

Abstract
AIM: To investigate the influence of Trichuris muris (T. muris) infection in a mouse model of genetic susceptibility to inflammatory bowel disease, Mdr1a-/-.
METHODS: Mdr1a-/- mice were housed under specific pathogen free conditions to slow the development of colitis and compared to congenic FVB controls. Mice were infected with approximately 200 embryonated ova from T. muris and assessed for worm burden and histological and functional markers of gut inflammation on day 19 post infection.
RESULTS: Mdr1a-/- mice exhibited a marked increase in susceptibility to T. muris infection with a 10-fold increase in colonic worm count by day 19 pi compared to FVB controls. Prior to infection, Mdr1a-/- exhibited low-level mucosal inflammation with evidence of an enhanced Th1 environment. T. muris infection accelerated the progression of colitis in Mdr1a-/- as evidenced by marked increases in several indicators including histological damage score, mucosal CD4+ T-cell and DC infiltration and dramatically increased production of pro-inflammatory cytokines.
CONCLUSION: These data provide further evidence of the complex interaction between T. muris and an inflammatory bowel disease (IBD)-susceptible host which may have relevance to the application of helminth therapy in the treatment of human IBD.
Keywords: Helminth, Colitis, Inflammatory bowel disease, P-glycoprotein, Mdr1a
Core tip: This study investigates the interaction between the helminth parasite Trichuris muris (T. muris) and Mdr1a-/- mice, a genetic model of inflammatory bowel disease linked to deficiency of a key transporter protein in the gut barrier. The main findings are that (1) Mdr1a mice exhibit dramatically increased susceptibility to worm infection compared to congenic controls and (2) challenge with T. muris induces severe pathological changes consistent with a marked exacerbation of colitis in this model with preliminary evidence pointing to worm persistence as a driver of this effect. These findings will be of interest in the emerging field of helminth therapy in inflammatory bowel disease (IBD) providing further evidence of the complexity of worm interaction with an IBD-susceptible host.
INTRODUCTION
The outcome of infection with the gastrointestinal nematode worm Trichuris, which occurs in a range of mammalian species, is genetically determined and involves subversion of host Th1 and Th2 immune responses to allow the worm to survive and colonise the gut[1,2]. This has led to the suggestion that helminths may have the potential to reduce dysregulated gut inflammation in human diseases such as Crohn’s disease[3]. Clinical trials using the pig worm Trichuris suis (T. muris) in patients with active Crohn’s disease or ulcerative colitis have reported a significant improvement in disease symptoms in a majority of patients receiving this treatment although the numbers of subjects tested remains relatively small[4,5]. There is currently a paucity of information on the mechanisms by which helminths regulate inflammation in man and, therefore, the possible impact of genetic variability on susceptibility and response to helminth challenge, including the potential for adverse effects remains uncertain[6]. Studies in a range of murine models of colitis have shown variable responses to infection with helminth species such as H. polygyrus, H. diminuta and T. spiralis including amelioration and exacerbation of disease[7]. To date, there is little information on the effects of Trichuris species in murine colitis with a single study showing severe inflammation in IL-10-/- mice infected with T. muris[8], although the precise contribution of the infection to the inflammation that develops spontaneously in this model was unclear.The aim of the present study was to explore the impact of T. muris infection on the intestinal inflammation seen in the Mdr1a-/- mouse model of inflammatory bowel disease (IBD). These mice develop spontaneous colitis as a result of a primary defect in the gut epithelial barrier caused by the deletion of the Mdr1a gene which encodes P-glycoprotein, a xenobiotic transporter located in the apical membrane of epithelial cells[9,10]. Recent studies have shown similarities between the transcriptomic changes occurring during early development of colitis in Mdr1a-/-[11] and those associated with T. muris infection in susceptible mouse strains[12] making this an interesting model in which to investigate interaction of the parasite with an IBD-susceptible host.
MATERIALS AND METHODS
Animals
Mdr1a-/- mice (KO) and the congenic background strain FVB (WT) were obtained from Taconic Farms (Germantown, New York) and bred and maintained under specific pathogen free condition in the Biological Services Facility of the University of Manchester. Mice had been backcrossed for 7 generations onto the FVB background and were periodically genotyped by polymerase chain reaction. Mice were housed individually in isolator cages with access to filtered water and standard chow and routinely monitored for outward signs of bowel disease (diarrhea, weight loss and rectal bleeding). Mice used in these studies were between 5 and 13 wk of age on infection with T. muris. A total of 43 animals (21 FVB and 22 Mdr1a-/-) were used in these studies. All animal studies conformed to current United Kingdom Home Office legislation.
Parasites
The maintenance of T. muris and the method used for infection were as described by Wakelin[13]. Mice were infected with approximately 200 embryonated eggs by oral gavage at day 0. Worm burden was assessed at day 19 post infection (pi) by counting the number of worms present in the caecum and first 4 cm of large intestine as described previously[14]. For a low dose, 20 egg infection, embryonated eggs were counted individually into Eppendorfs and dispensed to mice at day 0. T. muris excretory/secretory protein (T. muris E/S) was produced as previously described[13].
Isolation of mesenteric lymph nodes
Mesenteric lymph nodes (MLNs) were removed on day 19 pi and placed in 10 mL complete RPMI-1640 (PAA) containing 10% foetal calf serum (FCS) (PAA, gold- inactivated), 100 U/mL penicillin (Gibco), 100 μg/mL streptomycin (Gibco) and 1% L-glutamine (Gibco) on ice. MLNC were washed three times and resuspended at 5 × 106 cells/mL in RPMI 1640 medium (Gibco) containing 10% fetal calf serum, antibiotics as above, 2 mmol/L L-glutamine (Gibco). The final cell suspension (1 mL) was placed in duplicate wells of a 48 well culture plate (Costar). Cell suspensions were stimulated with concanavalin A (ConA) 2.5 μg/mL or 50 μg/mL 4 h T. muris E/S and incubated at 37 °C in 5% CO2 . Con A stimulated mesenteric lymph node cell culture was recovered after 24 h. Similarly, after 48 h the T. muris E/S stimulated cell culture was harvested and stored at -20 °C for cytokine analysis.
Measurement of parasite specific antibody
Parasite specific IgG1 and IgG2a levels in mouse serum was determined by ELISA as described previously[15]. Briefly, microplates were coated with adult T. muris excretory/secretory (E/S) antigen at 5 μg/mL (50 μL/well) in carbonate buffer pH 9.6. After washing with 0.05% Tween 20 in phosphate buffered saline (PBS-T), plates were blocked with PBS-T containing 2% bovine serum albumin. Serial dilutions of mouse serum from 1:20 to 1:2560 in PBS-T were added and plates were incubated for 1 h at 37 °C. After washing 50 μL/well of biotinylated rat anti-IgG1 (Serotec Ltd, Oxford, United Kingdom) or rat anti-IgG2a (BD Pharmingen, Oxford, United Kingdom) were added followed by streptavidin conjugated peroxidase and TMB detection at 405 nm.
Cytokine analysis
The cytokines interleukin (IL)-5, IL-12p40 and interferon (IFN)-γ were quantified by an in-house sandwich ELISA using capture and detection antibodies from BD Pharmingen (Oxford, United Kingdom). Plates were washed with PBS-T and blocked with 10% fetal calf serum in phosphate buffered saline. Standard curves prepared with recombinant murine cytokine standards were used to quantify cytokines in experimental samples with OD at 405 nm on a MRXII microplate reader. IL-13 and tumor necrosis factor (TNF)-α were analysed using commercial ELISA kits obtained from R and D Systems (Abingdon, United Kingdom) according to the manufacturer’s instructions.
Colonic epithelial cell isolation
Whole mouse colon was isolated, split longitudinally and washed in RPMI 1640 medium (PAA Laboratories, Yeovil, United Kingdom) supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin. Colon was cut into small pieces (approximately 5 mm) and incubated in 10 mL Dulbecco’s modified Eagle medium (Invitrogen, Paisley, United Kingdom) containing 20% FCS (PAA Laboratories, Yeovil, United Kingdom), 100 U/mL penicillin, 100 μg/mL streptomycin and 1 mg/mL Dispase (Sigma-Aldrich) with gentle rotation at 37 °C for 90 min. Epithelial cells were released by gentle aspiration with a pipette. Following brief centrifugation (750 g; 5 min), the epithelial cell pellet was washed in RPMI 1640 medium and re-pelleted. One milliliter of TRIzol reagent (Invitrogen, Paisley, United Kingdom) was added and RNA isolated according to the manufacturer’s instructions.
Gene expression analysis
Colonocyte RNA was reverse transcribed using a commercial first-strand cDNA synthesis kit (Amersham Biosciences, Little Chalfont, United Kingdom) using random hexamers. Relative quantification of gene expression was performed using assays designed by ProbeFinder software (www.universalprobelibrary.com); this gives gene-specific primer pairs combined with the appropriate hydrolysis probes [5-fluorescein (FAM)-labeled; 3-dark quencher dye] from the Universal Probe Library (UPL; Roche Diagnostics, Lewes, United Kingdom). Assays were performed using a Lightcycler480 (Roche Diagnostics) in a 20 μL volume with 200 nmol/L of each primer, 100 nmol/L of the UPL probe and approximately 0.3 μg of cDNA. Gene expression was quantified relative to β-actin.
Histology
Colon from mdr1a-/- or FVB controls was assessed histologically for evidence of inflammation and tissue injury using a previously described grading system[11]. Three main grading criteria with 4 different subgrades of severity (0-3) in each were applied. The resulting combined score (maximum 9) indicates disease severity with 0-normal, 1-3-mild changes, 3-6-moderate changes, 7-9-severe changes.
Immunohistochemistry
Samples of large intestine near to the caecum were obtained from naïve or T. muris infected (day 19) mdr1a-/- and FVB mice were snap-frozen and stored in liquid nitrogen until analysis. Cryostat frozen sections (6 μm) were fixed in 4% paraformaldehyde (for CD4+ staining) or acetone (for dendritic cells, DC). For CD4+, sections were incubated with 5 μg/mL biotinylated CD4 antibody (Clone H129.19; BD Pharmingen), followed by Vectastain ABC kit (Vector Laboratories, Peterborough, United Kingdom) and colour development with 3,3’-diaminobenzidine. Sections were counterstained with haematoxylin QS. For DC, sections were rehydrated in PBS and incubated with biotinylated anti-CD11c (BD Biosciences) and anti-cytokeratin (Sigma-Aldrich). Following incubation with Vectastain ABC kit (Vector Laboratories, Peterborough, United Kingdom), slides were treated with tyramide amplification reagents (PerkinElmer) and washed and counterstained with the nuclear stain 4′,6′-diamidino-2-phenylindole.
Statistical analysis
All data are presented as mean ± SD with n indicating the number of mice used. Statistical comparisons were performed by either t test or ANOVA with Dunnett’s post test for multiple comparisons using GraphPad Prism 3.02 software.
RESULTS
Mdr1a-/- mice exhibit enhanced susceptibility to T. muris infection
Following infection of Mdr1a- mice or the congenic background strain FVB with approximately 200 embryonated T. muris eggs on day 0, the worm burden in the large intestine was analysed in both groups on day 19 pi (Figure 1). Previous evidence suggests that resistant mouse strains generally expel T. muris by day 19 pi while susceptible strains will retain worms in the large intestine[16]. The worm count in FVB control mice was relatively low (11 ± 7) consistent with this strain having a resistant phenotype. In marked contrast, mice lacking the Mdr1a gene showed a 10-fold higher worm count (134 ± 28, P < 0.01) indicative of worm retention and a significantly increased susceptibility to T. muris infection (Figure 1). Infection with T. muris is associated with a strong anti-parasite antibody response which is suggested as a useful indicator of the Th1/Th2 balance and, therefore the resistance or susceptibility to parasite infection[17]. Figure 2 shows the parasite-specific IgG1 and IgG2a antibody response in the serum of naïve and T. muris infected FVB control and mdr1a-/- mice on day 19 pi Sera from infected FVB and mdr1a-/- mice contained parasite-specific IgG1 at equivalent levels. However, consistent with the increased susceptibility to T. muris infection seen in Mdr1a-/-, levels of anti-Trichuris IgG2a were only increased in the Mdr1a-/- mouse.
Figure 1.
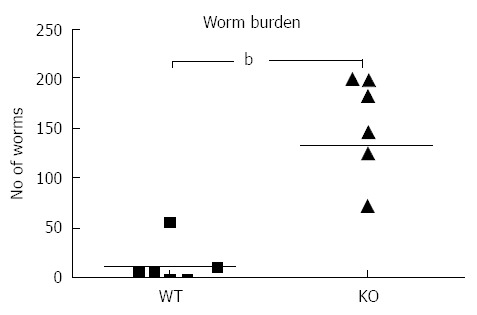
Mdr1a-/- (KO) mice show increased susceptibility to Trichuris muris infection compared to FVB controls (WT). Data shows large intestinal worm burden measured on day 19 following infection with approximately 200 embryonated Trichuris muris eggs. Each data point represents a single mouse (n = 6 in each group); the horizontal bar denotes the mean for each group. bP < 0.01.
Figure 2.
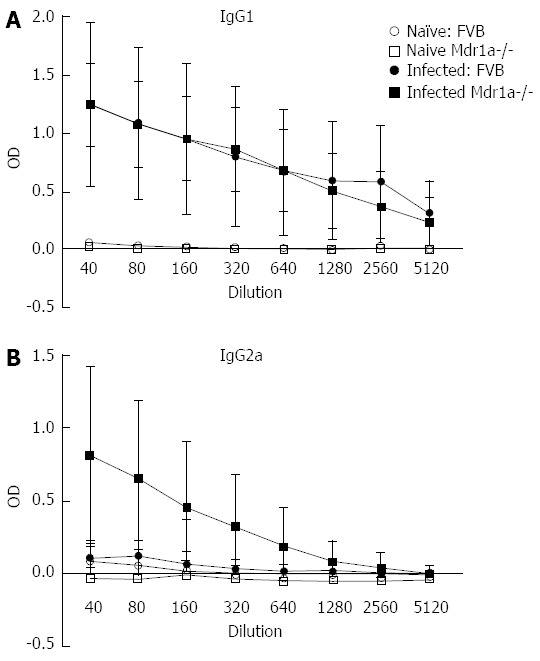
Parasite-specific IgG1 and IgG2a antibody responses in the serum of naïve and Trichuris muris infected FVB (WT) and mdr1a-/- (KO) mice. Serum was taken from mice at day 19 post infection. Data shows mean ± SD for n = 5-7 animals in each group. OD: Optical density.
The increased retention of worms in Mdr1a-/- compared to FVB control could be the result of an enhanced Th1 environment. Production of the Th1 cytokine, IFNγ by MLN from naïve animals was higher in Mdr1a-/- than FVB (562 ± 561 pg/mL vs 149 ± 108 pg/mL, n = 5; see Figure 3) but this did not reach statistical significance (P > 0.05). However, analysis of colonocytes isolated from these mice showed significant up-regulation of genes that are known to be IFNγ-dependent. Expression of Indoleamine 2, 3 dioxygenase (IDO), a tryptophan catabolizing enzyme linked to susceptibility to T. muris infection[18] and interferon gamma inducible protein-10, (IP-10; CXCL-10) were markedly higher (30- and 7-fold respectively) in naïve Mdr1a-/- mice compared to FVB controls (Figure 4). These data provide evidence that Mdr1a-/- mice have a pre-existing Th1 environment prior to infection with T. muris.
Figure 3.
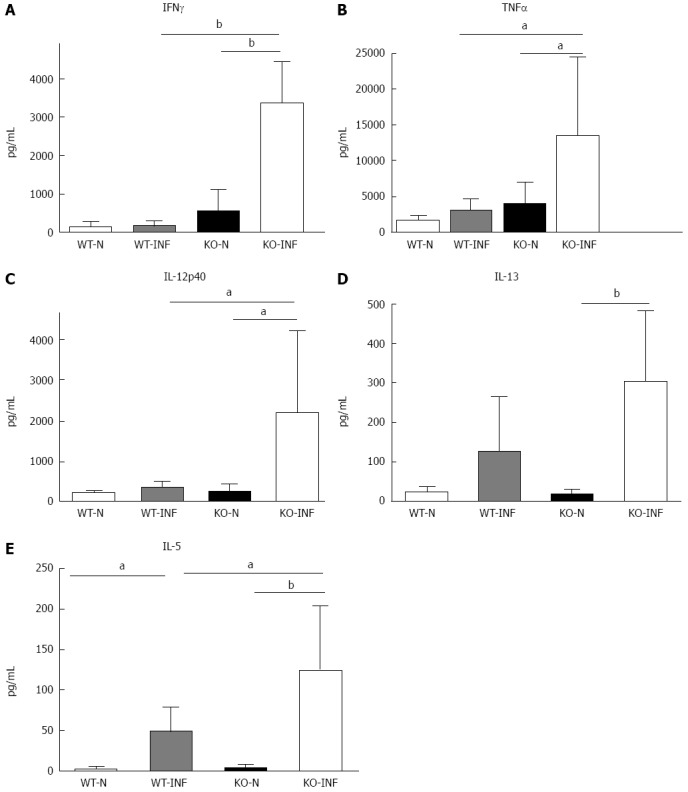
Cytokine production by mesenteric lymph nodes from naïve and Trichuris muris infected FVB (WT) and mdr1a-/- (PGP-KO) mice. Levels of Th1 [interferon (IFN)γ, A; interleukin (IL)-12p40, C] and Th2 (IL-13, D; IL-5, E) and the pro-inflammatory cytokine [tumor necrosis factor (TNF)α, B] were measured in mesenteric lymph nodes (MLN) following in vitro stimulation with Trichuris muris (T. muris) E/S antigen. MLN were isolated on day 19 post infection in T. muris infected groups. Data is shown as mean + SD for n = 5 animals in each group aP < 0.05; bP < 0.01.
Figure 4.
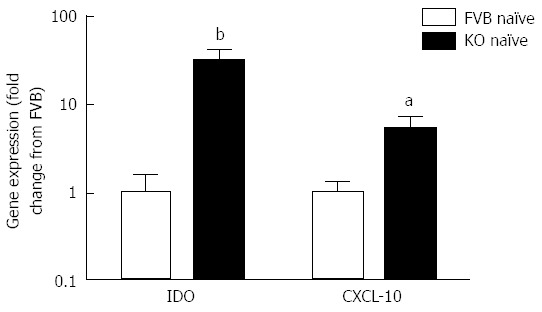
Expression of interferon γ-dependent genes IDO and CXCL-10 in colonocytes from naïve Mdr1a-/- and FVB mice. mRNA expression was measured relative to β-actin in each case. Data is expressed as fold change in expression in Mdr1a-/- colonocytes relative to FVB controls and is shown as mean ± SD for n = 4 preparations for each group. aP < 0.05, bP < 0.01 vs FVB groups.
T. muris infection increases the severity of colitis in Mdr1a-/- mice
We next investigated the impact of T. muris infection on colitis development in Mdr1a-/- mice. T. muris infection in FVB mice produced no significant clinical or histological evidence of colitis (Figure 5); both naïve and infected animals had normal colonic mucosal morphology with the exception of one mouse with a minor inflammatory infiltrate on day 19 post infection (Figure 5B). In contrast, there was a major difference in colitis indicators in age-matched Mdr1a-/-. Naïve Mdr1a-/- showed a modest disease score (1.5) indicative of an existing low-level mucosal inflammation and consistent with the spontaneous, age-dependent development of colitis in this model (Figure 5C). However, infection of Mdr1a-/- with T. muris, was associated with a dramatic increase in disease score (3.6) with the colon showing loss of architecture, a severe mixed inflammatory infiltrate and evidence of crypt abscess (Figure 5D and E). The pro-inflammatory effect of T. muris infection in Mdr1a-/- was also evident in the cytokine response of MLN isolated at day 19 pi (Figure 3). Production of the Th1 cytokine IFNγ in FVB controls was not significantly increased by T. muris infection (Figure 3A) consistent with the ability of this mouse strain to expel T. muris. However, infection of Mdr1a-/- mice led to a dramatic increase in IFNγ production with levels approximately 7 fold higher than naïve animals (P < 0.01). A second Th1-associated cytokine, IL-12p40 and the pro-inflammatory cytokine TNFα showed a similar pattern with marked increases in infected Mdr1a-/- but not infected FVB mice. (Figure 3B and C). A different pattern was observed for Th2 cytokines (Figure 3D and E). IL-13 and IL-5 were elevated in both FVB and Mdr1a-/- following T. muris infection (Figure 3D and E) and while the increase was somewhat greater in Mdr1a-/-, this was significant only for IL-5 (Mdr1a-/-: 124.3 ± 78.7 pg/mL vs FVB: 48.3 ± 29.7 pg/mL, P < 0.05). Taken together, these data shows that whilst both FVB and Mdr1a-/- mount a Th2 reponse pi a significantly heightened Th1/pro-inflammatory response is only seen in the Mdra1-/- mouse. Consistent with the significantly greater histological disease score seen in Figure 5, infected Mdr1a-/- mice exhibited a significantly greater intestinal CD4+ T cell infiltrate compared to both infected wild type mice and uninfected Mdr1a-/- mice (Figure 6). Likewise dendritic cells appeared more numerous in the gut tissue from infected Mdr1a-/- mice compared to uninfected mice and infected wild type mice, indicative of a more inflamed environment (Figure 7). One possibility to explain these findings is that the worsened pathology in infected Mdr1a-/- mice may simply be a reflection of the inflammatory response associated with greater exposure to worms rather an increase in severity of the underlying colitis present in this model. To investigate this possibility, mdr1a-/- and FVB mice were infected with a low dose T. muris infection. Low level infections established from 20 eggs are not expelled from any mouse strain[19] thus ensuring more equivalent antigen exposure between mouse strains with different susceptibility to T. muris infection. In this situation, both FVB and Mdr1a-/- strains harboured worms (4 ± 3 and 10 ± 3; n = 5 respectively) at day 19 post infection. This is in marked contrast to high dose infection where only Mdr1a-/- mice harboured worms at this time point (Figure 1). Importantly, despite the similarity in worm burden, gut pathology in Mdr1a-/- was still significantly worse as evidenced by the high disease score (mean score: infected Mdr1a-/- 4.1; infected FVB 1.4) consistent with T. muris accelerating the disease process in these animals.
Figure 5.
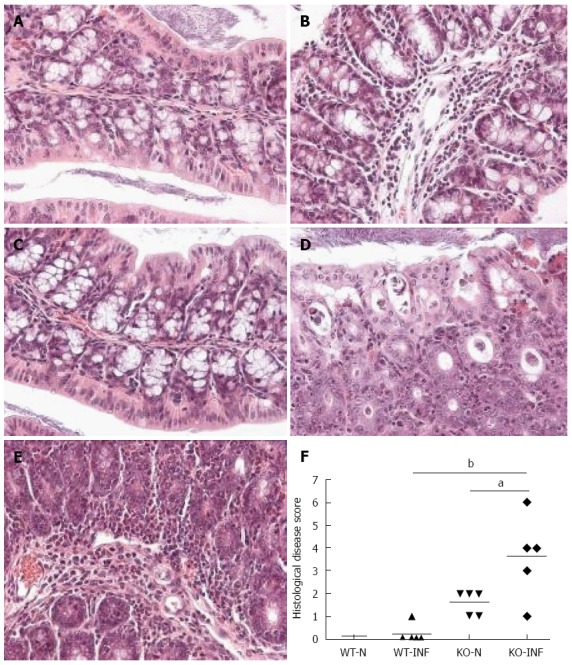
Histological analysis of proximal colon from FVB (WT) and mdr1a-/- (PGP-KO) mice. A-E: H and E stained sections from proximal colon of WT-N (A), WT-INF (B), KO-N (C) and KO-INF (D-E). Tissues were isolated on day 19 post-infection in Trichuris muris (T. muris) infected mice; F: Histological disease scores for colon from naïve (WT-N; KO-N) and T. muris infected (WT-INF; KO-INF) mice based on the grading system described by Collett et al[11], 2008. Bar shows mean values for each group aP < 0.05, bP < 0.01, n = 5 for each group.
Figure 6.
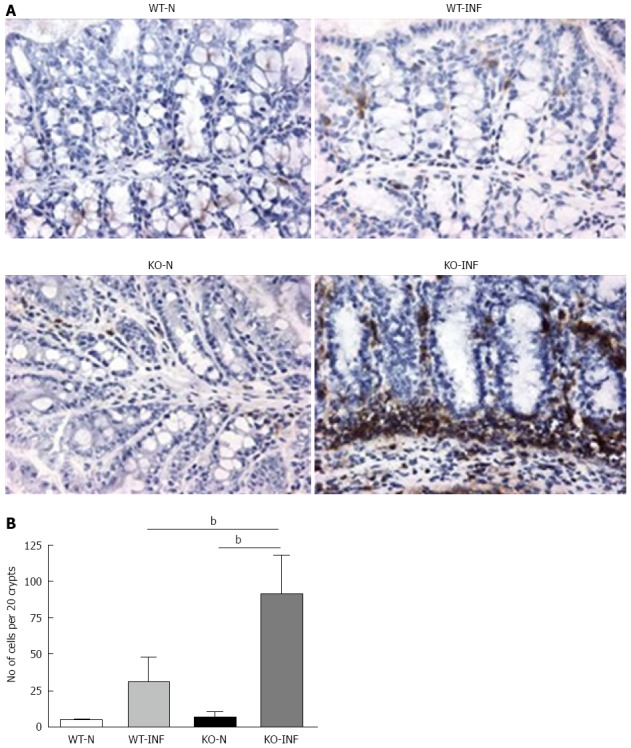
CD4+ T-cell infiltration associated with Trichuris muris infection in the proximal colon of FVB (WT) and mdr1a-/- (PGP-KO) mice. A: Immunohistochemical staining for CD4+ T cells in naïve (WT-N; KO-N) and Trichuris muris (T. muris) infected (WT-INF; KO-INF) WT and PGP-KO mice; B: Mean data (± SD) from 5 animals in each group showing CD4+ T cells present per 20 crypts units. Tissues were isolated on day 19 post-infection in T. muris infected mice, bP < 0.01.
Figure 7.
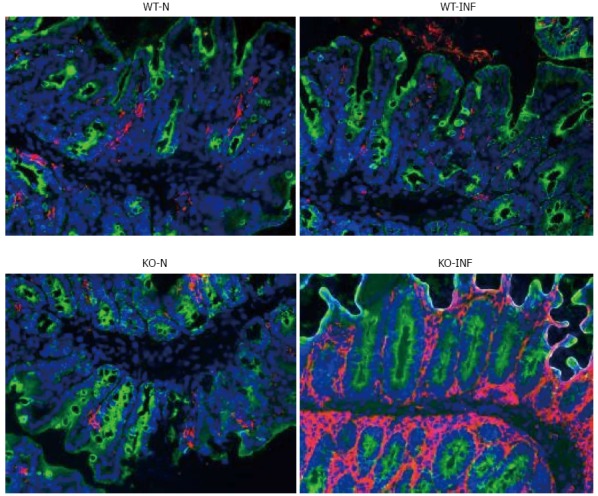
Dendritic cell infiltration associated with Trichuris muris infection in the proximal colon of FVB (WT) and mdr1a-/- (PGP-KO) mice. Fluorescence staining (pink) for dendritic cells in frozen sections of proximal colon from naïve (WT-N; KO-N) and Trichuris muris (T. muris) infected (WT-INF; KO-INF) mice. Tissues were removed on day 19 post infection in T. muris infected mice. Original magnification × 20. Images are representative of tissues from 5 mice in each group.
DISCUSSION
This study provides further evidence of the complex interaction between T. muris and IBD which may have relevance to the proposed use of helminth therapy in the treatment of IBD. Mice with a genetic predisposition to IBD, mediated by deletion of an epithelial transporter (p-glycoprotein) that forms a key part of the gut barrier, were shown to have a markedly increased susceptibility to infection with T. muris compared to the congenic background strain that was resistant to infection. The most likely driver of increased susceptibility to infection in Mdr1a-/- would seem to be an enhanced Th1 environment. A recent study by Collett et al[11] showed upregulation in expression of a range of IFNγ-dependent genes in Mdr1a-/- colon including IDO, T-cell specific GTPase and MHC Class II subtypes that precedes the development of active colonic inflammation. This is consistent with the present observation that expression of IDO and CXCL-10 is elevated in colonocytes from Mdr1a-/- mice exhibiting little or no gut pathology. Similar increases in expression of IFNγ-dependent genes are found in T. muris-susceptible mouse strains[12] while neutralisation of IFNγ effectively reversed susceptibility to infection by promoting worm expulsion[20-22].
The Mdr1a-/- strain is characterised by spontaneous and relatively slow development of colitis[9,11] and the animals used in the present study were at an early stage with evidence of modest inflammatory changes. The finding that challenge with T. muris rapidly induced severe pathological changes consistent with an advanced stage of colitis in these animals was somewhat unexpected in light of evidence that infection with different parasitic helminths can prevent or ameliorate colonic inflammation[23].
Nevertheless, while a majority of studies report beneficial effects, there have been reports of enhanced disease following helminth infection. Infection with Hymenolepis diminuta resulted in a significant exacerbation of oxalazone-induced colitis in mice[24,25] despite evidence that the parasite prevents DNBS-induced colitis[26]. Similarly, Heligmosomoides polygyrus bakeri enhances gut inflammation induced by enteric infection with Citrobacter rodentium[27] but has protective effects in a number of other colitis models[28,29]. Surprisingly, there is little information on T. muris in mouse models despite the fact that the porcine strain of this parasite (T. suis) has been used in the majority of human trials of helminth therapy[30]. Wilson et al[8] showed development of a lethal colitis in IL-10-/- mice infected with T. muris although the extent to which the parasite enhanced the colitis that develops spontaneously in these mice is unclear. A recent study showing close transcriptional similarities between human colitis and the response to T. muris infection, including changes in gene expression that could exacerbate inflammation, has urged caution in the therapeutic use of helminths[6]. The present study adds some support to this view suggesting interdependence between genetic predisposition to colitis and susceptibility to T. muris infection, most likely linked to the prevailing immune environment that determines the degree to which worms persist and their overall impact on mucosal inflammation. This could at best compromise the efficacy of worm therapy but, at worst, potentially result in exacerbation of the disease as demonstrated here. For example, the prevailing immune profile will clearly depend on the type of IBD present; Crohn’s disease is characteristically a Th1 driven, IFNγ-dominated inflammation while ulcerative colitis is primarily Th2-mediated with little evidence of an increased IFNγ response[31].
Key questions remain regarding the precise mechanism by which T. muris exacerbates colitis in Mdr1a-/-. These relate to whether the effects of T. muris infection vary according to the stage of the disease (e.g., early development or established colitis) and whether the exacerbation of colitis depends on worm persistence, number or both. The involvement of the IL-17/IL-23 pathway in the responses observed is also worthy of further investigation given recent evidence of its role in both IBD pathogenesis and host response to helminth infection[32,33].
The relevance of these findings to the use of worm therapy in the treatment of IBD in man is difficult to determine at this stage. The pigworm T. suis may not be able to establish for any length of time in the human colon due to species incompatibility and the relatively few clinical trials conducted so far have not reported significant safety issues[7,34]. However, the potential for pathological effects in man continues to be raised[35] and information on the level and persistence of worms following human administration and whether this varies across individuals is necessary but not currently available. The present study in Mdr1a-/- is relevant in that it shows that a predisposition to T. muris infection is associated with increased inflammation rather than amelioration. In this respect, the preliminary observation that low dose challenge with T. muris, which produces a persistent infection but with low worm burden, produced a similar enhancement of colitis to that seen with high dose in Mdr1a is intriguing. Further studies are clearly required but this could indicate that persistence of worms rather than worm burden per se may be a key factor in determining whether effects on host inflammation are beneficial or detrimental.
In conclusion, these data, taken together with similar studies in man and other animal model of IBD support the view that the impact of helminths on IBD in a given situation may be difficult to predict because the balance between disease amelioration and exacerbation will depend on a range of factors including species compatibility, the prevailing immune environment and host genetic susceptibility.
COMMENTS
Background
Preliminary clinical trials suggest that helminths such as Trichuris spp. may have therapeutic potential in the treatment inflammatory bowel disease (IBD) in man. The precise mechanisms are unclear although this may be related to subversion of host Th1 and Th2 responses which allows the worm to survive and colonise the gut.
Research frontiers
The success and acceptance of helminths as a treatment for IBD in man will depend on the ability to target therapy to individuals where the risk/benefit is clearly defined. Currently, there is a lack of information on the impact of host genetic variability on susceptibility and response to infection with Trichuris and the potential for adverse effects. In this study, the authors investigate the interaction of Trichuris with mice that are predisposed to IBD due to lack of the epithelial transporter gene Mdr1a.
Innovations and breakthroughs
A novel finding was that Mdr1a-/- mice have a much greater susceptibility to Trichuris infection than congenic controls resulting in higher numbers of worms that persist for longer. This was associated with a marked exacerbation of gut inflammation rather than amelioration of disease as might have been expected. Preliminary evidence using worm infection at a lower doses points to worm persistence rather than worm number as being the main driver of increased disease severity, although further studies are needed to confirm this observation.
Applications
These findings will be of interest in the emerging field of helminth therapy in IBD providing further evidence of the complexity of worm interaction with an IBD-susceptible host. This study in mice highlights the unpredictability of the effects of helminth infection in hosts that have a genetic susceptibility to inflammatory bowel disease. The relevance of the current findings to the therapeutic use of Trichuris in human inflammatory bowel disease is unclear, however they indicate the need for further studies to determine how genetic factors influence the level and persistence of Trichuris infection and its impact on gut inflammation in man.
Terminology
IBD is characterized by dysregulation of mucosal immunity associated with an inappropriate response to the gut microbiota and has a significant genetic component. Trichuris is an intestinal helminth parasite that infects a range of mammalian species, modulating the host immune system to survive and colonise the intestine. The Mdr1a gene encodes a common xenobiotic transporter, P-glycoprotein which is an important component of the gut epithelial barrier and appears to be important in regulating interactions with the gut flora. Mdr1a-/- mice lack P-glycoprotein in the intestinal epithelium and spontaneously develop inflammatory bowel disease.
Peer review
The investigation has profound therapeutical implication highlighting the new approaches to treatment of autoimmune diseases such as Crohn’s disease. Exploring the effect of helminth therapy in this disease by following the cytokine expression of Th1 and Th2 immune responses as well as CD4+ and dendritic cells they showed the complexity of processes in worm infection especially in IBD-susceptible host.
Footnotes
P- Reviewers: Iijima H, Liu ZJ, Maric I S- Editor: Cui XM L- Editor: A E- Editor: Wang CH
References
- 1.Cliffe LJ, Grencis RK. The Trichuris muris system: a paradigm of resistance and susceptibility to intestinal nematode infection. Adv Parasitol. 2004;57:255–307. doi: 10.1016/S0065-308X(04)57004-5. [DOI] [PubMed] [Google Scholar]
- 2.Else KJ. Have gastrointestinal nematodes outwitted the immune system? Parasite Immunol. 2005;27:407–415. doi: 10.1111/j.1365-3024.2005.00788.x. [DOI] [PubMed] [Google Scholar]
- 3.Wang LJ, Cao Y, Shi HN. Helminth infections and intestinal inflammation. World J Gastroenterol. 2008;14:5125–5132. doi: 10.3748/wjg.14.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Summers RW, Elliott DE, Urban JF, Thompson R, Weinstock JV. Trichuris suis therapy in Crohn’s disease. Gut. 2005;54:87–90. doi: 10.1136/gut.2004.041749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Summers RW, Elliott DE, Urban JF, Thompson RA, Weinstock JV. Trichuris suis therapy for active ulcerative colitis: a randomized controlled trial. Gastroenterology. 2005;128:825–832. doi: 10.1053/j.gastro.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Levison SE, McLaughlin JT, Zeef LA, Fisher P, Grencis RK, Pennock JL. Colonic transcriptional profiling in resistance and susceptibility to trichuriasis: phenotyping a chronic colitis and lessons for iatrogenic helminthosis. Inflamm Bowel Dis. 2010;16:2065–2079. doi: 10.1002/ibd.21326. [DOI] [PubMed] [Google Scholar]
- 7.Weinstock JV, Elliott DE. Translatability of helminth therapy in inflammatory bowel diseases. Int J Parasitol. 2013;43:245–251. doi: 10.1016/j.ijpara.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson MS, Ramalingam TR, Rivollier A, Shenderov K, Mentink-Kane MM, Madala SK, Cheever AW, Artis D, Kelsall BL, Wynn TA. Colitis and intestinal inflammation in IL10-/- mice results from IL-13Rα2-mediated attenuation of IL-13 activity. Gastroenterology. 2011;140:254–264. doi: 10.1053/j.gastro.2010.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol. 1998;161:5733–5744. [PubMed] [Google Scholar]
- 10.Maggio-Price L, Shows D, Waggie K, Burich A, Zeng W, Escobar S, Morrissey P, Viney JL. Helicobacter bilis infection accelerates and H. hepaticus infection delays the development of colitis in multiple drug resistance-deficient (mdr1a-/-) mice. Am J Pathol. 2002;160:739–751. doi: 10.1016/S0002-9440(10)64894-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collett A, Higgs NB, Gironella M, Zeef LA, Hayes A, Salmo E, Haboubi N, Iovanna JL, Carlson GL, Warhurst G. Early molecular and functional changes in colonic epithelium that precede increased gut permeability during colitis development in mdr1a(-/-) mice. Inflamm Bowel Dis. 2008;14:620–631. doi: 10.1002/ibd.20375. [DOI] [PubMed] [Google Scholar]
- 12.Datta R, deSchoolmeester ML, Hedeler C, Paton NW, Brass AM, Else KJ. Identification of novel genes in intestinal tissue that are regulated after infection with an intestinal nematode parasite. Infect Immun. 2005;73:4025–4033. doi: 10.1128/IAI.73.7.4025-4033.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wakelin D. Acquired immunity to Trichuris muris in the albino laboratory mouse. Parasitology. 1967;57:515–524. doi: 10.1017/s0031182000072395. [DOI] [PubMed] [Google Scholar]
- 14.Else KJ, Wakelin D, Wassom DL, Hauda KM. The influence of genes mapping within the major histocompatibility complex on resistance to Trichuris muris infections in mice. Parasitology. 1990;101 Pt 1:61–67. doi: 10.1017/s0031182000079762. [DOI] [PubMed] [Google Scholar]
- 15.Else KJ, Entwistle GM, Grencis RK. Correlations between worm burden and markers of Th1 and Th2 cell subset induction in an inbred strain of mouse infected with Trichuris muris. Parasite Immunol. 1993;15:595–600. doi: 10.1111/pim.1993.15.10.595. [DOI] [PubMed] [Google Scholar]
- 16.Else K, Wakelin D. The effects of H-2 and non-H-2 genes on the expulsion of the nematode Trichuris muris from inbred and congenic mice. Parasitology. 1988;96(Pt 3):543–550. doi: 10.1017/s0031182000080173. [DOI] [PubMed] [Google Scholar]
- 17.Blackwell NM, Else KJ. A comparison of local and peripheral parasite-specific antibody production in different strains of mice infected with Trichuris muris. Parasite Immunol. 2002;24:203–211. doi: 10.1046/j.1365-3024.2002.00452.x. [DOI] [PubMed] [Google Scholar]
- 18.Bell LV, Else KJ. Regulation of colonic epithelial cell turnover by IDO contributes to the innate susceptibility of SCID mice to Trichuris muris infection. Parasite Immunol. 2011;33:244–249. doi: 10.1111/j.1365-3024.2010.01272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bancroft AJ, Else KJ, Grencis RK. Low-level infection with Trichuris muris significantly affects the polarization of the CD4 response. Eur J Immunol. 1994;24:3113–3118. doi: 10.1002/eji.1830241230. [DOI] [PubMed] [Google Scholar]
- 20.Else KJ, Finkelman FD, Maliszewski CR, Grencis RK. Cytokine-mediated regulation of chronic intestinal helminth infection. J Exp Med. 1994;179:347–351. doi: 10.1084/jem.179.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hepworth MR, Grencis RK. Disruption of Th2 immunity results in a gender-specific expansion of IL-13 producing accessory NK cells during helminth infection. J Immunol. 2009;183:3906–3914. doi: 10.4049/jimmunol.0900577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor BC, Zaph C, Troy AE, Du Y, Guild KJ, Comeau MR, Artis D. TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. J Exp Med. 2009;206:655–667. doi: 10.1084/jem.20081499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elliott DE, Summers RW, Weinstock JV. Helminths and the modulation of mucosal inflammation. Curr Opin Gastroenterol. 2005;21:51–58. [PubMed] [Google Scholar]
- 24.Hunter MM, Wang A, McKay DM. Helminth infection enhances disease in a murine TH2 model of colitis. Gastroenterology. 2007;132:1320–1330. doi: 10.1053/j.gastro.2007.01.038. [DOI] [PubMed] [Google Scholar]
- 25.Wang A, Fernando M, Leung G, Phan V, Smyth D, McKay DM. Exacerbation of oxazolone colitis by infection with the helminth Hymenolepis diminuta: involvement of IL-5 and eosinophils. Am J Pathol. 2010;177:2850–2859. doi: 10.2353/ajpath.2010.100537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hunter MM, Wang A, Hirota CL, McKay DM. Neutralizing anti-IL-10 antibody blocks the protective effect of tapeworm infection in a murine model of chemically induced colitis. J Immunol. 2005;174:7368–7375. doi: 10.4049/jimmunol.174.11.7368. [DOI] [PubMed] [Google Scholar]
- 27.Chen CC, Louie S, McCormick B, Walker WA, Shi HN. Concurrent infection with an intestinal helminth parasite impairs host resistance to enteric Citrobacter rodentium and enhances Citrobacter-induced colitis in mice. Infect Immun. 2005;73:5468–5481. doi: 10.1128/IAI.73.9.5468-5481.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elliott DE, Setiawan T, Metwali A, Blum A, Urban JF, Weinstock JV. Heligmosomoides polygyrus inhibits established colitis in IL-10-deficient mice. Eur J Immunol. 2004;34:2690–2698. doi: 10.1002/eji.200324833. [DOI] [PubMed] [Google Scholar]
- 29.Blum AM, Hang L, Setiawan T, Urban JP, Stoyanoff KM, Leung J, Weinstock JV. Heligmosomoides polygyrus bakeri induces tolerogenic dendritic cells that block colitis and prevent antigen-specific gut T cell responses. J Immunol. 2012;189:2512–2520. doi: 10.4049/jimmunol.1102892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elliott DE, Weinstock JV. Where are we on worms? Curr Opin Gastroenterol. 2012;28:551–556. doi: 10.1097/MOG.0b013e3283572f73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1756–1767. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geremia A, Jewell DP. The IL-23/IL-17 pathway in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol. 2012;6:223–237. doi: 10.1586/egh.11.107. [DOI] [PubMed] [Google Scholar]
- 33.Fasnacht N, Greweling MC, Bollati-Fogolín M, Schippers A, Müller W. T-cell-specific deletion of gp130 renders the highly susceptible IL-10-deficient mouse resistant to intestinal nematode infection. Eur J Immunol. 2009;39:2173–2183. doi: 10.1002/eji.200838710. [DOI] [PubMed] [Google Scholar]
- 34.Sandborn WJ, Elliott DE, Weinstock J, Summers RW, Landry-Wheeler A, Silver N, Harnett MD, Hanauer SB. Randomised clinical trial: the safety and tolerability of Trichuris suis ova in patients with Crohn’s disease. Aliment Pharmacol Ther. 2013;38:255–263. doi: 10.1111/apt.12366. [DOI] [PubMed] [Google Scholar]
- 35.Tilp C, Kapur V, Loging W, Erb KJ. Prerequisites for the pharmaceutical industry to develop and commercialise helminths and helminth-derived product therapy. Int J Parasitol. 2013;43:319–325. doi: 10.1016/j.ijpara.2012.12.003. [DOI] [PubMed] [Google Scholar]