

Abstract
Background
GATA-5, a zinc-finger transcription factor and member of the GATA family proteins 1–6, is known to be involved in cellular differentiation. We recently found that tumor-specific hypermethylation of the GATA5 CpG island (CGI) occurs in renal cell carcinoma (RCC) and is associated with an adverse clinical outcome. In this study, we investigated whether epigenetic GATA5 alterations may result in changes in GATA5 mRNA expression levels and correlate with the observed prognostic impact of epigenetic changes in GATA5 in RCC.
Methods
Quantitative real-time reverse-transcribed polymerase chain reaction was applied to measure relative GATA5 mRNA expression levels in 135 kidney tissue samples, including 77 clear cell RCC (ccRCC) tissues and 58 paired adjacent normal renal tissue samples. Relative GATA5 expression levels were determined using the ΔΔCt method and detection of three endogenous control genes then compared to previously measured values of relative methylation.
Results
The mean relative GATA5 mRNA expression level exhibited an approximately 31-fold reduction in tumor specimens compared with corresponding normal tissues (p < 0.001, paired t-test). Decreased GATA5 mRNA expression was inversely correlated with increased GATA5 CGI methylation (p < 0.001) and was associated with shortened recurrence-free survival in ccRCC patients (p = 0.023, hazard ratio = 0.25).
Conclusion
GATA5 mRNA expression is decreased in ccRCC, likely due to gene silencing by methylation of the GATA5 CGI. Moreover, reduced GATA5 mRNA levels were associated with a poor clinical outcome, indicating a possible role of GATA5 for the development of aggressive ccRCC phenotypes.
Keywords: GATA5, Renal cell carcinoma, mRNA, Prognosis, DNA methylation
Background
Renal cell carcinoma (RCC) is one of the top ten causes of cancer deaths in industrialized countries, and its incidence has consistently increased during the past decades [1]. Clear cell renal cell carcinomas (ccRCCs) are the most frequently occurring histological entity, comprising approximately 75% of all RCC.
As a member of the GATA family of transcription factors, GATA5 is known to be functionally involved in cellular lineage and cell differentiation during embryonic development of the heart, lung, urogenital tract, and gut epithelium [2]. Altered expression of GATA5 has been associated with intestinal epithelial cell differentiation [3]. GATA5 is assumed to be a selective transcriptional regulator of mucin genes in gastrointestinal tissues [4], and regulates the promoter of the sodium-hydrogen exchanger isoform 3 that is expressed in intestinal and renal epithelium via Sp-family transcription factors [5].
Of note, a previous study found GATA5 hypermethylation and associated epigenetic silencing to be involved in carcinogenesis of gastric and colorectal cancers [6]. Epigenetic alterations of GATA5 were also described in other tumor tissues and were linked to the development of ovarian, lung, pancreatic, and esophageal cancer [7-10]. In a recent study aimed at identifying new DNA methylation targets in ccRCC, we detected for the first time a tumor-specific hypermethylation of the GATA5 CpG island (CGI) in RCC [11]. Hypermethylation was also associated with advanced disease and shortened recurrence-free survival (RFS) of patients.
In this study, we asked whether mRNA expression levels of GATA5 are reduced as suggested by our previous DNA methylation analysis, and if the mRNA levels are associated with adverse clinical parameters, further underlining the relevance of epigenetic GATA5 alterations in ccRCC carcinogenesis.
Material and methods
Tissue specimens
One hundred and thirty-five kidney tissues, including 77 ccRCCs and 58 paired adjacent normal renal tissue samples, were included in this study. RCC tissues were obtained from open or laparoscopic nephrectomies and partial resection. Paired tissue samples with adjacent normal tissue (adN) were obtained from a subgroup of our 77 ccRCCs cohort. Adjacent normal tissues, i.e. morphologically normal kidney were isolated with minimum of 0.5 cm to 2 cm distant from the primary tumor lesion. Samples were snap-frozen in liquid nitrogen immediately following surgery and stored at −80°C. Ethical approval of the university ethical committee (Prof. H. D. Tröger, Hannover Medical School, Carl-Neuberg-Str. 1, Hannover, Germany) and informed consent from all patients were obtained. Localized disease was defined as pT ≤ 2, lymph node involvement and metastasis negative (N0/M0), and grading (G) G1 and 1–2, whereas advanced RCC was defined as pT ≥ 3, N1 and/or M1, and G2-3 and G3. Patients with G2 were assigned to the intermediate risk group and were not considered as a parameter for low vs. high grade group comparisons. Follow up data were available for 35 patients, and RFS was defined as the interval up to the time that disease progression could be detected by computer tomography scans. Clinical and histopathological parameters are summarized in Table 1.
Table 1.
Clinicopathological parameters of ccRCC patients
| Clinicopathological parameters | n | % | |
|---|---|---|---|
| Cases in total |
|
77 |
100 |
|
Sex |
Female |
29 |
38 |
| |
Male |
48 |
62 |
|
Median age, years |
|
64 |
|
|
Median tumor size, cm |
|
5.5 |
|
|
Primary tumor classification |
pT1 |
6 |
8 |
| |
pT1a |
19 |
25 |
| |
pT1b |
14 |
18 |
| |
pT2 |
3 |
4 |
| |
pT3 |
2 |
3 |
| |
pT3a |
9 |
12 |
| |
pT3b/c |
22 |
29 |
| |
pT4 |
0 |
0 |
| |
not known |
2 |
3 |
|
Lymph node status |
N0 |
68 |
88 |
| |
N1 |
9 |
12 |
| |
N2 |
0 |
0 |
|
Status of metastasis |
M0 |
56 |
73 |
| |
M1 |
21 |
27 |
|
Grade |
|
|
|
| - Low risk |
G1 |
14 |
18 |
| |
G1-2 |
8 |
10 |
| - Intermediate risk |
G2 |
40 |
52 |
| - High risk |
G2-3 |
5 |
6 |
| |
G3 |
10 |
13 |
|
Localized disease |
pT ≤ 2, N0, M0 and G1; G1-2 |
34 |
44 |
|
Advanced disease |
pT ≥ 3 and/or N1, M1 or G2-3; G3 |
43 |
56 |
| Paired samples | 58 | 75 | |
Abbreviations: ccRCC clear cell renal cell carcinoma, n number.
RNA isolation, cDNA synthesis, and quantitative real-time PCR analysis
Isolation of total RNA from tissue specimens and from renal proximal tubular epithelial cells (RPTEC) as controls, cDNA synthesis, and quantitative real-time PCR analysis (qRT-PCR) were carried out as described previously [12]. Duplicate measurements for qRT-PCR analysis were performed using 384 sample plates, an automated liquid handling system (FasTrans, AnalyticJena, Jena, Germany), and the ABI 7900 Fast Sequence Detection System as described previously [12]. Experimenters were blinded to any patient clinicopathological or survival information. The TaqMan expression assays used were GATA-5 (Hs00388359_m1), HPRT1 (Hs99999909_m1), GUSB (Hs00939627_m1), and RPL13A (Hs03043885_g1) (all assays were from Life Technologies, Foster City, CA, USA). HPRT1, GUSB, and RPL13A were included as endogenous references. The cDNA obtained from RPTEC primary cell transcripts served as biological controls. For each qRT-PCR run, blank and no-template controls were included. Relative expression levels were calculated using the delta-deltaCT (ΔΔCt) method [13,14], and the SDS 2.3 Manager and dataAssist V2.0 software (Life technologies) as described previously [12]. The endogenous controls, HPRT1, RPL13A and GUSB, were combined by dataAssist V2.0 software and “arithmetic mean” was used as a method of normalization.
Statistics and survival analysis
Natural logarithms of relative expression (lnRQ) values were used for statistical calculations. All statistics were done using the statistical software, R 2.15.2 [15]. The paired t-test was used for statistical analyses of expression differences in paired tumor and adjacent normal tissues samples, whereas univariate Cox regression models were used for statistical analysis of RFS. The threshold for dichotomization of expression values was calculated using the selected rank statistics of R package, which provides the minimum p-value for log rank statistics [15]. P-values < 0.05 were considered to be statistically significant. The Kaplan-Meier method was used for survival analyses.
Results
GATA5 mRNA expression is decreased in ccRCC
The analyses of relative GATA5 mRNA expression levels revealed significantly decreased expression in tumor specimens (TU; mean lnRQ = −1.7; ±SD = 1.63) compared with the corresponding adN (mean lnRQ = 1.73; ±SD = 1.32; p < 0.001; paired t-test). Figure 1A illustrates the differences in expression values observed for paired tumor and adN, indicating a strong reduction of up to 31-fold in the expression levels, largely in tumor tissues. The comparison of the distribution of relative expression values between both tissue groups showed only a small overlap (Figure 1B).
Figure 1.
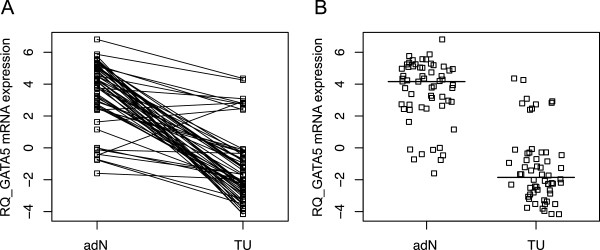
GATA5 mRNA expression in paired clear cell renal cell carcinoma and adjacent normal tissues. A) Comparison of the relative GATA5 expression (RQ) values in adjacent normal (adN) and tumor (TU) tissues from ccRCC patients (p < 0.001). B) Scatterplot analysis illustrating the distribution of relative expression values (RQ) observed for TU and adN in ccRCC specimens. Bold lines indicate the median of relative expression values.
Loss of GATA5 mRNA expression correlates with CGI hypermethylation
We compared lnRQ expression values and natural logarithms relative methylation (lnRML) values of GATA5 in all samples (Figure 2). Regression analysis revealed an inverse relationship between relative expression levels and methylation of GATA5 (coefficient of regression = −0.41, p < 0.001). The comparison of paired tissues, indicated by solid lines, shows that high methylation values with concurrent low expression is frequently observed in tumor tissues, whereas corresponding adN samples largely had high expression levels and low methylation.
Figure 2.

Association of GATA5 CGI methylation and relative mRNA expression in tumor and adjacent normal tissues. Solid lines connect the subgroup of paired tissues (tumor tissue = solid triangle; adjacent normal = solid squares). The regression line (dashed line) and 95% CI (grey shaded) are presented. Note that tissues exhibiting concurrent high methylation and low mRNA expression can be found in the upper left corner, whereas the occurrence of low methylation and higher relative expression in tissues is displayed in the lower right corner.
GATA5 mRNA expression is associated with tumor diameter
Logistic regression analysis for comparison of tumor subgroups detected a significant difference in mRNA expression levels only for the tumor diameter (p = 0.02, odds ratio = 0.63; 95% CI: 0.42–0.93) whereas other clinicopathological parameters like sex, gender, distant metastasis, lymph node metastasis and tumor grade exhibited no statistically significant association.
Loss of GATA5 mRNA expression is associated with decreased recurrence-free survival
Cox regression survival analyses using a statistically calculated optimum cut off value for relative GATA5 mRNA expression (lnRQ = −3.52) showed that a lower expression status was associated with increased risk for shorter time to disease recurrence (p = 0.023, hazard ratio (HR) = 0.25, 95% CI: 0.07–0.82; Table 2). Within 30 months, four out of five patients (80%) whose tumor specimens demonstrated expression values below the cut off value were identified with disease recurrence (Figure 3). The status of localized and advanced disease (p = 0.03, HR = 4.18; 95% CI: 1.15–15.2), status of metastasis (p = 0.009, HR = 4.27; 95% CI: 1.43–12.8), and tumor grade (p < 0.001, HR = 9.48; 95% CI: 2.92–30.8) were also shown to be associated with RFS (Table 2). Pairwise bivariate Cox regression analyses were first carried out to investigate whether an association between expression status and clinicopathological parameters and RFS exists. In bivariate statistical models, considering the statuses of advanced disease, metastasis, and tumor grade as covariates, we found in each case that GATA5 expression was not associated with RFS while mRNA levels were detected as a significant parameter in bivariate Cox regression models including the statuses of lymph node metastasis, age and gender (Table 3). Moreover, we carried out a multivariate analysis demonstrating that mRNA expression of GATA5 (p = 0.12, HR = 0.32; 95% CI: 0.07–1.34) is not significantly associated with RFS including the covariates status of advance disease (p = 0.29, HR = 0.18; 95% CI: 0.01-4.41), status of metastasis (p = 0.14, HR = 6.60; 95% CI: 0.55–78.68), tumor grade p = 0.002, HR = 0.09; 95% CI: 2.46–56.14, age (p = 0.36, HR = 2.54; 95% CI: 0.05–2.85) and gender (p = 0.39, HR = 2.03; 95% CI: 0.39–10.38).
Table 2.
Univariate statistical association of GATA5 mRNA expression and clinicopathological parameters with recurrence-free survival
| p-value° | HR | 95% CI | |
|---|---|---|---|
|
GATA5 mRNA expression |
0.023 |
0.25 |
0.07-0.82 |
| Localized vs. Advanced |
0.030 |
4.18 |
1.15-15.2 |
| Status of metastasis |
0.009 |
4.27 |
1.43-12.8 |
| Tumor grade |
<0.001 |
9.48 |
2.92-30.8 |
| Lymph node status |
0.398 |
1.920 |
0.42-8.72 |
| Age* |
0.155 |
0.420 |
0.13-1.38 |
| Gender | 0.489 | 1.510 | 0.47-0.82 |
°Univariate Cox regression analysis.
*Dichotomization by the median of parameter.
bold numbers: p-value < 0.05 (statistically significant).
hazard ratio (HR).
confidence interval (CI).
Figure 3.

Kaplan-Meier plot for illustrating recurrence-free survival. The solid line shows the Kaplan-Meier curve for 5 patients (pts.) with GATA5 mRNA expression lower than or equal to the cut off of −3.52 (natural logarithm), indicating patients with a shortened recurrence-free survival. The dashed line illustrates the Kaplan-Meier curve for patients with mRNA expression levels above the cut off including 30 pts. Disease progression of ccRCC within a period of approximately two years was observed in seven cases with a low relative mRNA expression level, whereas high GATA5 mRNA expression phenotypes showed only four progression events within that time interval.
Table 3.
Bivariate statistical association of GATA5 mRNA expression and clinicopathology with recurrence-free survival
| p-value° | HR | 95% CI | |
|---|---|---|---|
|
GATA5 mRNA expression |
0.137 |
0.390 |
0.11-1.35 |
| Localized vs Advanced |
0.080 |
3.340 |
0.87-12.9 |
|
GATA5 mRNA expression |
0.113 |
0.370 |
0.11-1.27 |
| Status of metastasis |
0.036 |
3.410 |
1.08-10.8 |
|
GATA5 mRNA expression |
0.511 |
0.640 |
0.17-2.41 |
| Tumor grade |
0.001 |
8.320 |
2.34-29.6 |
|
GATA5 mRNA expression |
0.032 |
0.260 |
0.08-0.90 |
| Lymph node metastasis |
0.657 |
1.420 |
0.29-6.77 |
|
GATA5 mRNA expression |
0.035 |
0.270 |
0.08-0.91 |
| Age* |
0.214 |
0.470 |
0.14-1.55 |
|
GATA5 mRNA expression |
0.023 |
0.250 |
0.08-0.83 |
| Gender | 0.512 | 1.480 | 0.46-4.84 |
°Bivariate Cox regression analysis.
*Dichotomization by the median of parameter.
bold numbers: p-value < 0.05 (statistically significant).
hazard ratio (HR).
confidence interval (CI).
Discussion
Members of the GATA1-6 transcription factor family contribute to stem cell differentiation in embryonic tissue, and GATA5 is involved in intestinal epithelial cell differentiation in adults [3]. Moreover, previous analyses found GATA5 hypermethylation in human malignancies such as gastric and colorectal cancers and demonstrated that epigenetic silencing of the gene occurred in various human cancer cell lines, providing evidence that GATA5 alterations may represent epigenetic alterations of wider relevance for carcinogenesis [4,6].
In a recent study aimed at identifying new DNA methylation targets in ccRCC, we detected tumor-specific hypermethylation of the GATA5 CGI in RCC [11]. Hypermethylation was also associated with advanced disease and shortened RFS of patients, which had not been previously reported for any other human cancer.
Hypermethylation of GATA5 in RCC indicated that the expression of GATA5 might be epigenetically silenced in tumor cells, leading to a biologically more aggressive tumor phenotype. In the current study, we demonstrate that GATA5 mRNA expression is strongly reduced in ccRCC and, moreover, that a subgroup of tissues shows a clear relationship between methylation of the GATA5 CGI and reduced mRNA expression, indicating that epigenetic silencing of GATA5 occurs in a substantial fraction of ccRCC. A significant relationship between GATA5 hypermethylation and reduced GATA5 mRNA expression within a human tissue, to the best of our knowledge, has not been previously demonstrated. Thus, our results support the notion that epigenetic silencing due to DNA methylation is a relevant process in RCC.
A subgroup of tissues showed only moderate reduction of expression, although GATA5 methylation was detectable, indicating that other biological mechanisms, e.g. histone alterations, play a role in tumor development in ccRCC. Additional functional investigations are required to clarify these aspects.
GATA5 methylation is associated with various clinicopathological parameters as well as RFS. Hence, we hypothesized that reduced mRNA expression levels in ccRCC would also show an association with unfavorable clinical parameters. Indeed, we found that decreased GATA5 mRNA expression is associated with the diameter of tumors and RFS in univariate Cox regression analysis, showing a hazard ratio of 0.25, which resembles the reciprocal hazard ratio observed for the corresponding methylation analysis.
Interestingly, a subset of patients with very low mRNA expression levels also demonstrated a shortened recurrence-free survival in univariate Cox regression analysis. However, taking into account that only a small number of tumors have been identified, these results require future extended evaluation studies including multivariate analyses.
Conclusion
Decreased GATA5 mRNA expression in ccRCC may be caused by epigenetic silencing, and is likely associated with a poor clinical outcome. Our results underline the need for further functional studies to characterize the interaction of GATA5 and cellular signaling in ccRCC with respect to the observed changes in expression and methylation levels, and its association with tumor progression.
Competing interest
The authors’ declare that they have no competing interest.
Authors’ contributions
IP wrote the manuscript, prepared the figures and participated in the study design. ND and MK carried out the methylation analyses and participated in the sequence analyses. JH and MK assembled histopathological, clinicopathological and survival data. JH performed isolation and characterization of tissue samples and assembly of patients. MA evaluated the histopathologies of given tissues. MK, CvK, AM and AS assisted with general scientific discussion. JS identified the candidate promoter, conceived of the study, developed the study design and analytical assays, constructed and ran the clinical database, performed statistical analyses together with RS and participated in manuscript preparation. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Inga Peters, Email: peters.inga@mh-hannover.de.
Natalia Dubrowinskaja, Email: dubrowinskaja.natalie@mh-hannover.de.
Michael Kogosov, Email: michael.kogosov@stud.mh-hannover.de.
Mahmoud Abbas, Email: abbas.mahmoud@mh-hannover.de.
Jörg Hennenlotter, Email: joerg.hennenlotter@med.uni-tuebingen.de.
Christoph von Klot, Email: Klot.Christoph@mh-hannover.de.
Axel S Merseburger, Email: merseburger.axel@mh-hannover.de.
Arnulf Stenzl, Email: stenzl.arnulf@med.uni-tuebingen.de.
Ralph Scherer, Email: scherer.ralph@mh-hannover.de.
Markus A Kuczyk, Email: kuczyk.markus@mh-hannover.de.
Jürgen Serth, Email: serth.juergen@mh-hannover.de.
Acknowledgements
We thank Christel Reese and Magrit Hepke for technical assistance.
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Morrisey EE, Ip HS, Tang Z, Lu MM, Parmacek MS. GATA-5: a transcriptional activator expressed in a novel temporally and spatially-restricted pattern during embryonic development. Dev Biol. 1997;183(1):21–36. doi: 10.1006/dbio.1996.8485. [DOI] [PubMed] [Google Scholar]
- Gao X, Sedgwick T, Shi YB, Evans T. Distinct functions are implicated for the GATA-4, -5, and −6 transcription factors in the regulation of intestine epithelial cell differentiation. Mol Cell Biol. 1998;18(5):2901–2911. doi: 10.1128/mcb.18.5.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren CY, Akiyama Y, Miyake S, Yuasa Y. Transcription factor GATA-5 selectively up-regulates mucin gene expression. J Cancer Res Clin Oncol. 2004;130(5):245–252. doi: 10.1007/s00432-003-0537-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiela PR, LeSueur J, Collins JF, Ghishan FK. Transcriptional regulation of the rat NHE3 gene. Functional interactions between GATA-5 and Sp family transcription factors. J Biol Chem. 2003;278(8):5659–5668. doi: 10.1074/jbc.M209473200. [DOI] [PubMed] [Google Scholar]
- Akiyama Y, Watkins N, Suzuki H, Jair K-W, van Engeland M, Esteller M, Sakai H, Ren C-Y, Yuasa Y, Herman JG. et al. GATA-4 and GATA-5 transcription factor genes and potential downstream antitumor target genes are epigenetically silenced in colorectal and gastric cancer. Mol Cell Biol. 2003;23(23):8429–8439. doi: 10.1128/MCB.23.23.8429-8439.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakana K, Akiyama Y, Aso T, Yuasa Y. Involvement of GATA-4/-5 transcription factors in ovarian carcinogenesis. Cancer Lett. 2006;241(2):281–288. doi: 10.1016/j.canlet.2005.10.039. [DOI] [PubMed] [Google Scholar]
- Guo M, Akiyama Y, House MG, Hooker CM, Heath E, Gabrielson E, Yang SC, Han Y, Baylin SB, Herman JG. et al. Hypermethylation of the GATA genes in lung cancer. Clin Cancer Res. 2004;10(23):7917–7924. doi: 10.1158/1078-0432.CCR-04-1140. [DOI] [PubMed] [Google Scholar]
- Guo M, House MG, Akiyama Y, Qi Y, Capagna D, Harmon J, Baylin SB, Brock MV, Herman JG. Hypermethylation of the GATA gene family in esophageal cancer. Int J Cancer. 2006;119(9):2078–2083. doi: 10.1002/ijc.22092. [DOI] [PubMed] [Google Scholar]
- Fu B, Guo M, Wang S, Campagna D, Luo M, Herman JG, Iacobuzio-Donahue CA. Evaluation of GATA-4 and GATA-5 methylation profiles in human pancreatic cancers indicate promoter methylation patterns distinct from other human tumor types. Cancer Biol Ther. 2007;6(10):1546–1552. doi: 10.4161/cbt.6.10.4708. [DOI] [PubMed] [Google Scholar]
- Peters I, Eggers H, Atschekzei F, Hennenlotter J, Waalkes S, Trankenschuh W, Grosshennig A, Merseburger AS, Stenzl A, Kuczyk MA. et al. GATA5 CpG island methylation in renal cell cancer: a potential biomarker for metastasis and disease progression. BJU Int. 2012;110(2 Pt 2):E144–E152. doi: 10.1111/j.1464-410X.2011.10862.x. [DOI] [PubMed] [Google Scholar]
- Waalkes S, Atschekzei F, Kramer MW, Hennenlotter J, Vetter G, Becker JU, Stenzl A, Merseburger AS, Schrader AJ, Kuczyk MA. et al. Fibronectin 1 mRNA expression correlates with advanced disease in renal cancer. BMC Cancer. 2010;10:503. doi: 10.1186/1471-2407-10-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Team RDC. R: A Language and Environment for Statistical Computing. Vienna Austria; 2011. http://www.r-project.org/ [Google Scholar]