

Abstract
Objective
Allelic variation in the α5 nicotinic acetylcholine receptor (nAChR) subunit gene, CHRNA5, increases vulnerability to tobacco addiction. Here, we investigated the role of α5* nAChRs in the effects of nicotine on brain reward systems.
Materials and methods
Effects of acute (0.03125-0.5 mg/kg SC) or chronic (24 mg/kg per day; osmotic minipump) nicotine, and mecamylamine-precipitated withdrawal, on intracranial self-stimulation (ICSS) thresholds were assessed in wildtype and α5 nAChR subunit knockout mice. Noxious effects of nicotine were further investigated using a conditioned taste aversion (CTA) procedure.
Results
Lower nicotine doses (0.03125-0.125 mg/kg) decreased ICSS thresholds in wildtype and α5 knockout mice. At higher doses (0.25-0.5 mg/kg), threshold-lowering effects of nicotine were diminished in wildtype mice, whereas nicotine lowered thresholds across all doses tested in α5 knockout mice. Nicotine (1.5 mg/kg) conditioned a taste aversion to saccharine equally in both genotypes. Mecamylamine (5 mg/kg) elevated ICSS thresholds by a similar magnitude in wildtype and α5 knockout mice prepared with minipumps delivering nicotine. Unexpectedly, mecamylamine also elevated thresholds in saline-treated α5 knockout mice.
Conclusion
α5* nAChRs are not involved in reward-enhancing effects of lower nicotine doses, the reward-inhibiting effects of nicotine withdrawal, or the general noxious effects of higher nicotine doses. Instead, α5* nAChRs regulate the reward-inhibiting effects nicotine doses that oppose the reward-facilitating effects of the drug. These data suggest that disruption of α5* nAChR signaling greatly expands the range of nicotine doses that facilitate brain reward activity, which may help explain the increased tobacco addiction vulnerability associated with CHRNA5 risk alleles.
Keywords: CHRNA5, α5 nicotinic receptors, nicotine, reward, aversion, habenula, interpeduncular nucleus, conditioned taste aversion
INTRODUCTION
Nicotine is the principal reinforcing component in tobacco smoke responsible for its addictive qualities (Stolerman and Jarvis 1995). Nicotine can enhance the activity of brain reward circuitries, and this action likely plays a key role in the development and persistence of the tobacco habit in human smokers (Kenny and Markou 2006). In addition to the rewarding effects of nicotine, escape from the aversive consequences of nicotine withdrawal is also likely to play a role in the persistence of the tobacco habit (Doherty et al. 1995; Kenny and Markou 2001). Indeed, smoking cessation precipitates an aversive nicotine withdrawal syndrome (Hughes et al. 1991; Shiffman and Jarvik 1976), with the duration and severity of withdrawal predicting relapse in abstinent smokers (Piasecki et al. 1998; Piasecki et al. 2003). Therefore, understanding the mechanisms by which nicotine enhances, and withdrawal diminishes, the activity of brain reward systems is likely to shed important insights into the neurobiology of tobacco addiction.
The addiction-relevant actions of nicotine are derived from its stimulatory and/or desensitizing actions on neuronal nicotinic acetylcholine receptors (nAChRs) in the central nervous system (CNS). Nicotinic receptors are composed of five distinct membrane-spanning subunits (α and β subunits) that combine to form a functional receptor (Albuquerque et al. 1995; Lena and Changeux 1998). The neuronal α subunit exists in nine isoforms (α2-α10), and the β subunit exists in three isoforms (β2-β4) (Elgoyhen et al. 1994; Elgoyhen et al. 2001; Le Novere et al. 2002). Nicotinic receptors containing α4 and β2 subunits (denoted as α4β2* nAChRs) are the predominant nAChR subtypes in the CNS and account for most of the high-affinity nicotine binding sites (Flores et al. 1992). Somewhat surprisingly, recent findings have shown that genetic variation in the lesser-expressed α5/α3/β4 nAChR subunit gene cluster significantly increases risk of tobacco addiction (Berrettini et al. 2008; Lips et al. 2009; Saccone et al. 2007). In particular, polymorphisms in the α5 subunit gene (CHRNA5) present compelling evidence for a genetic contribution to tobacco addiction vulnerability (Bierut 2010). A non-synonymous single nucleotide polymorphism (SNP) in CHRNA5 (rs16969968) that results in an aspartic acid to asparagine substitution at amino acid reside 398 (D398N), and decreases the function of α5* nAChRs incorporating this subunit (Bierut et al. 2008), increases the risk of tobacco dependence by ~30% in individuals carrying a single copy of the variant and more than doubles the risk in those carrying two risk alleles (Berrettini et al. 2008; Bierut et al. 2008; Grucza et al. 2008; Stevens et al. 2008; Wang et al. 2009). The D398N major risk allele is associated with heavy smoking (Berrettini et al. 2008; Bierut et al. 2008; Grucza et al. 2008; Stevens et al. 2008), early onset of smoking behavior (Weiss et al. 2008), and with “pleasurable buzz” from tobacco (Sherva et al. 2008).
Recently, we found that mice with null nutation in the α5 nAChR subunit gene consumed significantly more nicotine than their wildtype counterparts, as measured by intravenous self-administration of the drug (Fowler et al. 2011). Increased nicotine consumption in the knockout mice was most prominent when high unit doses of the drug were available for self-administration (Fowler et al. 2011). These findings are reminiscent of the increased tobacco addiction vulnerability and heavier smoking detected in individuals carrying the D398N risk allele. Intriguingly, virus-mediated re-expression of the otherwise absent subunit in the medial habenula (MHb), which projects almost exclusively to the interpeduncular nucleus (IPN), rescued this phenotype in the knockout mice (Fowler et al. 2011). We also found that virus-mediated knockdown of α5 nAChR subunits in the MHb-IPN pathway of rats increased their nicotine intake similar to the knockout mice. Moreover, this manipulation in rats did not alter the stimulatory effects of nicotine on brain reward systems, but attenuated the reward-inhibiting effects of higher units doses of the drug, as measured by nicotine-induced lowering and elevations of ICSS thresholds, respectively (Fowler et al. 2011).
The above finding suggests that α5* nAChRs in the MHb-IPN system regulate the aversion-related effects of higher doses of nicotine. Although the α5 nAChR subunit is most densely expressed in the MHb-IPN pathway, expression is also found in deep layers of the cortex, hippocampus, ventral tegmental area (VTA) and substantia nigra (Marks et al. 1992). Hence, a limitation of these prior studies is that reward-relevant actions of nicotine were examined in rats after α5 nAChR subunits where manipulated only in the MHb-IPN system. As such, it is unclear if α5* nAChRs expressed more broadly in other addiction-relevant brain circuits may also contribute to reward-related action of nicotine. Also, it is unclear if α5* nAChRs may regulate the reward-inhibiting effects of nicotine withdrawal or more generalized aversion-related effects of nicotine. Therefore, in the current study we assessed the effects of acutely administered nicotine, across a broad range of doses, on ICSS thresholds in wildtype and α5 nAChR subunit knockout mice. In this manner the contribution of α5 nAChRs throughout the entire brain, rather than only those expressed in the MHb-IPN system, to the effects of acutely administered nicotine on brain reward function could be assessed. In addition, we also examined the effects of mecamylamine-precipitated withdrawal from chronic nicotine treatment in wildtype and α5 subunit knockout mice on ICSS thresholds to determine whether α5* nAChRs may contribute to negative affective components of the nicotine withdrawal syndrome. Finally, to more fully investigate the involvement of the α5* nAChRs in noxious effects of nicotine, and to other noxious stimuli, we examined the development of a condition taste aversion (CTA) to in α5 knockout and wildtype mice.
METHODS
Animals
Male mice with null mutation in the α5 nAChR subunit and their wildtype littermates were bred in our animal facilities by mating heterozygous pairs. Subjects were genotyped as described previously (Fowler et al. 2011). Mice were at least 6 weeks of age at the beginning of each experiment. Subjects were maintained in an environmentally controlled vivarium on a 12h:12h reversed light:dark cycle, and food and water were provided ad libitum until behavioral training commenced. All procedures were conducted in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute - Florida.
Drugs
(-)-Nicotine hydrogen tartrate salt (Sigma) was dissolved in 0.9% sterile saline; the doses of nicotine refer to the free-base form. Mecamylamine hydrochloride (Tocris) and lithium chloride (LiCl, Sigma) were dissolved in 0.9% sterile saline. Systemically administered drugs were delivered in a volume of 10 ml/kg body weight.
Electrode Implantation and ICSS Procedure
Thirty-four mice were anesthetized with an isoflurane (1-3%)/oxygen vapor mixture and positioned in a stereotaxic frame in the ‘flat-skull’ position (Kopf Instruments). A stainless steel bipolar electrode (Plastics One) was implanted into the lateral hypothalamus (AP: -0.5mm from bregma; ML: ±1.3mm from midline; DV: -5.0mm from brain surface) (Paxinos 2001). All subjects were permitted at least 72 h recovery from surgery prior to initiating training in the behavioral procedure. Mice were trained to respond according to a modification of the discrete-trial current-threshold procedure of Kornetsky and Esposito, as previously described (Johnson et al. 2008). The overall threshold for the session is defined as the mean of the thresholds for the individual series. Because of large variation in threshold current intensities (μA) between animals, data for each animal was expressed as percent change from baseline, with group means also presented and analyzed as percent of baseline. These mice were tested in experiment 1, followed by experiment 2, and subsequently experiment 3, described below. Four mice were excluded due to sickness/death (1 following nicotine D-R). Three mice were excluded due to inability to acquire/maintain stable ICSS responding.
Experiment 1: Effects of acute nicotine treatments on ICSS thresholds
After establishing stable baseline levels of ICSS threshold responding, defined as <25 % variation in daily ICSS thresholds and requiring approximately 10 daily ICSS sessions, all mice received systemic injections of saline or nicotine ((0.03125-0.5 mg/kg SC) according to a within-subjects repeated-measures Latin-square experimental design. There was at least 2 days between each injection of the Latin-square during which ICSS thresholds were assessed daily to ensure that they returned to pre-injection baseline levels. The doses of nicotine were chosen based on preliminary studies (not shown) to identify doses that reliably lowered ICSS thresholds in mice.
Experiment 2: Effects of chronic nicotine treatment on ICSS thresholds
After reestablishing stable baseline levels of ICSS threshold responding, defined as <25% variation in daily ICSS thresholds, mice were allocated to two groups per genotype (four groups in total) such that there was no statistically significant differences in mean raw ICSS thresholds between groups. Mice were anesthetized with isoflurane and subcutaneously implanted with osmotic minipumps (Alzet; model 2004; 28 day pumps) delivering saline or nicotine. The concentration of the nicotine salt solution was adjusted according to animal body weight to deliver 24 mg/kg per day free-base nicotine. This dose was chosen based on previous studies demonstrating that this dose induces nicotine dependence in mice, reflected in elevations of ICSS thresholds during antagonist-precipitated or spontaneous withdrawal (Johnson et al. 2008). The surgical wound was sutured following implantation (or removal) of the minipump, and mice were administered the analgesic metacam (0.2 mg/kg, meloxicam, Boehringer Ingelheim). ICSS threshold assessments re-commenced 24 h after implantation of osmotic minipumps and continued during daily sessions.
Experiment 3: Effects of precipitated nicotine withdrawal on ICSS thresholds
After 7 consecutive days of ICSS threshold assessment following minipump implantation, the effects of mecamylamine-precipitated nicotine withdrawal on ICSS thresholds were assessed. To precipitate withdrawal, mice were injected with saline or mecamylamine (5 mg/kg, intraperitoneal [IP]) 10 min prior to the behavioral session, according to a balanced cross-over design. This dose of mecamylamine was chosen based on other reports examining the effects of mecamylamine-precipitated withdrawal on ICSS thresholds with chronic minipump administration of nicotine in mice (Hilario et al. 2012; Stoker et al. 2012). After injection, mice were placed into the ICSS chambers and post-injection ICSS thresholds were assessed. Thresholds were assessed daily for at least two days, until they returned to pre-injection baseline levels, before the next injection in the cross-over design.
Experiment 4: Conditioned Taste Aversion to nicotine and lithium
In new cohorts of wildtype and knockout mice, the development of a conditioned taste aversion to saccharin paired with nicotine (1.5 mg/kg, SC) or saline (control) (n=4-7 per group) was examined. Similarly, development of a CTA to lithium chloride (LiCl; 100 mg/kg, IP) or saline (control) (n=6-8 per group) was also examined. No mice were excluded from the data analysis. Drinking tubes were constructed as previously reported (Bachmanov et al. 2002), and the procedure was based on previously published reports studies (Rauhut et al. 2008; Shoaib et al. 2002). The mice were initially water restricted for 24 h and were then permitted 20 min access to tap water. 24 h later, the mice were permitted 20 min access to 0.15% saccharin diluted in tap water. The following day, mice were permitted 60 min access to saccharin and five min later received a subcutaneous injection of either saline (control groups) or test drug (nicotine or lithium) dissolved in physiological saline (drug groups). The following day was a nondrug session in which all of the mice received 60 min access to tap water. The mice underwent three additional saccharin/drug sessions (injections of either saline or nicotine), which alternated with nondrug sessions of tap water access. Finally, on the test day, mice were presented with two drinking tubes containing either tap water or the saccharin solution. Data are presented as percentage of the total volume of intake.
Statistical Analyses
For ICSS, percentage of baseline reward thresholds scores were calculated by expressing the absolute threshold scores as a percentage of the scores obtained during baseline. The baseline values were the mean thresholds on the three sessions prior to minipump implantation or the session before mecamylamine. For percentage change from baseline, 100 was subtracted from the percentage of baseline scores. Data were analyzed by a t-test, or one- or two-way repeated-measures analyses of variance (ANOVA), as appropriate. Following significant main or interaction effects, groups were compared by Bonferroni post-hoc tests. In all cases, the level of significance was set at 0.05. All statistical analyses were performed using GraphPad Prism software.
RESULTS
Experiment 1: Effects of acute nicotine treatments on ICSS thresholds
The wildtype and α5 knockout mice did not differ in their mean (±SEM) raw threshold values (WT: 128.9±20.66 μA; KO: 157.1±17.55 μA; p>0.05) or baseline response latencies (WT: 3.38±0.21 msec; KO: 3.27±0.16 msec; p>0.05). Two-way repeated-measures ANOVA showed that nicotine significantly altered ICSS thresholds in wildtype and α5 knockout mice: Dose (F(5,25)=10.3, p<0.001); Dose × Genotype interaction (F(5,25)=2.5, p<0.05). As seen in Fig. 1a, nicotine had a bimodal action on ICSS thresholds in wildtype mice, with lower doses (0.03125-0.125 mg/kg) decreasing ICSS thresholds, and higher doses returning thresholds towards baseline values. In α5 knockout mice, nicotine lowered thresholds at all doses tested, with the magnitude of lowering similar across all doses. The Bonferroni multiple comparisons post-hoc test showed that thresholds were significantly lower at the highest nicotine dose tested (0.5 mg/kg) in α5 knockout mice compared with wildtype mice (p<0.05) (Fig. 1a). Two-way repeated-measures ANOVA showed that wildtype and α5 knockout mice did not differ in their latency to respond at any dose tested (Fig. 1b).
Figure 1.
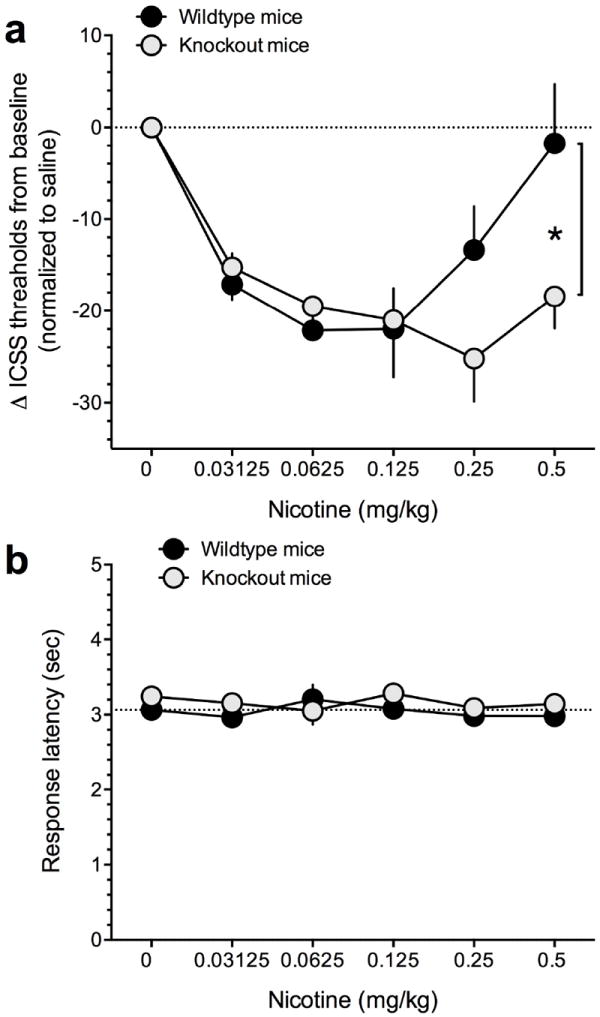
The effects of acutely administered nicotine injections on ICSS in wildtype and α5 knockout mice. (a) Nicotine injections elicited a U-shaped dose response function across a range of doses. Data are expressed as mean (± SEM) percentage chance from baseline ICSS thresholds. *P<0.05 (b) Response latencies did not differ across nicotine injections or between genotypes. Data are expressed as mean (± SEM) seconds to respond for stimulation.
Experiment 2: Effects of chronic nicotine treatment on ICSS thresholds
As seen in Fig. 2, ICSS thresholds remained stable across the 7 consecutive days of testing after implantation of osmotic minipumps in wildtype (Fig. 2a) and α5 knockout mice (Fig. 2b). Interestingly, thresholds were elevated in the saline-treated wildtype mice on the first day of assessment after pump implantation. Therefore, we compared thresholds across all groups and genotypes of mice on this day. As seen in Fig. 2c, there was a significant Genotype × Treatment interaction effects, F(3,22)=5.1, p<0.05, which reflected the elevated ICSS thresholds in wildtype mice treated with saline compared with the other groups on this day.
Figure 2.

The effects of chronic administration of saline or nicotine via osmotic minipump on ICSS thresholds were examined in wildtype and α5 knockout mice. Data are expressed as mean (± SEM) percentage chance from baseline ICSS thresholds. (a) ICSS thresholds were elevated in saline-treated wildtype mice compared with nicotine-treated wildtype mice on day 1 after minipump implantation. Thereafter, ICSS thresholds remained similar and altered across sessions in both groups of mice. (b) ICSS thresholds were similar and unaltered in saline-treated and nicotine-treated knockout mice across 7 consecutive days. (c) Comparison of ICSS in wildtype and knockout mice on day 1 after minipump implantation demonstrated that thresholds were transiently elevated only in saline-treated wildtype mice. *P<0.05.
Experiment 3: Effects of precipitated nicotine withdrawal on ICSS thresholds
Injections of saline did not alter ICSS thresholds in wildtype mice with minipumps continuously delivering nicotine or saline (Fig. 3a, p>0.05). Mecamylamine injections, however, significantly altered ICSS thresholds in the wildtype mice (Fig. 3a). When threshold data in wildtype mice were analyzed by two-way repeated-measures ANOVA, we found a statistically significant main effect of Mecamylamine (F(1, 12) = 22.9, p<0.0005); and a Mecamylamine × Pump (Saline or Nicotine) interaction effect (F(1, 12) = 18.2, p<0.005). The Bonferroni multiple comparisons test demonstrated that mecamylamine treatment significantly elevated ICSS thresholds compared with saline in wildtype mice prepared with nicotine-delivering pumps (p<0.001; Fig. 3a). In α5 knockout mice, injections of saline also did not alter ICSS thresholds. However, mecamylamine significantly altered ICSS thresholds in these mice (Fig. 3b). This was reflected in a main effect of Mecamylamine (F(1, 10) = 24.7, p<0.001). Interestingly, in α5 knockout mice there was no statistically significant Mecamylamine × Pump interaction effect (p>0.05). Consistent with this finding, Bonferroni test demonstrated that mecamylamine treatment significantly elevated ICSS thresholds compared with saline in mice prepared with saline- or nicotine-delivering pumps (p<0.05 in both cases; Fig. 3b). Two-way repeated-measures ANOVA shoed that the latency to respond for stimulation following mecamylamine was unaltered in wildtype or knockout mice with saline or nicotine minipumps (Fig. 3c).
Figure 3.

The effects of mecamylamine (5 mg/kg) on ICSS thresholds were examined in nicotine-dependent wildtype and α5 knockout mice. Data are expressed as mean (± SEM) percentage chance from baseline ICSS thresholds. (a) ICSS thresholds were elevated by mecamylamine in nicotine-treated but not in saline-treated wildtype mice. ***P<0.001 (b) ICSS thresholds were elevated by mecamylamine in both saline-treated and nicotine-treated knockout mice. *P<0.05 (c) Response latencies did not differ based on minipump (saline or nicotine) or injection (saline or mecamylamine) between genotypes. Data are expressed as mean (± SEM) seconds to respond for stimulation.
As thresholds were unexpectedly elevated by mecamylamine in the saline-treated α5 knockout mice (Fig. 2b), we directly compared the effects of mecamylamine on thresholds in wildtype and α5 knockout mice prepared with saline-delivering minipumps. We found that mecamylamine had statistically significant effects on ICSS thresholds, reflected in a main effect of Mecamylamine (F(1, 12) = 9.7, p<0.01) and a significant Mecamylamine × Genotype interaction effect (F(1, 12) = 7.8, p<0.05). Bonferroni test again demonstrated that mecamylamine significantly elevated ICSS thresholds in saline-treated α5 knockout mice compared with saline-treated α5 knockout mice (p<0.01), with thresholds also elevated in this group when compared with the mecamylamine-treated wildtype mice (p<0.01).
Experiment 4: Conditioned Taste Aversion to nicotine and lithium
Wildtype and α5 knockout mice did not differ in their baseline consumption of 0.15% saccharin in a one-bottle pre-test (p>0.05) (Fig. 4a). When saccharin was paired with nicotine injections, both the wildtype and α5 knockout mice demonstrated statistically significant reductions in their consumption of the nicotine-paired solution: Drug (F(1,19)= 101, p<0.0001); Dose × Genotype interaction (F(1,19)= 1.21, NS) (Fig. 4b). Similarly, when saccharin consumption was paired with LiCl injections, both the wildtype and α5 knockout mice drank considerably less saccharin solution than their saline-injected littermates: Drug (F(1,23)=55.9, p<0.0001); Dose × Genotype interaction (F(1,23)= 0.60, NS) (Fig. 4c). To investigate whether the same learned aversion could be elicited with nicotine, a separate cohort of mice were examined for their ability to develop a CTA with a high dose of nicotine (1.5 mg/kg, free base).
Figure 4.
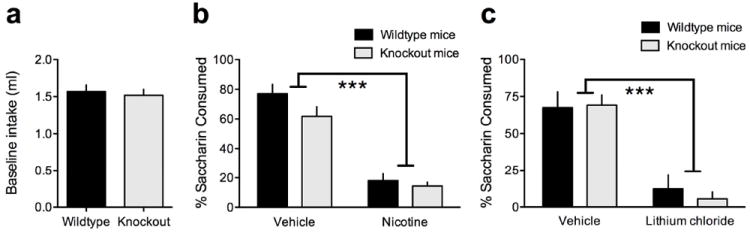
Nicotine and lithium condition a taste aversion to saccharine similarly in wildtype and α5 knockout mice. (a) Wildtype and α5 knockout mice did not differ in their baseline consumption of 0.15% saccharin. Data are expressed as mean (± SEM) volume (ml) consumed. (b) Nicotine (1.5 mg/kg) injections paired with saccharin drinking resulted in decreased consumption of saccharin on the test day for both wildtype and α5 knockout mice, as compared to vehicle-injected controls. Data are expressed as mean (± SEM) percentage of saccharin consumed in a two-bottle choice test. *P<0.05 (c) Pairing LiCl injections with saccharin drinking resulted in decreased consumption of saccharin on the test day for both wildtype and α5 knockout mice, as compared to vehicle-injected controls. Data are expressed as mean (± SEM) percentage of saccharin consumed in a two-bottle choice test. *P<0.05.
DISCUSSION
We found that the magnitude by which acutely administered nicotine injections lowered ICSS thresholds was similar in α5 nAChR subunit knockout mice and their wildtype littermates. We also found that the magnitude by which mecamylamine-precipitated elevations of ICSS thresholds was similar in nicotine-dependent wildtype and knockout mice. These findings suggest that the reward-enhancing effects of acutely administered nicotine, and the reward-inhibiting effects of precipitated nicotine withdrawal, are not regulated by α5* nAChRs. Interestingly, however, as the dose of acutely administered nicotine increased we found that the lowering effects of the drug on ICSS thresholds were greatly diminished in wildtype mice. This likely reflects the fact that reward-inhibiting (aversive) effects of higher nicotine doses oppose the reward-enhancing effects of the drug, resulting in ICSS thresholds returning to baseline levels. Higher doses of nicotine than those tested here would likely have had more intense reward-inhibiting effects and raised thresholds above baseline levels in wildtype mice, consistent with recently published observations in rats (Fowler et al. 2011). Importantly, higher nicotine doses continued to lower ICSS thresholds in the α5 knockout mice. Wildtype and knockout mice were equally sensitive to noxious effects of a high nicotine dose and to lithium, as measured using a conditioned taste aversion procedure. These findings suggest that deficits in α5* nAChR signaling do not abolish general sensitivity to noxious effects of nicotine. Instead, deficient α5* nAChR signaling renders mice resistant to the reward-inhibiting actions of nicotine at higher doses that oppose its reward-enhancing effects and broadens the range of nicotine doses that facilitate brain reward activity. Obtaining the reward-enhancing properties of nicotine is considered a major source of motivation that drives the development and persistence of the tobacco habit in human smokers (Kenny and Markou 2006). Therefore, the current observations may explain the high rates to tobacco dependence in individual carrying CHRNA5 risk alleles, and the fact that these individuals are also likely to be heavier smokers.
The lowering effects of acute experimenter-administered nicotine injections on ICSS thresholds in wildtype and α5 knockout mice are similar in magnitude to those observed in rats that volitionally self-administered the drug by intravenous injections (Kenny et al. 2009; Kenny and Markou 2006). Rats regulate their nicotine self-administration behavior to a level that achieves maximal nicotine-induced lowering of ICSS thresholds (Kenny et al. 2009; Kenny and Markou 2006). Hence, the lowering effects of nicotine on ICSS thresholds, induced by experimenter- or self-administered nicotine infusions likely reflect the stimulatory actions of the drug on brain reward systems that are responsible for establishing and maintaining the nicotine-taking habit in rodents and perhaps the tobacco habit in human smokers (Kenny 2007). As such, the present findings suggest that the reward-enhancing effects of nicotine that drive the establishment of the tobacco habit are unaltered by genetic ablation of α5* nAChRs.
This is consistent with recent observations from our laboratory demonstrating that virus-mediated knockdown of α5 nAChR subunits specifically in the MHb-IPN system did not impact the lowering effects of systemic nicotine injections on ICSS thresholds in rats (Fowler et al. 2011). Hence, the present findings generalize these previous observations suggesting a lack of involvement of α5* nAChRs in the MHb-IPN system in nicotine reward to global brain reward circuitry with relevance to addiction. Based on these data, we conclude that α5* nAChRs are unlikely to play any role in the reward-enhancing properties of nicotine.
In contrast to the lack of effect of α5 nAChR subunit ablation on the magnitude of nicotine-induced lowering of ICSS thresholds, we observed that higher doses of nicotine that no longer lowered ICSS thresholds in wildtype mice continued to lower thresholds in the knockout mice. Recently, we showed that instead of the lowered ICSS thresholds detected at lower doses, high nicotine doses elevate ICSS thresholds above baseline levels in rats (Fowler et al. 2011). Elevations of ICSS thresholds induced by higher doses of nicotine reflect an inhibitory action of the drug on brain reward systems, which drives avoidance behavior. Indeed, higher doses of nicotine that elevate ICSS thresholds are known to be aversive in rodents, with animals avoiding actions or environments associated with these concentrations of the drug (Fowler et al. 2011; Jensen et al. 1990; Risinger and Oakes 1995). Therefore, it is likely that higher nicotine doses failed to lower ICSS thresholds in wildtype mice because of the opposing reward-inhibiting actions of the drug at these doses. The fact that higher nicotine doses continued to lower ICSS thresholds in the knockout mice suggests that the reward-inhibiting effects of the drug are selectively ablated by disruption of α5* nAChR signaling. Nicotine, including that contained in tobacco smoke, has aversive effects, with non-human primates and human tobacco smokers demonstrating avoidance of the drug particularly when higher unit doses are available for consumption. It is important to note that even doses of nicotine that are actively self-administered by non-human primates or humans can be aversive and trigger avoidance behaviors (Goldberg and Spealman 1982; 1983; Goldberg et al. 1981; Goldberg et al. 1983; Henningfield and Goldberg 1983; Henningfield et al. 1986; Spealman and Goldberg 1982). This suggests that the reward-enhancing and reward-inhibiting effects of nicotine occur concurrently, with the reward-inhibiting effects that suppress nicotine intake becoming more marked as the unit dose of nicotine available for consumption increases (Fowler et al. 2011). As the reward-inhibiting effects of nicotine act to diminish the motivation to consume the drug (Fowler et al. 2011), the present findings suggest that increased vulnerability to tobacco dependence in those carrying CHRNA5 risk alleles may be explained not by alterations in the reward-enhancing effects of nicotine, but instead by diminished sensitivity to reward-inhibiting actions of the drug that support avoidance behaviors. Consistent with this interpretation, we recently reported that α5 nAChR subunit knockdown in the MHb-IPN system resulted in greater amounts of nicotine consumption in rats with concurrent reductions in sensitivity to the reward-inhibiting effects of the drug, without impacting its reward-enhancing effects (Fowler et al. 2011). When interpreting the current data within this context, it seems that the major contribution of α5* nAChR throughout the entire brain is to control aversion-related actions of nicotine, as global knockout of the subunit results in a pattern of nicotine effects on ICSS very similar to that seen when knockdown was restricted to the MHb-IPN system; see also (Jackson et al. 2010). A similar role has recently been ascribed to β4 nAChRs in the MHb-IPN system in regulating the aversive properties of nicotine (Frahm et al. 2011). This suggests that the entire CHRNA5-CHRNA3-CHRNB4 gene cluster, expression of which is enriched in the MHb-IPN system, is critical for regulating sensitivity to the aversive effects of nicotine and for avoidance of the drug. As such, deficits in the function of nAChRs incorporating these subunits likely results in reduced sensitivity to nicotine aversion and consequently greater consumption of the drug and vulnerability to tobacco addiction. Nevertheless, it is important to note that we found that α5 knockout mice developed conditioned taste aversions to nicotine and lithium of similar magnitude to their wildtype counterparts. This observation suggests that α5* nAChRs are not involved in all aspects of the noxious effects of higher nicotine doses that may trigger avoidance of the drug, but instead are specifically involved in regulating the inhibitory effects of the drug on brain reward systems.
It has been shown that direct infusion of mecamylamine into the IPN of nicotine-dependent wildtype mice precipitates the expression of somatic withdrawal signs (Salas et al. 2009). This suggests that the IPN may play a prominent role in the nicotine withdrawal syndrome. Indeed, α5 knockout mice that were rendered nicotine dependent via osmotic minipumps administration did not show somatic signs of nicotine withdrawal when precipitated with mecamylamine (Jackson et al. 2008; Salas et al. 2009). These findings suggest that α5* nAChRs, particularly those in the MHb-IPN tract, regulate aspects of the nicotine withdrawal syndrome. We previously reported that spontaneous or mecamylamine-precipitated withdrawal from chronic nicotine elevates ICSS thresholds in nicotine-dependent wildtype mice (Johnson et al. 2008), observations that are consistent with those from other laboratories (Stoker et al. 2008). Avoidance and alleviation of the reward-inhibiting effects of nicotine withdrawal is hypothesized to serve as an important substrate for negative reinforcement that motivates continued nicotine use and relapse to use during periods of attempted abstinence (Kenny 2007; Kenny and Markou 2001). Considering the role for α5* nAChRs in the reward-inhibiting effects of acutely administered nicotine described above, and the potential involvement of α5* nAChR signaling in the IPN in regulating aspects of the nicotine withdrawal syndrome, we next sought to investigate the contribution of these receptors to the reward-inhibiting effects of nicotine withdrawal. To induce nicotine dependence, we implanted wildtype and α5 knockout mice with osmotic minipumps delivering nicotine or saline continuously. Interestingly, we found that thresholds were elevated in wildtype mice one day after implantation of saline-delivering minipumps compared with thresholds measured in wildtype mice receiving nicotine-delivering pumps. This may reflect that fact that nicotine is known to have pain-alleviating effects (Marubio et al. 1999), which may have attenuated some post-surgical discomfort experienced by the animals during this relatively non-invasive surgery (see Methods). Interestingly, the α5 knockout mice receiving saline treatments did not demonstrate similarly elevated ICSS thresholds one day after osmotic minipumps implantation surgery. This is consistent with recent findings suggesting that α5* nAChRs may regulate pain sensitivity, with increases in α5 subunit expression perhaps contributing to states of allodynia (Marubio et al. 1999; Vincler and Eisenach 2005; Young et al. 2008).
After nicotine dependence had been induced in wildtype and α5 knockout mice, we found that the magnitude by which the nAChR antagonist mecamylamine elevated ICSS thresholds was similar in nicotine-dependent wildtype and α5 knockout mice. This suggests that separate neurobiological systems regulate the aversion-related effects of acute nicotine or withdrawal from chronic treatment, and that these systems can be dissociated by genetic deletion of the α5 nAChR subunit. From a conceptual perspective, such dissociation in function may not be such a surprise. It has been hypothesized that neuroadaptative responses to nicotine that occur during prolonged exposure to drugs of abuse, which give rise to the reward-inhibiting effects of nicotine withdrawal, may reside in the same brain circuits that regulate the acute reward-enhancing actions of these drugs (Le Moal and Koob 2007). Furthermore, it has been proposed that the mesoaccumbens system, particularly the VTA, is the key substrate that regulates the reward-enhancing effects of nicotine (Caille et al. 2009; Farquhar et al. 2012; Kenny et al. 2009), whereas the MHb-IPN systems seems to be primarily involved in the reward-inhibiting effects of nicotine (see above). Therefore, it may be predicted that molecular adaptations that reside in the VTA or other areas involved in nicotine reward are likely to play a prominent role in the emergence of a state of negative reward during withdrawal from chronic nicotine treatment and the development of nicotine dependence. Indeed, it has been shown that disruption of nAChR signaling in the VTA can precipitate withdrawal in nicotine-dependent rodents (Bruijnzeel and Markou 2004; Hildebrand et al. 1999; Liu and Jin 2004), however, see (Salas et al. 2009). A major caveat to the interpretation that α5* nAChRs are not involved in reward-inhibiting effects of nicotine withdrawal is the fact that we precipitated withdrawal using mecamylamine, and the effects of spontaneous nicotine withdrawal on ICSS thresholds were not assessed. Therefore, it will be important in future studies to assess the effects of spontaneous nicotine withdrawal on ICSS thresholds in α5 subunit knockout mice and their wildtype counterparts.
We were surprised to find that mecamylamine precipitated withdrawal-like elevations of ICSS thresholds in saline-treated α5 knockout mice but not in wildtype mice. Mecamylamine at doses used in this study and higher typically do not induce behavioral deficits, such as ICSS threshold elevations, in non-nicotine treated mice (Collins et al. 1994; Hilario et al. 2012; Johnson et al. 2008; Singh et al. 2013; Stoker et al. 2012). We observed no differences response latency for self-stimulation in response to mecamylamine administration in either wildtype or knockout mice, suggesting that the elevating effect of mecamylamine on ICSS thresholds in the knockout mice was a specific action on brain reward function in these mice. This finding could be interpreted in at least two ways. First, the α5 knockout mice are more sensitive to the aversive effects of mecamylamine that would also be detected in wildtype mice if they had been treated with substantially higher doses of the antagonist. In this case, the intrinsic aversive properties of mecamylamine seen in the α5 knockout mice would be distinct from the state of negative reward precipitated by the drug in nicotine-dependent animals. Second, another explanation is that the α5 knockout mice are already in a behavioral state that resembles nicotine dependence, perhaps due to adaptions in non-α5* nAChRs in response to genetic deletion of the α5 subunit. In this case, the elevated ICSS thresholds induced by mecamylamine in saline-treated α5 knockout mice would be mechanistically indistinguishable from the elevated thresholds precipitated by the drug in nicotine-dependent wildtype mice. Further studies will be required to test these possibilities.
Finally, nicotine continued to lower ICSS thresholds in the knockout mice at doses that no longer lowered thresholds in wildtype mice. This suggests that α5* nAChRs are likely involved in reward-inhibiting (aversive) effects of nicotine that oppose its rewarding actions. However, it is currently unclear if α5* nAChRs regulate the general noxious effects of nicotine (malaise, sickness-like behaviors etc.) such that the knockout mice are simply insensitive to the effects of higher nicotine doses, or if the role for α5* nAChRs is more specifically involved in reward-inhibiting effects of the drug. To investigate this issue further, we examined whether α5* nAChRs regulate the development of a conditioned taste aversion to nicotine. In addition, we also assessed the development of a conditioned taste aversion to lithium in order to investigate if α5 knockout mice may have altered sensitivity to noxious stimuli other than nicotine. The conditioned taste aversion (CTA) procedure provides an index of the aversive effects of drugs of abuse that serve to limit intake. Unlike ICSS thresholds, which provide a specific measure of the reward-enhancing or reward-inhibiting effects of a drug, CTA can provide a more general measure of noxious drug effects (Cappell and Le Blanc 1975). Interestingly, wildtype and α5 knockout mice displayed robust CTA to saccharin paired with nicotine or lithium injections. This suggests that α5* nAChRs do not regulate the general aversive actions of nicotine, such as sickness-like behaviors or malaise, but rather the reward-inhibiting effects of the drug that oppose its reward-enhancing effects. It should be noted that controversy exists regarding the interpretation of the conditioned taste aversion elicited with drugs of abuse; the reduction in drug-paired solution has been proposed to reflect conditioned suppression of intake because of a negative contrast of saccharine effects to positive drug effects, rather than bona fide aversion towards the drug (Grigson 1997). However, in our studies, both nicotine and lithium induced a similar CTA in the mice, suggesting that both were similarly aversive. It has been proposed that the negative physiological state induced by nicotine is due to activation of ganglionic nAChRs, particularly those containing α3* and/or β4 subunits (Marubio et al. 1999; Shoaib et al. 2002). More recently, Shoaib and colleagues have shown that the β2* nAChR-preferring antagonist DHβE, but not the α7 nAChR antagonist methyllycaconitine, decreased the expression of a CTA (Gommans et al. 2000) and ablation of β2 nAChR subunits attenuates the formation of a CTA across a range of nicotine doses (0.4-2.0 mg/kg) (Shoaib et al. 2002). As such, it is likely that nAChRs containing α3, β4 and/or β2 subunits regulate the general noxious effects of nicotine, as measured by CTA, and that α5 subunits make a minimal contribution to this state. Together, these findings suggest that α5* nAChRs regulate the reward-inhibiting effects of nicotine that oppose its reward-enhancing effects, but are not involved in the more global aversive effects of the drug when delivered at high doses.
In summary, the present findings demonstrate that disruption of α5* nAChR signaling extends the range of nicotine doses that can facilitate the activity of brain reward systems. However, α5* nAChRs are unlikely to play a role in the reward-enhancing effects of nicotine, the reward-inhibiting effects of nicotine withdrawal, or the general noxious effects of high-dose nicotine. Together, these findings enhance our understanding of the role of α5 nAChR signaling in regulating the actions of nicotine on brain reward systems and thereby contribute to our understanding of the factors likely influencing tobacco dependence.
Acknowledgments
This work was supported by a grant from the National Institute on Drug Abuse (D032543 to C.D.F. and DA020686 to P.J.K.). This is manuscript number 23093 from The Scripps Research Institute.
Footnotes
CONFLICT OF INTEREST:
The authors have no conflicts of interest to disclose.
References
- Albuquerque EX, Pereira EF, Castro NG, Alkondon M, Reinhardt S, Schroder H, Maelicke A. Nicotinic receptor function in the mammalian central nervous system. Ann N Y Acad Sci. 1995;757:48–72. doi: 10.1111/j.1749-6632.1995.tb17464.x. [DOI] [PubMed] [Google Scholar]
- Bachmanov AA, Reed DR, Beauchamp GK, Tordoff MG. Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behavior genetics. 2002;32:435–43. doi: 10.1023/a:1020884312053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrettini W, Yuan X, Tozzi F, Song K, Francks C, Chilcoat H, Waterworth D, Muglia P, Mooser V. Alpha-5/alpha-3 nicotinic receptor subunit alleles increase risk for heavy smoking. Molecular psychiatry. 2008;13:368–73. doi: 10.1038/sj.mp.4002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ. Convergence of genetic findings for nicotine dependence and smoking related diseases with chromosome 15q24-25. Trends Pharmacol Sci. 2010;31:46–51. doi: 10.1016/j.tips.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, Saccone NL, Saccone SF, Bertelsen S, Fox L, Horton WJ, Breslau N, Budde J, Cloninger CR, Dick DM, Foroud T, Hatsukami D, Hesselbrock V, Johnson EO, Kramer J, Kuperman S, Madden PA, Mayo K, Nurnberger J, Jr, Pomerleau O, Porjesz B, Reyes O, Schuckit M, Swan G, Tischfield JA, Edenberg HJ, Rice JP, Goate AM. Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry. 2008;165:1163–71. doi: 10.1176/appi.ajp.2008.07111711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijnzeel AW, Markou A. Adaptations in cholinergic transmission in the ventral tegmental area associated with the affective signs of nicotine withdrawal in rats. Neuropharmacology. 2004;47:572–9. doi: 10.1016/j.neuropharm.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Caille S, Guillem K, Cador M, Manzoni O, Georges F. Voluntary nicotine consumption triggers in vivo potentiation of cortical excitatory drives to midbrain dopaminergic neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:10410–5. doi: 10.1523/JNEUROSCI.2950-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappell H, Le Blanc AE. Conditioned aversion by psychoactive drugs: does it have significance for an understanding of drug dependence? Addictive behaviors. 1975;1:55–64. doi: 10.1016/s0306-4603(75)80018-9. [DOI] [PubMed] [Google Scholar]
- Collins AC, Luo Y, Selvaag S, Marks MJ. Sensitivity to nicotine and brain nicotinic receptors are altered by chronic nicotine and mecamylamine infusion. The Journal of pharmacology and experimental therapeutics. 1994;271:125–33. [PubMed] [Google Scholar]
- Doherty K, Kinnunen T, Militello FS, Garvey AJ. Urges to smoke during the first month of abstinence: relationship to relapse and predictors. Psychopharmacology. 1995;119:171–8. doi: 10.1007/BF02246158. [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S. Alpha 9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell. 1994;79:705–15. doi: 10.1016/0092-8674(94)90555-x. [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Vetter DE, Katz E, Rothlin CV, Heinemann SF, Boulter J. alpha10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:3501–6. doi: 10.1073/pnas.051622798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquhar MJ, Latimer MP, Winn P. Nicotine self-administered directly into the VTA by rats is weakly reinforcing but has strong reinforcement enhancing properties. Psychopharmacology. 2012;220:43–54. doi: 10.1007/s00213-011-2452-8. [DOI] [PubMed] [Google Scholar]
- Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up-regulated by chronic nicotine treatment. Molecular pharmacology. 1992;41:31–7. [PubMed] [Google Scholar]
- Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ. Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011;471:597–601. doi: 10.1038/nature09797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frahm S, Slimak MA, Ferrarese L, Santos-Torres J, Antolin-Fontes B, Auer S, Filkin S, Pons S, Fontaine JF, Tsetlin V, Maskos U, Ibanez-Tallon I. Aversion to Nicotine Is Regulated by the Balanced Activity of beta4 and alpha5 Nicotinic Receptor Subunits in the Medial Habenula. Neuron. 2011;70:522–35. doi: 10.1016/j.neuron.2011.04.013. [DOI] [PubMed] [Google Scholar]
- Goldberg SR, Spealman RD. Maintenance and suppression of behavior by intravenous nicotine injections in squirrel monkeys. Federation proceedings. 1982;41:216–20. [PubMed] [Google Scholar]
- Goldberg SR, Spealman RD. Suppression of behavior by intravenous injections of nicotine or by electric shocks in squirrel monkeys: effects of chlordiazepoxide and mecamylamine. The Journal of pharmacology and experimental therapeutics. 1983;224:334–40. [PubMed] [Google Scholar]
- Goldberg SR, Spealman RD, Goldberg DM. Persistent behavior at high rates maintained by intravenous self-administration of nicotine. Science. 1981;214:573–5. doi: 10.1126/science.7291998. [DOI] [PubMed] [Google Scholar]
- Goldberg SR, Spealman RD, Risner ME, Henningfield JE. Control of behavior by intravenous nicotine injections in laboratory animals. Pharmacology, biochemistry, and behavior. 1983;19:1011–20. doi: 10.1016/0091-3057(83)90408-2. [DOI] [PubMed] [Google Scholar]
- Gommans J, Stolerman IP, Shoaib M. Antagonism of the discriminative and aversive stimulus properties of nicotine in C57BL/6J mice. Neuropharmacology. 2000;39:2840–7. doi: 10.1016/s0028-3908(00)00130-1. [DOI] [PubMed] [Google Scholar]
- Grigson PS. Conditioned taste aversions and drugs of abuse: a reinterpretation. Behavioral neuroscience. 1997;111:129–36. [PubMed] [Google Scholar]
- Grucza RA, Wang JC, Stitzel JA, Hinrichs AL, Saccone SF, Saccone NL, Bucholz KK, Cloninger CR, Neuman RJ, Budde JP, Fox L, Bertelsen S, Kramer J, Hesselbrock V, Tischfield J, Nurnberger JI, Jr, Almasy L, Porjesz B, Kuperman S, Schuckit MA, Edenberg HJ, Rice JP, Goate AM, Bierut LJ. A Risk Allele for Nicotine Dependence in CHRNA5 Is a Protective Allele for Cocaine Dependence. Biological psychiatry. 2008 doi: 10.1016/j.biopsych.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henningfield JE, Goldberg SR. Control of behavior by intravenous nicotine injections in human subjects. Pharmacology, biochemistry, and behavior. 1983;19:1021–6. doi: 10.1016/0091-3057(83)90409-4. [DOI] [PubMed] [Google Scholar]
- Henningfield JE, Goldberg SR, Herning RI, Jasinski DR, Lukas SE, Miyasato K, Nemeth-Coslett R, Pickworth WB, Rose JE, Sampson A, et al. Human studies of the behavioral pharmacological determinants of nicotine dependence. NIDA research monograph. 1986;67:54–65. [PubMed] [Google Scholar]
- Hilario MR, Turner JR, Blendy JA. Reward sensitization: effects of repeated nicotine exposure and withdrawal in mice. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2012;37:2661–70. doi: 10.1038/npp.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand BE, Panagis G, Svensson TH, Nomikos GG. Behavioral and biochemical manifestations of mecamylamine-precipitated nicotine withdrawal in the rat: role of nicotinic receptors in the ventral tegmental area. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 1999;21:560–74. doi: 10.1016/S0893-133X(99)00055-X. [DOI] [PubMed] [Google Scholar]
- Hughes JR, Gust SW, Skoog K, Keenan RM, Fenwick JW. Symptoms of tobacco withdrawal. A replication and extension. Arch Gen Psychiatry. 1991;48:52–9. doi: 10.1001/archpsyc.1991.01810250054007. [DOI] [PubMed] [Google Scholar]
- Jackson KJ, Marks MJ, Vann RE, Chen X, Gamage TF, Warner JA, Damaj MI. Role of alpha5 nicotinic acetylcholine receptors in pharmacological and behavioral effects of nicotine in mice. The Journal of pharmacology and experimental therapeutics. 2010;334:137–46. doi: 10.1124/jpet.110.165738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson KJ, Martin BR, Changeux JP, Damaj MI. Differential role of nicotinic acetylcholine receptor subunits in physical and affective nicotine withdrawal signs. The Journal of pharmacology and experimental therapeutics: in press. 2008 doi: 10.1124/jpet.107.132977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RA, Gilbert DG, Meliska CJ, Landrum TA, Szary AB. Characterization of a dose-response curve for nicotine-induced conditioned taste aversion in rats: relationship to elevation of plasma beta-endorphin concentration. Behavioral and neural biology. 1990;53:428–40. doi: 10.1016/0163-1047(90)90310-3. [DOI] [PubMed] [Google Scholar]
- Johnson PM, Hollander JA, Kenny PJ. Decreased brain reward function during nicotine withdrawal in C57BL6 mice: evidence from intracranial self-stimulation (ICSS) studies. Pharmacology, biochemistry, and behavior. 2008;90:409–15. doi: 10.1016/j.pbb.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PJ. Brain reward systems and compulsive drug use. Trends Pharmacol Sci. 2007;28:135–41. doi: 10.1016/j.tips.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Kenny PJ, Chartoff E, Roberto M, Carlezon WA, Jr, Markou A. NMDA receptors regulate nicotine-enhanced brain reward function and intravenous nicotine self-administration: role of the ventral tegmental area and central nucleus of the amygdala. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2009;34:266–81. doi: 10.1038/npp.2008.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PJ, Markou A. Neurobiology of the nicotine withdrawal syndrome. Pharmacology, biochemistry, and behavior. 2001;70:531–49. doi: 10.1016/s0091-3057(01)00651-7. [DOI] [PubMed] [Google Scholar]
- Kenny PJ, Markou A. Nicotine self-administration acutely activates brain reward systems and induces a long-lasting increase in reward sensitivity. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2006;31:1203–11. doi: 10.1038/sj.npp.1300905. [DOI] [PubMed] [Google Scholar]
- Le Moal M, Koob GF. Drug addiction: pathways to the disease and pathophysiological perspectives. European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology. 2007;17:377–93. doi: 10.1016/j.euroneuro.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Le Novere N, Corringer PJ, Changeux JP. The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J Neurobiol. 2002;53:447–56. doi: 10.1002/neu.10153. [DOI] [PubMed] [Google Scholar]
- Lena C, Changeux JP. Allosteric nicotinic receptors, human pathologies. J Physiol Paris. 1998;92:63–74. doi: 10.1016/S0928-4257(98)80140-X. [DOI] [PubMed] [Google Scholar]
- Lips EH, Gaborieau V, McKay JD, Chabrier A, Hung RJ, Boffetta P, Hashibe M, Zaridze D, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, Field JK, Liloglou T, Xinarianos G, McLaughlin J, Liu G, Skorpen F, Elvestad MB, Hveem K, Vatten L, Study E, Benhamou S, Lagiou P, Holcatova I, Merletti F, Kjaerheim K, Agudo A, Castellsague X, Macfarlane TV, Barzan L, Canova C, Lowry R, Conway DI, Znaor A, Healy C, Curado MP, Koifman S, Eluf-Neto J, Matos E, Menezes A, Fernandez L, Metspalu A, Heath S, Lathrop M, Brennan P. Association between a 15q25 gene variant, smoking quantity and tobacco-related cancers among 17 000 individuals. Int J Epidemiol. 2009 doi: 10.1093/ije/dyp288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZH, Jin WQ. Decrease of ventral tegmental area dopamine neuronal activity in nicotine withdrawal rats. Neuroreport. 2004;15:1479–81. doi: 10.1097/01.wnr.0000126218.25235.b6. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1992;12:2765–84. doi: 10.1523/JNEUROSCI.12-07-02765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marubio LM, del Mar Arroyo-Jimenez M, Cordero-Erausquin M, Lena C, Le Novere N, de Kerchove d’Exaerde A, Huchet M, Damaj MI, Changeux JP. Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature. 1999;398:805–10. doi: 10.1038/19756. [DOI] [PubMed] [Google Scholar]
- Paxinos G. The mouse brain in stereotaxic coordinates. Academic Press; San Diego, California: 2001. [Google Scholar]
- Pescatore KA, Glowa JR, Riley AL. Strain differences in the acquisition of nicotine-induced conditioned taste aversion. Pharmacology, biochemistry, and behavior. 2005;82:751–7. doi: 10.1016/j.pbb.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Piasecki TM, Fiore MC, Baker TB. Profiles in discouragement: two studies of variability in the time course of smoking withdrawal symptoms. J Abnorm Psychol. 1998;107:238–51. doi: 10.1037//0021-843x.107.2.238. [DOI] [PubMed] [Google Scholar]
- Piasecki TM, Jorenby DE, Smith SS, Fiore MC, Baker TB. Smoking withdrawal dynamics: II. Improved tests of withdrawal-relapse relations. J Abnorm Psychol. 2003;112:14–27. [PubMed] [Google Scholar]
- Rauhut AS, Hawrylak M, Mardekian SK. Bupropion differentially alters the aversive, locomotor and rewarding properties of nicotine in CD-1 mice. Pharmacology, biochemistry, and behavior. 2008;90:598–607. doi: 10.1016/j.pbb.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risinger FO, Oakes RA. Nicotine-induced conditioned place preference and conditioned place aversion in mice. Pharmacology, biochemistry, and behavior. 1995;51:457–61. doi: 10.1016/0091-3057(95)00007-j. [DOI] [PubMed] [Google Scholar]
- Saccone SF, Hinrichs AL, Saccone NL, Chase GA, Konvicka K, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau O, Swan GE, Goate AM, Rutter J, Bertelsen S, Fox L, Fugman D, Martin NG, Montgomery GW, Wang JC, Ballinger DG, Rice JP, Bierut LJ. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Human molecular genetics. 2007;16:36–49. doi: 10.1093/hmg/ddl438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Sturm R, Boulter J, De Biasi M. Nicotinic receptors in the habenulo-interpeduncular system are necessary for nicotine withdrawal in mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:3014–8. doi: 10.1523/JNEUROSCI.4934-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherva R, Wilhelmsen K, Pomerleau CS, Chasse SA, Rice JP, Snedecor SM, Bierut LJ, Neuman RJ, Pomerleau OF. Association of a single nucleotide polymorphism in neuronal acetylcholine receptor subunit alpha 5 (CHRNA5) with smoking status and with ‘pleasurable buzz’ during early experimentation with smoking. Addiction. 2008;103:1544–52. doi: 10.1111/j.1360-0443.2008.02279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiffman SM, Jarvik ME. Smoking withdrawal symptoms in two weeks of abstinence. Psychopharmacology. 1976;50:35–9. doi: 10.1007/BF00634151. [DOI] [PubMed] [Google Scholar]
- Shoaib M, Gommans J, Morley A, Stolerman IP, Grailhe R, Changeux JP. The role of nicotinic receptor beta-2 subunits in nicotine discrimination and conditioned taste aversion. Neuropharmacology. 2002;42:530–9. doi: 10.1016/s0028-3908(01)00194-0. [DOI] [PubMed] [Google Scholar]
- Singh TG, Rehni AK, Arora S. Ro 32-0432 attenuates mecamylamine-precipitated nicotine withdrawal syndrome in mice. Naunyn-Schmiedeberg’s archives of pharmacology. 2013;386:197–204. doi: 10.1007/s00210-012-0825-0. [DOI] [PubMed] [Google Scholar]
- Spealman RD, Goldberg SR. Maintenance of schedule-controlled behavior by intravenous injections of nicotine in squirrel monkeys. The Journal of pharmacology and experimental therapeutics. 1982;223:402–8. [PubMed] [Google Scholar]
- Stevens VL, Bierut LJ, Talbot JT, Wang JC, Sun J, Hinrichs AL, Thun MJ, Goate A, Calle EE. Nicotinic receptor gene variants influence susceptibility to heavy smoking. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2008;17:3517–25. doi: 10.1158/1055-9965.EPI-08-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoker AK, Olivier B, Markou A. Role of alpha7- and beta4-containing nicotinic acetylcholine receptors in the affective and somatic aspects of nicotine withdrawal: studies in knockout mice. Behavior genetics. 2012;42:423–36. doi: 10.1007/s10519-011-9511-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoker AK, Semenova S, Markou A. Affective and somatic aspects of spontaneous and precipitated nicotine withdrawal in C57BL/6J and BALB/cByJ mice. Neuropharmacology. 2008;54:1223–32. doi: 10.1016/j.neuropharm.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolerman IP, Jarvis MJ. The scientific case that nicotine is addictive. Psychopharmacology. 1995;117:2–10. doi: 10.1007/BF02245088. discussion 14-20. [DOI] [PubMed] [Google Scholar]
- Vincler MA, Eisenach JC. Knock down of the alpha 5 nicotinic acetylcholine receptor in spinal nerve-ligated rats alleviates mechanical allodynia. Pharmacology, biochemistry, and behavior. 2005;80:135–43. doi: 10.1016/j.pbb.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Wang JC, Grucza R, Cruchaga C, Hinrichs AL, Bertelsen S, Budde JP, Fox L, Goldstein E, Reyes O, Saccone N, Saccone S, Xuei X, Bucholz K, Kuperman S, Nurnberger J, Jr, Rice JP, Schuckit M, Tischfield J, Hesselbrock V, Porjesz B, Edenberg HJ, Bierut LJ, Goate AM. Genetic variation in the CHRNA5 gene affects mRNA levels and is associated with risk for alcohol dependence. Molecular psychiatry. 2009;14:501–10. doi: 10.1038/mp.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss RB, Baker TB, Cannon DS, von Niederhausern A, Dunn DM, Matsunami N, Singh NA, Baird L, Coon H, McMahon WM, Piper ME, Fiore MC, Scholand MB, Connett JE, Kanner RE, Gahring LC, Rogers SW, Hoidal JR, Leppert MF. A candidate gene approach identifies the CHRNA5-A3-B4 region as a risk factor for age-dependent nicotine addiction. PLoS genetics. 2008;4:e1000125. doi: 10.1371/journal.pgen.1000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T, Wittenauer S, Parker R, Vincler M. Peripheral nerve injury alters spinal nicotinic acetylcholine receptor pharmacology. European journal of pharmacology. 2008;590:163–9. doi: 10.1016/j.ejphar.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]