

Abstract
The Protein Kinase Inhibitor (Pki) gene family inactivates nuclear PKA and terminates PKA-induced gene expression. We previously showed that Pkig is the primary family member expressed in osteoblasts and that Pkig knockdown increases the effects of parathyroid hormone and isoproterenol on PKA activation, gene expression, and inhibition of apoptosis. Here, we determined whether endogenous levels of Pkig regulate osteoblast differentiation. Pkig is the primary family member in MEFs, murine marrow-derived mesenchymal stem cells, and human mesenchymal stem cells. Pkig deletion increased forskolin-dependent nuclear PKA activation and gene expression and Pkig deletion or knockdown increased osteoblast differentiation. PKA signaling is known to stimulate adipogenesis; however, adipogenesis and osteogenesis are often reciprocally regulated. We found that the reciprocal regulation predominates over the direct effects of PKA since adipogenesis was decreased by Pkig deletion or knockdown. Pkig deletion or knockdown simultaneously increased osteogenesis and decreased adipogenesis in mixed osteogenic/adipogenic medium. Pkig deletion increased PKA-induced expression of Leukemia Inhibitory Factor (Lif) mRNA and LIF protein. LIF neutralizing antibodies inhibited the effects on osteogenesis and adipogenesis of either Pkig deletion in MEFs or PKIγ knockdown in both murine and human mesenchymal stem cells. Collectively, our results show that endogenous levels of Pkig reciprocally regulate osteoblast and adipocyte differentiation and that this reciprocal regulation is mediated in part by LIF.
Keywords: Protein kinase inhibitor, osteogenesis, adipogenesis, Leukemia Inhibitory Factor, mesenchymal stem cell
Introduction
The cAMP/Protein Kinase A (PKA) signaling pathway is activated by Gαs–coupled receptors and potently regulates multiple cellular processes. Because of the importance of this pathway, its activity is precisely controlled by the interaction of multiple mechanisms (1–4), including inactivation of PKA by the Protein Kinase Inhibitor (Pki) gene family (5–8). We previously showed that inactivation of nuclear PKA by PKIγ is a primary mechanism that reduces PKA signaling induced by Gαs–coupled receptors in mesenchymal cells (9, 10). Consequently, inactivation of nuclear PKA by Pkig reduces PKA-dependent activation of transcription factors and the resultant expression of rapidly and transiently induced mRNAs (9, 10). There have been extensive and elegant investigations of the biochemistry and structure of the PKI family and of the inactivation of PKA by PKI overexpression (5–8), but prior to our studies (9–11), there were only two publications that showed effects of endogenous PKI expressed at physiological levels (12, 13). In this regard, genetic deletion of the other two members of the Pki family, either separately or in combination, caused little detectable phenotype in the resultant mice because of compensatory regulation by other components of the PKA pathway (14, 15).
PKA signaling stimulates bone formation due to effects on the osteoblast lineage, including increased proliferation and differentiation of osteoblast precursors, activation of quiescent lining cells, and prevention of apoptosis (16, 17). We previously showed that inactivation of nuclear PKA by PKIγ is a primary mechanism that reduces the anti-apoptotic effects of Gαs–coupled receptors in osteoblasts (11). Moreover, overexpression of PKIγ was reported to reduce BMP2-induced osteoblast differentiation (18). One of the goals of the current study was therefore to determine whether endogenous PKIγ, expressed at physiological levels, reduces PKA-induced osteoblast differentiation.
Adipocyte differentiation is also stimulated by PKA signaling (19–22) and therefore might be expected to be reduced by PKIγ. However, despite the preponderance of evidence that PKA signaling stimulates adipocyte differentiation (19–22), there are also reports that PKA signaling can have the opposite effect (23–26). As such, timing and magnitude of the PKA signal may be important and the effects of PKI may be complex. It is also unknown whether the direct effects of PKA on adipocyte differentiation will predominate over the reciprocal regulation of adipocyte and osteoblast differentiation that frequently occurs because both lineages differentiate from the same mesenchymal precursor cells (27, 28). Examples of reciprocal regulation that are particularly relevant to the current study are the reduction of adipocyte differentiation and stimulation of osteoblast differentiation in response to PKA activation by forskolin (29), parathyroid hormone (PTH) (30, 31), or transgenic expression of constitutively active PTH receptors (32). The second goal of this study was therefore to determine whether endogenous PKIγ, expressed at physiological levels, stimulates or reduces adipocyte differentiation and to resolve whether the direct effects of PKA predominate over effects due to the reciprocal regulation of adipocyte and osteoblast differentiation.
Leukemia inhibitory factor (LIF), a member of the IL6 family of gp130-dependent cytokines, can stimulate osteoblast differentiation (33–38) and reduce adipocyte differentiation (34, 37, 39, 40). We previously found that Lif is one of the mRNAs that are rapidly and transiently induced by PKA signaling in osteoblasts in cell culture and in vivo (41–43). The third goal of this study was therefore to determine whether downregulation of LIF expression by PKIγ mediates regulation of osteoblast and adipocyte differentiation.
Methods
Murine Embryonic Fibroblasts (MEFs)
Pkig−/− mice generated in our lab were backcrossed with C57BL/6J mice for four generations. Wild type and Pkig−/− mice were obtained by breeding of the resultant Pkig+/− mice and MEFs were prepared from embryos 12.5 to 13.5 days postcoitum using standard techniques (44). MEFs were maintained in growth medium consisting of α-MEM medium (SH30265.01, HyClone, Logan, UT) supplemented with 10% fetal bovine serum (SH30071.03, HyClone), 2 mM L-glutamine (25-005-Cl, Cellgro, Manassas, VA), non-essential amino acids (25-025-Cl, Cellgro), 100 units/ml penicillin and 100 ug/ml streptomycin (30-002-Cl, Cellgro). For experiments, MEFs that had been passaged less than five times were plated at 1×104/cm2 in growth medium.
Osteoblast differentiation by MEFs was induced as previously described (45). Briefly, confluent MEFs were incubated in osteogenic medium consisting of the growth medium supplemented with 50 ug/ml 2-phospho-L-ascorbic acid (49752, Sigma-Aldrich, St. Louis, MO) and 10 mM β-glycerophosphate (G-9891, Sigma-Aldrich) plus 1 uM forskolin (344270, Calbiochem, Gibbstown, NJ) or 0.1% DMSO as a vehicle control with medium changes every 3 or 4 days. Specificity of the effects of forskolin was confirmed by showing that the inactive analogue, dideoxy-forskolin (46), does not induce osteoblast differentiation (Supplementary Figure 1). Indicated cultures also received 20% conditioned medium from CHO cells that stably express BMP4, a kind gift from Dr. Tatsuya Koike, Osaka City University Medical School (47, 48). The ability of this conditioned medium to stimulate osteoblast differentiation is shown in Supplementary Figure 2.
Adipocyte differentiation was induced by incubating two day post-confluent MEFs with multiple cycles of three days in induction medium and one day in maintenance medium as described (49). The maintenance medium was the growth medium supplemented with 10 ug/ml insulin (I5500, Sigma-Aldrich). The induction medium was the maintenance medium further supplemented with 1 uM dexamethasone (D8893, Sigma-Aldrich), either 200 uM indomethacin (I7378, Sigma-Aldrich) or 0.4% ethanol, and either 0.5 mM 3-isobutyl-1-methylxanthine (IBMX, I701, Sigma-Aldrich) or 0.1% DMSO.
To simultaneously induce both osteoblast and adipocyte differentiation, confluent MEFs were incubated with a 1:1 mixture of the osteogenic and adipogenic induction media as recommended (49, 50). For this purpose, the adipogenic induction medium described above was supplemented with both indomethacin and IBMX, but neither forskolin nor BMP4 was added to the osteogenic medium. Media were changed every 3 or 4 days.
Murine marrow-derived Mesenchymal Stem Cells (mdMSCs)
The mdMSC cell line was obtained from bone marrow of adult male C57BL/6 mice and was previously described (51). mdMSCs were maintained in the same growth medium described above for MEFs. For reverse transfection, 5×104 mdMSCs in 1 ml of growth medium were added to individual wells of a 12-well plate containing 200 pmoles of murine Pkig siRNA (Dharmacon On-TARGET plus J-049150-10, Thermo Scientific, Waltham, MA) or non-targeting siRNA (Dharmacon D-001810-10-20, Thermo Scientific) that had been pre-incubated for 30 minutes at room temperature with DharmaFECT 3 transfection reagent (T-2003-03, Thermo Scientific) and DharmaFECT cell culture reagent (B-004500-100, Thermo Scientific). Transfection reagents were replaced with growth medium after four hours of incubation at 37°C in 5% CO2. Twenty hours later, mdMSCs were incubated with a 1:1 mixture of the osteogenic and adipogenic media previously described for these cells (51) further supplemented with 0.25 mM IBMX. Media were changed every 3 days.
Human Mesenchymal Stem Cells (hMSCs)
The hMSCs were obtained from Lonza (PT-2501, Walkersville, MD) and maintained in growth medium consisting of DMEM/high glucose medium (SH30243.01, HyClone) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, non-essential amino acids, 100 units/ml penicillin and 100 ug/ml streptomycin. For reverse transfection, 4×104 hMSCs in 400 ul of growth medium were added to individual wells of a 24-well plate containing 100 pmoles of human PKIG siRNA (Dharmacon On-TARGET plus J-017218-06, Thermo Scientific) or non-targeting siRNA that had been pre-incubated as described in the previous paragraph. Transfection reagents were replaced with growth medium after four hours of incubation at 37°C in 5% CO2. Twenty hours later, the hMSCs were then used in osteogenic or adipogenic experiments. Osteogenic medium consisted of the growth medium supplemented with 50 ug/ml 2-phospho-L-ascorbic acid and 10 mM β-glycerophosphate and was changed every 3 days. Adipocyte differentiation was induced by 3 cycles of 3 days of adipogenic induction medium (growth medium supplemented with 5 ug/ml insulin, 100 nM dexamethasone, 50 uM indomethacin, and 0.5 mM IBMX) and 2 days of adipogenic maintenance medium (growth medium supplemented with 5 ug/ml insulin) as recommended by the manufacturer.
Quantitative real-time RT-PCR
Gene expression was measured by quantitative real-time RT-PCR using the linear portion of standard curves as we previously described (52). All PCR primers were designed to overlap exon-exon junctions at their 3′-termini using primer3 software (53) except for Pkig and those mRNAs that are encoded by a single exon (Gapdh and C/ebpa). All primers were validated by sequencing of PCR amplicons. Murine and human primers are listed in Supplementary Table 1 and Supplementary Table 2, respectively. All assays included negative controls without RNA, analysis of melting curves, and agarose gel electrophoresis to confirm the presence of single PCR amplicons.
PKA activity assay
Nuclear fractions were prepared as we previously described (10). PKA activities were assayed using fluorescently labeled-kemptide as substrate (Nonradioactive PepTag Assay kit, V5340, Promega, Madison, WI). The fraction of phosphorylated kemptide was quantified by measuring fluorescence (excitation at 535 nm and emission at 600 nm) on a 4000MM In-Vivo F Image Station (Kodak, Rochester, NY) after separation from non-phosphorylated kemptide by electrophoresis on 0.8% agarose gels in Tris-Cl (pH 8.0) buffer. >90% of kemptide phosphorylation by nuclear fractions from forskolin-treated MEFs is inhibited by PKIα (Supplementary Figure 3) and is therefore due to PKA activity. PKA activity was expressed as umol/min/ug protein. Total protein was measured using the BCA protein assay kit (23227, Pierce, Rockford, IL).
Western blotting
MEFs were lysed in SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, and 10% glycerol) plus protease inhibitors (cocktail tablet 04693159001, Roche, Mannheim, Germany) and phosphatase inhibitors (1 mM sodium pyrophosphate, 1 mM sodium orthovanadate, and 1 mM β-glycerophosphate). Equivalent amounts of protein (BCA protein assay) per lane were electrophoresed on 12% Tris-Glycine gels (59503, Lonza, Rockland, ME) and transferred to PVDF membranes. Membranes were blocked in Tris-buffered saline (pH 8.0) containing 0.05% Tween 20 and 5% non-fat dry milk and incubated with 1:1000 dilutions of primary antibody (pSer133-CREB, 9191; CREB, 9197; ATF4, 11815; β-actin, 4970; or GAPDH, 2118; all from Cell Signaling, Danvers, MA) or with 1:500 dilutions of primary antibody (Runx2, 5356-1, Epitomics, Burlingame, California; or Osterix, ab22552, Abcam, Cambridge, MA). The membranes were then incubated with horseradish peroxidase-conjugated anti-rabbit IgG (Cell Signaling, 7074). Bands were detected by chemiluminescence (Amersham ECL Plus, RPN2132, GE Health Care, Piscataway, NJ).
Alkaline phosphatase, Alizarin Red S, and Oil Red O assays
Alkaline phosphatase activity was measured biochemically as previously described (54). Alkaline phosphatase staining was performed using a kit (86C-1KT, Sigma-Aldrich) following the recommendations of the manufacturer. Staining of mineralization with Alizarin Red S (A5533, Sigma-Aldrich) was performed and quantified following extraction with 10% cetylpyridinium chloride as previously described (55). Staining of lipid accumulation with Oil Red O (O0625, Sigma-Aldrich) was performed and quantified following extraction with isopropanol as previously described (56). For co-staining, alkaline phosphatase staining was performed first and the cells were then rinsed with water twice before the Oil Red O staining.
LIF ELISA and neutralizing antibodies
LIF protein in cell culture supernatants was measured using a kit (MLF00, R & D Systems) following the recommendations of the manufacturer. To determine whether LIF mediates the effects of Pkig deletion or knockdown, indicated concentrations of either a neutralizing affinity-purified goat anti-murine LIF antibody (AF449, R & D Systems, Minneapolis, MN) or a normal goat control immunoglobulin (AB-108-C, R & D Systems) was incubated with MEFs or mdMSCs experiments. To determine whether LIF mediates the effects of PKIG knockdown, 10 ug/ml of either a neutralizing mouse anti-human LIF monoclonal antibody (MAB250, R & D Systems) or an isotype control mouse IgG2B (MAB004, R & D Systems) was incubated with hMSCs.
Statistical analysis
Unless otherwise indicated, all quantitative data are expressed as mean ± SD of three independent experiments, each containing triplicate culture wells assayed in triplicate. Statistical analysis was by Student’s t-test for experiments with two groups and by One Way ANOVA with Bonferroni post hoc tests of the indicated comparisons for experiments with three or more groups. All statistical tests were performed with SigmaStat 3.0 software (Systat Software, Inc, Point Richmond, CA). P values less than 0.05 were considered statistically significant. All non-quantitative results were also derived from three independent experiments. Images from representative experiments were selected based on the results from the quantitative portions of the same experiments. For example, the images in Figure 1C are from the experiment that was closest to the means in Figure 1B.
Figure 1.
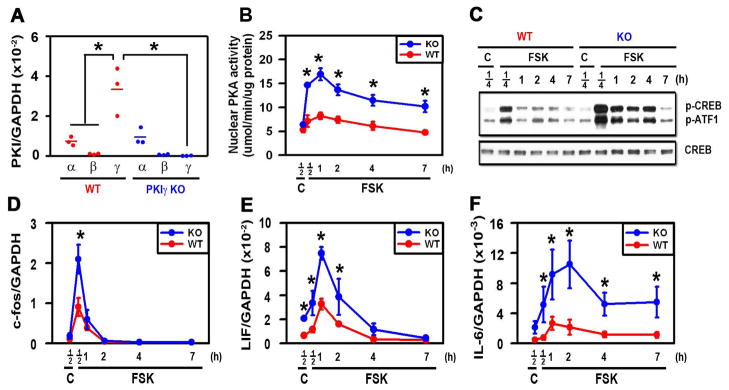
PKIγ deletion substantially extends PKA signaling following stimulation with FSK. MEFs from wild type (WT) and PKIγ−/− (KO) mice were incubated for 12 hours in osteogenic medium (A) or for the indicated times (B–F) in osteogenic medium containing 1 uM forskolin (FSK) or 0.1% DMSO as a vehicle control (C). mRNAs encoding members of the PKI family (A) and PKA-dependent genes (D–F) were measured by quantitative real-time RT-PCR, nuclear PKA activity was assessed biochemically (B), phosphorylation of CREB and ATF-1 and total CREB were assessed by Western blotting (C). * denotes p < 0.05 as compared with the indicated groups (A) or the WT groups at the same time points (B, D–F). In (A), the horizontal bars represent the means of the 3 independent experiments indicated by the individual symbols that represent means of 3 culture wells/group, each assayed in triplicate. In (B, D–F), the symbols represent the mean ± SD of 3 independent experiments, each consisting of 3 culture wells/group, each assayed in triplicate.
Results
Pkig deletion increases PKA signaling
Pkig mRNA is the dominant Pki family member expressed in osteoblasts and other mesenchymal cells (6, 10). Consistent with those findings, Pkig mRNA expression is 3.8-fold higher than Pkia mRNA expression in wild type MEFs and Pkib mRNA is barely detectable (first three groups in Fig. 1A). We previously showed that Pkig knockdown by either siRNA or antisense approaches substantially increases PKA signaling (10, 11). To determine whether off-target effects might be responsible for the effects of Pkig knockdown, we compared wild type and Pkig−/− MEFs. Pkig deletion does not induce detectable compensatory changes in mRNAs encoding either of the other PKI family members (last three groups in Fig. 1A). Pkig deletion substantially increases the magnitude and extends the time course of both nuclear PKA activation and phosphorylation of CREB and ATF1 transcription factors induced by forskolin in MEFs (Figs. 1B–C). Similarly, Pkig deletion substantially increases stimulation by forskolin of c-fos, Lif, and IL6 mRNAs (Figs. 1D–F). Taken together, these results demonstrate that PKA signaling is increased by Pkig deletion in MEFs. They therefore confirm our previous studies of Pkig knockdown (10, 11) and validate MEFs as a cellular model system for investigating the effects of Pkig deletion.
Pkig deletion increases osteoblast differentiation
PKA signaling induced by the adenylate cyclase activator forskolin stimulates osteoblast differentiation in vitro (29, 48, 57). To examine whether the enhanced PKA signaling induced by Pkig deletion also increases osteoblast differentiation, we took advantage of the ability of MEFs to model MSCs by undergoing osteoblastic differentiation (45, 58–61) as well as adipocytic and chondrocytic differentiation (62, 63). In osteogenic medium, Pkig remains the dominant Pki family member expressed in MEFs and Pkig deletion does not induce detectable compensatory changes in the other Pki family members (Figs. 2A–C). The transcription factors, Runx2 and osterix were expressed at high levels by both wild type and Pkig−/− MEFs at the beginning of the culture period at both the mRNA (1st group in Figs. 2D–E) and protein levels (Supplementary Figure 4). Runx2 and osterix mRNA levels decreased in the wild type MEFs during culture (red symbols in Figs. 2D–E) but remained elevated in the Pkig−/− MEFs (blue symbols in Figs. 2D–E). As expected, ATF4 was induced during osteoblast differentiation and was modestly increased at the mRNA level by Pkig deletion at day 7 (Fig. 2F).
Figure 2.

PKIγ deletion substantially increases osteoblast differentiation. MEFs from wild type (WT) and PKIγ−/− (KO) mice were incubated for the indicated times in osteogenic medium supplemented with 1 uM forskolin (FSK) or 0.1% DMSO as a vehicle control in the presence or absence of BMP-4. mRNAs encoding members of the PKI family (A–C) and osteoblastic marker genes (D–H) were measured by quantitative real-time RT-PCR. Osteoblast differentiation was also assessed by examining alkaline phosphatase (ALP) biochemically and cytotochemically (I), and by Alizarin Red S staining of mineral (J), after 7 or 14 days, respectively, in osteogenic medium. * denotes p < 0.05 and NS denotes not significant. The horizontal bars represent the means of the 3 (A–H, J) or 4 (I) independent experiments indicated by the individual symbols that represent means of 3 culture wells/group, each assayed in triplicate.
Alkaline phosphatase was used as a marker of the middle stage of osteoblast differentiation. Culture in osteogenic medium containing forskolin increases alkaline phosphatase mRNA levels (red symbols in Fig. 2G). Pkig deletion further increases forskolin-induced alkaline phosphatase mRNA levels by 3.7 and 5.3-fold respectively after three and seven days (blue symbols in Fig. 2G). As expected, for a late stage marker of osteoblast differentiation, osteocalcin mRNA levels are only modestly affected after three days of incubation in osteogenic medium containing forskolin (middle pair of groups in Fig. 2H). However, Pkig deletion increases osteocalcin mRNA levels by 16.5-fold after seven days (last pair of groups in Fig. 2H).
To confirm the effects observed on the mRNA markers, alkaline phosphatase activity and mineralization were assessed. These experiments were conducted in the absence and presence of BMP4 because osteoblast differentiation induced by BMPs is enhanced by forskolin (48, 57) and blocked by PKIγ overexpression (18). Forskolin increases alkaline phosphatase activity and mineralization both in the absence and presence of BMP4 (red symbols in Figs. 2I–J). Pkig deletion further increases forskolin-induced alkaline phosphatase activity assessed biochemically both in the absence and presence of BMP4 by 2.3 and 3.9-fold respectively and increases forskolin-independent alkaline phosphatase activity both in the absence and presence of BMP4 by 4.3 and 4.9-fold respectively (blue symbols in Fig. 2I). Staining of alkaline phosphatase activity confirms these results (lower panel of Fig. 2I). Pkig deletion also increases mineralization in cultures continuously stimulated with BMP4 by 6.4 and 1.6-fold respectively in the absence and presence of forskolin (3rd and 4th pairs of groups in Fig. 2J) and there was a trend towards increased mineralization by the Pkig−/− MEFs in the absence of BMP4 (1st and 2nd pairs of groups in Fig. 2J). These results collectively demonstrate that Pkig deletion substantially increases osteoblast differentiation by MEFs.
Pkig deletion decreases adipocyte differentiation
Osteoblast and adipocyte differentiation are often regulated reciprocally but both lineages are stimulated by PKA signaling (see Introduction for details). We therefore next asked which of these mechanisms would predominate during adipogenesis when PKA signaling is enhanced by Pkig deletion. In adipogenic medium, Pkig remains the dominant Pki family member expressed in MEFs and Pkig deletion does not induce detectable compensatory changes in the other Pki family members (Figs. 3A–C). As expected (19–22), induction of PKA signaling with the phosphodiesterase inhibitor IBMX increases adipocyte differentiation as assessed by measuring expression of both early and late adipogenic marker genes (red symbols in Figs. 3D–E for early markers and in Figs. 3F–G for late markers) or by staining for lipid accumulation with Oil Red O (red symbols in Fig. 3H). In contrast, further enhancement of PKA signaling by Pkig deletion decreases IBMX-induced expression of both the early and late adipogenic marker genes by 20–52% (blue symbols in Figs. 3D–G). Moreover, Pkig deletion inhibits Oil Red O staining by 33% in the presence of IBMX and indomethacin (last pair of groups in Fig. 3H) and there was a trend toward inhibition by Pkig deletion in all other tested conditions (first three pairs of groups in Fig. 3H). Taken together, these results demonstrate that Pkig deletion decreases adipocyte differentiation.
Figure 3.

PKIγ deletion decreases adipocyte differentiation. MEFs from wild type (WT) and PKIγ−/− (KO) mice were incubated for the indicated times in repeated cycles of adipogenic induction medium and maintenance medium as described in the Methods. mRNAs encoding members of the PKI family (A–C) and adipocyte marker genes (D–G) were measured by quantitative real-time RT-PCR. Adipocyte differentiation was also assessed by staining lipid accumulation with Oil Red O after 14 days (H). * denotes p < 0.05 and NS denotes not significant, The horizontal bars represent the means of the 3 independent experiments indicated by the individual symbols that represent means of 3 culture wells/group, each assayed in triplicate.
Pkig deletion simultaneously increases osteoblast differentiation and decreases adipocyte differentiation
We next determined the effect of Pkig deletion on the simultaneous differentiation of osteoblasts and adipocytes in mixed osteogenic/adipogenic medium. Pkig deletion increases osteoblast differentiation as alkaline phosphatase activity, alkaline phosphatase staining, and mineralization are increased (Figs. 4A–B). Pkig deletion decreases adipocyte differentiation as assessed by Oil Red O staining by 30% on both day 4 and day 14 (Figs. 4C–D). Co-staining for alkaline phosphatase and Oil Red O confirmed that the Pkig−/− MEFs are more osteogenic (blue staining in Fig. 4E) and substantially less adipogenic (red staining in Fig. 4E) than the wild type MEFs. These images also confirm that the two lineages are distinct from each other since almost no individual cells exhibited positive staining for both lineages (Fig. 4E). In summary, the results with the mixed osteogenic/adipogenic medium are consistent with the lineage-specific media reported above as Pkig deletion simultaneously increases osteoblast differentiation and inhibits adipocyte differentiation and the magnitude of the effect on osteoblast differentiation is larger than the effect on adipocyte differentiation.
Figure 4.

PKIγ deletion simultaneously increases osteoblast differentiation and decreases adipocyte differentiation in mixed osteogenic/adipogenic medium. MEFs from wild type (WT) and PKIγ−/− (KO) mice were incubated for the indicated times in the mixed osteogenic/adipogenic medium. Osteoblast differentiation was assessed by examining alkaline phosphatase (ALP) biochemically and histochemically (A) and by Alizarin Red S staining of mineral (B). Adipocyte differentiation was assessed by examining Oil Red O staining (C–D). In (A–D), the horizontal bars represent the means of the 3 independent experiments indicated by the individual symbols that represent means of 3 culture wells/group, each assayed in triplicate. In (E), cultures were co-stained for alkaline phosphatase activity (blue) and Oil Red O (red) after 4 days in the mixed osteogenic/adipogenic medium. Scale bars: 100 mm. * denotes p < 0.05.
The effects of Pkig knockdown in murine and human MSCs are similar to the effects of Pkig deletion in MEFs
To determine whether the results with MEFs are generalizable to mesenchymal precursor cells, we first measured the effects of Pkig knockdown on mdMSCs cultured in mixed osteogenic/adipogenic medium. Pkig is also the dominant Pki family member expressed in the mdMSCs and Pkig knockdown does not induce detectable compensatory changes in the other Pki family members (1st pair of groups in Figs. 5A–C). Pkig knockdown increases expression of alkaline phosphatase and osteocalcin mRNAs by 2.2 and 1.6-fold compared to control groups transfected with non-targeting siRNA duplexes (1st pair of groups in Figs. 5D–E). Simultaneously, Pkig knockdown decreases expression of both the early and late adipogenic marker genes by 30–37% (1st pair of groups in Figs. 5F–H).
Figure 5.

LIF mediates the effects of PKIγ knockdown on osteogenesis and adipogenesis in mdMSCs. mdMSCs reverse transfected with non-targeting siRNA duplexes as a control (C) or siRNA targeting PKIγ (KD) were incubated for 6 days in mixed osteogenic/adipogenic medium. Culture medium was supplemented with 0.3 ug/ml of either control immunoglobulin (IgG) or the anti-murine LIF Antibody (LIF Ab). mRNAs encoding members of the PKI family (A–C), osteoblastic marker genes (D–E), and adipogenic marker genes (F–H) were measured by quantitative real-time RT-PCR. Leptin mRNA was undetectable in all groups (not shown). * denotes p < 0.05 and NS denotes not significant. In (A–H), the horizontal bars represent the means of the 3 independent experiments indicated by the individual symbols that represent means of 3 culture wells/group, each assayed in triplicate.
To further determine whether the results are generalizable to human mesenchymal precursor cells, we measured the effects of PKIG knockdown on hMSCs cultured in either osteogenic or adipogenic medium. PKIG is also the dominant PKI family member expressed in the hMSCs and PKIG knockdown does not induce detectable compensatory changes in other PKI family members (1st pair of groups in Figs. 6A–C and Supplementary Figure 5). PKIG knockdown increases expression of both alkaline phosphatase and osteocalcin mRNAs by 1.8-fold compared to control groups transfected with non-targeting siRNA duplexes (1st pair of groups in Figs. 6D–E). In contrast, PKIG knockdown decreases mRNA expression of PPARG by 38% and adiponectin by 46% (1st pair of groups in Figs. 6F–G). The effects of PKIγ knockdown with mdMSCs and hMSCs therefore confirm the results with MEFs (Figs. 2–4) that Pkig deletion reciprocally increases osteoblast differentiation and decreases adipocyte differentiation.
Figure 6.

LIF mediates the effects of PKIγ knockdown on osteogenesis and adipogenesis in hMSCs. hMSCs reverse transfected with non-targeting siRNA duplexes as a control (C) or siRNA targeting PKIγ (KD) were incubated for 6 days in osteogenic medium (A–E) or 3 cycles of adipogenic induction medium (3 days) and adipogenic maintenance medium (2 days) (F–G). Culture medium was supplemented with 10 ug/ml of either control immunoglobulin (IgG2B) or the anti-human LIF monoclonal antibody (LIF MAb). mRNAs encoding members of the PKI family (A–C), osteoblastic marker genes (D–E), and adipogenic marker genes (F–G) were measured by quantitative real-time RT-PCR. C/EBPA and leptin mRNA were undetectable in all groups with adipogenic media (not shown). * denotes p < 0.05 and NS denotes not significant. Horizontal bars represent the means of the 3 independent experiments indicated by the individual symbols that represent means of 3 culture wells/group, each assayed in triplicate.
Reciprocal regulation of osteoblast and adipoctye differentiation by Pkig is mediated by LIF
Lif expression is induced by PKA activation (Fig. 1E and references (41–43)) and LIF increases osteoblast differentiation (33–38) and decreases adipocyte differentiation (34, 37, 39, 40). We therefore asked whether LIF mediates the effects of Pkig deletion. Consistent with the results that Pkig deletion increases basal and forskolin-induced expression of Lif mRNA (Fig. 1E), Pkig deletion also increases basal (2.7-fold), IBMX-induced (9.6-fold), and forskolin-induced (7.6-fold) secretion of LIF protein (Fig. 7A). Concomitantly, Pkig deletion increases basal (3.5-fold), IBMX-induced (3.9-fold), and forskolin-induced (5.9-fold) alkaline phosphatase activity (Fig. 7B) as expected based on the results in Figures 2 and 4.
Figure 7.

LIF mediates the effects of PKIγ deletion on osteogenesis and adipogenesis in MEFs (G). MEFs from wild type (WT) and PKIγ−/− (KO) mice were incubated for 1 day (A), 4 days (B–D), or 14 days (E–F) in mixed osteogenic/adipogenic medium. Culture medium was supplemented with 0.25 mM 3-isobutyl-1-methylxanthine (IBMX), 1 uM forskolin (FSK), or 0.1% DMSO (C) as a vehicle control (A–B). Culture medium was supplemented with IBMX plus the indicated concentrations of the neutralizing anti-murine LIF antibody (LIF Ab) in (C) and 0.3 ug/ml of either control immunoglobulin (IgG) or the LIF antibody (LIF Ab) in (D–F). LIF protein in the cell culture supernatants was measured by ELISA (A). Osteoblast differentiation was assessed by examining alkaline phosphatase (ALP) biochemically (B–D) and cytochemically (lower panel in D) and by Alizarin Red S staining of mineral (E). Adipocyte differentiation was assessed by examining Oil Red O staining (F). * denotes p < 0.05 and NS denotes not significant. In (A–B, D–F), the horizontal bars represent the means of the 3 independent experiments indicated by the individual symbols that represent means of 3 culture wells/group, each assayed in triplicate. In (C), the symbols represent the mean ± SD of 3 independent experiments, each consisting of 3 culture wells/group, each assayed in triplicate.
An antibody that neutralizes murine LIF was used to determine whether the effects of Pkig deletion are mediated by LIF. Initial experiments showed that 0.3 ug/ml of the antibody maximally inhibits osteoblast differentiation in Pkig−/− MEFs (Fig. 7C) and that concentration was therefore used for all subsequent experiments. That concentration of control immunoglobulin does not alter the effects of Pkig deletion on osteogenesis or adipogenesis (compare 1st pair of groups in Figs. 7D–F with Figs. 2–4). In contrast, the LIF antibody inhibits alkaline phosphatase activity, alkaline phosphatase staining, and mineralization in the PKIg−/− MEFs (blue symbols and lower panels in Figs. 7D–E). The LIF antibody also enhances lipid accumulation in Pkig−/− MEFs (blue symbols in Fig. 7F). As a result, in the presence of the LIF antibody, there are no significant differences between the wild type and the Pkig−/− MEFs in mineralization or lipid accumulation (last pair of groups in Figs. 7E–F). These results demonstrate that the LIF antibody inhibits the effects of Pkig deletion on both osteoblast and adipocyte differentiation by MEFs.
The anti-murine LIF antibody also inhibits the effects of Pkig knockdown on the mRNA expression of the osteoblast marker genes by the mdMSCs (blue symbols in Figs. 5D–E). As a result, in the presence of the LIF antibody, there are no significant differences between the control and the Pkig knockdown groups in osteocalcin mRNA levels (2nd pair of group in Fig. 5E). There are also no significant differences between the control and the Pkig knockdown groups in levels of the adipocyte marker genes in the presence of the LIF antibody (2nd pair of groups in Figs. 5F–H).
To determine whether LIF also mediates the effects of PKIG knockdown in human cells, we used a monoclonal antibody that neutralizes human LIF. 10 ug/ml of that monoclonal antibody was incubated with hMSCs based on the results in Fig. 7C and the manufacturer’s measurements of the relative potencies of the two antibodies. The anti-LIF monoclonal antibody inhibits the effects of PKIG knockdown on the mRNA expression of osteoblast and adipocyte marker genes (blue symbols in Figs. 6D–G). As a result, in the presence of the LIF monoclonal antibody, there are no significant differences between the control and the PKI knockdown groups in ALP and osteocalcin mRNA levels (2nd pair of groups in Figs. 6D–E). There are also no significant differences between the control and the PKI knockdown hMSCs in levels of the adipocyte marker genes in the presence of the LIF monoclonal antibody (2nd pair of groups in Figs. 6F–G). The mRNA expression results with both mdMSCs and hMSCs therefore confirm and extend the biochemical and cytochemical results with MEFs (Fig. 7) that the effects of Pkig deletion on the reciprocal regulation of osteoblast and adipocyte differentiation are mediated by LIF.
Discussion
This study showed that activation of nuclear PKA by Pkig deletion or knockdown increases osteoblast differentiation (right side of Fig. 7G). This occurred both in the presence of exogenous activators of the PKA pathway (forskolin and IBMX) and in their absence. The results in the absence of exogenous PKA activation likely reflect inhibition of basal PKA activity by PKIγ. These results, together with our previous demonstration that PKIγ is a primary mechanism that reduces the anti-apoptotic effects of PKA signaling in osteoblasts (11), indicate that PKIγ is likely to be a major constraint on bone formation induced by Gαs–coupled receptors. Downregulation of PKIγ may therefore provide a useful co-therapy in combination with PKA agonists, such as intermittent PTH, for bone loss in conditions such as osteoporosis. Downregulation of PKIγ may be particularly useful for the approximately 20% patients who are unresponsive to therapy with intermittent PTH (64, 65). Downregulation of PKIγ may also be useful to enhance repair of bone defects by mesenchymal precursor cells by programming their fate towards the osteoblastic lineage (66–68). For example, our demonstration of relatively long term knockdown of Pkig may provide a feasible approach to downregulate PKIγ ex vivo in mesenchymal precursor cells prior to their implantation in bone defects. The primary limitation of our study with regard to these potential clinical applications is that all of our experiments were conducted in vitro and future studies will therefore be needed to assess the in vivo outcomes of Pkig deletion or knockdown.
This study also showed that activation of nuclear PKA by Pkig deletion or knockdown decreases adipocyte differentiation simultaneously with the increase of osteoblast differentiation (left side of Fig. 7G). Thus, the reciprocal regulation between differentiation of mesenchymal precursor cells into osteoblasts and adipocytes (27, 28) predominates over the direct effects of PKA signaling to stimulate adipocyte differentiation (19–22). However, the direct effects of PKA signaling (grey arrow in Fig. 7G) likely partially counterbalance the reciprocal regulation, which may explain why Pkig deletion or knockdown has generally more modest effects on adipoctye differentiation than on osteoblast differentiation. Nonetheless, the decrease in adipocyte differentiation gives further credence to the possibility that downregulation of PKIγ may be useful to enhance repair of bone defects, especially in situations where adipogenesis impairs defect repair (67, 68).
This study further showed that the reciprocal regulation of osteoblast and adipocyte differentiation by PKIγ is mediated, at least in part, by LIF (middle of Fig. 7G). Thus, Pkig deletion increased PKA-induced Lif mRNA and LIF protein. Moreover, the effects of Pkig deletion or knockdown on osteogenesis and adipogenesis were inhibited by two different antibodies that respectively neutralize either murine or human LIF. Our results are consistent with prior studies that LIF can increase osteoblast differentiation (33–38) and inhibit adipogenesis (34, 37, 39, 40). Interestingly, the effects of LIF in those studies are primarily on early events in differentiation (34, 36–38), perhaps reflecting regulation of mesenchymal lineage specification. It is likely that LIF acts together with other factors to mediate the reciprocal regulation of osteogenesis and adipogenesis by PKIγ. Future studies will be needed to identify those other factors and elucidate the details of how they interact with LIF to reciprocally regulate osteoblast and adipocyte differentiation.
This study also confirmed our previous results (10, 11) that PKIγ reduces PKA signaling as well as PKA-dependent phosphorylation of CREB and the resultant PKA-dependent expression of rapidly induced mRNAs. We previously characterized c-fos, Lif, and IL6 as immediate-early genes induced by PKA signaling (11, 42). However, the quantitative real-time RT-PCR measurements of gene expression in this study distinguished between the mRNAs. c-fos showed a typical immediate-early pattern of gene expression both in the wild type and Pkig−/− MEFs. In contrast, Lif and IL6 mRNAs were induced and declined more slowly and this was especially pronounced in the Pkig−/− MEFs.
In conclusion, our results show that Pkig reciprocally favors adipogenesis over osteogenesis and, together with our previous studies (9–11), demonstrate that PKIγ is a major regulator of PKA signaling in mesenchymal cells. PKA activity was recently shown to be regulated by miRNA-27a or miRNA-155 that targets Pkia in hematopoietic cells (69–71). Intriguingly, miRNA-155 can induce osteoblast differentiation (72). A transient increase in PKIβ level was also shown to be required for neuronal differentiation in cell culture (73). Those results, together with our studies and the earlier publications on PKIα (12, 13), represent accumulating evidence that endogenous levels of the PKI family impact multiple cellular processes by regulating PKA activity.
Supplementary Material
Supplementary Figure 1. The effects of forskolin (FSK) on osteoblast differentiation are specific. Murine MSCs reverse transfected with non-targeting siRNA duplexes as a control (C) or siRNA targeting PKIγ (KD) were incubated for 7 days in osteogenic medium supplemented with 0.1% DMSO as control (C), 1 uM forskolin (FSK), or 1 uM dideoxy-forskolin (inactive analogue). Alkaline phosphatase activity was measured as described in the Methods. The horizontal bars represent the means of each group, which consisted of 3 culture wells, each assayed in triplicate in a single experiment. * denotes p = 0.008 and NS denotes not significant.
Supplementary Figure 2. Conditioned media from CHO cells that stably express BMP4 dose-dependently increase alkaline phosphatase activity. MC3T3-E1 cells were incubated for 3 days in osteogenic medium supplemented with the indicated concentrations of the conditioned medium. Alkaline phosphatase activity was measured as described in the Methods. Results are presented as mean ± SD of triplicate culture wells assayed in triplicate in a single experiment. * denotes p < 0.001 as compared with the control group.
Supplementary Figure 3. >90% of phosphorylation of the PKA substrate is inhibited by PKIα and is therefore due to PKA activity. Nuclear PKA activity of PKIγ−/− MEFs treated for 1 hour with 1 uM forskolin (FSK) was measured in the absence and presence of 1 uM PKIα-(6–22)-amide. Each assay included negative controls in the absence of nuclear fractions (c) and positive controls containing 25 units of the catalytic subunit of PKA (cPKA). The upper panel shows the separation of non-phosphorylated and phosphorylated substrate by electrophoresis. The lower panel shows quantification of PKA activity in the indicated groups. The horizontal bars represent the means of each group, which consisted of 3 reaction wells in a single experiment. * denotes p < 0.001 between the two groups.
Supplementary Figure 4. PKIγ deletion increases osteoblast differentiation (A) without increasing protein levels of the transcription factors Runx2, osterix, and ATF4 (B). MEFs from wild type (WT) and PKIγ−/− (KO) mice were incubated in osteogenic medium supplemented with 1 uM forskolin (FSK) or 0.1% DMSO as a vehicle control. Osteoblast differentiation was confirmed by examining alkaline phosphatase histochemically at day 7 (A). Protein levels of the transcription factors (Runx2, osterix, and ATF4) were assessed by Western blotting at the indicated time points (B).
Supplementary Figure 5. PKIγ is also the dominant PKI family member expressed by the hMSCs in adipogenic media and PKIγ knockdown does not induce detectable compensatory changes in other PKI family members. hMSCs reverse transfected with non-targeting siRNA duplexes as a control (C) or siRNA targeting PKIγ (KD) were incubated for 3 cycles of adipogenic induction medium (3 days) and adipogenic maintenance medium (2 days). Culture medium was supplemented with 10 ug/ml of either control immunoglobulin (IgG2B) or the anti-human LIF monoclonal antibody (LIF MAb). mRNAs encoding members of the PKI family were measured by quantitative real-time RT-PCR. * denotes p < 0.05 and NS denotes not significant. Horizontal bars represent the means of the 3 independent experiments indicated by the individual symbols that represent means of 3 culture wells/group, each assayed in triplicate.
Acknowledgments
This work was supported by NIH R21 AR055230 to EMG and the Harry E. Figgie III M.D. Professorship to EMG.
CHO cells that stably express BMP4 were a kind gift from Dr. Tatsuya Koike, Osaka City University Medical School. This work was supported by NIH R21 AR055230 to EMG and the Harry E. Figgie III M.D. Professorship to EMG.
Footnotes
Author contributions:
Xin Chen: Conception and design, Collection and/or assembly of data, Data analysis and interpretation, Manuscript writing, Final approval of manuscript
Bryan S. Hausman: Conception and design, Provision of study material or patients, Collection and/or assembly of data, Data analysis and interpretation, Manuscript writing, Final approval of manuscript
Guangbin Luo: Conception and design, Data analysis and interpretation, Manuscript writing, Final approval of manuscript
Guang Zhou: Conception and design, Data analysis and interpretation, Manuscript writing, Final approval of manuscript
Shunichi Murakami: Conception and design, Data analysis and interpretation, Manuscript writing, Final approval of manuscript
Janet Rubin: Provision of study material or patients, Data analysis and interpretation, Manuscript writing, Final approval of manuscript
Edward M. Greenfield: Conception and design, Data analysis and interpretation, Manuscript writing, Final approval of manuscript
References
- 1.Sassone-Corsi P. Coupling gene expression to cAMP signalling: role of CREB and CREM. Int J Biochem Cell Biol. 1998;30:27–38. doi: 10.1016/s1357-2725(97)00093-9. [DOI] [PubMed] [Google Scholar]
- 2.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 3.Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab. 2006;17:159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 4.Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci. 2010;35:91–100. doi: 10.1016/j.tibs.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Walsh DA, Angelos KL, Patten SMV, et al. The inhibitor protein of the cAMP-dependent protein kinase. In: Kemp BE, editor. Peptides and protein phosphorylation. Boca Raton, Fla: CRC Press; 1990. pp. 43–84. [Google Scholar]
- 6.Collins SP, Uhler MD. Characterization of PKIγ, a novel isoform of the protein kinase inhibitor of cAMP-dependent protein kinase. J Biol Chem. 1997;272:18169–18178. doi: 10.1074/jbc.272.29.18169. [DOI] [PubMed] [Google Scholar]
- 7.Taylor SS, Kim C, Vigil D, et al. Dynamics of signaling by PKA. Biochim Biophys Acta. 2005;1754:25–37. doi: 10.1016/j.bbapap.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 8.Dalton GD, Dewey WL. Protein kinase inhibitor peptide (PKI): A family of endogenous neuropeptides that modulate neuronal cAMP-dependent protein kinase function. Neuropeptides. 2006;40:23–34. doi: 10.1016/j.npep.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Chen X, Dai JC, Greenfield EM. Termination of immediate-early gene expression after stimulation by parathyroid hormone or isoproterenol. Am J Physiol Cell Physiol. 2002;283:C1432–C1440. doi: 10.1152/ajpcell.00221.2002. [DOI] [PubMed] [Google Scholar]
- 10.Chen X, Dai JC, Orellana SA, et al. Endogenous protein kinase inhibitorγ terminates immediate-early gene expression induced by cAMP-dependent protein kinase (PKA) signaling: Termination depends on PKA inactivation rather than PKA export from the nucleus. J Biol Chem. 2005;280:2700–2707. doi: 10.1074/jbc.M412558200. [DOI] [PubMed] [Google Scholar]
- 11.Chen X, Song I, Dennis J, et al. Endogenous PKIγ terminates the anti-apoptotic effect of parathyroid hormone and β-adrenergic agonists. J Bone Miner Res. 2007;22:656–664. doi: 10.1359/jbmr.070122. [DOI] [PubMed] [Google Scholar]
- 12.Lecea Ld, Criado J, Rivera S, et al. Endogenous protein kinase A inhibitor (PKIalpha) modulates synaptic activity. J Neurosci Res. 1998;53:269–278. doi: 10.1002/(SICI)1097-4547(19980801)53:3<269::AID-JNR1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 13.Kawakami M, Nakanishi N. The role of an endogenous PKA inhibitor, PKIalpha, in organizing left-right axis formation. Development. 2001;128:2509–2515. doi: 10.1242/dev.128.13.2509. [DOI] [PubMed] [Google Scholar]
- 14.Gangolli EA, Belyamani M, Muchinsky S, et al. Deficient gene expression in protein kinase inhibitor alpha Null mutant mice. Mol Cell Biol. 2000;20:3442–3448. doi: 10.1128/mcb.20.10.3442-3448.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belyamani M, Gangolli EA, Idzerda RL. Reproductive function in protein kinase inhibitor-deficient mice. Mol Cell Biol. 2001;21:3959–3963. doi: 10.1128/MCB.21.12.3959-3963.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jilka RL. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone. 2007;40:1434–1446. doi: 10.1016/j.bone.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenfield EM. Anabolic Effects of Intermittent PTH on Osteoblasts. Curr Mol Pharmacol. 2012;5:127–134. [PubMed] [Google Scholar]
- 18.Zhao L, Yang S, Zhou G, et al. Downregulation of cAMP-dependent protein kinase inhibitor gamma is required for BMP-2-induced osteoblastic differentiation. Int J Biochem Cell Biol. 2006;38:2064–2073. doi: 10.1016/j.biocel.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 19.MacDougald OA, Lane MD. Transcriptional regulation of gene expression during adipocyte differentiation. Annu Rev Biochem. 1995;64:345–373. doi: 10.1146/annurev.bi.64.070195.002021. [DOI] [PubMed] [Google Scholar]
- 20.Yeh WC, Cao Z, Classon M, et al. Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev. 1995;9:168–181. doi: 10.1101/gad.9.2.168. [DOI] [PubMed] [Google Scholar]
- 21.Fox KE, Fankell DM, Erickson PF, et al. Depletion of cAMP-response element-binding protein/ATF1 inhibits adipogenic conversion of 3T3-L1 cells ectopically expressing CCAAT/enhancer-binding protein (C/EBP) alpha, C/EBP beta, or PPAR gamma 2. J Biol Chem. 2006;281:40341–40353. doi: 10.1074/jbc.M605077200. [DOI] [PubMed] [Google Scholar]
- 22.Petersen RK, Madsen L, Pedersen LM, et al. Cyclic AMP (cAMP)-mediated stimulation of adipocyte differentiation requires the synergistic action of Epac- and cAMP-dependent protein kinase-dependent processes. Mol Cell Biol. 2008;28:3804–3816. doi: 10.1128/MCB.00709-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antras J, Lasnier F, Pairault J. Beta-adrenergic-cyclic AMP signalling pathway modulates cell function at the transcriptional level in 3T3-F442A adipocytes. Mol Cell Endocrinol. 1991;82:183–190. doi: 10.1016/0303-7207(91)90030-v. [DOI] [PubMed] [Google Scholar]
- 24.Rickard DJ, Wang FL, Rodriguez-Rojas AM, et al. Intermittent treatment with parathyroid hormone (PTH) as well as a non-peptide small molecule agonist of the PTH1 receptor inhibits adipocyte differentiation in human bone marrow stromal cells. Bone. 2006;39:1361–1372. doi: 10.1016/j.bone.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 25.Li F, Wang D, Zhou Y, et al. Protein kinase A suppresses the differentiation of 3T3-L1 preadipocytes. Cell Res. 2008;18:311–323. doi: 10.1038/cr.2008.12. [DOI] [PubMed] [Google Scholar]
- 26.Li H, Fong C, Chen Y, et al. Beta-adrenergic signals regulate adipogenesis of mouse mesenchymal stem cells via cAMP/PKA pathway. Mol Cell Endocrinol. 2010;323:201–207. doi: 10.1016/j.mce.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 27.Gimble JM, Zvonic S, Floyd ZE, et al. Playing with bone and fat. J Cell Biochem. 2006;98:251–266. doi: 10.1002/jcb.20777. [DOI] [PubMed] [Google Scholar]
- 28.Rosen CJ, Ackert-Bicknell C, Rodriguez JP, et al. Marrow fat and the bone microenvironment: developmental, functional, and pathological implications. Crit Rev Eukaryot Gene Expr. 2009;19:109–124. doi: 10.1615/critreveukargeneexpr.v19.i2.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kao R, Lu W, Louie A, et al. Cyclic AMP signaling in bone marrow stromal cells has reciprocal effects on the ability of mesenchymal stem cells to differentiate into mature osteoblasts versus mature adipocytes. Endocrine. 2012;42:622–636. doi: 10.1007/s12020-012-9717-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato M, Westmore M, Ma YL, et al. Teriparatide [PTH(1–34)] strengthens the proximal femur of ovariectomized nonhuman primates despite increasing porosity. J Bone Miner Res. 2004;19:623–629. doi: 10.1359/JBMR.040112. [DOI] [PubMed] [Google Scholar]
- 31.Kulkarni N, Wei T, Kumar A, et al. Changes in Osteoblast, Chondrocyte, and Adipocyte Lineages Mediate the Bone Anabolic Actions of PTH and Small Molecule GSK-3 Inhibitor. J Cell Biochem. 2007;102:1504–1518. doi: 10.1002/jcb.21374. [DOI] [PubMed] [Google Scholar]
- 32.Kuznetsov SA, Riminucci M, Ziran N, et al. The interplay of osteogenesis and hematopoiesis: expression of a constitutively active PTH/PTHrP receptor in osteogenic cells perturbs the establishment of hematopoiesis in bone and of skeletal stem cells in the bone marrow. J Cell Biol. 2004;167:1113–1122. doi: 10.1083/jcb.200408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cornish J, Callon K, King A, et al. The effect of leukemia inhibitory factor on bone in vivo. Endocrinology. 1993;132:1359–1366. doi: 10.1210/endo.132.3.8440191. [DOI] [PubMed] [Google Scholar]
- 34.Gimble JM, Wanker F, Wang CS, et al. Regulation of bone marrow stromal cell differentiation by cytokines whose receptors share the gp130 protein. J Cell Biochem. 1994;54:122–133. doi: 10.1002/jcb.240540113. [DOI] [PubMed] [Google Scholar]
- 35.Dazai S, Akita S, Hirano A, et al. Leukemia inhibitory factor enhances bone formation in calvarial bone defect. J Craniofac Surg. 2000;11:513–520. doi: 10.1097/00001665-200011060-00002. [DOI] [PubMed] [Google Scholar]
- 36.Malaval L, Aubin J. Biphasic effects of leukemia inhibitory factor on osteoblastic differentiation. J Cell Biochem. 2001;81:S36, 63–70. doi: 10.1002/jcb.1086. [DOI] [PubMed] [Google Scholar]
- 37.Poulton IJ, McGregor NE, Pompolo S, et al. Contrasting roles of leukemia inhibitory factor in murine bone development and remodeling involve region-specific changes in vascularization. J Bone Miner Res. 2012;27:586–595. doi: 10.1002/jbmr.1485. [DOI] [PubMed] [Google Scholar]
- 38.Guihard P, Danger Y, Brounais B, et al. Induction of osteogenesis in mesenchymal stem cells by activated monocytes/macrophages depends on oncostatin M signaling. Stem Cells. 2012;30:762–772. doi: 10.1002/stem.1040. [DOI] [PubMed] [Google Scholar]
- 39.Marshall MK, Doerrler W, Feingold KR, et al. Leukemia inhibitory factor induces changes in lipid metabolism in cultured adipocytes. Endocrinology. 1994;135:141–147. doi: 10.1210/endo.135.1.8013346. [DOI] [PubMed] [Google Scholar]
- 40.White UA, Stephens JM. The gp130 receptor cytokine family: regulators of adipocyte development and function. Curr Pharm Des. 2011;17:340–346. doi: 10.2174/138161211795164202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greenfield EM, Gornik SA, Horowitz MC, et al. Regulation of cytokine expression in osteoblasts by parathyroid hormone: rapid stimulation of interleukin-6 and leukemia inhibitory factor mRNA. J Bone Miner Res. 1993;8:1163–1171. doi: 10.1002/jbmr.5650081003. [DOI] [PubMed] [Google Scholar]
- 42.Greenfield EM, Horowitz MC, Lavish SA. Stimulation by parathyroid hormone of interleukin-6 and leukemia inhibitory factor expression in osteoblasts is an immediate-early gene response induced by cAMP signal transduction. J Biol Chem. 1996;271:10984–10989. doi: 10.1074/jbc.271.18.10984. [DOI] [PubMed] [Google Scholar]
- 43.Pollock JH, Blaha MJ, Gornik SA, et al. In vivo demonstration that parathyroid hormone and parathyroid hormone-related protein stimulate expression by osteoblasts of interleukin-6 and leukemia inhibitory factor. J Bone Miner Res. 1996;11:754–759. doi: 10.1002/jbmr.5650110606. [DOI] [PubMed] [Google Scholar]
- 44.Conner DA. Mouse embryo fibroblast (MEF) feeder cell preparation. Curr Protoc Mol Biol. 2001;Chapter 23(Unit 23):22. doi: 10.1002/0471142727.mb2302s51. [DOI] [PubMed] [Google Scholar]
- 45.Wang Y, Sul HS. Pref-1 regulates mesenchymal cell commitment and differentiation through Sox9. Cell Metab. 2009;9:287–302. doi: 10.1016/j.cmet.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seamon KB, Daly JW. Forskolin: its biological and chemical properties. Adv Cyclic Nucleotide Protein Phosphorylation Res. 1986;20:1–150. [PubMed] [Google Scholar]
- 47.Sugama R, Koike T, Imai Y, et al. Bone morphogenetic protein activities are enhanced by 3′,5′-cyclic adenosine monophosphate through suppression of Smad6 expression in osteoprogenitor cells. Bone. 2006;38:206–214. doi: 10.1016/j.bone.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 48.Tsutsumimoto T, Wakabayashi S, Kinoshita T, et al. A phosphodiesterase inhibitor, pentoxifylline, enhances the bone morphogenetic protein-4 (BMP-4)-dependent differentiation of osteoprogenitor cells. Bone. 2002;31:396–401. doi: 10.1016/s8756-3282(02)00839-6. [DOI] [PubMed] [Google Scholar]
- 49.McBeath R, Pirone DM, Nelson CM, et al. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6:483–495. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 50.Kilian KA, Bugarija B, Lahn BT, et al. Geometric cues for directing the differentiation of mesenchymal stem cells. Proc Natl Acad Sci U S A. 2010;107:4872–4877. doi: 10.1073/pnas.0903269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Case N, Xie Z, Sen B, et al. Mechanical activation of beta-catenin regulates phenotype in adult murine marrow-derived mesenchymal stem cells. J Orthop Res. 2010;28:1531–1538. doi: 10.1002/jor.21156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dai JC, He P, Chen X, et al. TNFα and PTH utilize distinct mechanisms to induce IL-6 and RANKL expression with markedly different kinetics. Bone. 2006;38:509–520. doi: 10.1016/j.bone.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 53.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 54.Bonsignore LA, Anderson JR, Lee Z, et al. Adherent lipopolysaccharide inhibits the osseointegration of orthopedic implants by impairing osteoblast differentiation. Bone. 2012;52:93–101. doi: 10.1016/j.bone.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stanford CM, Jacobson PA, Eanes ED, et al. Rapidly forming apatitic mineral in an osteoblastic cell line (UMR 106–01 BSP) J Biol Chem. 1995;270:9420–9428. doi: 10.1074/jbc.270.16.9420. [DOI] [PubMed] [Google Scholar]
- 56.Ramirez-Zacarias JL, Castro-Munozledo F, Kuri-Harcuch W. Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil red O. Histochemistry. 1992;97:493–497. doi: 10.1007/BF00316069. [DOI] [PubMed] [Google Scholar]
- 57.Ghayor C, Ehrbar M, San Miguel B, et al. cAMP enhances BMP2-signaling through PKA and MKP1-dependent mechanisms. Biochem Biophys Res Commun. 2009;381:247–252. doi: 10.1016/j.bbrc.2009.02.032. [DOI] [PubMed] [Google Scholar]
- 58.Thomas DM, Carty SA, Piscopo DM, et al. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell. 2001;8:303–316. doi: 10.1016/s1097-2765(01)00327-6. [DOI] [PubMed] [Google Scholar]
- 59.Garreta E, Genove E, Borros S, et al. Osteogenic differentiation of mouse embryonic stem cells and mouse embryonic fibroblasts in a three-dimensional self-assembling peptide scaffold. Tissue Eng. 2006;12:2215–2227. doi: 10.1089/ten.2006.12.2215. [DOI] [PubMed] [Google Scholar]
- 60.Kang S, Bennett CN, Gerin I, et al. Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma. J Biol Chem. 2007;282:14515–14524. doi: 10.1074/jbc.M700030200. [DOI] [PubMed] [Google Scholar]
- 61.Saeed H, Taipaleenmaki H, Aldahmash AM, et al. Mouse embryonic fibroblasts (MEF) exhibit a similar but not identical phenotype to bone marrow stromal stem cells (BMSC) Stem Cell Rev. 2012;8:318–328. doi: 10.1007/s12015-011-9315-x. [DOI] [PubMed] [Google Scholar]
- 62.Alexander DL, Ganem LG, Fernandez-Salguero P, et al. Aryl-hydrocarbon receptor is an inhibitory regulator of lipid synthesis and of commitment to adipogenesis. J Cell Sci. 1998;111 (Pt 22):3311–3322. doi: 10.1242/jcs.111.22.3311. [DOI] [PubMed] [Google Scholar]
- 63.Lengner CJ, Lepper C, van Wijnen AJ, et al. Primary mouse embryonic fibroblasts: a model of mesenchymal cartilage formation. J Cell Physiol. 2004;200:327–333. doi: 10.1002/jcp.20118. [DOI] [PubMed] [Google Scholar]
- 64.Finkelstein JS, Hayes A, Hunzelman JL, et al. The effects of parathyroid hormone, alendronate, or both in men with osteoporosis. N Engl J Med. 2003;349:1216–1226. doi: 10.1056/NEJMoa035725. [DOI] [PubMed] [Google Scholar]
- 65.Sellmeyer DE, Black DM, Palermo L, et al. Hetereogeneity in skeletal response to full-length parathyroid hormone in the treatment of osteoporosis. Osteoporos Int. 2007;18:973–979. doi: 10.1007/s00198-007-0336-x. [DOI] [PubMed] [Google Scholar]
- 66.Arthur A, Zannettino A, Gronthos S. The therapeutic applications of multipotential mesenchymal/stromal stem cells in skeletal tissue repair. J Cell Physiol. 2009;218:237–245. doi: 10.1002/jcp.21592. [DOI] [PubMed] [Google Scholar]
- 67.Schantz JT, Woodruff MA, Lam CX, et al. Differentiation potential of mesenchymal progenitor cells following transplantation into calvarial defects. J Mech Behav Biomed Mater. 2012;11:132–142. doi: 10.1016/j.jmbbm.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 68.Liu L, Aronson J, Huang S, et al. Rosiglitazone inhibits bone regeneration and causes significant accumulation of fat at sites of new bone formation. Calcif Tissue Int. 2012;91:139–148. doi: 10.1007/s00223-012-9623-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Piazzon N, Maisonneuve C, Guilleret I, et al. Bicc1 links the regulation of cAMP signaling in polycystic kidneys to microRNA-induced gene silencing. J Mol Cell Biol. 2012;4:398–408. doi: 10.1093/jmcb/mjs027. [DOI] [PubMed] [Google Scholar]
- 70.Fassi Fehri L, Koch M, Belogolova E, et al. Helicobacter pylori induces miR-155 in T cells in a cAMP-Foxp3-dependent manner. PLoS One. 2010;5:e9500. doi: 10.1371/journal.pone.0009500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ghorpade DS, Leyland R, Kurowska-Stolarska M, et al. MicroRNA-155 is required for Mycobacterium bovis BCG-mediated apoptosis of macrophages. Mol Cell Biol. 2012;32:2239–2253. doi: 10.1128/MCB.06597-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu T, Xie M, Wang X, et al. miR-155 modulates TNF-alpha-inhibited osteogenic differentiation by targeting SOCS1 expression. Bone. 2012;51:498–505. doi: 10.1016/j.bone.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 73.Huang HS, Turner DL, Thompson RC, et al. Ascl1-induced neuronal differentiation of P19 cells requires expression of a specific inhibitor protein of cyclic AMP-dependent protein kinase. J Neurochem. 2012;120:667–683. doi: 10.1111/j.1471-4159.2011.07332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. The effects of forskolin (FSK) on osteoblast differentiation are specific. Murine MSCs reverse transfected with non-targeting siRNA duplexes as a control (C) or siRNA targeting PKIγ (KD) were incubated for 7 days in osteogenic medium supplemented with 0.1% DMSO as control (C), 1 uM forskolin (FSK), or 1 uM dideoxy-forskolin (inactive analogue). Alkaline phosphatase activity was measured as described in the Methods. The horizontal bars represent the means of each group, which consisted of 3 culture wells, each assayed in triplicate in a single experiment. * denotes p = 0.008 and NS denotes not significant.
Supplementary Figure 2. Conditioned media from CHO cells that stably express BMP4 dose-dependently increase alkaline phosphatase activity. MC3T3-E1 cells were incubated for 3 days in osteogenic medium supplemented with the indicated concentrations of the conditioned medium. Alkaline phosphatase activity was measured as described in the Methods. Results are presented as mean ± SD of triplicate culture wells assayed in triplicate in a single experiment. * denotes p < 0.001 as compared with the control group.
Supplementary Figure 3. >90% of phosphorylation of the PKA substrate is inhibited by PKIα and is therefore due to PKA activity. Nuclear PKA activity of PKIγ−/− MEFs treated for 1 hour with 1 uM forskolin (FSK) was measured in the absence and presence of 1 uM PKIα-(6–22)-amide. Each assay included negative controls in the absence of nuclear fractions (c) and positive controls containing 25 units of the catalytic subunit of PKA (cPKA). The upper panel shows the separation of non-phosphorylated and phosphorylated substrate by electrophoresis. The lower panel shows quantification of PKA activity in the indicated groups. The horizontal bars represent the means of each group, which consisted of 3 reaction wells in a single experiment. * denotes p < 0.001 between the two groups.
Supplementary Figure 4. PKIγ deletion increases osteoblast differentiation (A) without increasing protein levels of the transcription factors Runx2, osterix, and ATF4 (B). MEFs from wild type (WT) and PKIγ−/− (KO) mice were incubated in osteogenic medium supplemented with 1 uM forskolin (FSK) or 0.1% DMSO as a vehicle control. Osteoblast differentiation was confirmed by examining alkaline phosphatase histochemically at day 7 (A). Protein levels of the transcription factors (Runx2, osterix, and ATF4) were assessed by Western blotting at the indicated time points (B).
Supplementary Figure 5. PKIγ is also the dominant PKI family member expressed by the hMSCs in adipogenic media and PKIγ knockdown does not induce detectable compensatory changes in other PKI family members. hMSCs reverse transfected with non-targeting siRNA duplexes as a control (C) or siRNA targeting PKIγ (KD) were incubated for 3 cycles of adipogenic induction medium (3 days) and adipogenic maintenance medium (2 days). Culture medium was supplemented with 10 ug/ml of either control immunoglobulin (IgG2B) or the anti-human LIF monoclonal antibody (LIF MAb). mRNAs encoding members of the PKI family were measured by quantitative real-time RT-PCR. * denotes p < 0.05 and NS denotes not significant. Horizontal bars represent the means of the 3 independent experiments indicated by the individual symbols that represent means of 3 culture wells/group, each assayed in triplicate.