

Abstract
Permeability of the endothelial monolayer is increased when exposed to the bacterial endotoxin LPS. Our previous studies have shown that heat shock protein (Hsp) 90 inhibitors protect and restore LPS-mediated hyperpermeability in bovine pulmonary arterial endothelial cells. In this study, we assessed the effect of Hsp90 inhibition against LPS-mediated hyperpermeability in cultured human lung microvascular endothelial cells (HLMVECs) and delineated the underlying molecular mechanisms. We demonstrate that Hsp90 inhibition is critical in the early phase, to prevent LPS-mediated hyperpermeability, and also in the later phase, to restore LPS-mediated hyperpermeability in HLMVECs. Because RhoA is a well known mediator of endothelial hyperpermeability, we investigated the effect of Hsp90 inhibition on LPS-mediated RhoA signaling. RhoA nitration and activity were increased by LPS in HLMVECs and suppressed when pretreated with the Hsp90 inhibitor, 17-allylamino-17 demethoxy-geldanamycin (17-AAG). In addition, inhibition of Rho kinase, a downstream effector of RhoA, protected HLMVECs from LPS-mediated hyperpermeability and abolished LPS-induced myosin light chain (MLC) phosphorylation, a target of Rho kinase. In agreement with these findings, 17-AAG or dominant-negative RhoA attenuated LPS-induced MLC phosphorylation. MLC phosphorylation induced by constitutively active RhoA was also suppressed by 17-AAG, suggesting a role for Hsp90 downstream of RhoA. Inhibition of Src family kinases also suppressed RhoA activity and MLC phosphorylation. Together, these data indicate that Hsp90 inhibition prevents and repairs LPS-induced lung endothelial barrier dysfunction by suppressing Src-mediated RhoA activity and signaling.
Keywords: endothelial permeability, LPS, heat shock protein 90, RhoA, Rho kinase
Clinical Relevance
Endothelial hyperpermeability is a serious complication of acute respiratory distress syndrome. We now present data revealing a new mechanism regulating endothelial barrier function, and suggest potential new targets for the prevention and repair of endothelial hyperpermeability.
The endotoxin LPS, a major component of the outer membrane of gram-negative bacteria, is responsible for stimulating systemic inflammation and vascular dysfunction (1, 2). LPS binds to the host Toll-like receptor 4/cluster of differentiation 14/lymphocyte antigen 96 receptor complex and activates a variety of signaling pathways (3, 4). Exposure of endothelial cells to LPS causes vascular hyperpermeability and potentiates the systemic inflammation that occurs in acute and chronic diseases, such as sepsis, atherosclerosis, acute respiratory distress syndrome, and diabetes.
In endothelial cells, LPS causes phosphorylation and activation of pp60Src, a member of the Src family kinase (SFK) (5). pp60src plays a critical role in LPS-mediated vascular dysfunction (6, 7). Activated pp60Src phosphorylates adherens junction components, such as vascular endothelial–cadherin, p120 catenin, and β-catenin, resulting in the disruption of cell–cell adhesion and increased vascular permeability (8). In addition, activation of pp60Src is a key event in Rho-mediated signaling (6, 7).
The Rho family of small GTPases plays an important role in a variety of cellular responses, including the assembly of adherens junctions and of cortical actin fibers (9, 10). RhoA, a member of the Rho family, is activated by the exchange of GDP for GTP, and translocates to the plasma membrane where it stimulates downstream effectors, such as Rho kinase (ROCK). ROCK, either directly or indirectly, via phosphorylation of myosin light chain (MLC) phosphatase, increases phosphorylation of MLC (11). MLC phosphorylation increases actin filament cross-linking and produces a contractile force that results in increased vascular permeability (12, 13).
LPS-induced activation of pp60Src is attenuated by inhibition of heat shock protein (Hsp) 90 (5). Hsp90 is a highly abundant molecular chaperone, essential for cell growth and survival; it regulates the function of various proteins that include several protein kinases, and transcription factors (14). Inhibition of Hsp90 using small-molecule inhibitors have been extensively studied for their therapeutic potential in targeting cancer cells and promoting apoptosis (15–17). A small-molecule inhibitor of Hsp90, 17-allylamino-17 demethoxy-geldanamycin (17-AAG), has been tested in preclinical and early clinical studies in a variety of solid tumors, and has been shown to also possess antiangiogenic and anti-inflammatory activities (18, 19).
We have previously shown that inhibition of Hsp90 protects and restores LPS-induced hyperpermeability in bovine pulmonary arterial endothelial cells (BPAECs) (5, 20). However, the underlying mechanisms involved in this protection by Hsp90 inhibitors are yet to be determined. In this study we investigated the mode of action of 17-AAG and NVP-AUY922 (AUY-922), two well-known Hsp90 inhibitors, in protecting against LPS-mediated hyperpermeability of human lung microvascular endothelial cells (HLMVECs). Our data show that, during LPS-induced endothelial hyperpermeability, Hsp90 inhibitors preserve endothelial barrier function by interacting with targets both upstream and downstream of RhoA.
Materials and Methods
Antibodies and Reagents
Antibodies against MLC, di-phospho-MLC, and nitrotyrosine were purchased from Cell Signaling (Danvers, MA). Anti–β-actin antibody was purchased from Sigma-Aldrich (St. Louis, MO) and anti-RhoA antibody (sc-418) was purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). The Hsp90 inhibitor, 17-AAG, was obtained from the National Cancer Institute (Bethesda, MD). The Hsp90 inhibitor, AUY-922 (S1069), and the ROCK inhibitors, Y-27632 (S1049) and GSK429286 (S1474), were obtained from Selleck Chemicals (Houston, TX). The SFK inhibitor, PP2 (1,407), was obtained from Tocris (Minneapolis, MN).
Adenovirus Construction
Constitutively active (enhanced green fluorescent protein–RhoA-Q63L; Addgene, Cambridge, MA) and dominant-negative (DN) RhoA (enhanced green fluorescent protein–RhoA-T19N; Addgene) were subcloned into an adenoviral vector (Invitrogen, Grand Island, NY). Replication-deficient adenoviruses expressing the gene of interest, under the control of the cytomegalovirus promoter, were generated in HEK293 cells, purified using CsCl, titered, and stored in Dulbecco’s phosphate-buffered saline (PBS) containing 10% glycerol, 0.5 mmol/L MgCl2, and 0.5 mmol/L CaCl2 (21, 22). Adenoviruses for expression of green fluorescent protein (GFP) and DN Hsp90 (DN-Hsp90) were generated in-house, as described previously (23).
Cell Culture
HLMVECs were isolated and cultured in-house as described previously (24).
Assays of Endothelial Permeability
Transendothelial resistance (TER) assay was performed as described previously (5). Briefly, roughly 60,000 cells were seeded on each well of an 8W10E+ electric cell-substrate impedance sensing (ECIS) array. After 24 hours, the media were changed (virus was also added at that time) and treatments began at 48 hours, when the resistance was stable between 1,000–1,300 ohms at a frequency of 4,000 Hz and the capacitance was between 22 and 29 nanofarads. Each experiment was performed at least in triplicate and repeated at least three times (n = 3). Resistance was measured using the ECIS model Zθ and normalized to each well’s value at t = 0.
Paracellular influx across the HLMVEC monolayer was also studied using the Transwell assay system in 24-well Millicell culture plates. A total of 200,000 cells were seeded apically in each insert and media were changed after 24 hours. At 48 hours after seeding, cells were treated with either vehicle (0.1% dimethyl sulfoxide) or the Hsp90 inhibitor, AUY-922 (2 μM). After 2 hours, cells were exposed to either PBS or LPS (5 EU/ml). At 15 minutes after the addition of LPS, FITC-dextran (2 million [2M] kD, 1 μg/μl) was added to the apical media. At 10 hours after LPS addition, 100 μl of basal media was removed and fluorescence intensity was measured.
RhoA Activity Assay
RhoA activity was determined using a Rho G-LISA assay kit in accordance with the manufacturer’s instructions (Cytoskeleton, Inc., Denver, CO) using HLMVEC cell lysates. Results were normalized to protein levels measured by the Precision Red protein assay reagent.
Animal Studies
Plasmids (40 μg) carrying either DN-Hsp90 cDNA or luciferase cDNA under cytomegalovirus promoter control were incubated with the nontoxic jetPEI reagent (Polyplus Transfection, Inc., New York, NY) for 15–30 minutes per manufacturer instructions. The DNA–jetPEI complex was then injected into male C57BL/6 mice (7–8 wk of age; Harlan, Indianapolis, IN) through the tail vein. After 48 hours, LPS (2 mg/kg) was administered intraperitoneally. At 24 hours after LPS injection immunohistochemical staining of myeloperoxidase and measurement of Evans blue dye extravasation was performed as described previously (25). All animal care and experimental procedures were approved by the Animal Care Committee of Georgia Health Sciences University.
Western Blotting and Immunoprecipitation
Western blot analyses and immunoprecipitation experiments were performed as described previously (5, 20). Densitometry was performed using Imagequant 5.1 (GE Healthcare Bio-Sciences, Pittsburgh, PA) and plotted as fold change from vehicle.
Statistical Analyses
Data are presented as mean values (± SEM). Comparisons among groups were performed using either one-way or two-way ANOVA with Bonferroni’s post-test, or using paired t tests, as appropriate. Differences were considered significant at P less than 0.05; n represents the number of experimental repeats.
Results
Hsp90 Inhibition Protects against LPS-Mediated HLMVEC Barrier Dysfunction
HLMVECs were grown on gold electrode arrays. TER was monitored until successive constant values were attained, confirming a confluent monolayer. Cells were then exposed to vehicle or the Hsp90 inhibitor, 17-AAG (2 μM; Figure 1A) or AUY-922 (2 μM; Figure 1B) for 2 hours, followed by PBS or LPS (1 EU/ml). LPS decreased TER values, suggesting increased permeability of the monolayer. Both 17-AAG and AUY-922 pretreatment prevented the LPS-mediated decrease in TER in HLMVECs. In addition, paracellular permeability across the HLMVECs was studied using the transwell assay system. HLMVEC monolayers grown on a transwell insert were exposed to vehicle or AUY-922 (2 μM, Figure 1C) for 2 hours, followed by PBS or LPS (5 EU/ml). LPS increased the influx of 2M kD FITC-dextran in the basal media, suggesting increased paracellular permeability; pretreatment with AUY-922 prevented the LPS-mediated increase in the influx of 2M kD FITC-dextran, suggesting that Hsp90 inhibition prevents LPS-mediated paracellular permeability in HLMVECs.
Figure 1.

Inhibition of heat shock protein (Hsp) 90 protects and restores the LPS-mediated human lung microvascular endothelial cell (HLMVEC) hyperpermeability. (A and B) HLMVECs were grown to confluence on electric cell-substrate impedance sensing (ECIS) arrays (8W10E+). Once a constant resistance was attained, the cells were treated with vehicle, 2 μM 17-allylamino-17 demethoxy-geldanamycin (17-AAG) (A) or 2 μM AUY-922 (B) for 2 hours. LPS (1 EU/ml) was then added as indicated by the arrow, and transendothelial resistance (TER) values were measured continuously. Resistance was normalized to time = 0 and plotted as a function of time. Data represent means (± SEM); n = 3 experiments, each performed in triplicate. *P < 0.01 versus vehicle, #P < 0.01 versus 17-AAG + LPS and, ##P < 0.01 versus AUY-922 + LPS. C) HLMVECs were added to each insert of a 24-well millicell culture plate. After 48 hours cells were treated with vehicle or 2 μM AUY-922 for 2 hours. LPS (5 EU/ml) was then added followed by 2M kD FITC-dextran (1 μg/μl). At 10 hours after LPS addition, 100 μl of basal media was removed, fluorescence intensity was measured and shown relative to vehicle (100%). Data represent means (± SEM); n = 3. *P < 0.05 versus vehicle, ##P < 0.05 versus AUY-922 + LPS. (D and E) Confluent monolayer was exposed to LPS (1 EU/ml). After 3 hours, 17-AAG (C) or AUY-922 (D) was added to a final concentration of 2 μM. Resistance was normalized to time = 0 and plotted as a function of time. Data represent means (± SEM); n = 3. *P < 0.01 versus vehicle, #P < 0.01 versus 17-AAG + LPS and, ##P < 0.01 versus AUY-922 + LPS.
Hsp90 Inhibition Restores LPS-Mediated HLMVEC Barrier Dysfunction
To test whether Hsp90 inhibition can restore TER that has already been disrupted by LPS, HLMVEC monolayer were exposed to PBS or LPS (1 EU/ml), followed 3 hours later by either 17-AAG or AUY-922 (2 μM). Post-treatment with either 17-AAG (Figure 1D) or AUY-922 (Figure 1E) completely restored the LPS-mediated decrease in TER.
ROCK Inhibition Protects against LPS-Mediated Decrease in TER
RhoA signaling, facilitated downstream by ROCK, is a major pathway of LPS-mediated inflammation. We used two small-molecule inhibitors of ROCK (Y27632 and GSK429286 at 10 μM) to study their effects on TER and MLC phosphorylation. HLMVEC monolayers were pretreated with either vehicle, Y27632 or GSK429286, followed by either PBS or LPS. TER was monitored continuously. Y27632 or GSK429286, alone, had no effect on TER values (data not shown). However, both ROCK inhibitors protected HLMVECs against LPS-mediated decrease in TER (Figure 2A) suggesting a major role for the RhoA-ROCK pathway in LPS-mediated barrier disruption in HLMVECs. Furthermore, the ROCK inhibitors provided complete abrogation of LPS-induced MLC phosphorylation, suggesting that LPS-mediated MLC phosphorylation is via the RhoA-ROCK pathway (Figure 2B).
Figure 2.
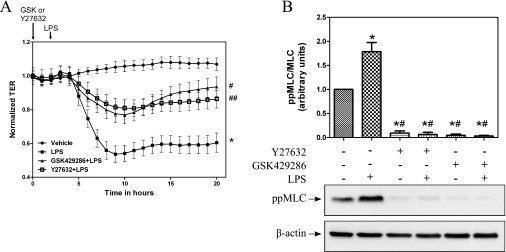
Inhibition of Rho kinase (ROCK) protects against the LPS-mediated decrease in TER. (A) HLMVECs were grown to confluence on ECIS arrays (8W10E+). Once a constant resistance was attained, the cells were treated with vehicle (0.1% dimethyl sulfoxide [DMSO]), Y27632 (10 μM), or GSK429286 (10 μM) for 2 hours. LPS (1 EU/ml) was then added as indicated by the arrow and TER values were measured continuously and normalized to the 0-hour value. Data represent means (± SEM); n = 4. *P < 0.0001 versus vehicle, #P < 0.0001 versus LPS, ##P < 0.01 versus LPS. (B) HLMVECs grown to confluence in six-well plates were preincubated with vehicle (0.1% DMSO), Y27632 (10 μM), or GSK429286 (10 μM) for 2 hours, followed by treatment with 1 EU/ml LPS for 2 hours. Western blot analysis was performed for diphosphorylated myosin light chain (ppMLC) and β-actin.
Hsp90 Inhibition Suppresses LPS-Mediated RhoA Activity and Nitration
We then tested the hypothesis that Hsp90 inhibition would affect LPS-mediated RhoA activation and signaling. LPS exposure for 2 hours increased RhoA activity in HLMVECs and 17-AAG pretreatment attenuated the LPS-mediated RhoA activation (Figure 3A). Recently, we demonstrated that RhoA nitration stimulates its activity (unpublished data). Therefore, we tested if 17-AAG pretreatment affected LPS-induced RhoA nitration as well. HLMVECs were pretreated with 17-AAG for 2 hours, followed by LPS exposure; immunoprecipitation of RhoA and Western blot analysis for nitrotyrosine and RhoA was then performed (Figure 3B). LPS increased nitrotyrosine levels of RhoA and, as shown in the histogram that depicts fold change in the ratio of nitrated to total RhoA band densities, 17-AAG completely blocked the LPS-mediated RhoA nitration (Figure 3B). Together, the above data indicate that Hsp90 inhibitors act upstream of RhoA to block LPS-induced nitration of RhoA, thereby suppressing its activation.
Figure 3.
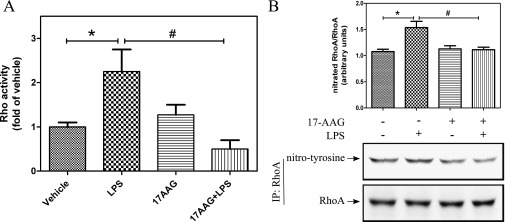
Hsp90 inhibition attenuates the LPS-mediated RhoA activity and nitration. (A) HLMVECs were grown in 100-mm dishes. Confluent cells were treated with 2 μM 17-AAG for 2 hours, followed by LPS (1 EU/ml) for 2 hours. Cells were then lysed and RhoA activity was measured using the G-LISA RhoA activation assay. Data represent means (± SEM); n = 4. *, #P < 0.05. (B) HLMVECs were treated as described previously here. Cells were then lysed, RhoA was immunoprecipitated using anti-RhoA antibody, and then immunoblotting was performed using antibodies against nitrotyrosine and RhoA. Normalized nitrotyrosine levels are shown. Data represent means (± SEM); n = 7. *P < 0.05 from vehicle; #P < 0.05 from LPS. IP, immunoprecipitated.
Hsp90 Inhibition Suppresses LPS-Mediated MLC Phosphorylation
MLC regulates actin–myosin contractility and is a known target of the RhoA-ROCK pathway. We tested the effect of Hsp90 inhibition on LPS-mediated MLC phosphorylation. LPS induced robust MLC phosphorylation at both 2 and 4 hours; this was totally blocked by either 17-AAG or AUY-922 (Figure 4). However, after 6 hours of LPS exposure, MLC phosphorylation was absent, suggesting that MLC phosphorylation is an early signaling event after LPS addition, and is not required to maintain LPS-induced hyperpermeability.
Figure 4.

Inhibition of Hsp90 suppresses the LPS-induced MLC phosphorylation. HLMVECs were grown in six-well plates. Confluent cells were treated with vehicle, 2 μM 17-AAG, or 2 μM AUY-922 for 2 hours, followed by exposure to LPS (1 EU/ml) for 2, 4, or 6 hours. Cells were then lysed and Western blot analysis was performed using antibodies against ppMLC and MLC. Densitometric analysis was performed, and the ratio of ppMLC to MLC was plotted. Data represent means (± SEM); n = 3. *P < 0.05 versus vehicle; #P < 0.05 versus 2-hour LPS alone treatment; ##P < 0.05 versus 4-hour LPS alone treatment.
Hsp90 Inhibition Attenuates the Decrease in TER Caused by Constitutively Active RhoA Expression
To investigate whether Hsp90 also functions downstream of RhoA, we utilized an adenovirus expressing a constitutively active form of RhoA (CA-RhoA). HLMVECs grown on ECIS arrays were exposed to adenoviruses expressing either GFP or CA-RhoA for 24 hours. Vehicle or 17-AAG was then added for another 24 hours. TER was recorded continuously throughout the experiment. HLMVECs infected with CA-RhoA exhibited a gradual decrease in TER values; however, 17-AAG restored TER to control values (Figure 5A). Western blot analysis of MLC phosphorylation demonstrated that expression of CA-RhoA increased MLC phosphorylation, and that 17-AAG suppressed the CA-RhoA-induced MLC phosphorylation (Figure 5B). Together, the above data suggest that Hsp90 also plays a significant role downstream of RhoA in mediating the LPS-induced barrier disruption and MLC phosphorylation.
Figure 5.
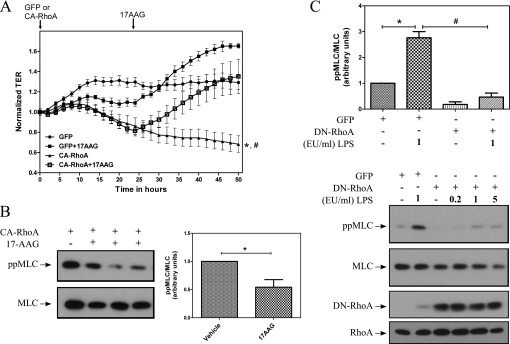
17-AAG restores LPS-mediated hyperpermeability caused by constitutively active RhoA (CA-RhoA). (A) HLMVECs were grown on ECIS arrays. Subconfluent HLMVECs were infected with adenoviruses expressing either GFP or CA-RhoA (50 multiplicity of infection [MOI]). After 24 hours, 17-AAG was added to a final concentration of 2 μM. TER values were measured continuously and normalized to time = 0 hours. Data represent means (± SEM); n = 3. *P < 0.001 versus GFP, #P < 0.001 versus CA-RhoA + 17-AAG. (B) Subconfluent HLMVECs were exposed to adenoviruses expressing either GFP (50 MOI) or CA-Rho (50 MOI). After 24 hours, transduction efficiency greater than 90% was noted by observing GFP-expressing cells. Cells were treated either with vehicle (0.1% DMSO) or with 2 μM 17-AAG for another 24 hours. Cell lysates were prepared and immunoblotted for ppMLC and MLC. Densitometric analysis was performed using Imagequant 5.1 and shown as fold change from vehicle. Data represent means (± SEM); n = 4. *P < 0.05. (C) Cells were grown on six-well plates. Subconfluent HLMVECs were infected with adenoviruses expressing either GFP or dominant-negative (DN)-RhoA (500 MOI). After 24 hours, cells were exposed to LPS (0–5 EU/ml) for 2 hours, lysed, and immunoblotted using antibodies against ppMLC, MLC, and RhoA. Densitometric analysis was performed, and the ratio of ppMLC to MLC was plotted. Data represent mean (± SEM); n = 3. *P < 0.05 from vehicle; #P < 0.05 from LPS.
Overexpression of DN-RhoA Suppresses LPS-Induced MLC Phosphorylation
Because LPS induced Rho activity and signaling, we also studied the effect of RhoA inhibition on MLC phosphorylation by overexpressing DN-RhoA. HLMVECs were infected with adenoviruses expressing either GFP or DN-RhoA. After 24 hours, cells were exposed to either PBS or LPS (1 EU/ml) for 2 hours, and Western blot analysis was performed on cell lysates. Overexpression of DN-RhoA completely blocked LPS-induced MLC phosphorylation (Figure 5C), suggesting that the RhoA-ROCK pathway is a major mediator of LPS-induced MLC phosphorylation in HLMVECs.
SFK Inhibition Suppresses LPS-Mediated RhoA Activity and Signaling
We previously reported that LPS-induced pp60Src phosphorylation and activity in bovine endothelial cells is suppressed by inhibition of Hsp90 (5). Therefore, we investigated if SFKs play a role in regulating RhoA activity and signaling. We first examined whether inhibition of SFKs affects LPS-induced RhoA activity. HLMVECs were pretreated for 2 hours with vehicle or the SFK inhibitor, PP2 (10 μM), followed by treatment with either PBS or LPS. After 2 hours of LPS treatment, RhoA activity was determined in cell lysates. PP2 pretreatment inhibited LPS-induced RhoA activity (Figure 6A). Furthermore, Western blot analysis showed that PP2 also suppressed LPS-mediated phosphorylation of MLC (Figure 6B). These data suggest that LPS induces RhoA activation and signaling via SFKs.
Figure 6.

Inhibition of Src family kinases (SFKs) attenuates the LPS-mediated RhoA activity and MLC phosphorylation. HLMVECs were grown to confluence in 60-mm dishes, treated with either vehicle (0.1% DMSO) or PP2 (10μM) for 2 hours, and then with LPS (2 EU/ml) for 90 minutes (A). RhoA activity was measured using the G-LISA RhoA activation assay. Data represent means (± SEM); n = 4. *P < 0.05 from vehicle, #P < 0.05 from LPS. (B) Western blot analysis was performed for ppMLC and MLC. Data represent means (± SEM); n = 3. *P < 0.05 from vehicle, #P < 0.05 from LPS.
Discussion
The molecular chaperone, Hsp90, is involved in a variety of cellular processes (14). Inhibition of Hsp90, therefore, affects a broad range of signaling molecules (14–17, 26). Our laboratory has previously shown that Hsp90 inhibition prolongs survival and attenuates inflammation in mice exposed to lethal doses of LPS (25), and protects and restores endothelial hyperpermeability in cultured BPAECs caused by various proinflammatory agents (5, 20). However, the mechanisms responsible for these effects remain unknown. Because RhoA activation has been shown to be a major factor in LPS-induced ALI, we then tested the hypotheses that hsp90 inhibitors ameliorate LPS-induced ALI by acting on targets both upstream and downstream of Rho. To enhance the translational nature of the studies, these experiments were performed on in-house–harvested HLMVECs.
Pretreatment of HLMVEC monolayers with Hsp90 inhibitors, either 17-AAG or AUY-922, effectively blocked the LPS-induced decrease in TER, suggesting that Hsp90 function is critical in mediating LPS-induced endothelial barrier disruption. In addition, the Hsp90 inhibitor, AUY-922, completely blocked the LPS-induced transport of 2M kD FITC-dextran across the HLMVEC monolayer, confirming the TER findings and suggesting that the effect of Hsp90 and its inhibitors is primarily on paracellular permeability. Furthermore, when Hsp90 inhibitors are added to cells that already exhibit a decreased TER due to LPS exposure, barrier function is restored, suggesting that the sustained disruption of the endothelial barrier by LPS downstream signaling also requires functional Hsp90. Together, the above data indicate that Hsp90 plays a role in two distinct phases of LPS-induced barrier disruption: the first phase involves the early processes wherein LPS binds the host Toll-like receptor 4 receptor complex and transmits signals that cause an immediate decrease in TER; and the second phase, where the barrier disruption is maintained.
LPS-mediated barrier disruption is associated with ROCK-dependent regulation of the actin cytoskeleton. Inhibitors of ROCK attenuate ROCK-mediated actin cytoskeleton changes (12, 27, 28). For example, inhibition of ROCK by fasudil attenuated LPS-induced hyperpermeability in guinea pigs (29). In addition, the ROCK inhibitor, Y27632, effectively blocked permeability caused by the gram-negative bacteria, Pseudomonas aeruginosa (30), but did not attenuate airway microvascular leakage induced by leukotriene D(4) or histamine (31). In contrast, in a recent study, pretreatment with the ROCK inhibitor, Y27632, failed to inhibit the LPS-induced hyperpermeability of commercially available HLMVECs (12). In light of this conflicting evidence, we evaluated the effect of ROCK inhibition on in-house–isolated and –characterized HLMVECs. Pretreatment with either of two ROCK inhibitors, Y27632 or GSK429286, at concentrations significantly higher (10 μΜ) and for much longer times (2 h) than those used in a previous study (0.5 and 5 μM for 15 min) (7), provided partial, but significant, protection against LPS-induced disruption of HLMVEC barrier function. These data suggest a major role for ROCK in the early phase of LPS-induced barrier disruption in human lung microvascular cells. Because Hsp90 also plays a crucial role in the early phase of LPS-mediated barrier disruption, the data point to a strong correlation between Hsp90 and ROCK-dependent actin cytoskeletal changes.
ROCK is a major effector kinase for the small-GTPase RhoA (32), the activity of which is induced by exposure of endothelial cells to LPS. Furthermore, inactivation of RhoA has been shown to improve barrier function (33, 34). Other studies have shown that vascular endothelial growth factor– and thrombin-induced RhoA activity was inhibited by the Hsp90 inhibitor geldanamycin (35, 36). S.M.B. and colleagues recently discovered that RhoA nitration stimulates RhoA activity (unpublished data). Therefore, we sought to evaluate whether Hsp90 inhibition by 17-AAG affects LPS-induced RhoA nitration and activation in HLMVECs. Our data demonstrate that LPS stimulated both RhoA tyrosine nitration and RhoA activity in HLMVECs. Furthermore, pretreatment with 17-AAG prevented both the LPS-induced RhoA activity and the LPS-mediated RhoA tyrosine nitration. This inhibition of RhoA activity also suppresses ROCK-dependent actin cytoskeleton changes and MLC phosphorylation.
MLC is a known downstream target of ROCK, and phosphorylation of MLC by ROCK causes remodeling of the actin cytoskeleton, leading to endothelial hyperpermeability (10, 12, 37, 38). In agreement with the studies cited previously here, LPS-induced MLC phosphorylation in HLMVECs is inhibited by both ROCK inhibitors, Y27632 and GSK429286. Furthermore, pretreatment with Hsp90 inhibitors completely blocked LPS-induced MLC phosphorylation, which supports our finding that hsp90 inhibitors block LPS-induced RhoA activation. Moreover, when HLMVECs were infected with adenovirus carrying DN form of RhoA, LPS-induced MLC phosphorylation was completely blocked. These data demonstrate that Hsp90 inhibitors block LPS-induced and RhoA-mediated signaling to MLC, and support the hypothesis that Hsp90 regulates key signaling steps upstream of RhoA. Therefore, we then investigated the role of Hsp90 downstream of RhoA, and tested the hypothesis that barrier disruption induced by the expression of CA-RhoA could be restored by Hsp90 inhibition. 17-AAG restored barrier function and attenuated MLC phosphorylation in cells expressing CA-RhoA, suggesting a barrier-disruptive role for Hsp90 downstream of RhoA as well. The precise target of Hsp90 regulation of MCL phosphorylation downstream of RhoA remains unclear. Recently, however, we demonstrated that 17-AAG prevents transforming growth factor-β–induced Hsp27 phosphorylation, and that Hsp27 is a client protein of Hsp90, in BPAECs (3). Hsp27 may thus be a possible Hsp90-sensitive target of RhoA downstream signaling.
Upstream of RhoA, we hypothesized that the kinase pp60Src, an Hsp90 client protein, mediates the LPS-induced RhoA activation and signaling. SFKs have been shown to contribute to endothelial cell hyperpermeability (6–8, 14). Inhibition of SFKs by PP2 or knockdown of SFK members, pp60Src, YES, or FYN, partially protected against LPS-mediated barrier disruption (8). In addition, expression of constitutively active pp60Src has been shown to decrease endothelial barrier function (39). Therefore, we tested whether inhibition of SFKs would affect the LPS-mediated RhoA activation. Our data clearly show that pretreatment with the SFK inhibitor, PP2, completely blocked the LPS-induced RhoA activation and suppressed the LPS-induced MLC phosphorylation in HLMVECs, suggesting that LPS-mediated RhoA activation is mediated by SFK, and raising the distinct possibility that Hsp90 inhibitors block RhoA activity by inhibiting the activation of SFK.
We have previously shown that inhibition of Hsp90 by 17-AAG prolongs survival, attenuates lung injury, and decreases neutrophil infiltration in a murine model of sepsis (25). Studies have also shown similar protective effect in the lung when mice are pretreated with either tyrosine kinase inhibitor (40–42), SFK inhibitor (8), RhoA inhibitor (40), or ROCK inhibitors (43, 44), followed by LPS exposure. In addition, down-regulation of SFK also reduces LPS-mediated endothelial hyperpermeability (8). Collectively, the data support the notion that Hsp90 inhibition prevents LPS-mediated barrier dysfunction by suppressing SFK-mediated RhoA-ROCK activation and signaling (Figure 7).
Figure 7.

Proposed regulation of LPS-mediated of Rho activation and signaling by Hsp90. LPS induces activation of Rho and its downstream effector kinase, ROCK, leading to MLC phosphorylation and endothelial hyperpermeability. Inhibition of Hsp90 prevents LPS-induced Rho activation and signaling by inhibiting pp60Src activity. In addition, downstream of Rho, inhibition of Hsp90 attenuates MLC phosphorylation, indicating a role of Hsp90 downstream of Rho. EC, endothelial cell.
Endothelial barrier dysfunction leads to a cascade of cytokine release resulting in systemic inflammation (1, 45, 46). Current therapeutic strategy of broad-spectrum antibiotics (47) and anti-cytokine antibodies (48) do not affect the underlying molecular machinery involved. The ATP-dependent molecular chaperone, Hsp90, regulates the stability and activity of over 200 proteins, including a number of protein kinases and transcription factors that are directly involved in inflammation and barrier protection (5, 14, 19, 20, 25, 49). These data, in combination with our studies, indicate that Hsp90 inhibition provides a multitargeted therapy that can play a major role in diseases affecting endothelial function, and support the growing belief that multitargeted rather than single-target approaches hold greater therapeutic promise (50).
Acknowledgments
Acknowledgments
The authors thank Bristol Myers Squibb and National cancer Institute for providing 17-allylamino-17 demethoxy-geldanamycin (17-AAG).
Footnotes
This work was supported by National Institutes of Health grants HL101902, HL066993, and HL093460.
Originally Published in Press as DOI: 10.1165/rcmb.2012-0496OC on August 23, 2013
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci. 2008;1123:134–145. doi: 10.1196/annals.1420.016. [DOI] [PubMed] [Google Scholar]
- 2.Oda M, Han JY, Nakamura M. Endothelial cell dysfunction in microvasculature: relevance to disease processes. Clin Hemorheol Microcirc. 2000;23:199–211. [PubMed] [Google Scholar]
- 3.Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest. 2006;86:9–22. doi: 10.1038/labinvest.3700366. [DOI] [PubMed] [Google Scholar]
- 4.Ostuni R, Zanoni I, Granucci F. Deciphering the complexity of Toll-like receptor signaling. Cell Mol Life Sci. 2010;67:4109–4134. doi: 10.1007/s00018-010-0464-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee A, Snead C, Yetik-Anacak G, Antonova G, Zeng J, Catravas JD. Heat shock protein 90 inhibitors attenuate LPS-induced endothelial hyperpermeability. Am J Physiol Lung Cell Mol Physiol. 2008;294:L755–L763. doi: 10.1152/ajplung.00350.2007. [DOI] [PubMed] [Google Scholar]
- 6.Hu G, Minshall RD. Regulation of transendothelial permeability by Src kinase. Microvasc Res. 2009;77:21–25. doi: 10.1016/j.mvr.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Hu G, Place AT, Minshall RD. Regulation of endothelial permeability by Src kinase signaling: vascular leakage versus transcellular transport of drugs and macromolecules. Chem Biol Interact. 2008;171:177–189. doi: 10.1016/j.cbi.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gong P, Angelini DJ, Yang S, Xia G, Cross AS, Mann D, Bannerman DD, Vogel SN, Goldblum SE. TLR4 signaling is coupled to SRC family kinase activation, tyrosine phosphorylation of zonula adherens proteins, and opening of the paracellular pathway in human lung microvascular endothelia. J Biol Chem. 2008;283:13437–13449. doi: 10.1074/jbc.M707986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Birukov KG. Small GTPases in mechanosensitive regulation of endothelial barrier. Microvasc Res. 2009;77:46–52. doi: 10.1016/j.mvr.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 11.Fukata Y, Amano M, Kaibuchi K. Rho–Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol Sci. 2001;22:32–39. doi: 10.1016/s0165-6147(00)01596-0. [DOI] [PubMed] [Google Scholar]
- 12.Bogatcheva NV, Zemskova MA, Poirier C, Mirzapoiazova T, Kolosova I, Bresnick AR, Verin AD. The suppression of myosin light chain (MLC) phosphorylation during the response to lipopolysaccharide (LPS): beneficial or detrimental to endothelial barrier? J Cell Physiol. 2011;226:3132–3146. doi: 10.1002/jcp.22669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Essler M, Staddon JM, Weber PC, Aepfelbacher M. Cyclic AMP blocks bacterial lipopolysaccharide–induced myosin light chain phosphorylation in endothelial cells through inhibition of Rho/Rho kinase signaling. J Immunol. 2000;164:6543–6549. doi: 10.4049/jimmunol.164.12.6543. [DOI] [PubMed] [Google Scholar]
- 14.Neckers L, Ivy SP. Heat shock protein 90. Curr Opin Oncol. 2003;15:419–424. doi: 10.1097/00001622-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 15.Barginear MF, Van Poznak C, Rosen N, Modi S, Hudis CA, Budman DR. The heat shock protein 90 chaperone complex: an evolving therapeutic target. Curr Cancer Drug Targets. 2008;8:522–532. doi: 10.2174/156800908785699379. [DOI] [PubMed] [Google Scholar]
- 16.Drysdale MJ, Brough PA, Massey A, Jensen MR, Schoepfer J. Targeting Hsp90 for the treatment of cancer. Curr Opin Drug Discov Devel. 2006;9:483–495. [PubMed] [Google Scholar]
- 17.Staufer K, Stoeltzing O. Implication of heat shock protein 90 (HSP90) in tumor angiogenesis: a molecular target for anti-angiogenic therapy? Curr Cancer Drug Targets. 2010;10:890–897. doi: 10.2174/156800910793357934. [DOI] [PubMed] [Google Scholar]
- 18.Kaur G, Belotti D, Burger AM, Fisher-Nielson K, Borsotti P, Riccardi E, Thillainathan J, Hollingshead M, Sausville EA, Giavazzi R. Antiangiogenic properties of 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin: an orally bioavailable heat shock protein 90 modulator. Clin Cancer Res. 2004;10:4813–4821. doi: 10.1158/1078-0432.CCR-03-0795. [DOI] [PubMed] [Google Scholar]
- 19.Rice JW, Veal JM, Fadden RP, Barabasz AF, Partridge JM, Barta TE, Dubois LG, Huang KH, Mabbett SR, Silinski MA, et al. Small molecule inhibitors of Hsp90 potently affect inflammatory disease pathways and exhibit activity in models of rheumatoid arthritis. Arthritis Rheum. 2008;58:3765–3775. doi: 10.1002/art.24047. [DOI] [PubMed] [Google Scholar]
- 20.Antonov A, Snead C, Gorshkov B, Antonova GN, Verin AD, Catravas JD. Heat shock protein 90 inhibitors protect and restore pulmonary endothelial barrier function. Am J Respir Cell Mol Biol. 2008;39:551–559. doi: 10.1165/rcmb.2007-0324OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Q, Church JE, Jagnandan D, Catravas JD, Sessa WC, Fulton D. Functional relevance of Golgi- and plasma membrane–localized endothelial NO synthase in reconstituted endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:1015–1021. doi: 10.1161/01.ATV.0000216044.49494.c4. [DOI] [PubMed] [Google Scholar]
- 22.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miao RQ, Fontana J, Fulton D, Lin MI, Harrison KD, Sessa WC. Dominant-negative Hsp90 reduces VEGF-stimulated nitric oxide release and migration in endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:105–111. doi: 10.1161/ATVBAHA.107.155499. [DOI] [PubMed] [Google Scholar]
- 24.Catravas JD, Snead C, Dimitropoulou C, Chang AS, Lucas R, Verin AD, Black SM. Harvesting, identification and barrier function of human lung microvascular endothelial cells. Vascul Pharmacol. 2010;52:175–181. doi: 10.1016/j.vph.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chatterjee A, Dimitropoulou C, Drakopanayiotakis F, Antonova G, Snead C, Cannon J, Venema RC, Catravas JD. Heat shock protein 90 inhibitors prolong survival, attenuate inflammation, and reduce lung injury in murine sepsis. Am J Respir Crit Care Med. 2007;176:667–675. doi: 10.1164/rccm.200702-291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimokawa H, Rashid M. Development of Rho-kinase inhibitors for cardiovascular medicine. Trends Pharmacol Sci. 2007;28:296–302. doi: 10.1016/j.tips.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 28.Satoh K, Fukumoto Y, Shimokawa H. Rho-kinase: important new therapeutic target in cardiovascular diseases. Am J Physiol Heart Circ Physiol. 2011;301:H287–H296. doi: 10.1152/ajpheart.00327.2011. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki K, Nemoto K, Ninomiya N, Kuno M, Kubota M, Yokota H. Fasudil, a Rho-kinase inhibitor, attenuates lipopolysaccharide-induced vascular hyperpermeability and colonic muscle relaxation in guinea pigs. J Surg Res. 2012;178:352–357. doi: 10.1016/j.jss.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 30.Ganter MT, Roux J, Su G, Lynch SV, Deutschman CS, Weiss YG, Christiaans SC, Myazawa B, Kipnis E, Wiener-Kronish JP, et al. Role of small GTPases and αvβ5 integrin in Pseudomonas aeruginosa–induced increase in lung endothelial permeability. Am J Respir Cell Mol Biol. 2009;40:108–118. doi: 10.1165/rcmb.2007-0454OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tokuyama K, Nishimura H, Iizuka K, Kato M, Arakawa H, Saga R, Mochizuki H, Morikawa A. Effects of Y-27632, a Rho/Rho kinase inhibitor, on leukotriene D(4)- and histamine-induced airflow obstruction and airway microvascular leakage in guinea pigs in vivo. Pharmacology. 2002;64:189–195. doi: 10.1159/000056170. [DOI] [PubMed] [Google Scholar]
- 32.Loirand G, Guérin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circ Res. 2006;98:322–334. doi: 10.1161/01.RES.0000201960.04223.3c. [DOI] [PubMed] [Google Scholar]
- 33.Carbajal JM, Schaeffer RC., Jr RhoA inactivation enhances endothelial barrier function. Am J Physiol. 1999;277:C955–C964. doi: 10.1152/ajpcell.1999.277.5.C955. [DOI] [PubMed] [Google Scholar]
- 34.Hippenstiel S, Soeth S, Kellas B, Fuhrmann O, Seybold J, Krüll M, Eichel-Streiber C, Goebeler M, Ludwig S, Suttorp N. Rho proteins and the p38-MAPK pathway are important mediators for LPS-induced interleukin-8 expression in human endothelial cells. Blood. 2000;95:3044–3051. [PubMed] [Google Scholar]
- 35.Le Boeuf F, Houle F, Huot J. Regulation of vascular endothelial growth factor receptor 2–mediated phosphorylation of focal adhesion kinase by heat shock protein 90 and Src kinase activities. J Biol Chem. 2004;279:39175–39185. doi: 10.1074/jbc.M405493200. [DOI] [PubMed] [Google Scholar]
- 36.Pai KS, Mahajan VB, Lau A, Cunningham DD. Thrombin receptor signaling to cytoskeleton requires Hsp90. J Biol Chem. 2001;276:32642–32647. doi: 10.1074/jbc.M104212200. [DOI] [PubMed] [Google Scholar]
- 37.Garcia JG, Verin AD, Schaphorst KL. Regulation of thrombin-mediated endothelial cell contraction and permeability. Semin Thromb Hemost. 1996;22:309–315. doi: 10.1055/s-2007-999025. [DOI] [PubMed] [Google Scholar]
- 38.Shen Q, Rigor RR, Pivetti CD, Wu MH, Yuan SY. Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc Res. 2010;87:272–280. doi: 10.1093/cvr/cvq144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adam AP, Sharenko AL, Pumiglia K, Vincent PA. Src-induced tyrosine phosphorylation of VE-cadherin is not sufficient to decrease barrier function of endothelial monolayers. J Biol Chem. 2010;285:7045–7055. doi: 10.1074/jbc.M109.079277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Nieuw Amerongen GP, Draijer R, Vermeer MA, van Hinsbergh VW. Transient and prolonged increase in endothelial permeability induced by histamine and thrombin: role of protein kinases, calcium, and RhoA. Circ Res. 1998;83:1115–1123. doi: 10.1161/01.res.83.11.1115. [DOI] [PubMed] [Google Scholar]
- 41.van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res. 2000;87:335–340. doi: 10.1161/01.res.87.4.335. [DOI] [PubMed] [Google Scholar]
- 42.Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul Pharmacol. 2002;39:213–223. doi: 10.1016/s1537-1891(03)00010-7. [DOI] [PubMed] [Google Scholar]
- 43.Tasaka S, Koh H, Yamada W, Shimizu M, Ogawa Y, Hasegawa N, Yamaguchi K, Ishii Y, Richer SE, Doerschuk CM, et al. Attenuation of endotoxin-induced acute lung injury by the Rho-associated kinase inhibitor, Y-27632. Am J Respir Cell Mol Biol. 2005;32:504–510. doi: 10.1165/rcmb.2004-0009OC. [DOI] [PubMed] [Google Scholar]
- 44.Li Y, Wu Y, Wang Z, Zhang XH, Wu WK. Fasudil attenuates lipopolysaccharide-induced acute lung injury in mice through the Rho/Rho kinase pathway. Med Sci Monit. 2010;16:BR112–BR118. [PubMed] [Google Scholar]
- 45.Bannerman DD, Goldblum SE. Direct effects of endotoxin on the endothelium: barrier function and injury. Lab Invest. 1999;79:1181–1199. [PubMed] [Google Scholar]
- 46.Lucas R, Verin AD, Black SM, Catravas JD. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol. 2009;77:1763–1772. doi: 10.1016/j.bcp.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Esper AM, Martin GS. Evolution of treatments for patients with acute lung injury. Expert Opin Investig Drugs. 2005;14:633–645. doi: 10.1517/13543784.14.5.633. [DOI] [PubMed] [Google Scholar]
- 48.Dinarello CA. Inflammation in human disease: anticytokine therapy. Biol Blood Marrow Transplant. 2009;15(1) Suppl:134–136. doi: 10.1016/j.bbmt.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 49.Poulaki V, Iliaki E, Mitsiades N, Mitsiades CS, Paulus YN, Bula DV, Gragoudas ES, Miller JW. Inhibition of Hsp90 attenuates inflammation in endotoxin-induced uveitis. FASEB J. 2007;21:2113–2123. doi: 10.1096/fj.06-7637com. [DOI] [PubMed] [Google Scholar]
- 50.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101:3765–3777. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]