

Abstract
Recent interest has focused on the importance of the nucleus and associated nucleoskeleton in regulating changes in cardiac gene expression in response to biomechanical load. Mutations in genes encoding proteins of the inner nuclear membrane and nucleoskeleton, which cause cardiomyopathy, also disrupt expression of a biomechanically responsive gene program. Furthermore, mutations in the outer nuclear membrane protein Nesprin 1 and 2 have been implicated in cardiomyopathy. Here, we identify for the first time a role for the outer nuclear membrane proteins, Nesprin 1 and Nesprin 2, in regulating gene expression in response to biomechanical load. Ablation of both Nesprin 1 and 2 in cardiomyocytes, but neither alone, resulted in early onset cardiomyopathy. Mutant cardiomyocytes exhibited altered nuclear positioning, shape, and chromatin positioning. Loss of Nesprin 1 or 2, or both, led to impairment of gene expression changes in response to biomechanical stimuli. These data suggest a model whereby biomechanical signals are communicated from proteins of the outer nuclear membrane, to the inner nuclear membrane and nucleoskeleton, to result in changes in gene expression required for adaptation of the cardiomyocyte to changes in biomechanical load, and give insights into etiologies underlying cardiomyopathy consequent to mutations in Nesprin 1 and 2.
Author Summary
Cardiomyopathy is one of the major causes of heart failure, and the leading cause of mortality in the western world. A number of cardiomyopathies are caused by genetic mutations in structural proteins within the cardiomyocyte. Recent interest has turned to structural proteins that link the nucleoskeleton to the cytoskeleton (The LINC complex). Disruption of these factors cause both mechanical and gene response changes that cause varying degrees of pathology. In particular two genes, Nesprins 1 and 2 are of key interest as they share a high degree of homology and both have been found in Emery-Dreifuss muscular dystrophy (EDMD) patients with cardiomyopathy. Studies of these factors in the heart; however, have been limited due to early lethality of dual ablation of both Nesprins 1 and 2 in global knockout mice. Here, we examine targeted genetic ablation of Nesprin 1 and/or Nesprin 2 in a novel set of mouse lines. These studies found that loss of these factors caused early onset of cardiomyopathy, changes in nuclear position and morphology, as well as inhibition of the biomechanical gene response. Together, these data demonstrate that Nesprin 1 and 2 play critical and overlapping roles in the heart during homeostasis and pathology.
Introduction
Mechanical cues from the extracellular matrix (ECM) transmitted through the cytoskeleton to the nucleoskeleton are regulated by a unique set of structural protein complexes [1], [2]. These mechanotransducers function as a platform to translate mechanical cues into biochemical signals and effect a wide range of biological functions (development, migration, cell specification) [2]. Recent attention has turned to protein complexes at the nucleus, their regulation of mechanotransduction and how these structures affect the biomechanical properties of the cell and tissue [1]. Indeed, a number of pathologies, including dilated cardiomyopathy (DCM) and Emery-Dreifuss muscular dystrophy (EDMD), arise from mutations within cardiomyocyte structural proteins affecting mechanical signal integration [3], [4]. Given this, better understanding of structural proteins within the nucleus involved in sensing biomechanical load and enabling adaptive cellular responses during cardiovascular development and pathology can lend significant insight into our understanding of cardiac function [5].
The nucleus itself is separated from the cytoplasm via the nuclear envelope (NE) which is comprised of two membranes; the Outer Nuclear Membrane (ONM) and the Inner Nuclear Membrane (INM) [6]–[8]. Both the ONM and INM contain elements of a protein complex that mechanically link the nucleoskeleton to the cytoskeleton termed the LINC complex (linker of nucleoskeleton and cytoskeleton). The LINC complex is comprised of a number of proteins from two families, the SUN proteins (Sad1p/UNC-84) anchored within the INM, and the Nesprins at the ONM [9]–[13].
The Nesprin family is comprised of four members (Nesprin 1–4) that anchor nuclei to the actin cytoskeleton, intermediate filaments and the microtubule cytoskeleton. Two members, Nesprin 1 and 2, link the nuclear lamina to the actin cytoskeleton and share 65% homology [9], [11], [13]–[16]. Both Nesprin 1 and Nesprin 2 use alternative promoters, RNA splicing and termination to give rise to multiple isoforms, ranging from 61 kDa–1.01 MDa, with the two largest being the giant Nesprins 1G (1.01 MDa) and Nesprin 2G (796 KDa) [9], [12], [17]. Structurally, Nesprin 1G and 2G contain two N-terminal Calponin-homology (CH) domains responsible for linkage to the actin cytoskeleton, a number of Spectrin-repeat (SR) rod domains and a C-terminal KASH (Klarsicht/Anc/Syne Homology) domain [11], [12], [14]–[16], [18]. The KASH domain is key in the interaction of Nesprins with their LINC complex partners, SUN proteins. Recent crystal structures have shown that Nesprin 1 or 2 function in the LINC complex by localizing to the ONM and linking to a homotrimeric SUN domain containing protein complex (either SUN1 or SUN2) via the KASH domain at the INM [19], [20].
In addition to Nesprin and SUN proteins, the INM contains a number of LINC associated factors, including the nuclear lamina, comprised of both A- and B- type Lamins and Emerin. The Lamins are type V intermediate filaments that comprise the protein meshwork that lines the INM and physically associate with the LINC complex via direct interactions with SUN proteins. A-type lamins (Lamin A and C) are alternatively spliced isoforms of the LMNA gene, whereas B-type lamins (Lamin B1 and B2) are encoded by two separate genes LMNB1 and LMNB2 [21]. Emerin, encoded by EMD, is an integral INM protein that can interact with both SUN and Lamin A/C and affects mechanical load responses in cells [22]. Both of these LINC associated factors were found at the cellular level to be critical for mechanotransduction, cellular stiffness, nuclear positioning and nuclear envelope integrity in a number of cell types [21]–[25]. These data suggest that other factors in the nucleus could also affect mechanical loading and nuclear structure/positioning.
Mutations in both LMNA and EMD have been observed to cause a number of diseases including autosomal dominant EDMD and X-linked EDMD (X-EDMD), respectively [26]. EDMD is characterized by skeletal muscle wasting, cardiac conduction defects and cardiomyopathy [26]. Interestingly, 60% of cases of EDMD do not have mutations in LMNA or EMD. Recently, mutations in both Nesprin 1 and 2 were observed to cause EDMD phenotypes [27]. Furthermore, Nesprin 1α mutations were observed in patients with non-ischemic dilated cardiomyopathy (DCM) [28]. Isolated skin fibroblasts from these patients were also observed to have altered nuclear shape [27], [28]. These data indicate that Nesprins may also play a role in cardiac pathology.
Taken together, these data lead to the intriguing possibility that Nesprins could play a role in nuclear function, mechanotransduction, cardiac function and pathology. However, the roles of Nesprins, in particular Nesprin 1 and 2, in cardiomyocyte mechanotransduction and overall cardiac function are largely unknown. Studies as to the function of Nesprin 1 and 2 in cardiomyocytes have been limited, given that dual global ablation of both factors results in postnatal lethality with respiratory defects [29]. Thus, to elucidate the role of Nesprin 1 and 2 in cardiomyocytes, we generated a number of knockout mouse lines in which Nesprin 1 and/or Nesprin 2 are ablated in cardiomyocytes. We demonstrate that ablation of both Nesprin 1 and 2 causes an early onset cardiomyopathy. Moreover we show that Nesprin 1 and 2 play a key role in the integration of biomechanical loading in cardiomyocytes. Taken together, analysis of these targeted ablation models have revealed that Nesprin 1 and 2 play critical roles in cardiomyocyte mechanobiology.
Results
Establishment of Nesprin 1 and 2 global null or cardiomyocyte specific knockout mice
To examine the role(s) of Nesprin 1 and 2, we generated a number of mouse lines in which all KASH-domain containing isoforms were floxed. Nesprin 1 global knockout mice were generated as previously described [13]. These mice were observed to not have a cardiac phenotype at one year of age, but did have severe skeletal muscle defects and some postnatal lethality [13]. Nesprin 2 global knockout mice were developed as previously described [13]. Briefly, we floxed the 7th exon counting backwards from the last exon of Nesprin 2 (Supplemental Figure. S1A). Targeted ES cells were confirmed using Southern blot analyses (Supplemental Figure. S1B). Chimeric males were bred to Black Swiss females to generate nesprin2f/+;neo+ mice. These mice were then crossed to Protamine-cre mice and bred to homozygosity as previously described to generate global knockout mice (nesprin 2−/−) [30]. Deletion of Nesprin 2 was confirmed via PCR from tails, semi-quantitative PCR from hearts and Real Time PCR from isolated cardiomyocytes (Supplemental Figure. S1C,D,G). Deletion was confirmed at the protein level via immunostaining in both cardiac fibroblasts and myocytes (Supplemental Figure. S1E,F). These mice were viable and did not present cardiac defects (see below), skeletal muscle defects (data not shown) or lethality at 1 year of age.
It has been reported the global ablation of both Nesprin 1 and Nesprin 2 results in neonatal lethality with respiratory defects [29]. To investigate the specific role of Nesprin 1 and/or 2 in myocytes, we established mouse lines in which one or both factors were ablated in cardiomyocytes. To minimize incomplete Cre-mediated excision in the presence of four floxed alleles, we generated Nesprin 1 and 2 double knockout mice with cardiac specific Nesprin 1 deletion, using mice which were positive for Nkx2.5Cre [31], [32], homozygous floxed for Nesprin 1, and germline homozygous null for Nesprin 2 (nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre). Excision of Nesprin 1 was observed to be ∼88±5% (n = 4 hearts) using isolated adult cardiomyocytes (Supplemental Figure S2). nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre knockout mice were observed to be viable and born at normal Mendelian ratios. These mice also did not have any significant lethality at 1 year of age, compared to Wild Type controls. nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre and nesprin 1f/f;nesprin 2−/− mice were used to model single deletion of Nesprin 1 or 2 respectively.
Dual Ablation of Nesprin 1 and 2 in myocardium results in early onset cardiomyopathy
As demonstrated by our previous studies, global Nesprin 1 knockout mice do not have a cardiac phenotype [13]. These data suggest that there is a potential overlap of function of both Nesprin 1 and 2. We first examined effects of ablation of both Nesprins 1 and 2 on cardiac function, via echocardiographic analyses. Mice from all four conditions were examined at 5, 10, 25 and 52 weeks of age. Wild Type, nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre and nesprin 1f/f;nesprin 2−/− mice did not have any functional defects over the time course examined (Figure 1). At 5 weeks of age, nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre were not observed to have any cardiac functional defects or myocardial wall thinning (Figure 1). Examination at 10 weeks of age found that nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre mice had decreased wall thickness; measured by left ventricular anterior wall during diastole and systole (LVAWd and LVAWs), left ventricular posterior wall during diastole and systole (LVPWd and LVPWs ) (Figure 1A–D) and diminished fractional shortening (Figure 1E). Examination at 25 weeks and 52 weeks found further decrease in fractional shortening with some decrease in wall thickness. Heart rates were not changed in these mice over the observed time course (Figure 1F).
Figure 1. Deletion of both Nesprin 1 and 2 causes decreased cardiac wall thickness and impaired cardiac function.

(A–D) Left ventricular anterior wall during diastole and systole (LVAWd and LVAWs), Left ventricular posterior wall during diastole and systole (LVPWd and LVPWs) at 5, 10, 25 and 52 weeks. (E) % Fractional Shortening (%FS) at 5, 10, 25, 52 weeks. (F) Heart Rate (HR) at 5, 10, 25, 52 weeks. N = 6–10 mice per condition/timepoint. ANOVA with a post hoc Bonferroni test *p<0.05, N/C = No significant change vs. Wild Type.
Given the early onset of cardiac dysfunction in nesprin 1f/f;nesprin2−/−;Nkx2.5Cre mice at 10 weeks of age, we performed detailed analyses of these hearts at 10 weeks. Examination of HW/BW and HW/TL ratios at this stage revealed significant increases in nesprin 1f/f;nesprin2−/−;Nkx2.5Cre hearts compared to Wild Type, nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre and nesprin 1f/f;nesprin 2−/− (Figure 2A,B). Real Time PCR analyses of fetal gene expression also found significant increases in ANP and ßMHC in nesprin 1f/f;nesprin2−/−;Nkx2.5Cre hearts (Figure 2C,D). No change in αMHC mRNA expression was observed (Figure 2E). Moreover, examination of pro-fibrotic genes procollagen1α1 and procollagen3α1 also observed significant increases in hearts from the latter group (Figure 2F,G). To further characterize cardiac pathology, we next performed Masson Trichrome and Picrosirius Red examination of nesprin 1f/f;nesprin2−/−;Nkx2.5Cre hearts. Histological analyses of hearts at 10 weeks of age also revealed fibrosis in nesprin 1f/f;nesprin2−/−;Nkx2.5Cre hearts when compared to Wild Type, nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre and nesprin 1f/f;nesprin 2−/− hearts (Figure 3B,C). We also probed our model to examine cell death and observed increased apoptosis as measured by TUNEL analyses at both 5 and 10 weeks in our nesprin 1f/f;nesprin2−/−;Nkx2.5Cre hearts (Figure 3D, Supplemental Figure. S3).
Figure 2. Ablation both Nesprin 1 and 2 alters cardiac morphology, reactivates fetal gene program and activates profibrotic genes.

(A) and (B), 10 week old mice hearts: Morphological analyses of heart weight (HW)/body weight (BW) or HW/tibia length(TL). N = 10–11samples per condition (C–F), Real Time PCR analyses of mRNA expression levels for (C) ANP (D) ßMHC (E) αMHC (F) Procol1α1 (G) Procol3α1 N = 3–4 per condition. N/C = No significant change, ANOVA with a post hoc Bonferroni test #p<0.01.
Figure 3. Histological and TUNEL analyses of Nesprin 1 and/or 2 ablation.

(A) Representative 100× H&E images of indicated hearts. Black arrows = Myocyte nuclei. Black bar = 80 µm. (B) and (C), Representative 40× images of (B), Picrosirius Red (Brown = nuclei, red = collagen) or (C) Masson Trichrome (Red = Myocardium, Blue = Collagen, Black = Nuclei). Black bar = 200 µm. (D) TUNEL analyses of Nesprin 1 and/or 2 knockout hearts at 5 Weeks or 10 Weeks. N = 3 per condition. N/C = No significant change, ANOVA with a post hoc Bonferroni test #p<0.01.
Deletion of both Nesprin 1 and 2 causes altered cardiac nuclear position and shape
Previous examination of nesprin 1 global knockout mice found altered skeletal muscle nuclear positioning [13]. In additional studies, mice with a 61 amino-acid substitution of the KASH domain of Nesprin 1 were observed to have elongated nuclei as examined by H&E sectioning [28]. Moreover, examination of nuclei in fibroblasts from patients with mutations in Nesprin 1 and/or 2 found significant morphological defects in nuclear shape [27]–[29]. Therefore, we performed detailed position and shape analyses of cardiomyocyte nuclei following ablation of Nesprin 1 and/or 2. Nesprin 1 or 2 global knockout mice were crossed into the Obscurin-H2B-GFP line, allowing for examination of all cardiomyocyte nuclei [33]. Qualitative vibratome thick section (∼30 µm) analyses revealed that loss of Nesprin 1 or 2 caused an elongation of cardiomyocyte nuclei (Supplemental Figure S4). Examination of H&E sections from Wild Type, nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre and nesprin 1f/f;nesprin 2−/− hearts also showed elongation and positional changes in myocyte nuclei, as indicated by black arrows (Figure. 3A). These changes were exacerbated in nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre hearts also as indicated by black arrows (Figure 3A).
To quantitate morphological differences observed in cardiomyocyte nuclei following ablation of Nesprin 1 and/or 2 we isolated cardiomyocytes from 10-week-old hearts and examined morphology using immunofluorescent and computational morphometric analyses (Figure 4A,D–F). Loss of either Nesprin 1 or 2 (nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre and nesprin 1f/f;nesprin 2−/−) resulted in increased nuclear area, perimeter and length when compared to nuclei in Wild Type controls (Figure 4D–F). This defect was intensified in nesprin 1f/f;nesprin2−/−;Nkx2.5Cre cells (Figure 4D–F). Moreover, nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre nuclei also appeared to be less circular than Wild Type controls. To quantify this we examined circularity of nuclei as previously described [34]. Here loss of either Nesprin 1 or 2 (nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre and nesprin 1f/f;nesprin 2−/−) was found to decrease nuclear circularity. Loss of both Nesprin 1 and 2 (nesprin 1f/f;nesprin2−/−;Nkx2.5Cre) also intensified loss of circularity in these nuclei (Figure 4C). No change in myocyte area was observed (Figure 4B).
Figure 4. Cardiomyocyte nuclear morphology is altered in response to loss of Nesprin 1 and/or 2.

(A) Representative 40× images of isolated cardiomyocytes (Blue = DAPI, Green = Phalloidin). White bar = 15 µm. (B–G) Morphological analyses of cardiomyocytes examining (B) Myocyte area (C) Nuclear circularity (D) Nuclear area (E) Nuclear perimeter (F) Nuclear length or (G) Nuclear distance. N = 200–300 cells per condition, from 3–4 mice. ANOVA with a post hoc Bonferroni test *p<0.05, #p<0.01.
Studies of the murine heart have shown that ∼95 to 98% of cardiomyocytes are binucleated [35], [36]. Loss of Nesprin 1 and/or 2 appeared to decrease the distance between the two nuclei (Figure 3A, Supplemental Figure S3). To quantify this, centroid position was determined for each nuclei and distance from the center of one nucleus to the center of the other was determined. Distance between nuclei was significantly decreased with loss of either Nesprin 1 or 2 (nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre and nesprin 1f/f;nesprin 2−/−). Inter-nuclear distance decreased from 41 µm±7.1 in Wild Type cells to 25 µm±2.8 or 30 µm±1.0 in nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre and nesprin 1f/f;nesprin 2−/− cells respectively (Figure 4A,G). Again, loss of both Nesprin alleles (nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre) resulted in a more significant decrease in inter-nuclear distance, to 12 µm±4.0 (Figure 4G).
Ultrastructural nuclear abnormalities in Nesprin 1 and 2 dual ablation hearts
Given observed defects in nuclear position and circularity, we further investigated the morphology of nesprin 1f/f;nesprin2−/−;Nkx2.5Cre nuclei. We first examined nuclei at postnatal day 1, prior to binucleation of cardiomyocytes [35]. Postnatal day 1 (P1) cardiomyocyte nuclei did not appear to be altered in nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre and nesprin 1f/f;nesprin 2−/− groups compared to Wild Type (Figure 5A–C). Nuclei appeared to be oval shaped with electron dense regions of similar size at the periphery. This suggests that chromatin localization was not altered at P1 in response to loss of Nesprin 1 or 2. Examination of P1 nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre nuclei found these nuclei did appear to have altered morphology with a more irregular shape compared to Wild Type controls (Figure 5D). Interestingly, nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre cardiomyocyte nuclei did not appear to have an increased number of electron dense regions, suggesting that chromatin localization was not altered at P1 in dual Nesprin 1 and 2 knockout hearts.
Figure 5. Examination of cardiomyocyte nuclear ultrastructure in response to loss of Nesprin 1 and/or 2.
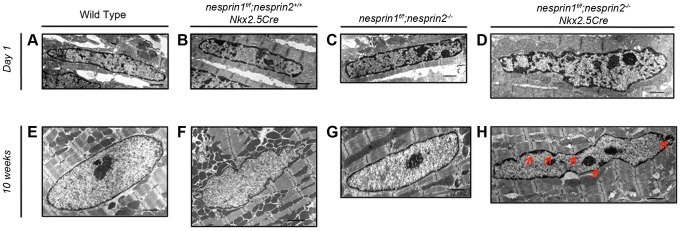
(A–D) Representative images of postnatal day 1 myocyte nuclei from (A) Wild Type (B) nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre (C),nesprin 1f/f;nesprin 2−/− or (D), nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre hearts. (E–H) Representative images of 10 week old myocyte nuclei from (E) Wild Type (F) nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre (G) nesprin 1f/f;nesprin 2−/− or (H) nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre hearts. Black Bars = 1 µm. Red Arrows highlight electron dense heterochromatin defects in 10 week old nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre hearts.
We then examined cardiomyocyte nuclei by electron microscopy at 10 weeks, after the onset of cardiac dysfunction and binucleation [35]. At 10 weeks of age, Wild Type cardiomyocyte nuclei appeared to be oval in shape with a thin peripheral electron dense heterochromatin proximal to the INM (Figure 5E). Nesprin 1 ablated (nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre) nuclei had an increased number of invaginations and increased perimeter, in agreement with immunofluorescent analyses. Again, these nuclei did not have altered electron dense heterochromatin at the INM (Figure 5F). Nesprin 2 knockout (nesprin 1f/f;nesprin 2−/−) nuclei also appeared elongated with increased perimeter, but not an increase number of invaginations (Figure 5G). These nuclei also did not have any alterations in electron dense heterochromatin at the INM (Figure 5G). Interestingly, ablation of both Nesprin 1 or 2 (nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre) resulted in significantly altered nuclear appearance. These nuclei were elongated compared to Wild Type controls, with regions of electron dense chromatin distal to the INM (red arrows, Figure 5H).
Dual ablation of Nesprin 1 and 2 did not alter expression, but did alter localization, of LINC complex or LINC complex associated factors
KASH domain containing isoforms of both Nesprin 1 and 2 have been shown to coimmunoprecipitate and colocalize with Lamin A/C and Emerin in fibroblasts and skeletal muscle [9], [15], [37], [38]. Conversely, loss of Lamin A/C in deletion and mutant fibroblasts and COS7 cells have been shown to disrupt localization of both Nesprin 1 and 2 from the nuclear envelope [15], [39], [40]. Moreover, EDMD fibroblasts with mutations in Nesprin 1α1, Nesprin 1 or Nesprin 2 were also observed to have disrupted Lamin A/C and Emerin localization [17], [41]. Given these data, we examined the LINC complex associated factors (Lamin A/C, B1,B2, Emerin) as well as the LINC complex factors (SUN 1 and 2) to look for changes in expression levels. Both Western blot and Real Time PCR analyses revealed that expression levels of LINC Complex and LINC complex associated factors were not changed at the protein (Supplemental Figure S5) or mRNA levels (Figure 6A–F). Analyses of localization of SUN 2 did not reveal any changes in localization (Supplemental Figure S6). However, loss of both Nesprin 1 and 2 had a significant impact on localization patterns of both Lamin A/C and Emerin (Supplemental Figures S7 and S8).
Figure 6. Real Time PCR analyses of LINC complex and LINC complex associated factors.

(A–E) Real Time PCR analyses of (A) Lamin A/C (B) Lamin B1 (C)Lamin B2 (D)Emerin (E)SUN1 (F)SUN2. Real Time PCR analyses are representative of N = 4–5 cardiomyocyte isolations. ANOVA with a post hoc Bonferroni test. N/C = No significant change.
Deletion of Nesprin 1 and 2 abrogated the nuclear biomechanical gene response
The nucleus has been observed to be a critical integration point of mechanotransduction, regulating gene expression and cellular function [42]. Previous studies have indicated that the mechanotransduction response is abrogated following mutation or deletion of LINC complex associated factors, Lamin A/C and Emerin [21], [22], [43]. However, the role(s) of Nesprin 1 and/or 2 in the integration of mechanotransduction within the cardiomyocyte is unknown. To examine this, we isolated day 1 to 2 neonatal cardiomyocytes from Wild Type, nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre, nesprin 1f/f;nesprin 2−/− and nesprin 1f/f;nesprin2−/−;Nkx2.5Cre hearts. These cells were plated onto micropatterned membranes to ensure in vivo-like cell morphology and rod-like orientation (Supplemental Figure S9). We then interrogated neonatal cardiomyocytes with a strain regime that recapitulated in vivo loading conditions, as per the literature [44], [45]. Indeed, previous research from our group has shown that anisotropic loading of neonatal rat cardiomyocytes induces increases in expression of the known hypertrophic markers [46]. Therefore, anisotropic biaxial loading was applied to 14% Lagrangian strain along the culture transverse axis, and 3.6% along the culture longitudinal axis using custom stretchers as previously described [47], [48].
Examination of known biomechanical response genes (egr-1, iex-1, c-jun, c-fos and c-myc) [21], [22], [49] via Real Time PCR found increased levels of these factors after 30 min of load in Wild Type cardiomyocytes (Figure 7A–E). Nesprin 1 loss (nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre) resulted in a blunted response in levels of egr-1, iex-1, c-jun and c-fos mRNAs, with no alterations in response of c-myc mRNA. Nesprin 2 loss (nesprin 1f/f;nesprin 2−/−) resulted in blunted response of egr-1, iex-1 and c-fos mRNAs with no alterations in response of c-myc or c-jun mRNAs. Interestingly, increased expression of all factors (egr-1, iex-1, c-jun, c-fos and c-myc) were significantly abrogated in nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre cardiomyocytes (Figure 7A–E). These data suggested that while the biomechanical response is impaired with the loss of Nesprin 1 or 2, it is strongly abrogated in the absence of both Nesprins. This indicates that either Nesprin 1 or 2 partially compensates for the loss of each factor in single knockouts. Furthermore, these data suggest that Nesprins 1 and/or 2 play a significant role in the biomechanical function of the cardiomyocyte nucleus.
Figure 7. Examination of biomechanical gene response in isolated neonatal cardiomyocytes from Wild Type, nesprin 1f/f;nesprin 2+/+; Nkx2.5Cre, nesprin 1f/f;nesprin 2−/− or nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre hearts.

(A–E)Real Time PCR analyses of mRNA expression levels for (A) egr-1 (B) c-jun (C) iex-1 (D) c-fos and (E) c-myc. N = 3–5 stretchers/isolations per condition. ANOVA with a post hoc Bonferroni test N/C = No significant change, *p<0.05, #p<0.01.
Discussion
A network of molecular factors act to transmit mechanical cues from the ECM to the cytoskeleton [2]. These mechanical forces are transduced throughout the cell by various structural proteins and affect the physiological functions of the organ, as well as organelle function and positioning; including the nucleus. Indeed, recent studies of the nucleus have suggested that it functions as a primary integration point for response and regulation of mechanical forces from the cell, and that alterations in nuclear shape or position could alter the global functions of the cell and organ [21], [22], [49], [50]. Thus structural factors that mediate the integration of biomechanical signals into the nucleus could lend significant insight into both the physiology and pathophysiology of the cardiomyocyte. Recent studies have highlighted the LINC complex, and in particular the Nesprins, as being important for nuclear mechanical function in isolated fibroblasts, clinical cardiac pathology and skeletal muscle function [13], [26]–[28]. Nesprins have also been implicated in mechanosensing in endothelial cells under shear stress [51]. Clinical studies have demonstrated that mutations in either Nesprins 1 or 2 can result in a number of cardiac pathologies and EDMD-like phenotypes [27], [28], [41]. Moreover, studies of isolated fibroblasts from Nesprins 1 or 2 mutations observed altered nuclear morphology and localization of the LINC complex associated factors [27], [28], [41]. Taken together, these studies have raised the intriguing possibility that Nesprins 1 and/or 2 could play critical role(s) in cardiac function, nuclear positioning and mechanotransduction. These types of studies in the heart have, however, been limited in ablation models due to early neonatal lethality in dual global Nesprin 1 and 2 double knockout mice [29]. In this study we examine the role of Nesprin 1 and/or 2 ablation(s) in a cardiac-targeted ablation model at the tissue and cellular level. Moreover, we sought to dissect the precise roles of these factors within the cardiomyocyte under in vitro biomechanical load.
Studies of Nesprin loss or mutation with regards to cardiac function have been focused on examination of Nesprin 1 KASH domain mutants and our global knockout mice. Mice where the KASH domain of Nesprin 1 was replaced by 61 alternate amino acids exhibited late onset (52 weeks) cardiomyopathy and conduction defects [27], [28]. Interestingly, we did not observe cardiac abnormalities in Nesprin 1 global knockouts at 52 weeks of age [13]. Despite the contradictory nature of these findings, both studies suggest that loss of Nesprin 1 alone is insufficient to cause early onset cardiomyopathy. These data also raise the possibility that another factor could compensate for the loss of Nesprin 1. Given the overlap in function as well as structure of both small and giant isoforms of Nesprins 1 and 2 [9], [15], [37], [38], we hypothesized that Nesprin 2 may act in a compensatory role in cardiac tissue function. Indeed, our data found that targeted cardiomyocyte ablation of Nesprin 1 and 2 caused an early onset of cardiac dysfunction with increased fibrosis and fetal gene re-expression (Figure 1–3). These data suggest that correct anchoring of the nucleus to the cytoskeleton is a key aspect of normal physiology and homeostasis.
Postnatal cardiomyocyte growth is characterized by unique growth patterns in response to increased hemodynamic load during postnatal life [52], [53]. During this process, cardiomyocytes transition from mononucleated to binucleated cells, primarily through karyokinesis without cytokinesis [35]. Given the dynamic nature of this process and the early onset of cardiomyopathy in nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre hearts, we hypothesized that loss of Nesprin 1 and/or 2 would cause morphological and positional defects in cardiomyocyte nuclei. Our study clearly showed that ablation of these factors alters nuclear positioning. Quantification of nuclear morphology showed that at 10 weeks of age (Figure 4A–G) cardiomyocyte nuclei were elongated, less circular and closer together with single ablation of Nesprin 1 or 2. This phenotype was exacerbated in dual ablation of Nesprin 1 and 2, wherein nuclear shape and position were significantly altered upon comparison to Wild Type controls. Moreover, TEM analyses indicated that loss of both Nesprin 1 and 2 resulted in an elongated, morphologically altered nucleus (Figure 5). The observed changes suggested that while loss of either Nesprin 1 or 2 causes some nuclear positioning and morphological defects, loss of both Nesprin 1 and 2 results in more severe aberrations in normal positioning and morphological features of cardiomyocyte nuclei. In addition, these data also show that loss of Nesprin 1 and/or 2 also alters localization of heterochromatin and thus may have an effect on the epigenetic status of myocyte nuclei. This in turn could alter the transcriptome of the cell in response to normal contractile activity or in response to stress. Further examination of this phenomenon is needed in future studies.
Observed changes in nuclear morphology suggested that these cells may have deficiencies in nuclear function. The “structural hypothesis” of disease suggests that mutations in genes involved in the position and tension of the nucleus can alter biomechanical properties of the cell [1]. Force transmission, via the cytoskeleton to the nucleus is mediated by the elements of the LINC complex. Components of the LINC complex associated factors (Lamin A/C, Emerin) have been shown to mediate cellular response to mechanotransduction, as measured by expression of candidate genes (egr-1, iex-1, c-fos, c-jun and c-myc) [21], [22], [49]. Given that dual ablation of Nesprin 1 and 2 resulted in cardiac defects, and severe abnormalities in nuclear positioning, we hypothesized that ablation of Nesprin 1 and 2 would cause abrogation of the mechanotransduction response in cardiomyocytes.
Biomechanical stimulation of cardiomyocytes results in upregulation of biomechanically responsive genes, including egr-1, iex-1, c-fos, c-jun and c-myc. In contrast to Wild Type controls, Nesprin 1 and 2 knockout cardiomyocytes did not exhibit upregulation of this biomechanically responsive gene program in response to stretch. Interestingly, single ablation of Nesprin 1 or 2 caused selective decreases in responsiveness of some of these factors to biomechanical load. Our data also suggested that while there are some defects in Nesprin 1 or 2 single ablation cardiomyocytes in both nuclei and gene responses to load, these defects may not be enough to cause significant alterations in cardiac function.
Together, these data clearly demonstrated that Nesprins 1 and 2 play overlapping and essential roles in cardiac physiology, maintenance and responsiveness to biomechanical load. These data also suggested a model (Figure 8) where the presence of both Nesprin 1 and 2 is required for cardiomyocyte nuclear positioning and linkage to the cytoskeleton/sarcomere via the LINC complex and LINC complex associated factors. Failing to correctly establish these structures affects normal cardiomyocyte development and the capacity to respond to mechanical stress, leading to impaired cardiac function. Ablation of Nesprin 1 and 2 results in abnormal localization of other members of the LINC complex, Emerin and Lamin A/C. It is possible that localization or functions of other cytoskeletal proteins, which connect to the LINC complex, are also perturbed, which will be the subject of future studies. It should also be pointed out that Nesprin 1 and 2 are ubiquitously expressed and therefore it is likely that the role we have uncovered for Nesprin in transducing biomechanical signals into gene expression in the context of cardiomyocytes is likely to be more generally applicable to the role of Nesprin in multiple cell types.
Figure 8. Model of Nesprin 1 and/or 2 loss in cardiomyocytes.

(A and B) Diagrammatic representation of (A) Wild Type and (B) Nesprin 1 and 2 double knockout cardiomyocyte nuclei. (A) Wild Type nuclei are anchored to the sarcomeric structure via Nesprin 1 and 2. Application of 14% anisotropic loading results in normal cellular biomechanical gene response (egr-1, c-jun, iex-1, c-fos and c-myc.). Cardiac development and nuclear positioning/morphology are also normal in these cells. (B) Loss of both Nesprin 1 and 2 causes ablation of biomechanical gene response upon application of strain. Hearts from these mice develop cardiomyopathy with fibrosis, and have altered nuclear positioning and morphology.
Materials and Methods
Animal care
The UCSD Animal Care Personnel maintained all animals and the UCSD Institutional Animal Care and Use Committee approved all experimental procedures.
Gene targeting and generation of Nesprin 2 knockout mice
Genomic DNA fragments of Nesprin 2 were amplified from R1 ES cells using polymerase chain reaction (PCR) and used to construct the Nesprin 2-targeting construct as previously described (Supplemental Figure. S1A) [31]. One LoxP site was inserted into intron 7, as counted backward from the last exon, and a second LoxP site, together with a neomycin cassette flanked by FRT sites, was inserted into intron 6, again as counted backward from the last exon. The targeting construct was verified by sequencing and linearized with the restriction enzyme NotI and electroporated into R1 ES cells derived from 129/SvJ mice (UCSD Transgenic and Gene Targeting Core, La Jolla, CA, USA). Targeted ES cells were identified by Sothern blotting analysis as previously described [54], [55]. Genomic DNA from G418-resistant ES cell clones was digested with the restriction enzyme EcoR V and hybridized with the radiolabeled 564 bp probe generated by PCR using mouse genomic DNAs and nesprin 2 specific primers (forward, 5′-TCCAGACAACAGGCTACACTTTACC-3′ and reverse, 5′-CCAATTAAAGAATTGTAAATTTGACC-3′). The WT allele is represented as a 12.1 kb band, whereas a 10.5 kb band represents the targeted allele. ES cells from a homologous recombinant clone were then microinjected into C57BL/6 blastocysts. Male chimeras were bred with female Black Swiss mice (Taconic Inc., Hudson, NY, USA) to generate germline-transmitted heterozygous mice with a neo cassette, which were further confirmed by PCR and Southern blot analysis of mouse-tail DNA. The global heterozygous Nesprin 2 knockout mice were generated by crossing floxed heterozygous mice with the protamine-Cre transgenic mouse line as previously described [30].
Semi-quantitative RT-PCR
Total RNA of various muscle tissues was isolated using Trizol (Invitrogen) according to the instructions of the manufacturer. RNA amounts were measured with a photospectrometer (Biorad) and equalized. Reverse transcriptase reaction was performed using oligo dT17 primer and the superscript II first-strand synthesis system (Invitrogen). Amplification of GAPDH for standardization was done using KOD DNA polymerase (EMD-Millipore). Bands were analyzed by agarose gel electrophoresis.
Transmission electron microscopy
For TEM, mice were anaesthetized with Ketamine with Xylene, and subsequently perfused through the left ventricle with Tryode buffer including 50 mM KCl, followed by fixative (2% paraformaldehyde, 2% glutaraldehyde in PBS, pH 7.4). The heart were removed, diced, and kept in fixative overnight. The tissues were washed 3 times by 0.15 M sodium cacodylate buffer and then post-fixed with 2% OsO4 in 0.15 M sodium cacodylate buffer for one hour. The following day, tissue was stained overnight in 2% uranyl acetate, dehydrated, and embedded into Durcupan resin (EMD, Gibbstown, NJ) using a standard method. Ultra-thin sections (60–70 nm) were stained with 2% uranyl acetate and statured lead citrate solution, and observed with a JEOL-1200EX transmission electron microscope at an accelerating voltage of 80 kV.
Nesprin 2 antibody
Rabbit antibody of Nesprin-2 was generated against the synthetic polypeptides: PREIQADSWRKRRES (6357–6371aa) (accession number NP_001005510) (Abgent, Ins., US). It is modified from human Nesprin 2 antibody [15]. Each polypeptide was conjugated to keyhole limpet haemocyanin and the conjugates injected into rabbits to produce polyclonal antibodies that were subsequently affinity purified and ELISA tested to confirm specificity.
Micropatterning of substrate, in vitro analyses of biomechanical loading
Cardiomyocytes were cultured on a flexible, microgrooved substrate and subjected to sustained mechanical loading according to methods derived from previous studies [46]–[48]. Micropatterned topography was created using soft photolithography and polydimethylsiloxane (PDMS) molding to provide mechanical and extracellular matrix cues to cells on three sides and permit cell coupling across groove walls. Silicon wafer master molds were micropatterned with SU-8 2005 negative photoresist (MicroChem Corp., Newton, MA) using a custom photomask (Advance Reproductions Corp., North Andover, MA). Sylgard 186 PDMS, prepared at 10 parts base to 1 part curing agent, was spin-coated on the molds, degassed, and thermoset at 70 degrees for 30 minutes and at room temperature overnight. The microgrooved PDMS was mounted in custom cell stretchers and murine laminin (Sigma-Aldrich, St. Louis, MO) was adsorbed under 350 nm wavelength radiation at 10 µg/ml in phosphate buffered saline (PBS) (MediaTech, Manassas, VA). Excess protein was removed by rinsing twice in PBS prior to plating cells. The resulting 10 µm wide microgrooves were defined by walls 5 µm high and 10 µm wide. Anisotropic biaxial loading was applied to 14% Lagrangian strain along the culture transverse axis, and 3.6% along the culture longitudinal axis. The micropatterned cultures were incubated at 37°C and 10% CO2 during the 30 minute mechanical loading. Controls were subjected to placebo manipulations.
Isolation of neonatal cardiomyocytes
Cardiomyocytes were isolated from 1–2 day old neonatal mouse hearts as previously described [56]. Cells were plated onto the microgrooved PDMS of the stretchers at a density of 1.5 million cardiomyocytes in an area of 1 square inch per stretcher. Myocytes were plated in a medium containing Dulbecco's modified Eagle medium and Medium 199 supplemented with 10% horse serum, 5% fetal bovine serum, 100 units/mL penicillin, and 100 µg/mL streptomycin, and incubated at 37°C and 10% CO2. At 72 hours after plating, media changed to a low-serum media containing 5% horse serum and 1% fetal bovine serum for another 24 hours prior to stretch.
Real Time-PCR (RT-PCR) analysis
Samples from Wild Type, nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre, nesprin 1f/f;nesprin 2−/− and nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre hearts, isolated cardiomyocytes or stretch samples were homogenized in Trizol reagent (Invitrogen) or RNeasy Mini Kit (QIAGEN)to isolate total RNA. First strand cDNA was generated utilizing random hexamers and the Invitrogen Superscript II kit (Invitrogen). Primers for RT-PCR were used as previously described [31] or as listed in Supplementary Table 1. These primers were optimized using control murine cDNA (data not shown). RT-PCR reactions were performed using Sso-Fast EvaGreen Real Time PCR (BioRad) master mix in 96 well low profile PCR plates in the CFX96 BioRad thermocycler (BioRad).
Paraffin processing, Histology analyses
Hearts were isolated from aged-matched Wild Type, nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre, nesprin 1f/f;nesprin 2−/− and I hearts (10 week old mice). Adult hearts were perfused with phosphate buffered saline (PBS) supplemented with 50 mM potassium chloride and subsequently perfused and fixed in fresh 4% paraformaldehyde supplemented with 50 mM potassium chloride. Hearts were removed and placed overnight in 4% paraformaldehyde supplemented with 50 mM potassium chloride. Hearts were then processed using standard procedures and embedded into paraffin blocks for sectioning (10 µm). Paraffin sections were cut and stained according to Masson Trichrome, Picrosirius Red, or hematoxylin and eosin standard protocols as previously described [31]. Sections on the Hamamatsu NanoZoomer 2.0HT Slide Scanning System (Hamamatsu).
Frozen processing, TUNEL staining and analyses
For frozen sections, hearts were fresh frozen using isopentane into frozen blocks (1∶1,OCT∶20%sucrose) and sections cut (7–10 µm). Frozen sections were then post fixed and samples stained first for α-Actinin (Abcam) and then for apoptosis using the In Situ Cell Death Detection kit (Roche) as per manufactures instructions. Images were taken using confocal microscopy (Olympus FV1000 Confocal) and images analyzed using ImagePro3D software analyses (MediaCybernetics).
Langendorff perfusion of adult hearts
Dissociation of heart by Langendorff perfusion was performed as in prior studies [57]. Briefly, hearts we perfused with collagenase type II (Worthington) to dissociate the individual cardiomyocytes. Isolated cells were then filtered through a 100 µm mesh nylon filter before performing sequential sedimentation to enrich for myocytes. Isolated myocytes were then plated onto glass-covered dishes coated with Laminin (Sigma) for 1–2 hours.
Echocardiographic analysis
Transthoracic echocardiography was performed on mice 5 to 52 weeks (adult) as previously described [31], [58]. Briefly, mice were anesthetized using 1% isoflurane. Mice were then moved to a biofeedback warming station. This was to maintained core body temperature. Mice were then placed under anesthesia (1 to .5% isoflurane). Ultrasound gel was applied to the chest of the animal. Echocardiography measurements then were obtained using the VEVO 2100 software package and ultrasound system with a linear transducer 45 MHz. (Visual Sonics, SonoSite). Images were processed and M-mode tracings were taken and measured for wall thicknesses and interdimensional space. This was done for both systole and diastole. Fractional shortening was calculated as previously described [58]. An average of six cardiac cycles per animal were analyzed. Six to ten animals were used per condition/per time point.
Western blot analysis
Isolated cardiomyocytes from Wild Type, nesprin 1f/f;nesprin 2+/+;Nkx2.5Cre, nesprin 1f/f;nesprin 2−/− and nesprin 1f/f;nesprin 2−/−;Nkx2.5Cre hearts were lysed into buffer, run up and down into a syringe, quantified and placed directly into SDS-sample buffer. Samples were run on Bis-Tris gels (Invitrogen) and transferred to membranes. Membrane was stained using Ponceau, destained and then UV cross-linked for 10 min. Membranes were then blocked for 1 hour with 5% milk in TBST. Membranes were then probed with rabbit anti-Lamin A/C (a gift from Larry Gerace), anti-Lamin B1 (a gift from Larry Gerace), anti-Lamin B2 (a gift from Larry Gerace), anti-SUN 1 (Epitomics), anti-SUN2 (Sigma), anti-Emerin (Santa Cruz) or mouse anti-GAPDH (Santa Cruz) for 1 hour at room temperature. Blots were then washed three times in TBST and probed for 1 hour with secondary anti-rabbit or anti-mouse antibody. After washing of membranes three times in TBST for 10 minutes, membranes were developed for expression analyses.
Immunofluorescent staining
For immunofluorescent staining, isolated adult cardiomyocytes were fixed for 30 min, room temperature with 2%PFA/PBS/0.1%Triton x-100. Samples were then stained with antibodies for Lamin A/C (A gift from Larry Gerace), Emerin (Santa Cruz), Sun2 (Sigma), α-actinin (Sigma) or Nesprin 2(Abgent). Samples were then stained with secondary antibodies conjugated to CY3 (Jackson ImmunoResearch Inc.). Samples were then incubated with DAPI (Invitrogen) and Phalloidin Alexa-488 (Invitrogen). Samples then imaged using standard confocal microscopy (Olympus FV1000 Confocal). Image analyses were then performed and samples quantified using either Velocity high performance 3D imaging software (PerkinElmer) or ImagePro3D software analyses (MediaCybernetics).
Statistics
The data obtained from all analyses were measured for significance via Graphpad Prism by either student-t test or ANOVA with a post hoc Bonferroni test, P<0.05 was considered significant. All data are ± SEM.
Supporting Information
Generation of Nesprin 2 all isoforms containing C-terminal domain knockout mice. (A) Targeting strategy. A restriction map of the relevant genomic region of Nesprin 2 is shown at the top, the targeting construct is shown in the middle, and the mutated locus after recombination is shown at the bottom. The grey box indicates an exon which is the 16th exon, as counted backward from the last exon, triangular black boxes indicate LoxP sites and rectangular grey boxes indicate frt sites. DTA, Diphtheria Toxin A chain gene, Neo, Neomycin resistance gene. (B) Detection of wildtype (WT) and knockout (KO) alleles by Southern blot analysis. DNA from electroporated ES cells was digested with EcoR V and analyzed by Southern Blot analysis with a probe as shown in A. The 12.1 kb and 10.5 kb bands represent WT and KO alleles, respectively. (C) PCR analysis of DNA isolated from tails of Nesprin 2 WT, heterozygous and homozygous KO mice. KO and WT mice show only one band using specific primers for the KO or WT allele, heterozygous mice show two bands. (D) RT-PCR analysis with RNA from muscle. No coding sequence of the deleted exon could be amplified from the KO sample. (E and F) Immunostaining from (E) Cardiac Fibroblasts and (F) Cardiomyocytes (Blue = DAPI, Red = Nesprin 2) Green bar = 10 µm, White Bar = 5 µm. Green arrows indicate nuclear membranes (G) Real Time PCR analyses of Nesprin 2 knockout from isolated cardiomyocytes. student-t test #P<0.01.
(TIF)
Validation of knockout of Nesprin 1 in cardiac specific deficient mice. Nesprin 1 Real Time PCR analyses of isolated cardiomyocytes from Wild Type, nesprin1f/f;Nkx2.5Cre and global nesprin1−/− mice. We observed an ∼88% decrease observed nesprin1f/f;Nkx2.5Cre isolated cardiomyocytes. student-t test #P<0.01.
(TIF)
Representative TUNEL stain in Nesprin 1 and/or 2 knockout hearts. Blue = DAPI, Green = TUNEL, Red = α-actinin White arrows indicate apoptotic nuclei.
(TIF)
Analyses of Nesprin 1 and Nesprin 2 knockout hearts. Representative 3D reconstruction, 30 µm thick images from (A) Wild Type;Obs-H2bGFP (B) nesprin1−/−;Obs-h2b-GFP and (C) nesprin2−/−;Obs-h2b-GFP Hearts. White arrows indicate cardiomyocyte nuclei. #p<0.01.
(TIF)
Western LINC complex and LINC complex associated factors. (A) Representative western blots for Lamin A/C, Lamin B1, Lamin B2, Emerin, SUN1, SUN2 and GAPDH. Western samples are representative of N = 3 cardiomyocyte isolations, (B–G) Quantification of Western analyses for LINC Complex and LINC Complex associated factors. ANOVA with a post hoc Bonferroni test N/C = No Change.
(TIF)
Loss of Nesprin 1 and 2 does not cause abnormal SUN 2 staining. (A) Representative immunofluorescence nuclear images from isolated adult cardiomyocytes (B) Quantification of percent cells with abnormal SUN 2 staining. Blue = DAPI, Red = SUN 2 ANOVA with a post hoc Bonferroni test #p<0.01. White Bar = 7 µm.
(TIF)
Loss of both Nesprin 1 and 2 had a significant impact on the localization patterns of Lamin A/C. (A) Representative immunofluorescence nuclear images from isolated adult cardiomyocytes (B) Quantification of percent cells with abnormal Lamin A/C staining. Blue = DAPI, Red = Lamin A/C ANOVA with a post hoc Bonferroni test #p<0.01. White Bar = 7 µm Yellow arrows = Abnormal Lamin A/C Stain.
(TIF)
Loss of both Nesprin 1 and 2 had a significant impact on the localization patterns of Emerin. (A) Representative immunofluorescence nuclear images from isolated adult cardiomyocytes (B) Quantification of percent cells with abnormal Emerin staining. Blue = DAPI, Red = Emerin ANOVA with a post hoc Bonferroni test #p<0.01. White Bar = 7 µm, Yellow arrows = Abnormal Emerin Stain.
(TIF)
Representative image of isolated cardiomyocytes on aligned, micropatterned substrates. Representative image of day 1–2 neonatal cardiomyocytes on aligned substrates. Cardiomyocytes appear rod-like and form cell-cell connections. (Green = α-actinin, Blue = DAPI, Red = Connexin-43).
(TIF)
Real Time PCR Primers for study.
(DOCX)
Acknowledgments
We would like to thank Jennifer Santini for technical assistance, Dr. Angela Peters for critical reading of the manuscript.
Funding Statement
JC and SME are funded by grants from the National Heart, Lung, and Blood Institute. LG is funded by a grant from National Institute for General Medical Sciences (RO1GM28521). AM is funded by grants from the National Heart, Lung, and Blood Institute and the National Institute for General Medical Sciences (1R01HL96544, 5P01HL098053, P50GM094503, 1R01HL105242). JC is the American Heart Association (AHA) Endowed Chair in Cardiovascular Research. TMM, MJS, and IB are supported by AHA postdoctoral fellowships (11POST7310066, 13POST17060120 and 12POST12030256 respectively). EP is supported by National Science Foundation Graduate Research Fellowship (DGE1144086). KO is supported by National Key Basic Research Program of China (2013CB531200). Microscopy work was performed at the UCSD Neuroscience Microscopy Shared Facility and was supported by the National Institutes of Health (grant P30 NS047101). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Dahl KN, Ribeiro AJS, Lammerding J (2008) Nuclear shape, mechanics, and mechanotransduction. Circulation Research 102 11: 1307–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Buxboim A, Ivanovska IL, Discher DE (2010) Matrix elasticity, cytoskeletal forces and physics of the nucleus: how deeply do cells “feel” outside and in? J Cell Sci 123 Pt 3: 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dellefave L, McNally EM (2010) The genetics of dilated cardiomyopathy. Current Opinion in Cardiology 25: 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Worman HJ, Ostlund C, Wang Y (2010) Diseases of the Nuclear Envelope. Cold Spring Harbor Perspectives in Biology 2: a000760–a000760 doi:10.1101/cshperspect.a000760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Evans SM, Yelon D, Conlon FL, Kirby ML (2010) Myocardial Lineage Development. Circulation Research 107: 1428–1444 doi:10.1161/CIRCRESAHA.111.258616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Razafsky D, Hodzic D (2009) Bringing KASH under the SUN: the many faces of nucleo-cytoskeletal connections. J Cell Biol 186: 461–472 doi:10.1083/jcb.200906068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Salina D, Bodoor K, Enarson P, Raharjo WH, Burke B (2001) Nuclear envelope dynamics. Biochem Cell Biol 79: 533–542 doi:10.1139/o01-130 [PubMed] [Google Scholar]
- 8. Horn HF, Brownstein Z, Lenz DR, Shivatzki S, Dror AA, et al. (2013) The LINC complex is essential for hearing. J Clin Invest 123: 740–750 doi:10.1172/JCI66911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang Q, Skepper JN, Yang F, Davies JD, Hegyi L, et al. (2001) Nesprin: a novel family of spectrin-repeat-containing proteins that localize to the nuclear membrane in multiple tissues. J Cell Sci 114 Pt 24: 4485–98. [DOI] [PubMed] [Google Scholar]
- 10. Starr DA, Han M (2002) Role of ANC-1 in tethering nuclei to the actin cytoskeleton. Science 298: 406–409 doi:10.1126/science.1075119 [DOI] [PubMed] [Google Scholar]
- 11. Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, et al. (2006) Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol 172: 41–53 doi:10.1083/jcb.200509124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Warren DT, Zhang Q, Weissberg PL, Shanahan CM (2005) Nesprins: intracellular scaffolds that maintain cell architecture and coordinate cell function? Expert Rev Mol Med 7: 1–15 doi:10.1017/S1462399405009294 [DOI] [PubMed] [Google Scholar]
- 13. Zhang J, Felder A, Liu Y, Guo LT, Lange S, et al. (2009) Nesprin 1 is critical for nuclear positioning and anchorage. Human Molecular Genetics 19: 329–341 doi:10.1093/hmg/ddp499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhen YY, Libotte T, Munck M, Noegel AA, Korenbaum E (2002) NUANCE, a giant protein connecting the nucleus and actin cytoskeleton. J Cell Biol 115: 3207–3222. [DOI] [PubMed] [Google Scholar]
- 15. Zhang Q, Ragnauth CD, Skepper JN, Worth NF, Warren DT, et al. (2005) Nesprin-2 is a multi-isomeric protein that binds lamin and emerin at the nuclear envelope and forms a subcellular network in skeletal muscle. J Cell Sci 118: 673–687 doi:10.1242/jcs.01642 [DOI] [PubMed] [Google Scholar]
- 16. Padmakumar VC, Abraham S, Braune S, Noegel AA, Tunggal B, et al. (2004) Enaptin, a giant actin-binding protein, is an element of the nuclear membrane and the actin cytoskeleton. Experimental cell research 295: 330–339 doi:10.1016/j.yexcr.2004.01.014 [DOI] [PubMed] [Google Scholar]
- 17. Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, et al. (2007) Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Human Molecular Genetics 16: 2816–2833 doi:10.1093/hmg/ddm238 [DOI] [PubMed] [Google Scholar]
- 18. Simpson JG, Roberts RG (2008) Patterns of evolutionary conservation in the nesprin genes highlight probable functionally important protein domains and isoforms. Biochem Soc Trans 36: 1359–1367 doi:10.1042/BST0361359 [DOI] [PubMed] [Google Scholar]
- 19. Sosa BA, Rothballer A, Kutay U, Schwartz TU (2012) LINC Complexes Form by Binding of Three KASH Peptides to Domain Interfaces of Trimeric SUN Proteins. Cell 149: 1035–1047 doi:10.1016/j.cell.2012.03.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang W, Shi Z, Jiao S, Chen C, Wang H, et al. (2012) Structural insights into SUN-KASH complexes across the nuclear envelope. Cell Res 22: 1440–1452 doi:10.1038/cr.2012.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, et al. (2004) Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 113: 370–378 doi:10.1172/JCI19670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lammerding J (2005) Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J Cell Biol 170: 781–791 doi:10.1083/jcb.200502148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hale CM, Shrestha AL, Khatau SB, Stewart-Hutchinson PJ, Hernandez L, et al. (2008) Dysfunctional connections between the nucleus and the actin and microtubule networks in laminopathic models. Biophys J 95: 5462–5475 doi:10.1529/biophysj.108.139428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee JSH, Hale CM, Panorchan P, Khatau SB, George JP, et al. (2007) Nuclear Lamin A/C Deficiency Induces Defects in Cell Mechanics, Polarization, and Migration. Biophys J 93: 2542–2552 doi:10.1529/biophysj.106.102426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stewart-Hutchinson PJ, Hale CM, Wirtz D, Hodzic D (2008) Structural requirements for the assembly of LINC complexes and their function in cellular mechanical stiffness. Experimental cell research 314: 1892–1905 doi:10.1016/j.yexcr.2008.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Worman HJ (2011) Nuclear lamins and laminopathies. J Pathol 226: 316–325 doi:10.1002/path.2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Puckelwartz MJ, Kessler E, Zhang Y, Hodzic D, Randles KN, et al. (2008) Disruption of nesprin-1 produces an Emery Dreifuss muscular dystrophy-like phenotype in mice. Human Molecular Genetics 18: 607–620 doi:10.1093/hmg/ddn386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Puckelwartz MJ, Kessler EJ, Kim G, DeWitt MM, Zhang Y, et al. (2010) Nesprin-1 mutations in human and murine cardiomyopathy. J Mol Cell Cardiol 48: 600–608 doi:10.1016/j.yjmcc.2009.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X, Xu R, Zhu B, Yang X, Ding X, et al. (2007) Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development 134: 901–908 doi:10.1242/dev.02783 [DOI] [PubMed] [Google Scholar]
- 30. Liang X, Sun Y, Ye M, Scimia MC, Cheng H, et al. (2009) Targeted ablation of PINCH1 and PINCH2 from murine myocardium results in dilated cardiomyopathy and early postnatal lethality. Circulation 120: 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Banerjee I, Zhang J, Moore-Morris T, Lange S, Shen T, et al. (2012) Thymosin Beta 4 Is Dispensable for Murine Cardiac Development and Function. Circulation Research 110: 456–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McFadden DG, Barbosa AC, Richardson J, Schneider MD, Srivastava D, et al. (2004) The Hand1 and Hand2 transcription factors regulate expansion of the embryonic cardiac ventricles in a gene dosage-dependent manner. Development 132: 189–201 doi:10.1242/dev.01562 [DOI] [PubMed] [Google Scholar]
- 33. Lange S, Ouyang K, Meyer G, Cui L, Cheng H, et al. (2009) Obscurin determines the architecture of the longitudinal sarcoplasmic reticulum. J Cell Sci 122: 2640–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Choi S, Wang W, Ribeiro AJS, Kalinowski A, Gregg SQ, et al. (2011) Computational image analysis of nuclear morphology associated with various nuclear-specific aging disorders. Nucleus 2: 570–579 doi:10.4161/nucl.2.6.17798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ (1996) Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol Heart Circ Physiol 271: H2183–H2189. [DOI] [PubMed] [Google Scholar]
- 36. Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA (2007) Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol 293: H1883–H1891 doi:10.1152/ajpheart.00514.2007 [DOI] [PubMed] [Google Scholar]
- 37. Mislow JMK, Kim MS, Davis DB, McNally EM (2002) Myne-1, a spectrin repeat transmembrane protein of the myocyte inner nuclear membrane, interacts with lamin A/C. J Cell Sci 115: 61–70. [DOI] [PubMed] [Google Scholar]
- 38. Mislow JMK, Holaska JM, Kim MS, Lee KK, Segura-Totten M, et al. (2002) Nesprin-1α self-associates and binds directly to emerin and lamin A in vitro. FEBS Letters 525: 135–140 doi:10.1016/S0014-5793(02)03105-8 [DOI] [PubMed] [Google Scholar]
- 39. Muchir A, van Engelen BG, Lammens M, Mislow JM, McNally E, et al. (2003) Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Experimental cell research 291: 352–362. [DOI] [PubMed] [Google Scholar]
- 40. Libotte T, Zaim H, Abraham S, Padmakumar VC, sCHNEIder M, et al. (2005) Lamin A/C–dependent localization of Nesprin-2, a giant scaffolder at the nuclear envelope. Molecular biology of the cell 16: 3411–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taranum S, Vaylann E, Meinke P, Abraham S, Yang L, et al. (2012) LINC complex alterations in DMD and EDMD/CMT fibroblasts. Eur J Cell Biol 91: 614–628 doi:10.1016/j.ejcb.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bray M-AP, Adams WJ, Geisse NA, Feinberg AW, Sheehy SP, et al. (2010) Nuclear morphology and deformation in engineered cardiac myocytes and tissues. Biomaterials 31: 5143–5150 doi:10.1016/j.biomaterials.2010.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zwerger M, Jaalouk DE, Lombardi ML, Isermann P, Mauermann M, et al. (2013) Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Human Molecular Genetics 22 12: 2335–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McCulloch AD, Smaill BH, Hunter PJ (1989) Regional left ventricular epicardial deformation in the passive dog heart. Circulation Research 64: 721–733. [DOI] [PubMed] [Google Scholar]
- 45. Gupta V, Grande-Allen KJ (2006) Effects of static and cyclic loading in regulating extracellular matrix synthesis by cardiovascular cells. Cardiovasc Res 72: 375–383 doi:10.1016/j.cardiores.2006.08.017 [DOI] [PubMed] [Google Scholar]
- 46. Gopalan SM, Flaim C, Bhatia SN, Hoshijima M, Knoell R, et al. (2003) Anisotropic stretch-induced hypertrophy in neonatal ventricular myocytes micropatterned on deformable elastomers. Biotechnol Bioeng 81: 578–587 doi:10.1002/bit.10506 [DOI] [PubMed] [Google Scholar]
- 47. Camelliti P, Gallagher JO, Kohl P, McCulloch AD (2006) Micropatterned cell cultures on elastic membranes as an in vitro model of myocardium. Nat Protoc 1: 1379–1391 doi:10.1038/nprot.2006.203 [DOI] [PubMed] [Google Scholar]
- 48. Camelliti P, McCulloch AD, Kohl P (2005) Microstructured cocultures of cardiac myocytes and fibroblasts: a two-dimensional in vitro model of cardiac tissue. Microsc Microanal 11: 249–259 doi:10.1017/S1431927605050506 [DOI] [PubMed] [Google Scholar]
- 49. Mammoto A, Mammoto T, Ingber DE (2012) Mechanosensitive mechanisms in transcriptional regulation. J Cell Sci 125: 3061–3073 doi:10.1242/jcs.093005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Caille N, Thoumine O, Tardy Y, Meister J-J (2002) Contribution of the nucleus to the mechanical properties of endothelial cells. Journal of Biomechanics 35: 177–187 doi:10.1016/S0021-9290(01)00201-9 [DOI] [PubMed] [Google Scholar]
- 51. Morgan JT, Pfeiffer ER, Thirkill TL, Kumar P, Peng G, et al. (2011) Nesprin-3 regulates endothelial cell morphology, perinuclear cytoskeletal architecture, and flow-induced polarization. Molecular biology of the cell 22: 4324–4334 doi:10.1091/mbc.E11-04-0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jurko A (2004) Echocardiographic evaluation of left ventricle postnatal growth in newborns and infants. Bratisl Lek Listy 105: 78–85. [PubMed] [Google Scholar]
- 53. Tiemann K, Weyer D, Djoufack PC, Ghanem A, Lewalter T, et al. (2003) Increasing myocardial contraction and blood pressure in C57BL/6 mice during early postnatal development. Am J Physiol Heart Circ Physiol 284: H464–H474 doi:10.1152/ajpheart.00540.2002 [DOI] [PubMed] [Google Scholar]
- 54. Chen J, Kubalak SW, Minamisawa S, Price RL (1998) Selective requirement of myosin light chain 2v in embryonic heart function. J Biol Chem 273 2: 1252–6. [DOI] [PubMed] [Google Scholar]
- 55. Liang X, Zhou Q, Li X, Sun Y, Lu M, et al. (2005) PINCH1 plays an essential role in early murine embryonic development but is dispensable in ventricular cardiomyocytes. Mol Cell Biol 25 8: 3056–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhou Q, Chu P-H, Huang C, Cheng C-F, Martone ME, et al. (2001) Ablation of Cypher, a PDZ-LIM domain Z-line protein, causes a severe form of congenital myopathy. J Cell Biol 155: 605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Moore-Morris T, Varrault A, Mangoni ME, Le Digarcher A, Negre V, et al. (2009) Identification of potential pharmacological targets by analysis of the comprehensive G protein-coupled receptor repertoire in the four cardiac chambers. Mol Pharmacol 75: 1108–1116 doi:10.1124/mol.108.054155 [DOI] [PubMed] [Google Scholar]
- 58. Banerjee I, Fuseler JW, Intwala AR, Baudino TA (2009) IL-6 loss causes ventricular dysfunction, fibrosis, reduced capillary density, and dramatically alters the cell populations of the developing and adult heart. Am J Physiol Heart Circ Physiol 296: H1694–H1704 doi:10.1152/ajpheart.00908.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Generation of Nesprin 2 all isoforms containing C-terminal domain knockout mice. (A) Targeting strategy. A restriction map of the relevant genomic region of Nesprin 2 is shown at the top, the targeting construct is shown in the middle, and the mutated locus after recombination is shown at the bottom. The grey box indicates an exon which is the 16th exon, as counted backward from the last exon, triangular black boxes indicate LoxP sites and rectangular grey boxes indicate frt sites. DTA, Diphtheria Toxin A chain gene, Neo, Neomycin resistance gene. (B) Detection of wildtype (WT) and knockout (KO) alleles by Southern blot analysis. DNA from electroporated ES cells was digested with EcoR V and analyzed by Southern Blot analysis with a probe as shown in A. The 12.1 kb and 10.5 kb bands represent WT and KO alleles, respectively. (C) PCR analysis of DNA isolated from tails of Nesprin 2 WT, heterozygous and homozygous KO mice. KO and WT mice show only one band using specific primers for the KO or WT allele, heterozygous mice show two bands. (D) RT-PCR analysis with RNA from muscle. No coding sequence of the deleted exon could be amplified from the KO sample. (E and F) Immunostaining from (E) Cardiac Fibroblasts and (F) Cardiomyocytes (Blue = DAPI, Red = Nesprin 2) Green bar = 10 µm, White Bar = 5 µm. Green arrows indicate nuclear membranes (G) Real Time PCR analyses of Nesprin 2 knockout from isolated cardiomyocytes. student-t test #P<0.01.
(TIF)
Validation of knockout of Nesprin 1 in cardiac specific deficient mice. Nesprin 1 Real Time PCR analyses of isolated cardiomyocytes from Wild Type, nesprin1f/f;Nkx2.5Cre and global nesprin1−/− mice. We observed an ∼88% decrease observed nesprin1f/f;Nkx2.5Cre isolated cardiomyocytes. student-t test #P<0.01.
(TIF)
Representative TUNEL stain in Nesprin 1 and/or 2 knockout hearts. Blue = DAPI, Green = TUNEL, Red = α-actinin White arrows indicate apoptotic nuclei.
(TIF)
Analyses of Nesprin 1 and Nesprin 2 knockout hearts. Representative 3D reconstruction, 30 µm thick images from (A) Wild Type;Obs-H2bGFP (B) nesprin1−/−;Obs-h2b-GFP and (C) nesprin2−/−;Obs-h2b-GFP Hearts. White arrows indicate cardiomyocyte nuclei. #p<0.01.
(TIF)
Western LINC complex and LINC complex associated factors. (A) Representative western blots for Lamin A/C, Lamin B1, Lamin B2, Emerin, SUN1, SUN2 and GAPDH. Western samples are representative of N = 3 cardiomyocyte isolations, (B–G) Quantification of Western analyses for LINC Complex and LINC Complex associated factors. ANOVA with a post hoc Bonferroni test N/C = No Change.
(TIF)
Loss of Nesprin 1 and 2 does not cause abnormal SUN 2 staining. (A) Representative immunofluorescence nuclear images from isolated adult cardiomyocytes (B) Quantification of percent cells with abnormal SUN 2 staining. Blue = DAPI, Red = SUN 2 ANOVA with a post hoc Bonferroni test #p<0.01. White Bar = 7 µm.
(TIF)
Loss of both Nesprin 1 and 2 had a significant impact on the localization patterns of Lamin A/C. (A) Representative immunofluorescence nuclear images from isolated adult cardiomyocytes (B) Quantification of percent cells with abnormal Lamin A/C staining. Blue = DAPI, Red = Lamin A/C ANOVA with a post hoc Bonferroni test #p<0.01. White Bar = 7 µm Yellow arrows = Abnormal Lamin A/C Stain.
(TIF)
Loss of both Nesprin 1 and 2 had a significant impact on the localization patterns of Emerin. (A) Representative immunofluorescence nuclear images from isolated adult cardiomyocytes (B) Quantification of percent cells with abnormal Emerin staining. Blue = DAPI, Red = Emerin ANOVA with a post hoc Bonferroni test #p<0.01. White Bar = 7 µm, Yellow arrows = Abnormal Emerin Stain.
(TIF)
Representative image of isolated cardiomyocytes on aligned, micropatterned substrates. Representative image of day 1–2 neonatal cardiomyocytes on aligned substrates. Cardiomyocytes appear rod-like and form cell-cell connections. (Green = α-actinin, Blue = DAPI, Red = Connexin-43).
(TIF)
Real Time PCR Primers for study.
(DOCX)