-propeptide mediated amelioration of dystrophic pathophysiology. FASEB J. 2005;19:543–549. doi: 10.1096/fj.04-2796com. [DOI] [PubMed] [Google Scholar]
- Borgstein N, Condon C, Yang Y, Wilson D, Haltom E, Lachey J, Seehra J, Sherman M. Preliminary results from single subcutaneous administration of ACE-031, a form of the soluble activin typeII B receptor, in healthy postmenopausal volunteers. Neuromusc Disorders. 2009;19:546. [Google Scholar]
- Bowser M, Herberg S, Arounleut P, Shi X, Fulzele S, Hill WD, Isales CM, Hamrick MW. Effects of the activin A-myostatin-follistatin system on aging bone and muscle progenitor cells. Exp Gerontol. 2013;48:290–97. doi: 10.1016/j.exger.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CS, Peekhaus N, Weber H, Adamski S, Murray EM, Zhang HZ, Zhao JZ, Ernst R, Lineberger J, Huang L, Hampton R, Arnold BA, Vitelli S, Hamuro L, Wang WR, Wei N, Dillon GM, Miao J, Alves SE, Glantschnig H, Wang F, Wilkinson HA. Increased Muscle Force Production and Bone Mineral Density in ActRIIB-Fc-Treated Mature Rodents. J Gerontol A Biol Sci Med Sci. 2013 Mar 22; doi: 10.1093/gerona/glt030. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Digirolamo D, Singhal V, Clemens T, Lee S-J. Systemic administration of soluble activin receptors produces differential anabolic effects in muscle and bone in mice. J Bone Miner Res suppl. 2011:1167. [Google Scholar]
- Elkasrawy MN, Hamrick MW. Myostatin (GDF-8) as a key factor linking muscle mass and bone structure. J Musculoskelet Neuronal Interact. 2010;10:56–63. [PMC free article] [PubMed] [Google Scholar]
- Elkasrawy MN, Fulzele S, Bowser M, Wenger K, Hamrick MW. Myostatin (GDF-8) inhibits chondrogenesis and chondrocyte proliferation in vitro by suppressing Sox-9 expression. Growth Factors. 2011;29:253–62. doi: 10.3109/08977194.2011.599324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkasrawy M, Immel D, Wen X, Liu X, Liang L, Hamrick MW. Immunolocalization of myostatin (GDF-8) following musculoskeletal injury and the effects of exogenous myostatin on muscle and bone healing. J Histochem Cytochem. 2012;60:22–30. doi: 10.1369/0022155411425389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girgis CM, Clifton-Bligh R, Hamrick MW, Holick MF, Gunton JE. The roles of vitamin D in skeletal muscle: form, function and metabolism. Endocrine Reviews. 2013;34:33–83. doi: 10.1210/er.2012-1012. [DOI] [PubMed] [Google Scholar]
- Hamrick MW, Ding KH, Pennington C, Chao YJ, Wu YD, Howard B, Immel D, Borlongan C, McNeil PL, Bollag WB, Curl WW, Yu J, Isales CM. Age-related loss of muscle mass and bone strength in mice is associated with a decline in physical activity and serum leptin. Bone. 2006a;39:845–853. doi: 10.1016/j.bone.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Hamrick MW, Samaddar T, Pennington C, McCormick J. Increased muscle mass with myostatin deficiency improves gains in bone strength with exercise. J Bone Miner Res. 2006b;21:477–483. doi: 10.1359/JBMR.051203. [DOI] [PubMed] [Google Scholar]
- Hamrick MW, Ding KH, Ponnala S, Ferrari SL, Isales CM. Caloric restriction decreases cortical bone mass but spares trabecular bone in the mouse skeleton: implications for the regulation of bone mass by body weight. J Bone Miner Res. 2008;23:870–879. doi: 10.1359/jbmr.080213. [DOI] [PubMed] [Google Scholar]
- Hamrick MW. Invited Perspective: Myostatin (GDF8) as a therapeutic target for the prevention of osteoporotic fractures. IBMS BoneKey. 2010;7:8–17. [Google Scholar]
- Hamrick MW, McNeil PL, Patterson SL. Role of muscle-derived growth factors in bone formation. J Musculoskelet Neuronal Interact. 2010a;10:64–70. [PMC free article] [PubMed] [Google Scholar]
- Hamrick MW, Arounleut P, Kellum E, Cain M, Immel D, Liang L. Recombinant myostatin (GDF-8) propeptide enhances the repair and regeneration of both muscle and bone in a model of deep penetrant musculoskeletal injury. J Trauma. 2010b;69:579–83. doi: 10.1097/TA.0b013e3181c451f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamrick MW. A role for myokines in muscle-bone interactions. Ex Sports Sci Revs. 2011;39:43–47. doi: 10.1097/JES.0b013e318201f601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamrick MW. The skeletal muscle secretome: an emerging player in muscle-bone crosstalk. Nature Bonekey. 2012;60:1–5. doi: 10.1038/bonekey.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvinen T, Sievanen H, Khan K, Heinonen A, Kannus P. Shifting the focus in fracture prevention from osteoporosis to falls. BMJ. 2008;336:124–126. doi: 10.1136/bmj.39428.470752.AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang MS, Liang LF, Wang S, Ratovitski T, Holmstrom J, Barker C, Stotish R. Characterization and identification of the inhibitory domain of GDF-8 propeptide. Biochem Biophys Res Commun. 2004;315:525–31. doi: 10.1016/j.bbrc.2004.01.085. [DOI] [PubMed] [Google Scholar]
- Kellum E, Starr H, Arounleut P, Immel D, Fulzele S, Wenger K, Hamrick MW. Myostatin (GDF-8) deficiency increases fracture callus size, Sox-5 expression, and callus bone volume. Bone. 2009;44:17–23. doi: 10.1016/j.bone.2008.08.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBrasseur NK, Schelhorn TM, Bernardo BL, Cosgrove PG, Loria PM, Brown TA. Myostatin inhibition enhances the effects of exercise on performance and metabolic outcomes in aged mice. J Gerontol A Biol Sci Med Sci. 2009;64:940–8. doi: 10.1093/gerona/glp068. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Reed LA, Davies MV, Girgenrath S, Goad ME, Tomkinson KN, Wright JF, Barker C, Ehrmantraut G, Holmstrom J, Trowell B, Gertz B, Jiang MS, Sebald SM, Matzuk M, Li E, Liang LF, Quattlebaum E, Stotish RL, Wolfman NL. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci USA. 2005;102:18117–22. doi: 10.1073/pnas.0505996102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K, Koopman R, Naim T, Leger B, Trieu J, Ibebunjo C, Lynch GS. Antibody-directed myostatin inhibition in 21-mo-old mice reveals novel roles for myostatin signaling in skeletal muscle structure and function. FASEB J. 2010;24:4433–42. doi: 10.1096/fj.10-159608. [DOI] [PubMed] [Google Scholar]
- Sànchez-Riera L, Wilson N, Kamalaraj N, Nolla JM, Kok C, Li Y, Macara M, Norman R, Chen JS, Smith EU, Sambrook PN, Hernández CS, Woolf A, March L. Osteoporosis and fragility fractures. Best Pract Res Clin Rheumatol. 2010;24:793–810. doi: 10.1016/j.berh.2010.10.003. [DOI] [PubMed] [Google Scholar]
- Souza TA, Chen X, Guo Y, Sava P, Zhang J, Hill JJ, Yaworsky PJ, Qiu Y. Proteomic identification and functional validation of activins and bone morphogenetic protein 11 as candidate novel muscle mass regulators. Mol Endocrinol. 2008;22:2689–702. doi: 10.1210/me.2008-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, et al. A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63:561–71. doi: 10.1002/ana.21338. [DOI] [PubMed] [Google Scholar]
- Wenger K, Fulzele S, Immel D, Chao Y, Freeman D, Elsalanty M, Powell B, Hamrick MW, Isales CM, Yu J. Effect of whole body vibration on bone properties in aging mice. Bone. 2010;47:746–55. doi: 10.1016/j.bone.2010.07.014. [DOI] [PubMed] [Google Scholar]