

Abstract
Supplemental oxygen is frequently used in an attempt to improve oxygen delivery; however, prolonged exposure results in damage to the pulmonary endothelium and epithelium. Although NF-κB has been identified as a redox-responsive transcription factor, whether NF-κB activation exacerbates or attenuates hyperoxic lung injury is unclear. We determined that sustained NF-κB activity mediated by IκBβ attenuates lung injury and prevents mortality in adult mice exposed to greater than 95% O2. Adult wild-type mice demonstrated evidence of alveolar protein leak and 100% mortality by 6 days of hyperoxic exposure, and showed NF-κB nuclear translocation that terminated after 48 hours. Furthermore, these mice showed increased expression of NF-κB–regulated proinflammatory and proapoptotic cytokines. In contrast, mice overexpressing the NF-κB inhibitory protein, IκBβ (AKBI), demonstrated significant resistance to hyperoxic lung injury, with 50% surviving through 8 days of exposure. This was associated with NF-κB nuclear translocation that persisted through 96 hours of exposure. Although induction of NF-κB–regulated proinflammatory cytokines was not different between wild-type and AKBI mice, significant up-regulation of antiapoptotic proteins (BCL-2, BCL-XL) was found exclusively in AKBI mice. We conclude that sustained NF-κB activity mediated by IκBβ protects against hyperoxic lung injury through increased expression of antiapoptotic genes.
Keywords: NF-κB, IκB, hyperoxic lung injury, apoptosis, inflammation
Clinical Relevance
Oxygen therapy is commonly employed in an attempt to improve oxygen delivery, and yet prolonged exposure results in lung injury. No pharmacologic strategies to attenuate hyperoxic lung injury have been identified. We show that enhancing hyperoxia-induced NF-κB activity attenuates lung injury, and thus represents a potential therapeutic target.
The first studies demonstrating the lethal effect of inspiring high concentrations of oxygen were published over a century ago (1). Due to this toxicity, hyperoxic exposure has been widely used in animal studies to induce and study acute lung injury (2). Despite this, no pharmacologic therapies to attenuate hyperoxic lung injury have been identified.
Hyperoxia induces proinflammatory cytokine expression (3), and results in pulmonary endothelial and epithelial cell death (4). Given these facts, both anti-inflammatory and antiapoptotic strategies have been employed to attenuate hyperoxic lung injury. Although anti-inflammatory interventions have met variable success (5–9), increasing the expression of antiapoptotic factors in transgenic murine models results in attenuated hyperoxic lung injury (10–12). These studies suggest that preventing hyperoxia-induced apoptosis can prevent lung injury, but the transcriptional mechanism mediating expression of these factors is largely unknown.
Hyperoxia-induced NF-κB activation has been documented in both pulmonary epithelial and endothelial cells (13). Whether this response is protective or injurious is unclear. Some in vivo studies suggest that intact NF-κB signaling confers protection against hyperoxia-induced lung injury (14, 15), whereas inhibition of oxidant stress-induced NF-κB activity exacerbates cell death (16–19). In contrast, NF-κB activity can potentiate a proinflammatory response to hyperoxia (20). Overall, the role of NF-κB in modulating the pulmonary response to hyperoxia is complicated. For example, NF-κB confers a protective response to oxidative stress through increased expression of antioxidant enzymes (e.g., manganese superoxide dismutase [MnSOD], glutathione peroxidase]), anti-inflammatory cytokines (IL-6, IL-11), and antiapoptotic genes (21). However, NF-κB may exacerbate lung injury through expression of both pro-oxidant enzymes (e.g., cyclo-oxygenase [COX]-2, reduced nicotinamide adenine dinucleotide phosphate oxidase NOX2 [gp91 phox]), and proinflammatory proteins (e.g., IL-1α, CD40, CXCL1, and intercellular adhesion molecule [ICAM]-1) (21). It is important to clarify whether NF-κB is cytoprotective or detrimental in hyperoxia to devise therapeutic strategies to obviate lung injury.
The penultimate step of NF-κB nuclear translocation and DNA binding involves the inhibitory proteins, IκBα and IκBβ (22). In quiescent cells, IκB proteins sequester NF-κB in the cytoplasm. Exposure to inflammatory stress (LPS, TNF-α) results in phosphorylation and degradation of these proteins, allowing NF-κB nuclear translocation. Importantly, although both IκBα and IκBβ inhibit NF-κB activation, signaling mediated by these proteins results in specific target gene expression. Because IκBα and IκBβ preferentially bind unique NF-κB dimer combinations, and these dimer combinations bind to unique DNA sequences, different downstream genes are targeted (23). In addition, after degradation, both newly synthesized IκBα and IκBβ enter the nucleus. A nuclear export sequence found on IκBα allows it to export DNA-bound NF-κB complexes from the nucleus (24). In contrast, IκBβ lacks a nuclear export sequence (25), and remains in the nucleus, facilitating sustained binding of NF-κB dimers to DNA (26). These differences have important implications for the expression of NF-κB target genes, as the duration of NF-κB nuclear localization plays a role in determining which target genes are expressed (27). Despite the known differences between IκBα and IκBβ, the unique roles of these proteins in modulating hyperoxia-induced NF-κB activation in the lung remains unexplored. In addition, although studies of hyperoxia-induced NF-κB activation have investigated the role of IκBα, no studies have evaluated the role of IκBβ.
Using AKBI mice, in which IκBβ cDNA has been inserted behind the IκBα promoter, and thus overexpress IκBβ and do not express IκBα (28), we demonstrate that prolonged NF-κB signaling enhances improved survival after exposure to hyperoxia. This protection occurs despite enhanced expression of proinflammatory cytokines, and is likely secondary to increased expression of protective cytokines and antiapoptotic genes. These findings suggest that strategies aimed at enhancing NF-κB activity may represent a therapeutic target to prevent hyperoxic lung injury.
Materials and Methods
Animal Model
ICR mice were purchased from Taconic and AKBI, or IκBβ knockin, mice were a gift of Richard Cohen (Harvard University, Cambridge, MA). Adult mice (6–8 wk old) were exposed to room air or hyperoxia (O2 > 95%) in an A-chamber (BioSpherix, Redfield, NY). Ambient carbon dioxide was maintained at under 1,200 ppm. All procedures were approved by the Institutional Animal Care and Use Committee at the Children’s Hospital of Philadelphia (Philadelphia, PA).
Bronchoalveolar Lavage Analysis of Leukocytes and Differential, Protein, and Cytokines
After exposure to hyperoxia, mice were anesthetized, killed, bronchoalveolar lavage fluid (BALF) obtained, and fluid analyzed (29). Total protein content was determined by the Bradford method with bovine serum albumin as a standard. BALF was assessed for IL-6, IL-1β, and IL-11 content using ELISA (IL-6, 88-7064; IL-1β, 88-7013 [eBioscience, San Diego, CA]; IL-11, RAB0251 [Sigma-Aldrich, St. Louis, MO]).
Preparation of Cytosolic and Nuclear Extracts
Freshly collected whole lungs were collected, and cytosolic and nuclear extracts prepared (15).
Immunoblot Analysis
Cytosolic and nuclear extracts were electrophoresed on a 4–12% polyacrylamide gel (Invitrogen, Carlsbad, CA) and proteins were transferred to an Immobilon membrane (Millipore, Billerica, MA). Membranes were blotted with anti-IκBα (sc-371), anti-IκBβ (sc-9130), anti-BAX (sc-493), anti–BCL-2 (sc-492), or anti–lamin B (sc-6216) antibody (Santa Cruz Biotechnology, Dallas, TX); anti–c-Rel (4,774), anti-IKKα (2,682), anti-IKKβ (2,370), anti–vascular endothelial growth factor receptor (VEGFR) 2 (2,479), or anti–BCL-XL (2,762) antibody (Cell Signaling Technology, Danvers, MA); anti-p50 (ab7971) antibody (Abcam, Cambridge, MA); anti–Mn-SOD (06-984) or anti-tubulin antibody (05-829; Millipore). Densitometric analysis was performed using ImageLab (Bio-Rad, Hercules, CA).
Evaluation of NF-κB Binding by Electrophoretic Mobility Shift Assay
A 32P-labeled oligonucleotide with the consensus sequence for NF-κB (5′-AGTTGAGGGGACTTTCCCAGGC-3′; Promega, Madison, WI) was used to evaluate NF-κB binding ability (30). To identify nonspecific binding of nuclear proteins, competition reactions were performed by addition of 50-fold excess of the nonradiolabeled NF-κB consensus sequence to the reaction mixtures before electrophoresis.
Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling Staining
Lung sections were assessed for apoptosis using the DeadEndFluorometric terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) system (Promega). Digital images were obtained using an Olympus IX71 fluorescence microscope (Olympus America, Center Valley, PA), and quantitation performed on five high-magnification fields per section, with sections from three separate animals assessed per time point.
Quantitative Real-Time PCR
Relative mRNA levels were evaluated by quantitative real-time PCR using the TaqMan gene expression system (Applied Biosystems, Carlsbad, CA). Total lung RNA was extracted with TRIzol Plus RNA Purification System (Ambion, Carlsbad, CA). RNA was assessed for purity and concentration using the NanoDrop (ThermoFisher Scientific, Waltham, MA), and cDNA synthesized using the Verso cDNA Synthesis Kit (ThermoFisher Scientific). Gene expression of CXCL1, COX-2, ICAM, MnSOD, BAX, BCL-XL, BCL-2, VEGFR2, and IL-11 was assessed using predesigned exon-spanning primers (Mm04207460_m1, Mm00478374_m1, Mm00516023_m1, Mm00449726_m1, Mm00432050_m1*, Mm00437783_m1, Mm00477631_m1, Mm01222421_m1, and Mm00434162_m1*; Applied Biosystems) using the StepOnePlus Real Time PCR System (Applied Biosystems). Relative quantitation was performed via normalization to the endogenous control β-2-microglobulin (cat. no. 433766F; Applied Biosystems) using the cycle threshold (ΔΔCT) method.
Statistical Analysis
For comparison between treatment groups, the null hypothesis that no difference existed between treatment means was tested by ANOVA for multiple groups or t test for two groups (InStat; GraphPad Software, Inc., San Diego, CA). Statistical significance (P < 0.05) between and within groups was determined by means of Bonferroni’s method of multiple comparisons.
Results
AKBI Mice Are Resistant to Hyperoxia-Induced Mortality
To evaluate the specific role of the IκB family of proteins in NF-κB signaling, IκBβ knockin (AKBI) mice have been derived (Figure 1A). The AKBI mice have IκBβ cDNA inserted behind the IκBα promoter, and are phenotypically indistinct from wild-type (WT) littermate control mice (28). In this study, AKBI mice had significantly improved survival after exposure to hyperoxia when compared with WT mice (Figure 1B). Both groups had 100% survival in normoxia. However, by 5 days of hyperoxia, WT mice demonstrated 100% mortality, whereas AKBI mice had over 90% survival. Animals were weighed daily as a measure of intake and overall health (Figure 1C). Although WT and AKBI mice had similar weights before hyperoxic exposure, by 96 hours, the significant weight loss observed in WT mice was absent in AKBI mice. Given these findings, animals were exposed to 96 hours of hyperoxia for other assays.
Figure 1.
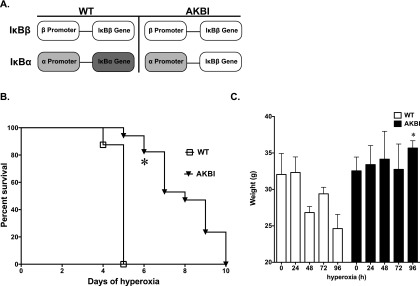
AKBI, or IκBβ knockin, mice have improved survival after hyperoxic exposure. (A) Schematic of IκB expression patterns in AKBI mice. The IκBα gene has been replaced by IκBβ cDNA. The IκBα promoter controls the expression of the IκBβ transgenic loci. Thus, AKBI overexpresses IκBβ, without expressing IκBα. (B) Kaplan-Meier survival analysis of wild-type (WT) and AKBI mice exposed to chronic hyperoxia (> 95%). Values are expressed as the percentage of surviving animals (n = 16/group). (C) Individual mouse weights through hyperoxic exposure. Data expressed as means ± SE (n = 4/time point); *P < 0.05 versus paired WT exposure.
AKBI Mice Have Less Vascular Leak after Hyperoxic Exposure
In normoxia, WT and AKBI mice had similar BALF protein content. Although WT mice had a 20-fold increase in BALF protein content after 96 hours of hyperoxic exposure, AKBI mice showed no significant increase (see Figure E1A in the online supplement). This suggested decreased oxygen toxicity in the AKBI compared with WT mice.
WT and AKBI Mice Have Similar BALF Cellular Content after Hyperoxic Exposure
In normoxia, WT and AKBI mice had similar BALF cellular content (Figure E1B). No significant difference in the total cellular BALF content or differential developed between WT and AKBI mice throughout the hyperoxic exposure (Figure E1B). Further evaluation demonstrated that there was no significant increase in the difference in BALF macrophage numbers between WT and AKBI mice (Figure E1C). Both WT and AKBI mice had similar and significantly increased neutrophil BALF counts after 96 hours of hyperoxia (Figure E1D).
WT and AKBI Mice Have Similar Pulmonary Expression of Key NF-κB Pathway Proteins
To evaluate the effect of the absence of IκBα and/or overexpression of IκBβ on proteins involved in NF-κB signaling, Western blots were performed on whole-lung lysates from WT and AKBI mice. As expected, AKBI mice did not express IκBα, and showed IκBβ overexpression when compared with WT (Figure 2A). No differences were found in the expression of p50, p65 c-Rel, IKKα, and IKKβ between WT and AKBI mice (Figure 2A).
Figure 2.

WT and AKBI mice have similar expression of key NF-κB pathway proteins, and hyperoxia induces IκB protein degradation in WT and AKBI lung. (A) Representative Western analysis of the NF-κB subunits, p50, p65, and c-Rel, the NF-κB inhibitory proteins, IκBα and IκBβ, and NF-κB kinases, IKKα and IKKβ, from WT and AKBI whole-lung homogenate. (B) Representative Western blot showing IκBα and IκBβ in whole-lung homogenate from WT and AKBI mice exposed to room air or hyperoxia (O2 > 95%, 24–96 h), with tubulin as loading control. Densitometric evaluation of (C) IκBα and (D) IκBβ. Values are means ± SEM (n = 4/time point); *P < 0.05 versus unexposed control.
Hyperoxia Decreases IκBβ Expression Both WT and AKBI Lung
To define the signaling events related to NF-κB activation, levels of immunoreactive IκBα and IκBβ were evaluated in cytosolic extracts from whole-lung homogenate. After exposure to hyperoxia, IκBα in WT mice demonstrated a cyclical pattern of degradation and reaccumulation through 96 hours of exposure (Figure 2B). Decreases in IκBβ seen in the lungs of WT mice after exposure to 96 hours of hyperoxia were not significant, but, similar to IκBα, decreased with prolonged exposure (Figures 2B and 2D). In contrast, in AKBI mice, IκBβ decreased significantly throughout the 96 hours of hyperoxic exposure (Figures 2B and 2D). These data suggest that hyperoxia promotes IκB degradation, and that AKBI mice have NF-κB signaling mediated solely through IκBβ degradation. This suggests that IκBβ-mediated NF-κB activation dictates a protective response to hyperoxia given the difference in survival between WT and AKBI mice.
AKBI Mice Have Prolonged Nuclear p65 Translocation after Hyperoxia
Having noted significant decreases in lung IκBβ levels in AKBI mice exposed to hyperoxia, nuclear extracts were assessed for translocation of the NF-κB subunit, p65. Western analysis revealed significant increases in nuclear p65 in the lungs of WT mice at 48 hours, which returned to baseline by 72 hours. In contrast, nuclear p65 increased in the lungs of AKBI mice after 48 hours of hyperoxia, and remained elevated through 96 hours of exposure (Figures 3A and 3B). Increased nuclear translocation of p65 at 96 hours of exposure was associated with increased NF-κB consensus sequence binding, as demonstrated by electrophoretic mobility shift assay (Figure 3C). These data suggest a prolonged and sustained level of hyperoxia-induced NF-κB activity in AKBI mice that was not present in WT mice.
Figure 3.

Hyperoxia-induced NF-κB activation is prolonged in AKBI mice. (A) Representative Western blot showing p65 in lung nuclear extracts from WT and AKBI mice exposed to room air or hyperoxia (O2 > 95%, 24–96 hr), with Lamin B as loading control. (B) Densitometric evaluation of nuclear p65. Values are means ± SEM (n = 4/time point); *P < 0.05 versus unexposed control; †P < 0.05 versus paired WT exposure. (C) Representative electrophoretic mobility shift assay of nuclear extracts from WT and AKBI lung after exposure to room air or hyperoxia (O2 > 95%, 96 h). Bands representing NF-κB consensus sequence binding and free probe are labeled. Lane 1, WT unexposed; lanes 2–5, WT exposed to 96 hours hyperoxia; lane 6, WT cold, WT exposed to 96-hour hyperoxia plus 50-fold excess of unlabeled oligonucleotide; lane 7, AKBI unexposed; lanes 8–11, AKBI exposed to 96-hour hyperoxia; lane 12, AKBI cold, AKBI exposed to 96-hour hyperoxia plus 50-fold excess of unlabeled oligonucleotide.
WT and AKBI Mice Have Similar Induction of NF-κB–Regulated Cytokines
Expression of IL-6 and IL-1β is known to be regulated by NF-κB activity (21), and elevated in murine models of hyperoxic lung injury (3). Interestingly, IL-6 expression attenuates hyperoxic lung injury (31), whereas expression of the proinflammatory IL-1β is thought to exacerbate lung injury (3). Levels of IL-1β and IL-6 were assessed in BALF obtained from WT and AKBI mice exposed to hyperoxia for 0, 24, 48, 72, and 96 hours. Both cytokines were significantly elevated by prolonged exposure to hyperoxia in the BALF obtained from AKBI mice when compared with WT mice (Figures 4A and 4B). In addition, we assessed whole-lung mRNA expression of the NF-κB–regulated proteins, CXCL1, COX-2, and ICAM, using quantitative real-time RT-PCR (21). Levels of CXCL1 and COX-2 mRNA were significantly elevated in both WT and AKBI mice exposed to hyperoxia (Figures 4C and 4D). In addition, there was a trend toward increased ICAM expression that did not reach significance (Figure 4E). These results are consistent with enhanced NF-κB activation, and suggest that increased expression of proinflammatory NF-κB–regulated targets does not exacerbate hyperoxic lung injury in AKBI mice.
Figure 4.

Hyperoxia induces cytokine expression in both WT and AKBI mice. (A) IL-1β and (B) IL-6 levels in bronchoalveolar lavage fluid (BALF) obtained from WT and AKBI mice exposed to hyperoxia assessed by ELISA (96 h, n = 4/time point). Data expressed as means ± SE; *P < 0.05 versus unexposed control; †P < 0.05 versus paired WT exposure. Expression of (C) CXCL1, (D) cyclo-oxygenase (COX)-2, and (E) intercellular adhesion molecule mRNA in WT and AKBI lung after exposure to hyperoxia (96 h, n = 4/time point). Data expressed as means ± SE; *P < 0.05 versus unexposed control.
Antiapoptotic and Antioxidant NF-κB Target Gene Expression Is Enhanced in AKBI Mice
The expression of NF-κB–regulated pro- and antiapoptotic proteins was assessed on the basis of previous reports linking them to hyperoxic exposure (10, 32, 33). Consistent with hyperoxia-induced NF-κB activation, both WT and AKBI mice demonstrated induction of BAX mRNA and protein expression at 96 hours of exposure (Figures 5A–5C). Whereas no increase in BCL-XL or BCL-2 mRNA or protein was observed in the lungs of WT mice exposed to hyperoxia (Figures 5D–5I), these were significantly increased in the lungs of AKBI mice (Figures 5D–5I).
Figure 5.

Hyperoxia induces NF-κB–regulated antiapoptotic gene expression only in AKBI mice. (A) Representative Western blot showing BAX from whole-lung homogenate from WT and AKBI mice exposed to hyperoxia (O2 > 95%, 96 h). (B) Densitometric evaluation of BAX. Values are means ± SEM (n = 4/time point); *P < 0.05 versus unexposed control. (C) Pulmonary BAX mRNA expression in WT and AKBI mice exposed to room air or hyperoxia (O2 > 95%, 96 h). Values are means ± SEM (n = 4/time point); *P < 0.05 versus unexposed control. (D) Representative Western blot showing BCL-XL from whole-lung homogenate from WT and AKBI mice exposed to hyperoxia (O2 > 95%, 96 h). (E) Densitometric evaluation of BCL-XL. Values are means ± SEM (n = 4/time point); *P < 0.05 versus unexposed control and paired WT exposure. (F) Pulmonary BCL-XL mRNA expression in WT and AKBI mice exposed to room air or hyperoxia (O2 > 95%, 96 h). Values are means ± SEM (n = 4/time point); *P < 0.05 versus unexposed control; †P < 0.05 versus paired WT exposure. (G) Representative Western blot showing BCL-2 from whole-lung homogenate from WT and AKBI mice exposed to hyperoxia (O2 > 95%, 96 h). (H) Densitometric evaluation of BCL-2. Values are means ± SEM (n = 4/time point); *P < 0.05 versus unexposed control and paired WT exposure. (I) Pulmonary BAX mRNA expression in WT and AKBI mice exposed to room air or hyperoxia (O2 > 95%, 96 h). Values are means ± SEM (n = 4/time point); *P < 0.05 versus unexposed control and paired WT exposure.
In addition to BCL-2 and BCL-XL, other NF-κB–regulated proteins are known to protect against hyperoxic lung injury. Specifically, NF-κB regulates the induction of IL-11 and MnSOD, as well as constitutive expression of VEGFR2 (21, 34). Expression of these factors attenuates hyperoxic lung injury (11, 12, 35, 36). Hyperoxia resulted in decreased expression of VEGFR2 and no induction of IL-11 expression in WT mice (Figures 6A–6E). In contrast, hyperoxia-exposed AKBI mice demonstrated a significant up-regulation of both VEGFR2 and IL-11 mRNA and protein (Figures 6A–6E). In addition, we found that hyperoxia significantly induced MnSOD mRNA and protein expression in AKBI compared with WT mice (Figures 6F–6H). Together with the increased expression of NF-κB–regulated antiapoptotic proteins, these results suggest that the prolonged hyperoxia-induced NF-κB nuclear translocation seen in AKBI mice tips the balance toward increased cell survival, and confers resistance to hyperoxic lung injury.
Figure 6.
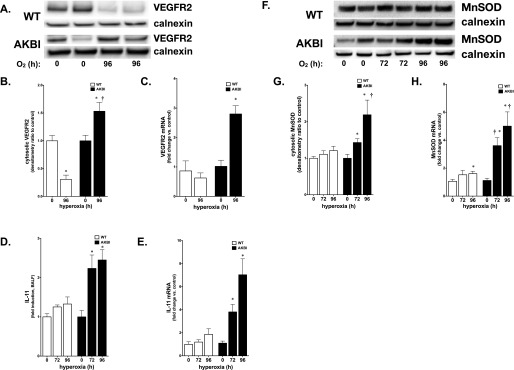
Hyperoxia induces expression of vascular endothelial growth factor receptor (VEGFR) 2, IL-11 and manganese superoxide dismutase (MnSOD) in AKBI mice. (A) Representative Western blot showing VEGFR2 from whole-lung homogenate from WT and AKBI mice exposed to hyperoxia (O2 > 95%, 96 h). (B) Densitometric evaluation of VEGFR2. Values are means ± SEM (n = 4/time point); *P < 0.05 versus unexposed control; †P < 0.05 versus paired WT exposure (C) Pulmonary VEGFR mRNA expression in WT and AKBI mice exposed to room air or hyperoxia (O2 > 95%, 96 h). Values are means ± SEM (n = 4/time point); *P < 0.05 versus unexposed control and paired WT exposure. (D) IL-11 fold increase in BALF obtained from WT and AKBI mice exposed to hyperoxia assessed by ELISA (72–96 h, n = 4/time point). Data expressed as means ± SE; *P < 0.05 versus unexposed control and paired WT exposure. (E) IL-11 mRNA in WT and AKBI lung after exposure to hyperoxia (96 h, n = 4/time point). Data expressed as means ± SE; *P < 0.05 versus unexposed control and paired WT exposure. (F) Representative Western blot showing MnSOD from lung cytosolic extracts from WT and AKBI mice exposed to room air or hyperoxia (O2 > 95%, 96 h), with calnexin as a loading control. (G) Densitometric evaluation of MnSOD. Values are means ± SEM (n = 4/time point); *P < 0.05 versus unexposed control; †P < 0.05 versus paired WT exposure. (H) Pulmonary MnSOD mRNA expression in WT and AKBI mice exposed to room air or hyperoxia (O2 > 95%, 96 h). Values are means ± SEM (n = 4/time point); *P < 0.05 versus unexposed control; †P < 0.05 versus paired WT exposure.
Hyperoxia Increases TUNEL Staining in WT Mice
Apoptosis was assessed by TUNEL staining of WT and AKBI adult mouse lungs at 96 hours of hyperoxia exposure. In agreement with the expression profile of antiapoptotic genes, hyperoxia induced apoptosis in the WT lung (Figure 7). In contrast, there was no significant increase in the number of cells staining positive in the AKBI lung exposed to hyperoxia (Figure 7). These results confirm increased apoptosis in the WT lung exposed to hyperoxia, a finding that was attenuated in the AKBI mice.
Figure 7.

Hyperoxia-induced apoptosis is attenuated in AKBI mice. (A) High-magnification (40×) images of representative terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)- (green) and 4′,6-diamidino-2-phenylindole (blue) -stained lung sections of WT and AKBI mice exposed to chronic hyperoxia (O2 > 95%, 0 or 96 h). (B) TUNEL-positive nuclei per high-powered field in lung sections from WT and AKBI mice exposed to hyperoxia. Values are means ± SEM (n = 4 animals/time point with 5 fields/animal); *P < 0.05 versus unexposed control; †P < 0.05 versus paired WT exposure.
Discussion
We found that hyperoxia-induced NF-κB activity occurs in both WT and AKBI mice. In WT mice, this induction occurs over the first 48 hours of exposure, and results in the expression of proinflammatory and proapoptotic proteins. This is associated with significant mortality that occurs within 5 days of exposure. In contrast, hyperoxia-induced NF-κB activity in AKBI mice persists through 96 hours of exposure. Although proinflammatory and proapoptotic genes are expressed at levels equivalent to and, in some cases, in excess of WT mice, up-regulation of antioxidant and antiapoptotic proteins occurs with prolonged exposure. This results in attenuated lung injury and improved survival. Thus, targeting hyperoxia-induced NF-κB signaling represents a potential therapeutic target to limit lung injury.
These results are interesting, because this is the first study showing that enhanced NF-κB activation mediated by IκBβ overexpression confers resistance to hyperoxic lung injury. Previous studies have shown that NF-κB activity prevents cell death induced by various types of oxidant stress. These include ischemia–reperfusion to the intestine and heart (16, 17, 19), oxidant stress induced by H2O2 in A549 cells (18), and glucose oxidase in murine embryonic fibroblast cells (37). In addition, enhanced hyperoxia-induced NF-κB activation explains the resistance to lung injury seen in neonatal mice, in part due to inhibition of apoptosis. Previous studies have demonstrated that hyperoxic exposure results in apoptosis of pulmonary endothelial and epithelial cells (32, 38). Consistent with this finding, pulmonary gene expression array analysis of mice exposed to hyperoxia shows induction of both pro- (BAX) and antiapoptotic (BCL-XL) proteins (33). Of note, BAX, BCL-XL, and BCL-2 expression is regulated by NF-κB (24). We found that the prolonged hyperoxia-induced NF-κB activation seen in AKBI mice was associated with increased expression of both BCL-2 and BCL-XL, and this was associated with attenuated apoptosis and improved survival. Although p53−/−, fas−/−, and tnfr−/− mice do not demonstrate resistance to hyperoxic lung injury (32, 39), specific overexpression of proteins that contain four Bcl-2 homology domains (BCL-2, BCL-XL) and prevent mitochondrial permeabilization attenuates hyperoxic lung injury, whereas their absence increases sensitivity to oxygen toxicity (10, 40). Our study supports the finding that enhancing expression of antiapoptotic proteins via NF-κB activation to prevent mitochondrial permeabilization prevents apoptosis and attenuates hyperoxic lung injury.
In this study, we evaluated the expression of various NF-κB–regulated proteins: pro- and anti-inflammatory cytokines and antioxidant enzymes. These targets were selected on the basis of previous reports linking these proteins to the pulmonary response to hyperoxic injury. Previous in vivo studies have shown that expression of MnSOD (36), IL-6 (31), and VEGF signaling mediated through VEGFR2 (11, 35) attenuate hyperoxic lung injury. Expression of each of these individual factors is regulated by NF-κB (21). Here, we have demonstrated that the enhanced hyperoxia-induced NF-κB activity seen in AKBI mice is associated with increased expression of all of these protective factors. Importantly, NF-κB regulates the expression of additional proteins known to attenuate hyperoxic lung injury, but not evaluated in the current study. These include hemeoxygenase and p21 (41, 42). Of note, the protection afforded by p21 was recently demonstrated to be mediated via BCL-XL, one of the NF-κB–regulated genes assessed in the current study (43). Thus, targeting events upstream of NF-κB activation to enhance duration of activity, and thus affecting the expression of multiple cytoprotective genes, represents a potential therapeutic target to be tested in future studies.
Furthermore, our results are consistent with other reports demonstrating that disrupting signaling events upstream of NF-κB activation exacerbates hyperoxic lung injury. The increased susceptibility to hyperoxic lung injury seen in TLR4−/− mice is associated with a more transient activation of NF-κB when compared with WT control animals, as well as attenuated Akt signaling and BCL-2 expression (14, 44). Importantly, Akt signaling plays a role in NF-κB activation (22), and constitutively active Akt prevents hyperoxic lung injury (45). Placed in context with these previous reports, our results argue for a central role played by NF-κB activation in attenuating hyperoxic lung injury.
Hyperoxia results in increased pulmonary expression of NF-κB–regulated proapoptotic proteins (BAX [46]), proinflammatory cytokines (IL-1β, CXCL1 [3, 47]), and pro-oxidant enzymes (COX-2 [48]). We found hyperoxia-induced induction of BAX, IL-1β, CXCL1, and COX-2 in both WT and AKBI mice. Our findings show that, despite expression of these proinflammatory, proapoptotic, and pro-oxidant factors, AKBI mice demonstrate resistance to hyperoxic lung injury. We speculate that, despite expression of proinflammatory signals, preventing apoptosis modulates lung injury and confers a survival advantage to hyperoxia-exposed mice. These findings are important, as targeting the transcriptional activity of NF-κB, rather than one specific gene target, represents a fundamentally simpler therapeutic intervention. It is clear that hyperoxia-induced NF-κB activity occurs in WT mice, and that maintaining this activity prevents lung injury and mortality. Our results demonstrate that, although hyperoxia-induced NF-κB activation affects the expression of both protective and injurious factors, the net result of prolonged NF-κB activity is attenuated lung injury.
The possible mechanism underlying our finding is that IκBβ-mediated, sustained NF-κB activity tips the balance of gene expression to include both antioxidant and antiapoptotic targets. In quiescent cells, NF-κB remains sequestered in the cytoplasm bound to members of the IκB family of inhibitory proteins (22). Exposure to inflammatory stimuli results in phosphorylation of two N-terminal serine residues on both IκBα and IκBβ, resulting in their degradation (22). IκBα is transcriptionally regulated by NF-κB, allowing for a well regulated negative feedback loop that is both sensitive to and rapidly influenced by NF-κB activation (49). Newly synthesized IκBα enters the nucleus and removes DNA-bound NF-κB complexes (24). In contrast to IκBα, IκBβ is degraded more slowly, it is not transcriptionally regulated by NF-κB, and, after degradation induced by inflammatory stimuli, reaccumulates as a hypophosphorylated form. Hypophosphorylated IκBβ chaperones NF-κB dimers and facilitates DNA binding (26). In contrast to the well defined NF-κB activation cascade that occurs after exposure to inflammatory stress, a definitive pathway after exposure to oxidant stress has not been established. Whether these same mechanisms are responsible for prolonged NF-κB activation seen in hyperoxia-exposed AKBI mice remains to be determined.
Our study is limited by the use of AKBI mice. From our work, it is clear that overexpression of IκBβ and lack of IκBα affects baseline expression of NF-κB targets, including IL-1β and IL-6 (Figures 6A and 6B). This is likely true for other NF-κB targets. These findings are in agreement with studies of IκBβ−/− mice that show increased constitutive NF-κB activity (23). Despite this difference, the AKBI mice are phenotypically indistinct from their WT control animals, and demonstrate normal lung architecture. In addition, we have not identified the specific cell types protected from apoptosis by prolonged NF-κB activation. Whether our findings are due to enhanced NF-κB activation in resident lung cells, or inflammatory cells recruited to the lung with ongoing hyperoxic injury, remains to be determined. Future studies identifying the specific cell type protected from apoptosis, and evaluating the effect of enhancing hyperoxia-induced NF-κB activity in WT mice, are needed to confirm our results.
We conclude that enhanced hyperoxia-induced NF-κB activity prevents hyperoxic lung injury and mortality in vivo. Specifically, NF-κB activation results in increased expression of cytoprotective factors, including MnSOD and antiapoptotic genes (BCL2 and BCL-XL). This protection occurs despite intact proinflammatory gene expression. Our results show that the toxic effects of hyperoxia can be attenuated by targeting the inhibitory proteins that dictate the duration of NF-κB activity and ultimately downstream target gene expression. We speculate that interventions aimed at enhancing hyperoxia-induced NF-κB activation could attenuate lung injury.
Footnotes
This work was supported by National Institutes of Health grants K08 HL098562 (C.J.W.) and HL058752-11 (P.A.D.).
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2013-0303OC on September 25, 2013
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Smith JL. The pathological effects due to increase of oxygen tension in the air breathed. J Physiol. 1899;24:19–35. doi: 10.1113/jphysiol.1899.sp000746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L379–L399. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnston CJ, Wright TW, Reed CK, Finkelstein JN. Comparison of adult and newborn pulmonary cytokine mRNA expression after hyperoxia. Exp Lung Res. 1997;23:537–552. doi: 10.3109/01902149709039242. [DOI] [PubMed] [Google Scholar]
- 4.Crapo JD. Morphologic changes in pulmonary oxygen toxicity. Annu Rev Physiol. 1986;48:721–731. doi: 10.1146/annurev.ph.48.030186.003445. [DOI] [PubMed] [Google Scholar]
- 5.Tsan MF, White JE, Michelsen PB, Wong GH. Pulmonary O2 toxicity: role of endogenous tumor necrosis factor. Exp Lung Res. 1995;21:589–597. doi: 10.3109/01902149509031761. [DOI] [PubMed] [Google Scholar]
- 6.Raj JU, Hazinski TA, Bland RD. Oxygen-induced lung microvascular injury in neutropenic rabbits and lambs. J Appl Physiol. 1985;58:921–927. doi: 10.1152/jappl.1985.58.3.921. [DOI] [PubMed] [Google Scholar]
- 7.Guthmann F, Wissel H, Rustow B. Early subcutaneous administration of etanercept (enbrel) prevents from hyperoxia-induced lung injury. Exp Lung Res. 2009;35:770–780. doi: 10.3109/01902140902887430. [DOI] [PubMed] [Google Scholar]
- 8.Husari AW, Khayat A, Awdeh H, Hatoum H, Nasser M, Mroueh SM, Zaatari G, El-Sabban M, Dbaibo GS. Activated protein C attenuates acute lung injury and apoptosis in a hyperoxic animal model. Shock. 2010;33:467–472. doi: 10.1097/SHK.0b013e3181c69213. [DOI] [PubMed] [Google Scholar]
- 9.Yamada M, Kubo H, Kobayashi S, Ishizawa K, Sasaki H. Interferon-gamma: a key contributor to hyperoxia-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1042–L1047. doi: 10.1152/ajplung.00155.2004. [DOI] [PubMed] [Google Scholar]
- 10.Waxman AB, Kolliputi N. IL-6 protects against hyperoxia-induced mitochondrial damage via BCL-2–induced BAK interactions with mitofusins. Am J Respir Cell Mol Biol. 2009;41:385–396. doi: 10.1165/rcmb.2008-0302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He CH, Waxman AB, Lee CG, Link H, Rabach ME, Ma B, Chen Q, Zhu Z, Zhong M, Nakayama K, et al. BCL-2–related protein A1 is an endogenous and cytokine-stimulated mediator of cytoprotection in hyperoxic acute lung injury. J Clin Invest. 2005;115:1039–1048. doi: 10.1172/JCI23004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waxman AB, Einarsson O, Seres T, Knickelbein RG, Warshaw JB, Johnston R, Homer RJ, Elias JA. Targeted lung expression of interleukin-11 enhances murine tolerance of 100% oxygen and diminishes hyperoxia-induced DNA fragmentation. J Clin Invest. 1998;101:1970–1982. doi: 10.1172/JCI1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gore A, Muralidhar M, Espey MG, Degenhardt K, Mantell LL. Hyperoxia sensing: from molecular mechanisms to significance in disease. J Immunotoxicol. 2010;7:239–254. doi: 10.3109/1547691X.2010.492254. [DOI] [PubMed] [Google Scholar]
- 14.Qureshi ST, Zhang X, Aberg E, Bousette N, Giaid A, Shan P, Medzhitov RM, Lee PJ. Inducible activation of TLR4 confers resistance to hyperoxia-induced pulmonary apoptosis. J Immunol. 2006;176:4950–4958. doi: 10.4049/jimmunol.176.8.4950. [DOI] [PubMed] [Google Scholar]
- 15.Yang G, Abate A, George AG, Weng YH, Dennery PA. Maturational differences in lung NF-kappaB activation and their role in tolerance to hyperoxia. J Clin Invest. 2004;114:669–678. doi: 10.1172/JCI19300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu X, Liu H, Wang L, Schaefer S. Activation of NF-kappaB is a critical element in the antiapoptotic effect of anesthetic preconditioning. Am J Physiol Heart Circ Physiol. 2009;296:H1296–H1304. doi: 10.1152/ajpheart.01282.2008. [DOI] [PubMed] [Google Scholar]
- 17.Tahepold P, Vaage J, Starkopf J, Valen G. Hyperoxia elicits myocardial protection through a nuclear factor kappaB–dependent mechanism in the rat heart. J Thorac Cardiovasc Surg. 2003;125:650–660. doi: 10.1067/mtc.2003.36. [DOI] [PubMed] [Google Scholar]
- 18.Franek WR, Horowitz S, Stansberry L, Kazzaz JA, Koo HC, Li Y, Arita Y, Davis JM, Mantell AS, Scott W, et al. Hyperoxia inhibits oxidant-induced apoptosis in lung epithelial cells. J Biol Chem. 2001;276:569–575. doi: 10.1074/jbc.M004716200. [DOI] [PubMed] [Google Scholar]
- 19.Chen LW, Egan L, Li ZW, Greten FR, Kagnoff MF, Karin M. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia–reperfusion. Nat Med. 2003;9:575–581. doi: 10.1038/nm849. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki Y, Nishio K, Takeshita K, Takeuchi O, Watanabe K, Sato N, Naoki K, Kudo H, Aoki T, Yamaguchi K. Effect of steroid on hyperoxia-induced ICAM-1 expression in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2000;278:L245–L252. doi: 10.1152/ajplung.2000.278.2.L245. [DOI] [PubMed] [Google Scholar]
- 21.Gilmore T. NF-kB target genes [internet]. Boston 9MA: Boston University; c2013 [accessed 2013 Oct 24]. Available from: http://www.bu.edu/nf-kb/gene-resources/target-genes/
- 22.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 23.Rao P, Hayden MS, Long M, Scott ML, West AP, Zhang D, Oeckinghaus A, Lynch C, Hoffmann A, Baltimore D, et al. Ikappabbeta acts to inhibit and activate gene expression during the inflammatory response. Nature. 2010;466:1115–1119. doi: 10.1038/nature09283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang TT, Miyamoto S. Postrepression activation of NF-kappaB requires the amino-terminal nuclear export signal specific to IkappaBalpha. Mol Cell Biol. 2001;21:4737–4747. doi: 10.1128/MCB.21.14.4737-4747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tam WF, Sen R. IkappaB family members function by different mechanisms. J Biol Chem. 2001;276:7701–7704. doi: 10.1074/jbc.C000916200. [DOI] [PubMed] [Google Scholar]
- 26.Suyang H, Phillips R, Douglas I, Ghosh S. Role of unphosphorylated, newly synthesized I kappa B beta in persistent activation of NF-kappa B. Mol Cell Biol. 1996;16:5444–5449. doi: 10.1128/mcb.16.10.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 28.Cheng JD, Ryseck RP, Attar RM, Dambach D, Bravo R. Functional redundancy of the nuclear factor kappa B inhibitors I kappa B alpha and I kappa B beta. J Exp Med. 1998;188:1055–1062. doi: 10.1084/jem.188.6.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Mei J, Gonzales L, Yang G, Dai N, Wang P, Zhang P, Favara M, Malcolm KC, Guttentag S, et al. IL-17A and TNF-alpha exert synergistic effects on expression of CXCL5 by alveolar type II cells in vivo and in vitro. J Immunol. 2011;186:3197–3205. doi: 10.4049/jimmunol.1002016. [DOI] [PubMed] [Google Scholar]
- 30.Yang G, Madan A, Dennery PA. Maturational differences in hyperoxic AP-1 activation in rat lung. Am J Physiol Lung Cell Mol Physiol. 2000;278:L393–L398. doi: 10.1152/ajplung.2000.278.2.L393. [DOI] [PubMed] [Google Scholar]
- 31.Ward NS, Waxman AB, Homer RJ, Mantell LL, Einarsson O, Du Y, Elias JA. Interleukin-6–induced protection in hyperoxic acute lung injury. Am J Respir Cell Mol Biol. 2000;22:535–542. doi: 10.1165/ajrcmb.22.5.3808. [DOI] [PubMed] [Google Scholar]
- 32.Barazzone C, Horowitz S, Donati YR, Rodriguez I, Piguet PF. Oxygen toxicity in mouse lung: pathways to cell death. Am J Respir Cell Mol Biol. 1998;19:573–581. doi: 10.1165/ajrcmb.19.4.3173. [DOI] [PubMed] [Google Scholar]
- 33.Perkowski S, Sun J, Singhal S, Santiago J, Leikauf GD, Albelda SM. Gene expression profiling of the early pulmonary response to hyperoxia in mice. Am J Respir Cell Mol Biol. 2003;28:682–696. doi: 10.1165/rcmb.4692. [DOI] [PubMed] [Google Scholar]
- 34.Iosef C, Alastalo TP, Hou Y, Chen C, Adams ES, Lyu SC, Cornfield DN, Alvira CM. Inhibiting NF-kappaB in the developing lung disrupts angiogenesis and alveolarization. Am J Physiol Lung Cell Mol Physiol. 2012;302:L1023–L1036. doi: 10.1152/ajplung.00230.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corne J, Chupp G, Lee CG, Homer RJ, Zhu Z, Chen Q, Ma B, Du Y, Roux F, McArdle J, et al. IL-13 stimulates vascular endothelial cell growth factor and protects against hyperoxic acute lung injury. J Clin Invest. 2000;106:783–791. doi: 10.1172/JCI9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ho YS, Vincent R, Dey MS, Slot JW, Crapo JD. Transgenic models for the study of lung antioxidant defense: enhanced manganese-containing superoxide dismutase activity gives partial protection to B6C3 hybrid mice exposed to hyperoxia. Am J Respir Cell Mol Biol. 1998;18:538–547. doi: 10.1165/ajrcmb.18.4.2959. [DOI] [PubMed] [Google Scholar]
- 37.Wright CJ, Agboke F, Muthu M, Michaelis KA, Mundy MA, La P, Yang G, Dennery PA. Nuclear factor-kappaB (NF-kappaB) inhibitory protein IkappaBbeta determines apoptotic cell death following exposure to oxidative stress. J Biol Chem. 2012;287:6230–6239. doi: 10.1074/jbc.M111.318246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mantell LL, Kazzaz JA, Xu J, Palaia TA, Piedboeuf B, Hall S, Rhodes GC, Niu G, Fein AF, Horowitz S. Unscheduled apoptosis during acute inflammatory lung injury. Cell Death Differ. 1997;4:600–607. doi: 10.1038/sj.cdd.4400278. [DOI] [PubMed] [Google Scholar]
- 39.Pryhuber GS, O’Brien DP, Baggs R, Phipps R, Huyck H, Sanz I, Nahm MH. Ablation of tumor necrosis factor receptor type I (p55) alters oxygen-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2000;278:L1082–L1090. doi: 10.1152/ajplung.2000.278.5.L1082. [DOI] [PubMed] [Google Scholar]
- 40.Staversky RJ, Vitiello PF, Yee M, Callahan LM, Dean DA, O’Reilly MA. Epithelial ablation of BCL-XL increases sensitivity to oxygen without disrupting lung development. Am J Respir Cell Mol Biol. 2010;43:376–385. doi: 10.1165/rcmb.2009-0165OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Otterbein LE, Kolls JK, Mantell LL, Cook JL, Alam J, Choi AM. Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung injury. J Clin Invest. 1999;103:1047–1054. doi: 10.1172/JCI5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Reilly MA, Staversky RJ, Watkins RH, Reed CK, de Mesy Jensen KL, Finkelstein JN, Keng PC. The cyclin-dependent kinase inhibitor p21 protects the lung from oxidative stress. Am J Respir Cell Mol Biol. 2001;24:703–710. doi: 10.1165/ajrcmb.24.6.4355. [DOI] [PubMed] [Google Scholar]
- 43.Wu YC, O’Reilly MA. BCL-X(L) is the primary mediator of p21 protection against hyperoxia-induced cell death. Exp Lung Res. 2011;37:82–91. doi: 10.3109/01902148.2010.521617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang X, Shan P, Qureshi S, Homer R, Medzhitov R, Noble PW, Lee PJ. Cutting edge: TLR4 deficiency confers susceptibility to lethal oxidant lung injury. J Immunol. 2005;175:4834–4838. doi: 10.4049/jimmunol.175.8.4834. [DOI] [PubMed] [Google Scholar]
- 45.Lu Y, Parkyn L, Otterbein LE, Kureishi Y, Walsh K, Ray A, Ray P. Activated Akt protects the lung from oxidant-induced injury and delays death of mice. J Exp Med. 2001;193:545–549. doi: 10.1084/jem.193.4.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Budinger GR, Mutlu GM, Urich D, Soberanes S, Buccellato LJ, Hawkins K, Chiarella SE, Radigan KA, Eisenbart J, Agrawal H, et al. Epithelial cell death is an important contributor to oxidant-mediated acute lung injury. Am J Respir Crit Care Med. 2011;183:1043–1054. doi: 10.1164/rccm.201002-0181OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sue RD, Belperio JA, Burdick MD, Murray LA, Xue YY, Dy MC, Kwon JJ, Keane MP, Strieter RM. CXCR2 is critical to hyperoxia-induced lung injury. J Immunol. 2004;172:3860–3868. doi: 10.4049/jimmunol.172.6.3860. [DOI] [PubMed] [Google Scholar]
- 48.Adawi A, Zhang Y, Baggs R, Finkelstein J, Phipps RP. Disruption of the CD40–CD40 ligand system prevents an oxygen-induced respiratory distress syndrome. Am J Pathol. 1998;152:651–657. [PMC free article] [PubMed] [Google Scholar]
- 49.Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. 1993;259:1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]