

Abstract
Like many steroid receptors, the glucocorticoid (GC) receptor (GR) is a phosphoprotein. Although there are multiple phosphorylation sites critical for GR transcriptional activity (i.e., serine [S]203, S211, and S226), their respective role in driving GR functions is highly cell specific. We have recently identified protein phosphatase 5 as an essential Ser/Thr phosphatase responsible for impairing GR function via S211 dephosphorylation in airway smooth muscle (ASM) cells. Because p38 mitogen-activated protein kinase (MAPK) directly phosphorylates GR in different cell types in a stimulus- and cell-dependent manner, we investigated the role of p38 MAPK on GR phosphorylation and function in ASM cells. Cells were transfected with 100 nM p38 MAPK small interfering RNA or 2 μg MAPK kinase 3 expression vector (a specific kinase that directly activates p38 MAPK) in the presence or absence of fluticasone (100 nM) and/or p38 MAPK pharmacological inhibitor SB203580. We found that p38 MAPK blockade positively regulates GR nuclear translocation and GR-dependent induction of the steroid-target gene GC-induced leucine zipper in a hormone-independent manner. We also found that p38 MAPK–dependent regulation of GR functions was associated with a differential action on GR phosphorylation at S203 and S211 residues. This study demonstrated that the inactive state of GR in resting conditions is not only ensured by the absence of the GC ligand but also by p38 MAPK–dependent phosphorylation of unliganded GR at specific residues, which appears to be important in determining the overall GC responsiveness of ASM cells.
Keywords: p38 MAPK, glucocorticoid receptor phosphorylation, airway smooth muscle, asthma, steroid resistance
Clinical Relevance
A number of different reports using peripheral blood mononuclear cells have described p38 mitogen-activated protein kinase (MAPK) as a potential player in driving glucocorticoid insensitivity seen in patients with severe asthma via the control of glucocorticoid receptor phosphorylation status. We report the first evidence in human airway smooth muscle cells that p38 MAPK acts as a negative regulator of glucocorticoid receptor transcriptional activity via the direct phosphorylation of serine 203 residues. The use of p38 MAPK inhibitors that are clinically safer would therefore lead to steroid-sparing effects that would benefit patients with severe asthma who require high doses of glucocorticoid.
Many nuclear receptors, including the glucocorticoid (GC) receptor (GR), are phosphoproteins (1). The phosphorylation of GR is a critical step in the receptor activation because it affects various essential functions of GR, including its ligand binding, subcellular localization, half-life, DNA binding, and biological activity (1). GR protein structure consists of an N-terminal domain with a potent transactivation domain (AF1), a DNA-binding domain, and a ligand-binding domain with an “activation functions” (AF2) subdomain (2, 3). In the AF1 domain, the major functionally important phosphorylation sites are serine (S)203, S211, and S226 (4). Ligand binding, which results in Hsp90 dissociation, allows kinases to interact with GR, which becomes phosphorylated. Evidence from different cell types, including airway smooth muscle (ASM) cells, suggests that GR-mediated transactivation positively correlates with the phosphorylation status of GR at S211 residues, whereas S226 decreases GR transcriptional activities (1, 5–8). In contrast to GR phosphorylation at S211 and S226 residues, which has received a lot of attention, the role of GR phosphorylation at S203 residue has not been fully investigated (7, 9, 10).
GR phosphorylation is regulated by a variety of Ser/Thr kinases and phosphatases. We recently found that the protein phosphatase (PP)5 was essential in driving cytokine-induced GC insensitivity by promoting GR dephosphorylation at S211 in ASM cells (8). However, the nature of kinases responsible for GR phosphorylation in ASM cells and in other cell types is controversial. Current reports show that the mitogen-activated protein kinase (MAPK) pathway is critical in determining the transcriptional activities of GR-mediated GC effects (11–16). p38 MAPK kinase is a Ser/Thr kinase involved in many processes thought to be important in inflammatory diseases (17–19). p38 MAPK is activated by upstream MAPK kinases (MKK3 or MKK6) in response to a number of inflammatory or environmental signals (17–19). Different reports have convincingly implicated p38 MAPK pathways in the pathogenesis of patients with asthma, in particular those with severe disease (20–24). The exact role of p38 MAPK in GR functions in human diseases has just started to emerge. Earlier studies in immune cells suggested that the p38 MAPK–GR interaction acts as a novel mechanism driving GC resistance in patients with severe asthma (11). Indeed, the blockade of p38 MAPK pathways by soluble inhibitors (20, 23) or by the long-acting β agonists (LABAs) formoterol and salmeterol (24) was associated with a restoration of cell sensitivity to GC. Those reports identified p38 MAPK as an important kinase in peripheral blood mononuclear cells (PBMCs) that reduces GR functions by impairing GR nuclear translocation and/or DNA binding affinity. Direct GR phosphorylation on S226 by p38 MAPK appears to be one possible pathway driving the loss of GC efficacy seen in patients with severe asthma. However, it is undetermined whether p38 MAPK regulates the sensitivity of structural lung tissues, such as ASM, to GC therapy. A similar role of p38 MAPK in regulating GC sensitivity has been described in mice overexpressing p38 MAPK, which have a reduced GC responsiveness to burn injury (25), or in Crohn’s disease, where p38 MAPK activity is significantly increased in colonic tissues from GC-resistant patients when compared with GC-sensitive patients (26). The implication of p38 MAPK in the pathogenesis of Crohn’s disease was further confirmed by the marked improvement of the disease in patients treated with p38 MAPK inhibitors (27). Although the use of p38 MAPK inhibitors in the treatment of human diseases has been compromised by the occurrence of serious side effects (e.g., liver toxicity) (28, 29), these studies confirmed that functional interactions exist between p38 MAPK and GR and are critical in the regulation of GR functions.
The purpose of the current study was to examine whether p38 MAPK in ASM cells regulates GR transcriptional activities under basal conditions and after GC treatment and to determine the GR phosphorylation sites that are affected by p38 MAPK.
Materials and Methods
ASM Cells Culture and Characterization
Human ASM cells were isolated from the tracheal tissue of lung transplants from deidentified subjects provided by the National Disease Research Interchange. All experiments involving human tissues were approved by the Thomas Jefferson University Institutional Review Board. The culture of human ASM cells was performed as described elsewhere (8, 30).
SDS-PAGE and Western Blot Analysis
Immunoblot analysis was performed as we have previously described (8) using anti–GR-S211, anti–p38, anti–p-p38, anti-MKK3, and anti-ATF2 (Cell Signaling, Beverly, MA) or antibodies specific for anti–GR-S203 and anti–GR-S226 (Abcam, Cambridge, MA) or anti-p38α, anti-p38β, anti-p38γ, and anti-PP5 (Santa Cruz Biotechnology, Santa Cruz, CA). To ensure equal loading, the membranes were stripped and reprobed with anti-total GR or anti–glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibodies (Santa Cruz Biotechnology). Scanning densitometry of three representative immunoblots with each condition normalized over the area density of the corresponding GAPDH content was then performed using a Gel-Pro Analyzer (Media Cybernetics, Silver Spring, MD) as we have previously described (8). Immunoprecipitation experiments using anti-p38 and anti-PP5 antibodies (Santa Cruz Biotechnology) were performed as described (31).
Transfection of ASM Cells
ASM cells were transfected using the Basic Nucleofector Kit for Primary Smooth Muscle Cells according to the manufacturer's instructions using an Amaxa Nucleofector II device (program U-25; Amaxa Biosystems, Cologne, Germany) (8). Small interfering RNA (siRNA) experiments were performed using the siRNA Test Kit for Cell Lines and Adherent Primary Cells according to the manufacturer's instructions (Amaxa Biosystems) (8). ASM cells were transfected using 2 μg of GC-responsive element (GRE)-dependent luciferase reporter (Clontech Laboratories Inc., Mountain View, CA) and/or 2 μg of GR mutated at S203 (S203A) (generously provided by Dr. Garabedian, New York University) (32), and/or 2 μg of constitutively active MKK3 constructs (generously provided by Dr. Burow, Tulane University) (33), and/or 100 nM of siRNA against p38 MAPK (SignalSilence p38 MAPK siRNA I; Cell Signaling, Danvers, MA), and/or 2 μg of NF-κb–dependent luciferase reporter (Agilent, Santa Clara, CA), and/or 1 μg of β-galactosidase vector (to normalize transfection efficiency) (Promega, Madison, WI). Controls included pcDNA3 empty vector (Stratagene, La Jolla, CA) and/or scrambled siRNA (Dharmacon, Lafayette, CO). The activities of luciferase and β-galactosidase were evaluated using luciferase and β-galactosidase detection kits (Promega), respectively, according to the manufacturer's instructions (8). The reporter luciferase activities were normalized to β-galactosidase activity and expressed as relative light units.
p38 MAPK Activity
MAPK p38 activities in cells were measured by nonradioactive MAPK-p38 assay kits (Cell Signaling Technology, Danvers, MA) using the protocols recommended by the manufacturer (34). Cells were directly lysed in the culture dishes. Cell lysates were then sonicated and centrifuged at 14,000 × g for 10 minutes at 4°C. The supernatant containing equivalent amounts of protein (260 μg) was incubated by gentle rocking with 20 μl of immobilized phospho-p38 MAPK monoclonal antibody overnight at 4°C. The immunoprecipitates were washed twice with the lysis buffer and pelleted by centrifugation. The p38 MAPK assay was performed using ATF-2 fusion protein (1 μg) as a substrate in the presence of 200 μM ATP and 1× kinase buffer following the manufacturer’s recommendation. Samples were resolved on 4 to 12% SDS-PAGE gel and visualized by chemiluminescence.
Phosphorylation of GR In Vitro
GR phosphorylation in vitro was performed as previously described (13) with the following modifications: (1) we used immune-purified GR from untreated-ASM cells, (2) only active p38 MAPK was used (Upstate, Lake Placid, NY), (3) samples were incubated in a 30°C water bath and harvested after 2 hours, and (4) the membranes were probed with anti–GR-S203, anti–GR-S211, or anti–GR-S226 specific antibodies. Proteins were prepared and assayed by Western blot analysis as described above.
Real-Time RT-PCR
Real-time RT-PCR was performed as we previously described (8) using GC-induced leucine zipper (GILZ) and GAPDH-specific primers (35) (Sigma, St. Louis, MO).
Materials and Reagents
Tissue culture reagents were obtained from Hyclone (Logan, UT). p38 MAPK inhibitor (SB203580) and fluticasone propionate (FP) were purchased from Sigma. Human recombinant TNF-α was provided by Roche Diagnostics (Indianapolis, IN).
Statistical Analysis
Data points from individual assays represent the mean values of triplicate measurements. Significant differences among groups were assessed with ANOVA (Bonferroni-Dunn test) or by t test, with values of P < 0.05 sufficient to reject the null hypothesis for all analyses. Each set of experiments was performed in triplicate with a minimum of three different human ASM cell lines.
Results
p38 MAPK Differentially Regulates GR Site-Specific Phosphorylation
We first examined the effect of p38 MAPK pathway modulation on GR site-specific phosphorylation in ASM cells. We found that SB203580 treatment increased basal and FP-induced GR-S211 phosphorylation by 50 and 30%, respectively (Figures 1A and 1B). In contrast, SB203580 treatment decreased basal and FP-induced GR-S203 phosphorylation by 50 and 35%, respectively (Figures 1A and 1B). GR-S226 phosphorylation was not affected by SB203580 treatment (Figures 1A and 1B). The level of basal GR phosphorylation on S211 residue, although low, varies between cell lines. In control experiments, we confirmed that SB203580 treatment markedly inhibits p38 MAPK activity, as shown by the significant inhibition of the phosphorylation of ATF2, a substrate for activated p38 MAPK, in basal and TNF-α–treated cells by 95 and 70%, respectively (Figure 1C). The presence of phospho-ATF2 under basal conditions indicates a basal activation of p38 MAPK in ASM cells (Figure 1C). The SB203580 concentration used in our experiments did not affect ERK and JNK phosphorylation or NF-κB activity (see Figure E1 in the online supplement).
Figure 1.

Effect of p38 mitogen-activated protein kinase (MAPK) inhibition on glucocorticoid receptor (GR) site-specific phosphorylation. (A) Top: Cells were pretreated with SB203580 (10 μM) for 1 hour before fluticasone propionate (FP) was added for an additional 2 hours. Cells were lysed, and whole cell lysate extracts were prepared and assayed for serine (S)203, S211, S226, GR, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) by immunoblot analysis. Results are representative of three separate blots. (B) Scanning densitometry of three representative immunoblots with each condition normalized over the area density of the corresponding GAPDH content. The results are expressed as the fold increase over basal values. *P < 0.05 compared with untreated cells; **P < 0.01 compared with untreated cells; ***P < 0.001 compared with untreated cells; #P < 0.05 compared with untreated cells; &P < 0.05 compared with fluticasone-treated cells. NS = not significant. (C) Cells were pretreated with SB203580 (10 μM) for 1 hour before TNF-α (10 ng/ml) was added for an additional 15 minutes. Cells were lysed, and whole cell lysate extracts were prepared and assayed for phospho-ATF2 (pATF2) or total ATF2 and GAPDH by immunoblot analysis. Results are representative of three separate blots. (D) Cells were transfected for 48 hours with 100 nM p38 MAPK small interfering RNA (siRNA) or control siRNA before FP was added for an additional 2 hours. Cells were lysed, and whole cell lysate extracts were prepared and assayed for S203, S211, S226, p38, GR, and GAPDH by immunoblot analysis. Results are representative of three separate blots. (E) Scanning densitometry of three representative immunoblots with each condition normalized over the area density of the corresponding GAPDH content. *P < 0.05 compared with untreated cells transfected with control siRNA; #P < 0.05 compared with untreated cells transfected with control siRNA; &P < 0.05 compared with FP-treated cells transfected with control siRNA.
Similar results were found when p38 MAPK was inhibited using silencing strategy (siRNA). Basal and FP-induced GR-S211 phosphorylation was augmented by 36 ± 1.93% and 47 ± 1.86%, respectively (Figures 1D and 1E) in siRNA p38 MAPK–transfected cells when compared with siRNA control–transfected cells. Inversely, basal and FP-induced GR-S203 phosphorylation were decreased by 45 ± 1.80% and 48 ± 6.5%, respectively (Figures 1D and 1E). GR-S226 phosphorylation remains unaffected by the p38 MAPK silencing (Figures 1D and 1E). In a control experiment, we found that p38 siRNA targeted specifically p38α and p38β isoforms (Figure E2).
Together, these data suggest that the basal p38 MAPK pathway differentially affects GR phosphorylation in a site-specific manner.
Basal p38 MAPK Negatively Regulates GR-Mediated Transactivation Activities
We next investigated whether the modulatory effects of p38 MAPK pathway on GR site-specific phosphorylation (Figure 1) was associated with an alteration of GR-mediated transactivation activities. To this end, GR-dependent gene transcription and the expression of a well-defined GC-inducible gene, GILZ, were assessed. Treatment with SB203580 significantly increased basal and FP-induced GRE reporter activities by 115 ± 2.8 and 95 ± 2.2%, respectively (Figure 2A, top). Similarly, real-time PCR analysis showed that treatment with SB203580 significantly increased basal and FP-induced GILZ mRNA expression by 89 ± 1.95% and 59 ± 1.79%, respectively (Figure 2A, bottom). Similar results were found when p38 MAPK was inhibited using silencing strategy (siRNA). Basal and FP-induced GRE reporter activity were increased in siRNA p38 MAPK–transfected cells by 130 ± 3.14% and 79 ± 1.84%, respectively, when compared with siRNA control–transfected cells (Figure 2B, top). Similarly, basal and FP-induced GILZ mRNA expression levels were increased in siRNA p38 MAPK–transfected cells by 69 ± 2.72% and 49 ± 1.23%, respectively, when compared with siRNA control–transfected cells (Figure 2B, bottom). Expression levels of p38 MAPK protein were down-regulated in siRNA p38 MAPK–transfected cells but not in siRNA control–transfected cells (Figure 2C). We next asked whether ectopic activation of the p38 MAPK pathway would similarly modulate GR function by transfecting cells with constitutively active MKK3 construct, a specific kinase that directly activates p38 MAPK (17–19, 33). MKK3 overexpression dramatically reduced basal and FP-induced GRE reporter activity by 53 ± 1.32% and 68 ± 1.63%, respectively, when compared with cells transfected with an empty vector (Figure 2D, top). Similarly, MKK3 overexpression decreased basal and FP-induced GILZ mRNA expression by 38 ± 0.95% and 45 ± 1.08%, respectively, when compared with cells transfected with an empty vector (Figure 2D, bottom). MKK3 overexpression significantly increased MKK3 protein levels (Figure 2E, top gel); this increase was paralleled by an enhanced phosphorylation of p38 MAPK (Figure 2E, middle gel). Collectively, these data suggest that the modulatory effects of p38 MAPK on GR site-specific phosphorylation seen in Figure 1 were associated with an impairment of GR-mediated transactivation activities (Figure 2).
Figure 2.


Effect of p38 pathway modulation on GR-mediated transactivation activities. (A, B, and D, top) Cells were transfected for 48 hours with 2 μg luciferase reporter plasmid driven by glucocorticoid-responsive element (GRE) motifs and 1 μg of β-galactosidase vector (A) and/or 100 nM p38 MAPK siRNA or control siRNA (B) and/or 2 μg MKK3 construct or the corresponding empty vector (D). Cells were pretreated with FP alone (100 nM) for 2 hours (A, B, and D) or with SB203580 (10 μM) for 1 hour before FP addition. Cells were lysed, and the luciferase activity was measured as described in Materials and Methods. The results are expressed as relative light unit (RLU). Data are representative of three separate experiments. (A, B, D, bottom) ASM cells were left untransfected (A) or transfected for 48 hours with 100 nM p38 MAPK siRNA or control siRNA (B) and/or 2 μg MKK3 construct or the corresponding empty vector (D). Cells were treated as above. Total mRNA was analyzed by real-time PCR. The values indicate expression of the steroid-inducible gene, glucocorticoid-induced leucine zipper (GILZ), normalized to GAPDH mRNA levels and presented relative to the expression of basal cells, which was set as 1. Data are representative of three separate experiments. *P < 0.05 compared with untreated cells (A) or with siRNA control–transfected cells (B) or with empty vector–transfected cells (D). **P < 0.01 compared with untreated cells (A), with siRNA control-transfected cells (B), or with empty vector–transfected cells (D). #P < 0.05 compared with empty vector–transfected cells left untreated (D). δP < 0.05 compared with empty vector–transfected cells treated with FP alone (D). (C, E) Cells were transfected for 48 hours with 100 nM p38 MAPK siRNA or control siRNA (C) or with 2 μg MKK3 construct or the corresponding empty vector (E) before FP (100 nM) was added for 2 hours. Cells were lysed, and whole cell lysate extracts were prepared and assayed for p38 MAPK and GAPDH (C) or MKK3, phospho-p38 MAPK, and total p38 MAPK (E) by immunoblot analysis. Results are representative of three separate blots.
p38 MAPK Activation Interferes with GR Nuclear Translocation
We further investigated whether the ability of p38 MAPK activation to impair GR-mediated transactivation activities (Figure 2) was due to a defect in GR nuclear translocation. Immunoblot analysis (Figure 3) showed that 2 hours of treatment with FP increased GR nuclear translocation, an effect that was further increased by 85 ± 2.83% when SB203580 was added 1 hour before. In the absence of FP, SB203580 treatment alone was able to significantly promote GR nuclear translocation by 150 ± 1.08%. Similar results were found when p38 MAPK was inhibited using silencing strategy (siRNA), although to lesser extent. Indeed, we found that GR nuclear translocation was increased in the absence or presence of the FP by 56 ± 3.1% and 39 ± 6.51%, respectively, in siRNA p38–transfected cells when compared with untransfected cells (Figure 3B). Taken together, these data suggest that p38 MAPK inhibition increases GR nuclear translocation.
Figure 3.

Effect of p38 MAPK inhibition on GR subcellular trafficking. (A) Top: Cells were treated with FP alone (100 nM) for 2 hours in the presence or absence of SB203580 (10 μM) added 1 hour before. (B) Top: Cells were transfected for 48 hours with 100 nM p38 siRNA or control siRNA before FP was added for an additional 2 hours. Cells were lysed, and cytosolic and nuclear extracts were prepared and assayed for GR and GAPDH by immunoblot analysis. Results are representative of three separate blots. Bottom: Scanning densitometry of three representative immunoblots with each condition normalized over the area density of the corresponding GAPDH content. The results are expressed as the fold increase over basal values. *P < 0.05 compared with untreated cells (A) or with untreated cells transfected with control siRNA (B). **P < 0.01 compared with untreated cells (A) or with untreated cells transfected with control siRNA (B). #P < 0.05 compared with untreated cells (A).
Critical Role of S203 Residues on GR Functions
The negative role of p38 MAPK on GR functions (Figure 2) was associated with an increase of GR phosphorylation at S203 residues (Figure 1). To test whether GR phosphorylation at S203 negatively regulates GR functions, we investigated whether reducing levels of GR phosphorylation at S203 residues enhances GR functions by transfecting cells with S203A, a nonphosphorylated mutant GR (32). Basal and FP-induced GRE-reporter activities were significantly increased in S203A-transfected cells by 73 ± 1.74% and 54 ± 1.18%, respectively, when compared with empty vector–transfected cells (Figure 4A, top). Similarly, real-time PCR analysis showed that basal and FP-induced GILZ mRNA expression was significantly increased in S203A-transfected cells by 56 ± 1.32% and 42 ± 0.94%, respectively, when compared with empty vector–transfected cells (Figure 4A, bottom). We further investigated whether the increase of GR-mediated transactivation activities observed in S203A-transfected cells was due to an increase of GR nuclear translocation. Basal and FP-induced GR nuclear translocation was significantly elevated in S203A-transfected cells by 120 ± 1.56% and 85 ± 1.75%, respectively, when compared with empty vector–transfected cells (Figure 4B). In control experiments, we examined the levels of GR site-specific phosphorylation in S203A-transfected cells. As expected, cell transfection with the GR-S203A mutant dramatically reduced GR-S203 phosphorylation (Figure 4C) while enhancing GR-S211 phosphorylation (Figure 4C). No change in GR-S226 phosphorylation was observed. Together, these data suggest that phosphorylation of GR at S203 residues negatively regulates GR transcriptional function.
Figure 4.
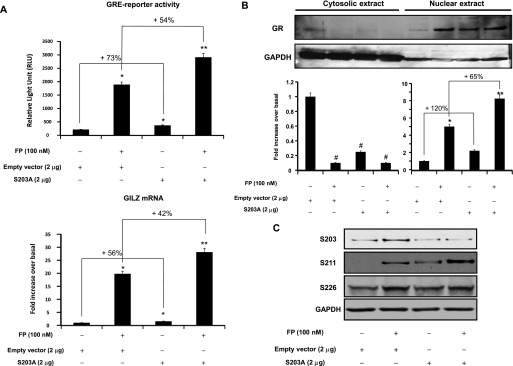
Overexpression of GR S203A mutant enhances GR-mediated transactivation activities and GR nuclear translocation. (A) Top: Cells were cotransfected for 48 hours with 2 μg of pcDNA3 empty vector or GR S203A mutant and 2 μg luciferase reporter plasmid driven by GRE motifs and 1 μg of β-galactosidase vector. Cells were treated with FP (100 nM) for 2 hours. Cells were lysed, and the luciferase activity was measured as described in Materials and Methods. The results are expressed as relative light unit. Data are representative of three separate experiments. Bottom: Cells were transfected for 48 hours with 2 μg of pcDNA3 empty vector or GR S203A mutant. Cells were treated as above. Total mRNA was analyzed by real-time PCR. The values indicate expression of the steroid-inducible gene, GILZ, normalized to GAPDH mRNA levels and presented relative to the expression of basal cells, was set as 1. Data are representative of three separate experiments. *P < 0.05 compared with untreated cells transfected with empty vector. **P < 0.01 compared with untreated cells transfected with empty vector. (B) Top: Cells were transfected and treated as above. Cells were lysed, and cytosolic and nuclear extracts were prepared and assayed for GR and GAPDH by immunoblot analysis. Results are representative of three separate blots. Bottom: Scanning densitometry of three representative immunoblots with each condition normalized over the area density of the corresponding GAPDH content. The results are expressed as the fold increase over basal values. *P < 0.05 compared with untreated cells in nuclear fraction Western blots. **P < 0.01 compared with untreated cells in nuclear fraction Western blots. #P < 0.05 compared with untreated cells in cytosolic fraction Western blots. (C) Cells were transfected and treated as above. Cells were lysed, and whole cell lysates were prepared and assayed for S203, S211, S226, GR, and GAPDH by immunoblot analysis. Results are representative of three separate blots.
p38 MAPK Phosphorylates GR at S203 Residues In Vitro
Because p38 MAPK regulates GR-S203 phosphorylation (Figure 1), we examined whether S203 residues of GR were a substrate for p38 MAPK. In vitro phosphorylation of GR was assayed using active p38 MAPK and immunopurified GR from untreated ASM cells. As expected, no basal GR phosphorylation was detected at S211 residues (Figure 5). A basal GR phosphorylation was detected at S226 and S203 residues. Although the addition of active p38 MAPK did not affect GR-S226 phosphorylation, it significantly increased GR-S203 phosphorylation by 1.85 ± 0.27-fold (Figure 5). In control experiments, buffer-only (unphosphorylated) samples and samples with calf intestine alkaline phosphatase added yielded no positive antibody reactions (data not shown). Together, these data indicate that S203 residues of GR are a substrate for p38 MAPK.
Figure 5.

p38 MAPK phosphorylates GR at S203 residues in vitro. Protein phosphorylation was performed as described in Materials and Methods. Top: Immunoprecipitated GR was incubated with assay buffer alone in the presence or absence of active p38 MAPK or calf intestine alkaline phosphatase. An ATP/Mg++ cocktail was added to all samples except the buffer control. Proteins were extracted and assayed for GR-S203, GR-S211, GR-S226, and total GR by immunoblot analysis. Data are representative of three separate experiments. Bottom: Scanning densitometry of three representative GR-S203 immunoblots with each condition normalized over the area density of the corresponding GR content. The results are expressed as the fold increase over basal values. *P < 0.05 compared with untreated cells.
GR Phosphorylation at S203 Residues Mediates p38 MAPK Inhibitory Effects on GR-Mediated Transactivation Activities
Because GR-S203 phosphorylation inhibits GR-mediated transactivation activities (Figure 4), we examined whether the negative role of p38 MAPK on GR-mediated transactivation activities (Figure 2) was mediated by GR phosphorylation at S203 residues (Figure 5) by investigating whether p38 MAPK inhibition is still able to increase GR-mediated transactivation activities in S203A-transfected cells. Although GRE-reporter activities and GILZ mRNA expression levels were significantly increased in S203A-transfected cells compared with empty vector–transfected cells irrespective of FP treatment, the addition of SB203580 failed to further increase such GR functions (Figure 6A). To further validate the above findings, p38 MAPK siRNA strategy was used to investigate whether p38 MAPK siRNA was still able to increase GR functions in S203A-transfected cells. Although GRE reporter activities and GILZ mRNA expression levels were significantly increased in S203A-transfected cells compared with empty vector–transfected cells irrespective of FP treatment, silencing p38 MAPK in S203A-transfected cells failed to further increase such GR functions (Figure 6B). To further confirm the latter siRNA p38 MAPK results, we examined whether ectopic activation of the p38 MAPK pathway inhibits GR function in S203A-transfected cells. Although FP-induced GRE reporter activity and GILZ mRNA expression levels were significantly increased in S203A-transfected cells compared with nontransfected cells treated with FP, MKK3 overexpression failed to inhibit GR functions in S203A-transfected cells when compared with empty vector–transfected cells treated with FP (Figure 6C). Together, our data suggest that p38 MAPK–induced impairment of GR transactivation activities is mediated, at least partially, by GR phosphorylation at S203 residues.
Figure 6.

Effect of p38 MAPK pathway modulation on GR-mediated transactivation activities in S203A-transfected cells. Top: Cells were cotransfected for 48 hours with 2 μg of pcDNA3 empty vector or GR S203A mutant and 2 μg luciferase reporter plasmid driven by GRE motifs and 1 μg of β-galactosidase vector (A) and/or 100 nM p38 MAPK siRNA or control siRNA (B) and/or 2 μg MKK3 construct or the corresponding empty vector (C). Cells were pretreated with FP (100 nM) for 2 hours (A–C) and/or SB203580 (10 μM) for 1 hour before FP addition (A). Cells were lysed, and the luciferase activity was measured as described in Materials and Methods. The results are expressed as RLU. Data are representative of three separate experiments. Bottom: Cells were cotransfected for 48 hours with 2 μg of pcDNA3 empty vector or GR S203A mutant (A) and/or 100 nM p38 MAPK siRNA or control siRNA (B) and/or 2 μg MKK3 construct or the corresponding empty vector (C). Cells were treated as above. Total mRNA was analyzed by real-time PCR. The values indicate expression of the steroid-inducible gene, GILZ, normalized to GAPDH mRNA levels and are presented relative to the expression of basal cells, which was set as 1. Data are representative of three separate experiments. *P < 0.05 compared with untreated cells (A) or with siRNA control–transfected cells (B) or with empty vector–transfected cells (C). **P < 0.01 compared with untreated cells (A) or with siRNA control–transfected cells (B) or with empty vector–transfected cells (C). #P < 0.05 compared with empty vector–transfected cells left untreated (C). δP < 0.05 compared with empty vector–transfected cells treated with FP alone (C). NS = not significant.
p38 MAPK Modulation of GR Signaling Is Independent of PP5
Previously, we showed that PP5 negatively modulates GR functions, and specifically GR phosphorylation, at S211 (8). Because we show here that p38 MAPK also negatively modulates GR functions, we sought to determine whether cross-talk between PP5 and p38 regulates GR functions. To this end, we examined the effect of p38 MAPK and PP5 inhibition alone and in combination on GR site-specific phosphorylation. We found that basal and FP-induced GR-S211 phosphorylation were significantly increased when cells were transfected with p38 siRNA (by 36 ± 1.81% and 47 ± 2.23%, respectively) but not with PP5 siRNA or control siRNA (Figure 7A). We also found that basal and FP-induced GR-S203 phosphorylation was decreased when cells were transfected with p38 siRNA (by 45 ± 2.15% and 47 ± 2.64%, respectively) but not with PP5 siRNA or control siRNA (Figure 7A). GR-S226 phosphorylation was not affected by siRNA p38 MAPK or by siRNA PP5 (Figure 7A). No further effect was observed when cells were transfected by p38 and PP5 siRNAs compared with cells transfected with p38 siRNA alone. We also examined the effect of p38 MAPK and PP5 inhibition alone or in combination on GR transcriptional activities. Cells transfected with p38 siRNA but not with PP5 siRNA or control siRNA showed a significant increase in basal and FP-induced GRE-reporter activities (by 41 ± 2.94% and 40 ± 2.23%, respectively) and GILZ mRNA expression (by 60 ± 1.77% and 57 ± 3.98%, respectively) (Figure 7B). No further effect was observed when cells were transfected by p38 and PP5 siRNAs compared with cells transfected with p38 siRNA alone. Additionally, we performed coimmunoprecipitation studies to determine whether p38 physically interacts with PP5. PP5 did not interact with p38 irrespective of GC treatment (Figure 7C). Together, our data suggest that p38 MAPK and PP5 may modulate GR signaling using different/parallel pathways.
Figure 7.

p38 MAPK and protein phosphatase (PP)5 pathways distinctively modulate GR-mediated transactivation activities. (A) Cells were transfected for 48 hours with 100 nM of p38 siRNA and/or PP5 siRNA or control siRNA before FP (100 nM) was added for 2 hours. Cells were lysed, and whole cell lysate extracts were prepared and assayed for S203, S211, S226, GR, p38 MAPK, PP5, and GAPDH by immunoblot analysis. Results are representative of three separate blots. (B) Top: Cells were transfected for 48 hours with 2 μg luciferase reporter plasmid driven by GRE motifs and 1 μg of β-galactosidase vector and 100 nM of p38 siRNA and/or PP5 siRNA or control siRNA. After that, FP was added for an additional 2 hours (top). Cells were lysed, and the luciferase activity was measured as described in Materials and Methods. The results are expressed as fold increase over basal. Data are representative of three separate experiments. (B) Bottom: ASM cells were transfected for 48 hours with 100 nM of p38 siRNA and/or PP5 siRNA or control siRNA. Cells were treated as above. Total mRNA was analyzed by real-time PCR. The values indicate expression of the steroid-inducible gene, GILZ, normalized to GAPDH mRNA levels and presented relative to the expression of untreated cells, which was set as 1. *P < 0.05 compared with untreated cells transfected with control siRNA. #P < 0.05 compared with FP-treated cells transfected with control siRNA. &P < 0.05 compared with FP-treated cells transfected with PP5 siRNA. (C) Cells were treated with FP (100 nM) for 2 hours and lysed, and whole cell lysate extracts were prepared and immunoprecipitated with anti-p38 or anti-PP5 antibodies and then assayed by immunoblot analysis. Results are representative of three separate blots. Data are representative of three separate experiments.
Discussion
In the present study, we identified p38 MAPK as a novel factor regulating GR key cellular responses in ASM cells, such as GR nuclear translocation and GRE-dependent expression of steroid-inducible genes such as GILZ. To our knowledge, this is the first study to demonstrate a negative role of p38 MAPK on GR functions in human ASM cells and to identify S203 residues as critical phosphorylation sites driving GR function in the absence or presence of GCs.
MAPK pathways are activated by different inflammatory cytokines, growth factors, or environmental stressors (17–19). However, there are examples showing the function of basal activity of MAPKs. Indeed, studies have demonstrated measurable levels of basal JNK, the disruption of which affects key fibroblast functions (36) and cell proliferation of the human carcinoma cell line (KB-3) (37). Basal activity of p38 MAPK was also detected in murine fibroblasts (38). We herein provide the first evidence that basal p38 MAPK activity maintains unliganded GR in an inactive state in ASM cells as evidenced by the increased phosphorylation at S211 (Figure 1) and nuclear translocation of GR (Figure 3) as well as GRE-dependent expression of GILZ (Figure 2) when p38 MAPK was blocked using different experimental strategies. Although the GR activation was originally thought to be GC dependent, some reports have demonstrated evidence of functional GR in the absence of GC. For instance, when overexpressed, unliganded GR has the capacity to bind GRE in vitro (39) and in vivo (40). Recently, Kotitschke and colleagues showed that the transcriptional activation of unliganded GR by gonadotropin-releasing hormone was dependent on MAPK-dependent phosphorylation of GR at S234 residues (the equivalent of S226 in human) (41). We found that S226 was not affected by p38 MAPK blockade, suggesting that GR regulation by MAPK is highly cell specific. In addition to the transactivation properties reported in previous studies, Verhoog and colleagues were the first to report ligand-independent transrepression by the GR that was dependent on phosphorylation of unliganded GR at S226 and recruitment of GR interacting protein-1 as a co-repressor (42). Whether the transcriptional activation of unliganded GR induced by p38 MAPK blockade showed in the present study requires the recruitment of GRIP-1 remains an interesting possibility. Our present study suggests that basal p38 MAPK signaling acts as a required “brake” to maintain unliganded GR in an inactive state. Consistent with this, Braz and colleagues showed that basal p38 MAPK signaling exerts a similar suppressive action on the ability of a different transcription factor, called NFAT, to translocate to the nucleus and to promote cardiac hypertrophic growth (43). Collectively, our findings suggest a role for basal p38 MAPK activity in maintaining different transcription factors, including GR, in the cytosol in an inactive state, although the underlying mechanisms have not been completely elucidated. We also found that a differential effect on GR phosphorylation on serine residues appears to be essential in p38 MAPK regulatory effects.
Our data showed that p38 MAPK directly phosphorylates S203 but not S211 residues (Figure 5), supporting the observation of a reduced GR-phosphorylation at S203 in ASM cells treated with the p38 MAPK inhibitor (Figure 1). This decrease in S203 phosphorylation induced by p38 MAPK blockade was associated with increased GR transcriptional activity as evidenced by the enhanced GILZ expression (Figure 2). The functional consequences of GR phosphorylation at S203 residues on receptor function remain controversial. For instance, earlier studies showed that the mutation of S203 reduced GR-dependent gene transcription (5, 44), suggesting the importance of S203 in GR transcriptional activities. In contrast, the evidence presented here suggests that mutation of GR at S203 residues enhanced GR-dependent transcription in basal and GC-treated cells (Figure 4A). This suggests that GR phosphorylation at S203 serves as a negative regulator of GR transcriptional activity in ASM cells. A similar observation was reported by Kino and colleagues, who found that the GR-S203A mutant dramatically increased GC-induced, GR-mediated transactivation activities in human colorectal carcinoma cell line (HCT116) (45). Other differences found in our study are the fact that GR phosphorylation at S203 was mostly located in the cytoplasm of ASM cells (data not shown) and that the transfection of the mutated form of GR at S203 residues was associated with increased GR nuclear translocation (Figure 4B). Although Takabe and colleagues showed a nuclear localization of GR-S203 phospho-isoform that is reduced by GC treatment with or without treatment with ERK inhibitors in Caco-2 cells (15), Wang and colleagues failed to detect any localization of GR-S203 isoform in the nuclear fractions of U2OS cells (4). Such discrepancies may be due to species-specific differences between murine, rat, and human GR, the reporter system used (MMTV-CAT versus GRE-reporter), and/or the cell lines used, transformed cells lacking endogenous GR (i.e., COS7 and U2OS cells) versus mammalian nontransformed primary cells (i.e., ASM cells). Nonetheless, our study demonstrates that the inactive state of GR in resting conditions is not only ensured by the absence of the GC ligand but also by p38 MAPK activity that regulates the levels of GR phosphorylation, specifically at S203 residues.
Our observation that p38 MAPK regulates GR phosphorylation at S203 combined with the fact that in other cell types p38 MAPK also phosphorylates GR at S211 (13, 46) suggests that this kinase can affect GR function by acting on multiple serine residues. In line with this, we showed that p38 MAPK can suppress GR function by exerting two opposite actions on GR key serine residues, namely directly phosphorylating S203 and interfering with GR phosphorylation at S211. Different groups have shown that GR phosphorylation at S226 is an important inhibitory mechanism of GC function, although some controversy exists regarding the nature of upstream kinases involved (6, 14). For example, in some reports, c-Jun N-terminal kinase (JNK) MAPK–induced GR phosphorylation at S226 was found to inhibit GR function (6, 12), whereas other studies identified p38 MAPK as another potential kinase regulating phosphorylation of GR at S226 and inhibiting its function (11, 23). In contrast to these latter studies, we found that p38 MAPK negatively regulates GR phosphorylation on S211 but had no effect on S226 (Figure 1). These controversial results may be explained by the use of different experimental conditions, such as different cell types used (immune cells in previous studies versus structural cells in the present study), the nature of the cells (transformed cells in previous studies versus primary cell in present study), and the nature of the stimuli used (cytokines in previous studies versus GC only in the present study). Our study completes and extends previous studies showing a negative role of inducible p38 MAPK on GR function in inflammatory conditions (cells treated with IL-2 and IL-4) and demonstrates the negative role of basal p38 on GR functions in the absence of inflammatory stimuli. Additional studies are needed to determine the mechanisms of the differential action of p38 MAPK on GR phosphorylation in ASM cells.
Earlier reports described the concept of intersite dependency where GR phosphorylation at one site may influence the ability of a neighboring site to be modified. This concept of intersite dependency has been shown to occur between S203 and S226 residues (47). Our experiments showed an unexpected relationship among GR phosphorylation sites. We found that p38 MAPK blockade not only leads to a decreased S203 phosphorylation but concomitantly increases S211 phosphorylation to the same extent (30%) (Figures 1B and 2B). Such intersite dependency between S203 and S211 residues was reinforced in studies using GR S203A mutants, which showed a considerable enhancement of phosphorylation at S211, with no effect on GR phosphorylation at S226 (Figure 4C). Although basal GR phosphorylation at S211 in U2OS cells seems to be independent of S203 phosphorylation status (47), basal GR phosphorylation at S211 in ASM cells seems to be influenced by S203 phosphorylation status because mutation of S203 increases basal S211 phosphorylation (Figure 4C). It is conceivable that phosphorylation of GR at S203 residue could result in the recruitment of a phosphatase that prevents hyperphosphorylation at the S211 residues (47). This mechanism is unlikely to be the case here because GC treatment reduces the physical interaction of PP with total GR in ASM cells (8) despite the fact that the physical interaction of PP with specific GR phospho-isoforms was not explored. Alternatively, the intersite dependency might reflect a conformational change induced by phosphorylation at S203 that renders the S211 a poor substrate for a kinase (47). Further investigation is needed to confirm this latter hypothesis in ASM cells.
A number of reports have reinforced the concept that changes in GR phosphorylation status could have a detrimental action on GC-resistant diseases. For instance, in GC-resistant hematological malignancies, restoration of GR phosphorylation at S211 via the inhibition of upstream kinases is associated with restored sensitivity to GC treatment (48). In severe asthma, there appear to be multiple mechanisms that affect GR phosphorylation status, which could explain GC insensitivity seen in these patients. Increased p38 MAPK expression in PBMCs (24) or activity in PBMCs and alveolar macrophages (23, 49) is associated with reduced patient sensitivity to GC therapy, possibly by driving GR phosphorylation on S226. The same group raised the hypothesis that decreased PP2A function could serve as a mechanism leading to GC insensitivity, in part via its failure to dephosphorylate GR on S226 residues (50). Our previous findings showed that increased PP5-dependent GR dephosphorylation at S211 was also associated with impaired GC sensitivity in ASM cells (8). Together, these studies strongly suggest that GR phosphorylation status at different serine residues is instrumental in determining GR activity; thus, an abnormal balance between upstream GR kinase and phosphatase represents one molecular mechanism driving GC insensitivity seen in different human conditions (11, 20, 23, 24).
In summary, our current study provides the first demonstration of a potential role of “constitutive” p38 MAPK activity in regulating GC responsiveness in ASM cells via the regulation of GR transactivation activities through direct and indirect regulation of GR phosphorylation at S203 and S211 residues, respectively, in a ligand-independent manner. A better understanding of the negative actions of p38 MAPK on GR functions could help in the design of novel drugs that will act as steroid-sparing agents, especially in patients requiring high doses of GC therapy.
Footnotes
This work was supported by National Institutes of Health grant R01 HL111541 (O.T.) and by the National Institute for Health Research Leicester Respiratory Biomedical Research (Y.A.).
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2012-0522OC on September 11, 2013
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Ismaili N, Garabedian MJ. Modulation of glucocorticoid receptor function via phosphorylation. Ann N Y Acad Sci. 2004;1024:86–101. doi: 10.1196/annals.1321.007. [DOI] [PubMed] [Google Scholar]
- 2.Godowski PJ, Rusconi S, Miesfeld R, Yamamoto KR. Glucocorticoid receptor mutants that are constitutive activators of transcriptional enhancement. Nature. 1987;325:365–368. doi: 10.1038/325365a0. [DOI] [PubMed] [Google Scholar]
- 3.Hollenberg SM, Evans RM. Multiple and cooperative trans-activation domains of the human glucocorticoid receptor. Cell. 1988;55:899–906. doi: 10.1016/0092-8674(88)90145-6. [DOI] [PubMed] [Google Scholar]
- 4.Wang Z, Frederick J, Garabedian MJ. Deciphering the phosphorylation “code” of the glucocorticoid receptor in vivo. J Biol Chem. 2002;277:26573–26580. doi: 10.1074/jbc.M110530200. [DOI] [PubMed] [Google Scholar]
- 5.Almlof T, Wright AP, Gustafsson JA. Role of acidic and phosphorylated residues in gene activation by the glucocorticoid receptor. J Biol Chem. 1995;270:17535–17540. doi: 10.1074/jbc.270.29.17535. [DOI] [PubMed] [Google Scholar]
- 6.Chen W, Dang T, Blind RD, Wang Z, Cavasotto CN, Hittelman AB, Rogatsky I, Logan SK, Garabedian MJ. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 2008;22:1754–1766. doi: 10.1210/me.2007-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar R, Calhoun WJ. Differential regulation of the transcriptional activity of the glucocorticoid receptor through site-specific phosphorylation. Biologics. 2008;2:845–854. doi: 10.2147/btt.s3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bouazza B, Krytska K, Debba-Pavard M, Amrani Y, Honkanen RE, Tran J, Tliba O. Cytokines alter glucocorticoid receptor phosphorylation in airway cells: role of phosphatases. Am J Respir Cell Mol Biol. 2012;47:464–473. doi: 10.1165/rcmb.2011-0364OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duma D, Jewell CM, Cidlowski JA. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J Steroid Biochem Mol Biol. 2006;102:11–21. doi: 10.1016/j.jsbmb.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 10.Weigel NL, Moore NL. Steroid receptor phosphorylation: a key modulator of multiple receptor functions. Mol Endocrinol. 2007;21:2311–2319. doi: 10.1210/me.2007-0101. [DOI] [PubMed] [Google Scholar]
- 11.Irusen E, Matthews JG, Takahashi A, Barnes PJ, Chung KF, Adcock IM. P38 mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: role in steroid-insensitive asthma. J Allergy Clin Immunol. 2002;109:649–657. doi: 10.1067/mai.2002.122465. [DOI] [PubMed] [Google Scholar]
- 12.Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, Imai K. Nuclear export of glucocorticoid receptor is enhanced by c-jun n-terminal kinase-mediated phosphorylation. Mol Endocrinol. 2002;16:2382–2392. doi: 10.1210/me.2002-0144. [DOI] [PubMed] [Google Scholar]
- 13.Miller AL, Webb MS, Copik AJ, Wang Y, Johnson BH, Kumar R, Thompson EB. p38 mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol Endocrinol. 2005;19:1569–1583. doi: 10.1210/me.2004-0528. [DOI] [PubMed] [Google Scholar]
- 14.Rogatsky I, Logan SK, Garabedian MJ. Antagonism of glucocorticoid receptor transcriptional activation by the c-Jun N-terminal kinase. Proc Natl Acad Sci USA. 1998;95:2050–2055. doi: 10.1073/pnas.95.5.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takabe S, Mochizuki K, Goda T. De-phosphorylation of GR at ser203 in nuclei associates with GR nuclear translocation and glut5 gene expression in caco-2 cells. Arch Biochem Biophys. 2008;475:1–6. doi: 10.1016/j.abb.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka T, Okabe T, Gondo S, Fukuda M, Yamamoto M, Umemura T, Tani K, Nomura M, Goto K, Yanase T, et al. Modification of glucocorticoid sensitivity by MAP kinase signaling pathways in glucocorticoid-induced T-cell apoptosis. Exp Hematol. 2006;34:1542–1552. doi: 10.1016/j.exphem.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 17.Adcock IM, Chung KF, Caramori G, Ito K. Kinase inhibitors and airway inflammation. Eur J Pharmacol. 2006;533:118–132. doi: 10.1016/j.ejphar.2005.12.054. [DOI] [PubMed] [Google Scholar]
- 18.Kumar S, Boehm J, Lee JC. p38 map kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov. 2003;2:717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- 19.Saklatvala J. The p38 map kinase pathway as a therapeutic target in inflammatory disease. Curr Opin Pharmacol. 2004;4:372–377. doi: 10.1016/j.coph.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 20.Bhavsar P, Khorasani N, Hew M, Johnson M, Chung KF. Effect of p38 MAPK inhibition on corticosteroid suppression of cytokine release in severe asthma. Eur Respir J. 2010;35:750–756. doi: 10.1183/09031936.00071309. [DOI] [PubMed] [Google Scholar]
- 21.Chang PJ, Bhavsar PK, Michaeloudes C, Khorasani N, Chung KF.Corticosteroid insensitivity of chemokine expression in airway smooth muscle of patients with severe asthma J Allergy Clin Immunol 2012130877–885.e875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung KF. p38 mitogen-activated protein kinase pathways in asthma and COPD. Chest. 2011;139:1470–1479. doi: 10.1378/chest.10-1914. [DOI] [PubMed] [Google Scholar]
- 23.Mercado N, Hakim A, Kobayashi Y, Meah S, Usmani OS, Chung KF, Barnes PJ, Ito K. Restoration of corticosteroid sensitivity by p38 mitogen activated protein kinase inhibition in peripheral blood mononuclear cells from severe asthma. PLoS ONE. 2012;7:e41582. doi: 10.1371/journal.pone.0041582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mercado N, To Y, Kobayashi Y, Adcock IM, Barnes PJ, Ito K. p38 mitogen-activated protein kinase-gamma inhibition by long-acting beta2 adrenergic agonists reversed steroid insensitivity in severe asthma. Mol Pharmacol. 2011;80:1128–1135. doi: 10.1124/mol.111.071993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D’Elia M, Patenaude J, Dupras C, Bernier J. T cells from burn-injured mice demonstrate a loss of sensitivity to glucocorticoids. Am J Physiol Endocrinol Metab. 2010;299:E299–E307. doi: 10.1152/ajpendo.00084.2010. [DOI] [PubMed] [Google Scholar]
- 26.Bantel H, Schmitz ML, Raible A, Gregor M, Schulze-Osthoff K. Critical role of NF-kappaB and stress-activated protein kinases in steroid unresponsiveness. FASEB J. 2002;16:1832–1834. doi: 10.1096/fj.02-0223fje. [DOI] [PubMed] [Google Scholar]
- 27.Hommes D, van den Blink B, Plasse T, Bartelsman J, Xu C, Macpherson B, Tytgat G, Peppelenbosch M, Van Deventer S. Inhibition of stress-activated MAP kinases induces clinical improvement in moderate to severe Crohn’s disease. Gastroenterology. 2002;122:7–14. doi: 10.1053/gast.2002.30770. [DOI] [PubMed] [Google Scholar]
- 28.Cohen P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr Opin Cell Biol. 2009;21:317–324. doi: 10.1016/j.ceb.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 29.Cohen SB, Cheng TT, Chindalore V, Damjanov N, Burgos-Vargas R, Delora P, Zimany K, Travers H, Caulfield JP. Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study of patients with active rheumatoid arthritis. Arthritis Rheum. 2009;60:335–344. doi: 10.1002/art.24266. [DOI] [PubMed] [Google Scholar]
- 30.Panettieri RA, Murray RK, DePalo LR, Yadvish PA, Kotlikoff MI. A human airway smooth muscle cell line that retains physiological responsiveness. Am J Physiol. 1989;256:C329–C335. doi: 10.1152/ajpcell.1989.256.2.C329. [DOI] [PubMed] [Google Scholar]
- 31.Tliba O, Damera G, Banerjee A, Gu S, Baidouri H, Keslacy S, Amrani Y. Cytokines induce an early steroid resistance in airway smooth muscle cells: novel role of interferon regulatory factor-1. Am J Respir Cell Mol Biol. 2008;38:463–472. doi: 10.1165/rcmb.2007-0226OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ismaili N, Blind R, Garabedian MJ. Stabilization of the unliganded glucocorticoid receptor by TSG101. J Biol Chem. 2005;280:11120–11126. doi: 10.1074/jbc.M500059200. [DOI] [PubMed] [Google Scholar]
- 33.Frigo DE, Basu A, Nierth-Simpson EN, Weldon CB, Dugan CM, Elliott S, Collins-Burow BM, Salvo VA, Zhu Y, Melnik LI, et al. p38 mitogen-activated protein kinase stimulates estrogen-mediated transcription and proliferation through the phosphorylation and potentiation of the p160 coactivator glucocorticoid receptor-interacting protein 1. Mol Endocrinol. 2006;20:971–983. doi: 10.1210/me.2004-0075. [DOI] [PubMed] [Google Scholar]
- 34.Kuo CH, Lin CH, Yang SN, Huang MY, Chen HL, Kuo PL, Hsu YL, Huang SK, Jong YJ, Wei WJ, et al. Effect of prostaglandin I2 analogs on cytokine expression in human myeloid dendritic cells via epigenetic regulation. Mol Med. 2012;18:433–444. doi: 10.2119/molmed.2011.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen W, Rogatsky I, Garabedian MJ. Med14 and Med1 differentially regulate target-specific gene activation by the glucocorticoid receptor. Mol Endocrinol. 2006;20:560–572. doi: 10.1210/me.2005-0318. [DOI] [PubMed] [Google Scholar]
- 36.Javelaud D, Laboureau J, Gabison E, Verrecchia F, Mauviel A. Disruption of basal JNK activity differentially affects key fibroblast functions important for wound healing. J Biol Chem. 2003;278:24624–24628. doi: 10.1074/jbc.M301942200. [DOI] [PubMed] [Google Scholar]
- 37.Du L, Lyle CS, Obey TB, Gaarde WA, Muir JA, Bennett BL, Chambers TC. Inhibition of cell proliferation and cell cycle progression by specific inhibition of basal JNK activity: evidence that mitotic Bcl-2 phosphorylation is JNK-independent. J Biol Chem. 2004;279:11957–11966. doi: 10.1074/jbc.M304935200. [DOI] [PubMed] [Google Scholar]
- 38.Wang X, Wu H, Miller AH. Interleukin 1alpha (IL-1alpha) induced activation of p38 mitogen-activated protein kinase inhibits glucocorticoid receptor function. Mol Psychiatry. 2004;9:65–75. doi: 10.1038/sj.mp.4001339. [DOI] [PubMed] [Google Scholar]
- 39.Srinivasan G, Patel NT, Thompson EB. Heat shock protein is tightly associated with the recombinant human glucocorticoid receptor:glucocorticoid response element complex. Mol Endocrinol. 1994;8:189–196. doi: 10.1210/mend.8.2.8170475. [DOI] [PubMed] [Google Scholar]
- 40.Siriani D, Mitsiou DJ, Alexis MN. Overexpressed glucocorticoid receptor negatively regulates gene expression under conditions that favour accumulation of non-hormone-binding forms of the receptor. J Steroid Biochem Mol Biol. 2003;84:171–180. doi: 10.1016/s0960-0760(03)00027-x. [DOI] [PubMed] [Google Scholar]
- 41.Kotitschke A, Sadie-Van Gijsen H, Avenant C, Fernandes S, Hapgood JP. Genomic and nongenomic cross talk between the gonadotropin-releasing hormone receptor and glucocorticoid receptor signaling pathways. Mol Endocrinol. 2009;23:1726–1745. doi: 10.1210/me.2008-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verhoog NJ, Du Toit A, Avenant C, Hapgood JP. Glucocorticoid-independent repression of tumor necrosis factor (TNF) alpha-stimulated interleukin (IL)-6 expression by the glucocorticoid receptor: a potential mechanism for protection against an excessive inflammatory response. J Biol Chem. 2011;286:19297–19310. doi: 10.1074/jbc.M110.193672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, Braunwart J, Glascock BJ, Klevitsky R, Kimball TF, et al. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest. 2003;111:1475–1486. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Webster JC, Jewell CM, Bodwell JE, Munck A, Sar M, Cidlowski JA. Mouse glucocorticoid receptor phosphorylation status influences multiple functions of the receptor protein. J Biol Chem. 1997;272:9287–9293. doi: 10.1074/jbc.272.14.9287. [DOI] [PubMed] [Google Scholar]
- 45.Kino T, Ichijo T, Amin ND, Kesavapany S, Wang Y, Kim N, Rao S, Player A, Zheng YL, Garabedian MJ, et al. Cyclin-dependent kinase 5 differentially regulates the transcriptional activity of the glucocorticoid receptor through phosphorylation: clinical implications for the nervous system response to glucocorticoids and stress. Mol Endocrinol. 2007;21:1552–1568. doi: 10.1210/me.2006-0345. [DOI] [PubMed] [Google Scholar]
- 46.Nader N, Ng SS, Lambrou GI, Pervanidou P, Wang Y, Chrousos GP, Kino T. AMPK regulates metabolic actions of glucocorticoids by phosphorylating the glucocorticoid receptor through p38 MAPK. Mol Endocrinol. 2010;24:1748–1764. doi: 10.1210/me.2010-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z, Chen W, Kono E, Dang T, Garabedian MJ. Modulation of glucocorticoid receptor phosphorylation and transcriptional activity by a c-terminal-associated protein phosphatase. Mol Endocrinol. 2007;21:625–634. doi: 10.1210/me.2005-0338. [DOI] [PubMed] [Google Scholar]
- 48.Garza AS, Miller AL, Johnson BH, Thompson EB. Converting cell lines representing hematological malignancies from glucocorticoid-resistant to glucocorticoid-sensitive: signaling pathway interactions. Leuk Res. 2009;33:717–727. doi: 10.1016/j.leukres.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhavsar P, Hew M, Khorasani N, Torrego A, Barnes PJ, Adcock I, Chung KF. Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax. 2008;63:784–790. doi: 10.1136/thx.2007.090027. [DOI] [PubMed] [Google Scholar]
- 50.Kobayashi Y, Mercado N, Barnes PJ, Ito K. Defects of protein phosphatase 2a causes corticosteroid insensitivity in severe asthma. PLoS ONE. 2011;6:e27627. doi: 10.1371/journal.pone.0027627. [DOI] [PMC free article] [PubMed] [Google Scholar]