

Abstract
Nonalcoholic fatty liver disease (NAFLD) has emerged as a common public health problem in recent decades. However, the underlying mechanisms leading to the development of NAFLD are not fully understood. The endoplasmic reticulum (ER) stress response has recently been proposed to play a crucial role in both the development of steatosis and progression to nonalcoholic steatohepatitis. ER stress is activated to regulate protein synthesis and restore homeostatic equilibrium when the cell is stressed due to the accumulation of unfolded or misfolded proteins. However, delayed or insufficient responses to ER stress may turn physiological mechanisms into pathological consequences, including fat accumulation, insulin resistance, inflammation, and apoptosis, all of which play important roles in the pathogenesis of NAFLD. Therefore, understanding the role of ER stress in the pathogenesis of NAFLD has become a topic of intense investigation. This review highlights the recent findings linking ER stress signaling pathways to the pathogenesis of NAFLD.
Keywords: Endoplasmic reticulum stress, Unfolded protein response, Nonalcoholic fatty liver disease, Nonalcoholic steatohepatitis
Core tip: Nonalcoholic fatty liver disease (NAFLD) is a progressive disorder that can lead to impaired liver function and, ultimately, liver failure. Chronic endoplasmic reticulum stress induces numerous intracellular pathways that can lead to hepatic steatosis, systemic inflammation, and hepatocyte cell death, all of which are keystones of NAFLD. This review highlights the recent findings linking ER stress signaling pathways with the pathogenesis of NAFLD, which may help identify novel therapeutic targets for the prevention and treatment of NAFLD.
INTRODUCTION
In recent decades, nonalcoholic fatty liver disease (NAFLD) has increasingly been recognized as one of the most common chronic liver diseases. The reported prevalence of NAFLD in Western countries ranges from 30% to 46%[1,2]. This disease has also become prevalent in Eastern countries and has become a significant public health concern in these areas[3,4]. NAFLD encompasses a spectrum of liver damage ranging from steatosis to nonalcoholic steatohepatitis (NASH), which can progress to fibrosis, cirrhosis, and ultimately to liver failure or hepatocellular carcinoma[5,6]. Individuals with NAFLD are also at an increased risk of cardiovascular disease, type 2 diabetes, and obesity-related mortality[7-9].
The precise mechanisms of NAFLD remain poorly understood. The “multiple-hits hypothesis” is currently the most recognized theory to explain disease development and progression. The initial hit leads to the development of simple steatosis, while the following hits, including mitochondrial dysfunction, oxidative stress, adipocytokine alteration, lipid peroxidation, Kupffer cell activation, etc. results in liver cell inflammation and apoptosis, which finally leads from simple steatosis to NASH. Recently, accumulating data have implicated disruption of endoplasmic reticulum (ER) homeostasis, or ER stress, in both the development of steatosis and progression to NASH[10-12]. ER stress may lead to activation of various intracellular stress pathways that can initiate or exacerbate insulin resistance (IR) and inflammation and, in some cases, culminate in hepatocyte cell death and liver damage, all of which are important in the pathogenesis of NAFLD. Therefore, there is an urgent need to better understand the mechanisms that disrupt ER homeostasis and lead to activation of the unfolded protein response (UPR) in NAFLD. The aim of this review is to highlight the recent findings linking activation of ER stress and NAFLD development.
UPR AND ER STRESS
The ER is a membrane-bound organelle that provides a specialized environment for the production and post-translational modification of secretory and membrane proteins, lipid biosynthesis, and homeostasis of intracellular Ca2+. In the ER lumen, newly synthesized proteins undergo post-translational modifications such as N-glycosylation, disulfide bond formation, and oligomerization, which require the presence of chaperone proteins. The ER is thus considered as a quality control checkpoint, and only correctly folded proteins can exit the ER and go through the secretory pathway.
Any event that disturbs ER folding capacity, such as excessive protein synthesis, accumulation of mutant misfolded proteins, ER calcium depletion, or a change in redox status, will induce a physiological reaction referred to as the UPR. These homeostatic responses trigger the production of additional chaperones to increase the folding capacity of the ER, enhance endoplasmic reticulum-associated protein degradation (ERAD), and reduce protein entry by altering the translation and synthesis of new proteins, thus bringing the organelle and the cell into a state of equilibrium[13-15].
The UPR is mediated by three integral proteins of the ER[16,17]: protein kinase RNA-like ER kinase (PERK), inositol-requiring enzyme-1 (IRE1), and activating transcription factor-6 (ATF6). These proteins are maintained in an inactive state as long as they are bound to binding immunoglobulin protein (Bip), which is an intraluminal chaperone. When the ER is stressed, Bip is displaced from these stress sensors, leading to their activation. Once activated, PERK phosphorylates and inhibits eukaryotic translation initiation factor 2α (eiF2α), resulting in a global decrease in protein translation. Moreover, p-eiF2α selectively promotes the translation of a growing number of mRNAs, including activating transcription factor 4 (ATF4) mRNA. Activation of IRE1 promotes the splicing of X-box-binding protein-1 (XBP1) mRNA and the subsequent transcription of molecular chaperones and genes involved in ERAD. Finally, activated ATF6 undergoes proteolytic cleavage in the Golgi, allowing its mature form to enter the nucleus and transactivate ER stress-related genes such as ER chaperones and foldases (Figure 1).
Figure 1.
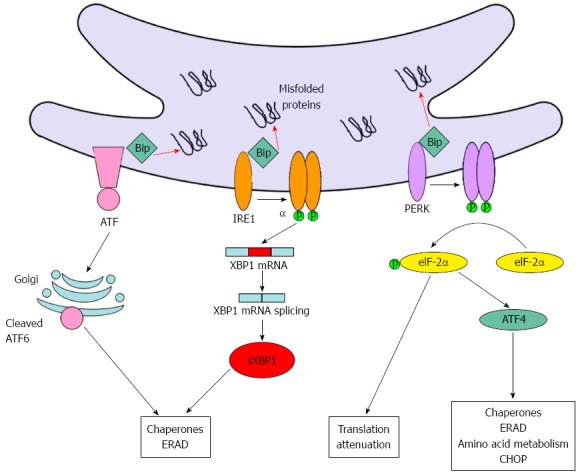
The unfolded protein response pathway. When the unfolded protein response is activated, the first event is the dissociation of the chaperone Bip from the three integral proteins PERK, IRE1, and ATF6, leading to their activation. When activated, PERK phosphorylates and inhibits eiF2α, leading to a global decrease in protein translation. Moreover, p-eiF2α activates ATF4, which induces the expression of several genes, including amino acid transporters, chaperones, and CHOP. Activation of IRE1 promotes the splicing of XBP1 mRNA and the subsequent transcription of molecular chaperones and genes involved in ERAD. Finally, activated ATF6 undergoes proteolytic cleavage in the Golgi, transactivating genes such as endoplasmic reticulum (ER) chaperones and foldases. Bip: Binding immunoglobulin protein; ATF6: Activating transcription factor-6; IRE1: Inositol requiring enzyme-1; PERK: Protein kinase RNA-like ER kinase; XBP1: X-box-binding protein-1; eiF2α: Eukaryotic translation initiation factor 2α; ATF4: Transcription factor 4; ERAD: Endoplasmic reticulum associated protein degradation; CHOP: C/EBP-homologous protein.
Thus, the UPR is primarily a cytoprotective survival response that aims to regulate protein folding and restore homeostatic balance. However, when the activation of the UPR fails to promote cell survival, the cell is directed down the pro-apoptotic ER stress response pathway, which can ultimately lead to apoptotic cell death[18-20].
ER STRESS AND NAFLD
Similar to other secretory cells, hepatocytes are rich in ER. Due to its high capacity for protein synthesis, the UPR/ER stress response plays important roles in both preventing and mediating pathological changes in various liver diseases[21,22]. The signaling pathways activated by ER stress have been linked to lipotoxicity, IR, inflammation, and apoptotic cell death, which are common to both obesity and NAFLD. Therefore, the role of ER stress in NAFLD has become a subject of considerable interest in recent years. The induction of ER stress was first described in the livers of genetic and diet-induced models of NASH[23]. Since then, these findings have been confirmed in other obese animal models[24-26] and in mice fed a methionine-choline-deficient (MCD) diet exhibiting hepatic steatosis without obesity[27]. Later, the activation of several UPR components was reported in the livers of patients with NAFLD or NASH[10,28]. Despite rapid growth in the field of ER stress research in the context of NAFLD, the exact contribution of the ER stress response to the pathogenesis of NAFLD remains to be fully elucidated. Here, we review and update the well-established associations between the ER stress response and NAFLD (Figure 2).
Figure 2.
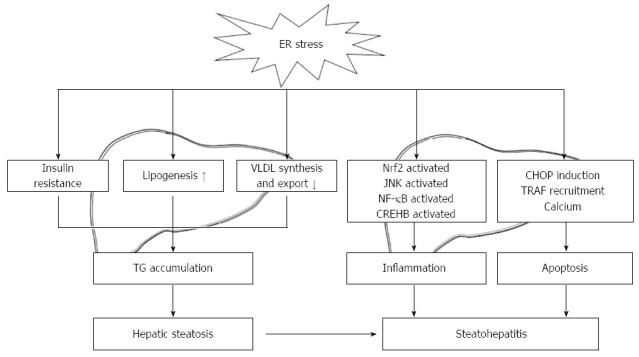
Roles of endoplasmic reticulum stress in the development of steatosis and progression to nonalcoholic steatohepatitis. ER stress interferes with hepatic lipid metabolism by activating lipogenesis and limiting VLDL formation and secretion. ER stress also acts indirectly on liver triglyceride accumulation by promoting insulin resistance in both the liver and adipose tissue. Furthermore, ER stress promotes the activation of Nrf2, JNK, NF-κB, CREBH, and CHOP, which actively participate in the inflammatory process and cell death. ER: Endoplasmic reticulum; VLDL: Very-low-density lipoprotein; Nrf2: Nuclear factor-erythroid-derived 2-related factor 2; JNK: c-Jun N-terminal kinase; NF-κB: Nuclear factor-κB; CREBH: Cyclic-AMP responsive element-binding protein H; CHOP: C/EBP-homologous protein; TRAF: Tumor-necrosis factor α-receptor-associated factor; TG: Triglyceride.
ER stress and lipid metabolism
The first step in the development of NAFLD is hepatic steatosis, which is characterized by macrovesicular accumulation of triglycerides in the cytoplasm of hepatocytes. Sources of increased hepatic lipids in NAFLD include (1) excess dietary chylomicron remnants; (2) increased de novo lipogenesis; (3) excess free fatty acids released from the lipolysis of adipose tissue; (4) diminished very-low-density lipoprotein (VLDL) secretion; and (5) reduced oxidation of fatty acids[29-31].
For approximately one decade, it has been known that ER stress can lead to altered lipid metabolism and hepatic steatosis. Recently, specific arms of the UPR and their downstream signaling molecules have been examined in cell culture and animal models to decipher their functions and roles in lipid metabolism. It is now well established that various components of the UPR signaling network play roles in the regulation of lipid metabolism.
First, the PERK-eiF2α-ATF4 pathway was reported to regulate lipogenesis and hepatic steatosis. PERK-dependent signaling has been found to contribute to lipogenic differentiation in the mammary epithelium, and deletion of PERK inhibits the sustained expression of the lipogenic enzymes fatty acid synthase (FAS), ATP-citrate lyase, and stearoyl-CoA desaturase-1 (SCD1)[32]. Oyadomari et al[33] reported that selectively compromising eiF2α-mediated signaling under ER stress conditions using GADD34, which acts to dephosphorylate eiF2α, results in diminished hepatosteatosis in animals fed a high-fat diet. Attenuated eiF2α phosphorylation is correlated with decreased expression of the adipogenic nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ) and its upstream regulators, the transcription factors CCAAT/enhancer-binding proteins α and β (C/EBPα, C/EBPβ)[33]. Protein kinase-mediated p-eiF2α increases ATF4 translation. ATF4-knockout mice are protected from diet-induced obesity, hypertriglyceridemia, and hepatic steatosis, as ATF4 depletion results in significantly decreased liver and white adipose tissue expression of lipogenic genes, such as PPARγ, sterol regulatory element-binding protein-1c (SREBP-1c), acetyl-CoA carboxylase (ACC), and FAS[34-36]. Taken together, these results suggest that the PERK-eiF2α-ATF4 pathway plays an important role in promoting lipogenesis, both in the liver and other tissues.
Second, the IRE1α-XBP1 pathway has been reported to be required for the maintenance of hepatic lipid homeostasis under ER stress conditions. Mice with a hepatocyte-specific deletion of IRE1α develop severe hepatic steatosis after treatment with an ER stress inducer due to the repressed expression of key metabolic transcriptional regulators, including C/EBPβ, C/EBPδ, and PPARγ, and enzymes involved in triglyceride biosynthesis[37]. IRE1α is also reportedly required for efficient apolipoprotein secretion upon disruption of ER homeostasis[37]. Moreover, the IRE1α-XBP1 pathway plays an essential role in the regulation of hepatic VLDL assembly and secretion, which is at least in part due to decreased microsomal triglyceride-transfer protein activity resulting from reduced protein disulfide isomerase expression[38]. In hepatocytes, XBP1 regulates hepatic lipogenesis by directly binding to the promoters of a subset of lipogenic genes, including SCD1, diacylglycerol acyltransferase-2 (DGAT2), and ACC2, and activating their expression[39]. Therefore, de novo lipid biosynthesis is reduced in the livers of mice with an XBP1 deletion[39].
Third, the ATF6 pathway also plays a role in stress-induced lipid accumulation. A close examination of the relationship between ATF6 activity and SREBP2-mediated lipogenesis has revealed that nuclear ATF6 interacts with the nuclear form of SREBP2, thereby antagonizing SREBP2-regulated transcription of lipogenic genes and lipid accumulation in cultured liver and kidney cells[40]. Moreover, ATF6α-knockout mice develop hepatic steatosis in response to an ER stress inducer as a result of reduced fatty acid β-oxidation and attenuated VLDL formation[41]. When fed a high-fat diet, ATF6α-deficient mice show a tendency to develop greater degrees of hepatic steatosis and glucose intolerance in association with increased expression of SREBP1c[42].
Taken together, all three proximal UPR sensors, including PERK, IRE1α, and ATF6α, can regulate lipid stores in the liver. Although the evidence summarized above provides strong support for ER stress response-induced steatosis, it remains uncertain whether a stressed UPR contributes to hepatic steatosis, or conversely, whether it is an adaptive response to restore hepatocyte function. Future studies should address this important issue.
ER stress and IR
IR is a disruption in insulin signaling in organs including the liver, fat, and muscle, and is a major characteristic of obesity, type 2 diabetes, and NAFLD. In the past decade, ER stress has emerged as an important factor that contributes to IR.
Several mechanisms can account for the impact of ER stress and UPR signaling on hepatic IR. Ozcan et al[23] have demonstrated that ER stress induces hepatic IR through IRE1α-mediated activation of c-Jun N-terminal kinase (JNK), which impairs insulin signaling through the serine phosphorylation of insulin receptor substrate-1. Moreover, tribbles 3 (TRB3), an ER stress-induced protein, was suggested to induce IR via inhibition of Akt/PKB signaling[43]. Furthermore, PERK-mediated FOXO phosphorylation is also involved in ER stress-induced IR. FOXO is the Forkhead transcription factor that mediates many of the effects of insulin on the phosphatidylinositol 3-kinase (PI3-kinase) → Akt cascade. Inhibiting FOXO1 activity has been shown to improve insulin sensitivity in genetic and diet-induced models of IR in mice[44,45]. Inhibition of PERK also improves cellular insulin responsiveness at the level of FOXO activity[46]. Further support is derived from in vivo studies in XBP+/- mice fed a high-fat diet, which exhibit IR and type 2 diabetes in conjunction with an increased ER stress response due to impaired UPR protection[23]. Db/db diabetic obese mice, which overexpress the ER chaperone oxygen-regulated protein 150 (ORP150), demonstrate improved IR, whereas silencing ORP150 in normal mice decreases insulin sensitivity[47].
NAFLD is strongly associated with hepatic IR as well as reduced whole-body insulin sensitivity[48-50]. Despite mounting evidence indicating that ER stress is responsible for hepatic IR, the contribution of ER stress to IR in NAFLD remains uncertain. Few studies have comprehensively examined these potential mechanisms in humans. One study reported that phosphorylation of eiF2α and C/EBP-homologous protein (CHOP) protein expression increased with worsening IR in 37 obese, nondiabetic individuals. However, there was no significant relationship between HOMA-IR and the expression of other ER stress factors, including spliced XBP1 mRNA and JNK phosphorylation, arguing against a causal role for ER stress in IR[48].
ER stress and inflammation
Approximately 10%-20% of patients who have NAFLD develop inflammation and fibrosis, termed NASH, which is a more progressive, inflammatory disease phenotype of NAFLD. ER stress and related signaling networks are emerging as central pathways that regulate the key features of NASH.
Several signaling pathways connect ER stress to inflammation, including (1) the production of reactive oxygen species (ROS); (2) the activation of the transcription factors nuclear factor-κB (NF-κB) and JNK; and (3) the induction of the acute-phase response.
ROS are important mediators of inflammation. Protein folding in the ER is linked to the generation of ROS and oxidative stress[51]. An increase in the protein-folding load in the ER can lead to the accumulation of ROS, which might initiate an inflammatory response. However, the PERK pathway of the UPR can activate an antioxidant program to limit the accumulation of ROS in response to ER stress via phosphorylation of nuclear factor-erythroid-derived 2-related factor 2 (Nrf2). Phosphorylated Nrf2 translocates to the nucleus and activates the transcription of a set of antioxidant and oxidant detoxifying enzymes[52,53]. Therefore, Nrf2 deletion results in the rapid onset and progression of steatohepatitis in mice fed an MCD diet or a high-fat diet[54,55]. Nrf2 activators can attenuate oxidative stress-induced liver injury in nutritional steatohepatitis[56-58]. Thus, Nrf2 activation by pharmaceutical intervention could be a new option for the prevention and treatment of steatohepatitis.
Activation of NF-κB and JNK is also involved in ER stress-induced inflammation. In response to ER stress, IRE1α binds to the adaptor protein tumor-necrosis factor-α (TNFα) receptor-associated factor 2 (TRAF2)[59]. The IRE1α-TRAF2 complex activates NF-κB and JNK, which in turn induce the synthesis of proinflammatory cytokines. Thus, IRE1α might provide a link between ER stress and inflammation[59]. Sustained ER stress can also activate NF-κB through the PERK and ATF6 branches[60-62]. Although the activation of NF-κB is detected in the MCD dietary model of steatohepatitis[63], it is presently unclear whether and how the ER stress-mediated activation of NF-κB is linked to NAFLD. Further studies will be needed to determine how ER-stress-induced signaling involving NF-κB and JNK might regulate inflammation, metabolism, cell survival, and apoptosis in NAFLD.
CREBH is another example of a protein that links ER stress to inflammatory processes. CREBH is a liver-enriched transcription factor that is activated by a regulated intramembrane proteolysis (RIP) process upon ER stress. This transcription factor transactivates genes of the acute phase response, such as C-reactive protein (CRP) and serum amyloid P-component (SAP)[64]. CREBH expression is itself strongly induced by inflammatory cytokines such as TNFα and interleukin 6 (IL-6) or by lipopolysaccharide (LPS)[64], which also contributes to the maintenance of an inflammatory state during ER stress. Thus, ER stress in the liver may be linked to systemic inflammation via the RIP-mediated mobilization of CREBH.
ER stress and apoptosis
Hepatocyte apoptosis is increased in patients with NASH, is correlated with disease severity[65], and has been proposed as a component of disease progression in NAFLD[66]. Chronic or unresolved ER stress can lead to apoptosis. Alteration in several signaling pathways are involved in ER stress-induced cell death, including (1) the induction of CHOP pathway; (2) the activation of the JNK pathway by IRE1-mediated recruitment of TRAF2; and (3) the disruption of the calcium homeostasis pathway.
CHOP, an ER-resident transcription factor that functions downstream of the transmembrane proteins PERK and ATF6, is perhaps the most well-characterized mediator of ER stress-induced cell death. Silencing CHOP reduces hepatocyte apoptosis in alcohol-induced liver disease and attenuates cholestasis-induced liver fibrosis[67,68]. However, the role of CHOP in NAFLD is paradoxical. One study demonstrated that CHOP depletion could reduce palmitate-induced apoptosis in hepatocyte cell lines, whereas MCD diet-induced liver injury was not reduced in CHOP knockout mice[69]. Moreover, another study showed that MCD diet-induced liver injury was even greater in CHOP-/- mice than in wild-type mice due to decreased cell death of activated macrophages[70]. Therefore, future studies in mice with tissue-specific CHOP deletions are needed to delineate the contribution of CHOP to the onset and progression of NASH.
The IRE1 branch of the UPR also plays an essential role in ER stress-induced apoptosis. Phosphorylated, activated mammalian IRE1 interacts with the adaptor protein TRAF2 and promotes a cascade of phosphorylation events that ultimately activates JNK[59]. Caspase-12 is also recruited by the IRE1-TRAF2 complex in ER stress-induced apoptosis in mice[71]. Moreover, IRE1 physically interacts with Bax and Bak, two proapoptotic Bcl-2 family members that promote mitochondrial-dependent cell death[72].
Perturbations in ER calcium levels are also linked to apoptosis effectors. The ER lumen is a major site of calcium storage, and calcium homeostasis is critical for maintaining both ER folding capacity and cell viability. A disruption of ER calcium homeostasis inhibits the sarco/endoplasmic reticulum ATPase (SERCA) uptake pump, reduces the folding capacity of the ER, induces ER stress, and is an important mediator of ER-associated apoptosis[73]. Moreover, sustained accumulation of calcium in the mitochondrial matrix induced by ER stress triggers mitochondrial membrane permeability and activates the apoptotic pathway[74].
Despite all of these potential and emerging mediators, the exact role of ER-induced hepatocyte apoptosis in the pathogenesis of NAFLD is not well defined. Further studies are needed to elucidate the exact pathways that mediate ER stress-induced apoptosis in the progression of NASH.
CONCLUSION
Despite the high prevalence of NAFLD in recent years, the mechanisms responsible for disease progression remains poorly understood. Chronic ER stress induces numerous intracellular pathways that can lead to NAFLD development and progression, including hepatic steatosis, systemic inflammation, and hepatocyte cell death. ER stress interferes with hepatic lipid metabolism by activating lipogenesis and limiting VLDL formation and secretion. ER stress also acts indirectly on liver triglyceride accumulation by promoting insulin resistance in both the liver and adipose tissue. Furthermore, ER stress promotes the activation of Nrf2, JNK, NF-κB, CREBH, and CHOP, which actively participate in the inflammatory process and cell death and provoke disease progression in NAFLD (Figure 2). Despite the many advances made in recent years, important questions remain. For example, is ER stress solely an adaptive response? Which of the UPR mediators are key players during the onset and progression of NAFLD? Which UPR pathway is linked with which specific cellular response? How does ER stress-induced signaling involving JNK, NF-κB, and CHOP regulate inflammation, metabolism, cell survival, and apoptosis in NAFLD? Studies that answer these questions could identify novel therapeutic strategies for the prevention and treatment of NAFLD.
Footnotes
Supported by National Key Basic Research Development Program, No. 2012CB524905; National Science and Technology Support Plan Project, No. 2012BAI06B04; National Natural Science Foundation of China, No. 30900677, No. 81070315, No. 81070366, No. 81100278, No. 81170378, No. 81230012 and No. 81270487; Zhejiang Provincial Natural Science Foundation of China, No. Y2090463 and No. Y2110026; International Science and Technology Cooperation Projects of Zhejiang Province, No. 2013C24010; Science Foundation of Health Bureau of Zhejiang Province, No. 2009A070 and No. 2012RCA026; and Specialized Research Fund for the Doctoral Program of Higher Education, No. 20090101120110
P- Reviewers: Assy N, Jun DW, Lu MY, Nowicki M, Pei ZH, Tiniakos DG, Zhang Y S- Editor: Ma YJ L- Editor: A E- Editor: Ma S
References
- 1.Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, Landt CL, Harrison SA. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–131. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 2.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 3.Fan JG. Epidemiology of alcoholic and nonalcoholic fatty liver disease in China. J Gastroenterol Hepatol. 2013;28 Suppl 1:11–17. doi: 10.1111/jgh.12036. [DOI] [PubMed] [Google Scholar]
- 4.Farrell GC, Wong VW, Chitturi S. NAFLD in Asia--as common and important as in the West. Nat Rev Gastroenterol Hepatol. 2013;10:307–318. doi: 10.1038/nrgastro.2013.34. [DOI] [PubMed] [Google Scholar]
- 5.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 6.Bugianesi E, Leone N, Vanni E, Marchesini G, Brunello F, Carucci P, Musso A, De Paolis P, Capussotti L, Salizzoni M, et al. Expanding the natural history of nonalcoholic steatohepatitis: from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology. 2002;123:134–140. doi: 10.1053/gast.2002.34168. [DOI] [PubMed] [Google Scholar]
- 7.Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med. 2010;363:1341–1350. doi: 10.1056/NEJMra0912063. [DOI] [PubMed] [Google Scholar]
- 8.Targher G, Byrne CD. Clinical Review: Nonalcoholic fatty liver disease: a novel cardiometabolic risk factor for type 2 diabetes and its complications. J Clin Endocrinol Metab. 2013;98:483–495. doi: 10.1210/jc.2012-3093. [DOI] [PubMed] [Google Scholar]
- 9.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51:679–689. doi: 10.1002/hep.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, Sanyal AJ. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134:568–576. doi: 10.1053/j.gastro.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 11.Rinella ME, Siddiqui MS, Gardikiotes K, Gottstein J, Elias M, Green RM. Dysregulation of the unfolded protein response in db/db mice with diet-induced steatohepatitis. Hepatology. 2011;54:1600–1609. doi: 10.1002/hep.24553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang DL, Wan Y, Shen W, Cao J, Sun ZX, Yu HH, Zhang Q, Cheng WH, Chen J, Ning B. Endoplasmic reticulum stress leads to lipid accumulation through upregulation of SREBP-1c in normal hepatic and hepatoma cells. Mol Cell Biochem. 2013;381:127–137. doi: 10.1007/s11010-013-1694-7. [DOI] [PubMed] [Google Scholar]
- 13.Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, Wang ZV, Zorzano A, Hill JA, Jaimovich E, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol. 2013;301:215–290. doi: 10.1016/B978-0-12-407704-1.00005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diehl JA, Fuchs SY, Koumenis C. The cell biology of the unfolded protein response. Gastroenterology. 2011;141:38–41, 41.e1-2. doi: 10.1053/j.gastro.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 16.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 17.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 18.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 21.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaplowitz N, Than TA, Shinohara M, Ji C. Endoplasmic reticulum stress and liver injury. Semin Liver Dis. 2007;27:367–377. doi: 10.1055/s-2007-991513. [DOI] [PubMed] [Google Scholar]
- 23.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 24.Wang D, Wei Y, Pagliassotti MJ. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 2006;147:943–951. doi: 10.1210/en.2005-0570. [DOI] [PubMed] [Google Scholar]
- 25.Yang L, Jhaveri R, Huang J, Qi Y, Diehl AM. Endoplasmic reticulum stress, hepatocyte CD1d and NKT cell abnormalities in murine fatty livers. Lab Invest. 2007;87:927–937. doi: 10.1038/labinvest.3700603. [DOI] [PubMed] [Google Scholar]
- 26.Sreejayan N, Dong F, Kandadi MR, Yang X, Ren J. Chromium alleviates glucose intolerance, insulin resistance, and hepatic ER stress in obese mice. Obesity (Silver Spring) 2008;16:1331–1337. doi: 10.1038/oby.2008.217. [DOI] [PubMed] [Google Scholar]
- 27.Rahman SM, Schroeder-Gloeckler JM, Janssen RC, Jiang H, Qadri I, Maclean KN, Friedman JE. CCAAT/enhancing binding protein beta deletion in mice attenuates inflammation, endoplasmic reticulum stress, and lipid accumulation in diet-induced nonalcoholic steatohepatitis. Hepatology. 2007;45:1108–1117. doi: 10.1002/hep.21614. [DOI] [PubMed] [Google Scholar]
- 28.Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, Klein S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58:693–700. doi: 10.2337/db08-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology. 2012;142:711–725.e6. doi: 10.1053/j.gastro.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Sozio MS, Liangpunsakul S, Crabb D. The role of lipid metabolism in the pathogenesis of alcoholic and nonalcoholic hepatic steatosis. Semin Liver Dis. 2010;30:378–390. doi: 10.1055/s-0030-1267538. [DOI] [PubMed] [Google Scholar]
- 31.Reddy JK, Rao MS. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol. 2006;290:G852–G858. doi: 10.1152/ajpgi.00521.2005. [DOI] [PubMed] [Google Scholar]
- 32.Bobrovnikova-Marjon E, Hatzivassiliou G, Grigoriadou C, Romero M, Cavener DR, Thompson CB, Diehl JA. PERK-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc Natl Acad Sci USA. 2008;105:16314–16319. doi: 10.1073/pnas.0808517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008;7:520–532. doi: 10.1016/j.cmet.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seo J, Fortuno ES, Suh JM, Stenesen D, Tang W, Parks EJ, Adams CM, Townes T, Graff JM. Atf4 regulates obesity, glucose homeostasis, and energy expenditure. Diabetes. 2009;58:2565–2573. doi: 10.2337/db09-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang C, Huang Z, Du Y, Cheng Y, Chen S, Guo F. ATF4 regulates lipid metabolism and thermogenesis. Cell Res. 2010;20:174–184. doi: 10.1038/cr.2010.4. [DOI] [PubMed] [Google Scholar]
- 36.Xiao G, Zhang T, Yu S, Lee S, Calabuig-Navarro V, Yamauchi J, Ringquist S, Dong HH. ATF4 protein deficiency protects against high fructose-induced hypertriglyceridemia in mice. J Biol Chem. 2013;288:25350–25361. doi: 10.1074/jbc.M113.470526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang K, Wang S, Malhotra J, Hassler JR, Back SH, Wang G, Chang L, Xu W, Miao H, Leonardi R, et al. The unfolded protein response transducer IRE1α prevents ER stress-induced hepatic steatosis. EMBO J. 2011;30:1357–1375. doi: 10.1038/emboj.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang S, Chen Z, Lam V, Han J, Hassler J, Finck BN, Davidson NO, Kaufman RJ. IRE1α-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab. 2012;16:473–486. doi: 10.1016/j.cmet.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeng L, Lu M, Mori K, Luo S, Lee AS, Zhu Y, Shyy JY. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 2004;23:950–958. doi: 10.1038/sj.emboj.7600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamamoto K, Takahara K, Oyadomari S, Okada T, Sato T, Harada A, Mori K. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol Biol Cell. 2010;21:2975–2986. doi: 10.1091/mbc.E09-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Usui M, Yamaguchi S, Tanji Y, Tominaga R, Ishigaki Y, Fukumoto M, Katagiri H, Mori K, Oka Y, Ishihara H. Atf6α-null mice are glucose intolerant due to pancreatic β-cell failure on a high-fat diet but partially resistant to diet-induced insulin resistance. Metabolism. 2012;61:1118–1128. doi: 10.1016/j.metabol.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 43.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–1577. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- 44.Nakae J, Biggs WH, Kitamura T, Cavenee WK, Wright CV, Arden KC, Accili D. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet. 2002;32:245–253. doi: 10.1038/ng890. [DOI] [PubMed] [Google Scholar]
- 45.Kim JJ, Li P, Huntley J, Chang JP, Arden KC, Olefsky JM. FoxO1 haploinsufficiency protects against high-fat diet-induced insulin resistance with enhanced peroxisome proliferator-activated receptor gamma activation in adipose tissue. Diabetes. 2009;58:1275–1282. doi: 10.2337/db08-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang W, Hietakangas V, Wee S, Lim SC, Gunaratne J, Cohen SM. ER stress potentiates insulin resistance through PERK-mediated FOXO phosphorylation. Genes Dev. 2013;27:441–449. doi: 10.1101/gad.201731.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, Ozawa K, Ogawa S, Hori M, Yamasaki Y, et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem. 2005;280:847–851. doi: 10.1074/jbc.M411860200. [DOI] [PubMed] [Google Scholar]
- 48.Kumashiro N, Erion DM, Zhang D, Kahn M, Beddow SA, Chu X, Still CD, Gerhard GS, Han X, Dziura J, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA. 2011;108:16381–16385. doi: 10.1073/pnas.1113359108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bugianesi E, Moscatiello S, Ciaravella MF, Marchesini G. Insulin resistance in nonalcoholic fatty liver disease. Curr Pharm Des. 2010;16:1941–1951. doi: 10.2174/138161210791208875. [DOI] [PubMed] [Google Scholar]
- 50.Utzschneider KM, Kahn SE. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2006;91:4753–4761. doi: 10.1210/jc.2006-0587. [DOI] [PubMed] [Google Scholar]
- 51.Santos CX, Tanaka LY, Wosniak J, Laurindo FR. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signal. 2009;11:2409–2427. doi: 10.1089/ars.2009.2625. [DOI] [PubMed] [Google Scholar]
- 52.Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol. 2006;38:317–332. doi: 10.1016/j.biocel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 53.Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem. 2004;279:20108–20117. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- 54.Okada K, Warabi E, Sugimoto H, Horie M, Gotoh N, Tokushige K, Hashimoto E, Utsunomiya H, Takahashi H, Ishii T, et al. Deletion of Nrf2 leads to rapid progression of steatohepatitis in mice fed atherogenic plus high-fat diet. J Gastroenterol. 2013;48:620–632. doi: 10.1007/s00535-012-0659-z. [DOI] [PubMed] [Google Scholar]
- 55.Chowdhry S, Nazmy MH, Meakin PJ, Dinkova-Kostova AT, Walsh SV, Tsujita T, Dillon JF, Ashford ML, Hayes JD. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic Biol Med. 2010;48:357–371. doi: 10.1016/j.freeradbiomed.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 56.Shimozono R, Asaoka Y, Yoshizawa Y, Aoki T, Noda H, Yamada M, Kaino M, Mochizuki H. Nrf2 activators attenuate the progression of nonalcoholic steatohepatitis-related fibrosis in a dietary rat model. Mol Pharmacol. 2013;84:62–70. doi: 10.1124/mol.112.084269. [DOI] [PubMed] [Google Scholar]
- 57.Okada K, Warabi E, Sugimoto H, Horie M, Tokushige K, Ueda T, Harada N, Taguchi K, Hashimoto E, Itoh K, et al. Nrf2 inhibits hepatic iron accumulation and counteracts oxidative stress-induced liver injury in nutritional steatohepatitis. J Gastroenterol. 2012;47:924–935. doi: 10.1007/s00535-012-0552-9. [DOI] [PubMed] [Google Scholar]
- 58.Gupte AA, Lyon CJ, Hsueh WA. Nuclear factor (erythroid-derived 2)-like-2 factor (Nrf2), a key regulator of the antioxidant response to protect against atherosclerosis and nonalcoholic steatohepatitis. Curr Diab Rep. 2013;13:362–371. doi: 10.1007/s11892-013-0372-1. [DOI] [PubMed] [Google Scholar]
- 59.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 60.Yamazaki H, Hiramatsu N, Hayakawa K, Tagawa Y, Okamura M, Ogata R, Huang T, Nakajima S, Yao J, Paton AW, et al. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J Immunol. 2009;183:1480–1487. doi: 10.4049/jimmunol.0900017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol. 2004;24:10161–10168. doi: 10.1128/MCB.24.23.10161-10168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol Cell Biol. 2003;23:5651–5663. doi: 10.1128/MCB.23.16.5651-5663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dela Peña A, Leclercq I, Field J, George J, Jones B, Farrell G. NF-kappaB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology. 2005;129:1663–1674. doi: 10.1053/j.gastro.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 64.Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 65.Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, Gores GJ. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 66.Wieckowska A, Zein NN, Yerian LM, Lopez AR, McCullough AJ, Feldstein AE. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44:27–33. doi: 10.1002/hep.21223. [DOI] [PubMed] [Google Scholar]
- 67.Ji C, Mehrian-Shai R, Chan C, Hsu YH, Kaplowitz N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol Clin Exp Res. 2005;29:1496–1503. doi: 10.1097/01.alc.0000174691.03751.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tamaki N, Hatano E, Taura K, Tada M, Kodama Y, Nitta T, Iwaisako K, Seo S, Nakajima A, Ikai I, et al. CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am J Physiol Gastrointest Liver Physiol. 2008;294:G498–G505. doi: 10.1152/ajpgi.00482.2007. [DOI] [PubMed] [Google Scholar]
- 69.Pfaffenbach KT, Gentile CL, Nivala AM, Wang D, Wei Y, Pagliassotti MJ. Linking endoplasmic reticulum stress to cell death in hepatocytes: roles of C/EBP homologous protein and chemical chaperones in palmitate-mediated cell death. Am J Physiol Endocrinol Metab. 2010;298:E1027–E1035. doi: 10.1152/ajpendo.00642.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Malhi H, Kropp EM, Clavo VF, Kobrossi CR, Han J, Mauer AS, Yong J, Kaufman RJ. C/EBP homologous protein-induced macrophage apoptosis protects mice from steatohepatitis. J Biol Chem. 2013;288:18624–18642. doi: 10.1074/jbc.M112.442954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem. 2001;276:13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- 72.Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, Brandt GS, Iwakoshi NN, Schinzel A, Glimcher LH, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- 73.Luciani DS, Gwiazda KS, Yang TL, Kalynyak TB, Bychkivska Y, Frey MH, Jeffrey KD, Sampaio AV, Underhill TM, Johnson JD. Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes. 2009;58:422–432. doi: 10.2337/db07-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Deniaud A, Sharaf el dein O, Maillier E, Poncet D, Kroemer G, Lemaire C, Brenner C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27:285–299. doi: 10.1038/sj.onc.1210638. [DOI] [PubMed] [Google Scholar]