

Abstract
Aggregation of the amyloid β protein (Aβ) peptide with 40 or 42 residues is one key feature in Alzheimer’s disease (AD). The 1,4-naphthoquinon-2-yl-l-tryptophan (NQTrp) molecule was reported to alter Aβ self-assembly and reduce toxicity. Though nuclear magnetic resonance experiments and various simulations provided atomic information about the interaction of NQTrp with Aβ peptides spanning the regions of residues 12–28 and 17–42, none of these studies were conducted on the full-length Aβ1–42 peptide. To this end, we performed extensive atomistic replica exchange molecular dynamics simulations of Aβ1–42 dimer with two NQTrp molecules in explicit solvent, by using a force field known to fold diverse proteins correctly. The interactions between NQTrp and Aβ1–42, which change the Aβ interface by reducing most of the intermolecular contacts, are found to be very dynamic and multiple, leading to many transient binding sites. The most favorable binding residues are Arg5, Asp7, Tyr10, His13, Lys16, Lys18, Phe19/Phe20, and Leu34/Met35, providing therefore a completely different picture from in vitro and in silico experiments with NQTrp with shorter Aβ fragments. Importantly, the 10 hot residues that we identified explain the beneficial effect of NQTrp in reducing both the level of Aβ1–42 aggregation and toxicity. Our results also indicate that there is room to design more efficient drugs targeting Aβ1–42 dimer against AD.
Keywords: Alzheimer’s disease, amyloid β dimer, NQTrp, replica exchange molecular dynamics simulation
One of the hallmarks of Alzheimer’s disease (AD) is the aggregation of amyloid β (Aβ) peptides into insoluble amyloid plaques.1,2 The Aβ peptide results from the cleavage of the amyloid precursor protein (APP) by the β- and γ-secretases. The predominant Aβ peptides found in the brain are essentially composed of 40 (Aβ1–40) and 42 (Aβ1–42) residues.3,4 The aggregation pathways from unstructured Aβ peptide monomers to amyloid plaques with cross-β structure have been extensively studied at a low-resolution level,5−8 and there is a growing body of evidence that the low-molecular weight oligomers are the main pathogenic agents in AD.9,10 Even the Aβ dimer, the smallest oligomer, is able to impair the normal functions of neuron cells.11,12
Characterizing the Aβ1–40/Aβ1–42 dimer at the molecular and atomic level is a crucial and important step toward understanding Aβ aggregation and toxicity. Thus far, only low-resolution experimental data for Aβ dimer are available. Using ion-mobility mass spectrometry, Bernstein et al. reported a collision cross section of 1256 Å2 for the Aβ1–42 dimer.13 Using different preparation methods and circular dichroism (CD) analysis, Teplow reported a β-strand content between 12 and 25% and an α-helix content between 3 and 9% at 295 K and pH 7.5 on day 0, for therefore a mixture of various aggregates.14,15 Experimental characterization of Aβ1–42 dimer at a higher level of resolution is narrowed because it is strongly aggregation-prone in aqueous solution.16 In addition, because of its poor solubility, Aβ1–42 dimer is not suitable for NMR analysis in aqueous solvent.
Computer simulations have been extensively used to provide atomistic-level information about the structures of Aβ1–40/Aβ1–42 monomers in aqueous solution17−20 or Aβ16–22, Aβ14–20, or Aβ18–24 oligomers with inhibitors.20−22 Several simulations have been conducted on dimers of Aβ1–40/Aβ1–42 using various coarse-grained force fields17,23,24 and all-atom force fields with implicit25,26 and explicit solvent.27−30 One common finding is that the interface of Aβ1–42 dimer is mainly composed of the central hydrophobic core (CHC, residues 17–21) and the C-terminal region (CT, residues 29–42), with predominant CHC–CHC, CT–CT, and CHC–CT interchain interactions, though the contact probabilities vary depending on the force field and conformational sampling method used.17,26,28−30 Other simulations of larger Aβ aggregates, such as the trimer of Aβ17–42 peptide using the coarse-grained OPEP force field31 and 32-mers of the N-terminally truncated Aβ3–40, Aβ3–42, Aβ11–40, and Aβ11–42 peptides using a square-well coarse-grained force field,32 have also reported similar binding interfaces. Overall, the interpeptide interface in Aβ1–42 dimer and oligomers mainly involves the CHC and CT regions and is independent of the force field used.
Finding an effective inhibitor of Aβ oligomerization is an important step in the treatment of AD. In the past decade, many molecules have been shown to inhibit Aβ oligomerization in vitro and reduce toxicity in cell assays. These include (1) proteins, such as Escherichia coli maltose binding protein (MBP);33 (2) short peptides, such as the “Ghanta peptide”34 and “Soto peptide”;35 (3) fragments of Aβ peptides, such as KLVFF,36 or fragments spanning the C-terminal region;37 (4) chemical compounds extracted from natural products, such as EGCG from green tea38 and curcumin from cinnamon;39 and (5) synthetic compounds, such as the 1,4-naphthoquinon-2-yl-l-tryptophan (NQTrp),40 based on the inhibitory capability of naphtoquinone on Aβ oligomerization41 and the strong amyloidogenic potential of tryptophan.42
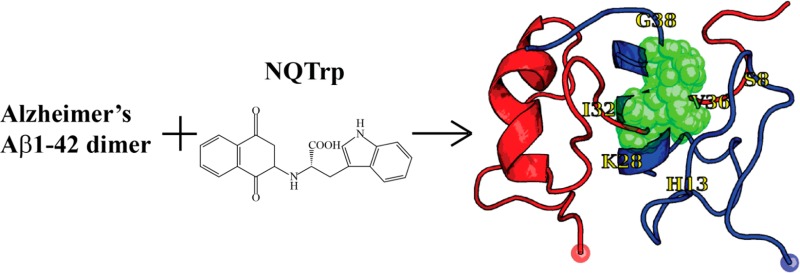
The structure of NQTrp is shown in Figure 1A. Experimentally, NQTrp has been reported to reduce the toxicity of Aβ oligomers and completely recover the phenotype on a transgenic AD Drosophila model.40 Its inhibition effect has also been discussed with respect to other amyloidogenic peptides, such as α-synuclein, islet amyloid polypeptide, and calcitonin.43 The NMR structure of Aβ12–28 monomer determined in the presence of a 0.25 molar ratio of NQTrp to Aβ12–28 showed the most prominent interactions in the region of residues 18–24.40 All-atom implicit solvent molecular dynamics (MD) simulations using the CHARMM/CMAP force field of Aβ12–28 monomer with one single NQTrp molecule reported there are no predominant binding modes, although NQTrp preferentially interacts with residues His13–Phe20 through its carboxyl group (labeled 15–17 in Figure 1A) and two aromatic moieties (labeled 1–12 for naphthoquinone and 19–27 for indole in Figure 1A).44 Replica exchange molecular dynamics (REMD) simulation using the coarse-grained OPEP force field followed by all-atom docking calculations also marked the NQTrp molecule as the best binder to Aβ17–42 trimeric structures among five small-molecule drugs. These simulations also showed that NQTrp has multiple binding modes with different affinities, although it interacts preferentially with the CHC region. Finally, all-atom implicit solvent MD simulations of the trimers of Aβ14–20, Aβ16–22, and Aβ18–24 peptides with one single NQTrp molecule and NQTrp derivatives emphasized the role of Phe19 and Phe20 side chains and the anilic NH group of NQTrp (labeled as 13 in Figure 1A) in binding.22 Overall, although these simulations provide mechanistic insight into the activity of NQTrp on Aβ fragments, how NQTrp interacts with the full-length Aβ protein remains to be determined.
Figure 1.
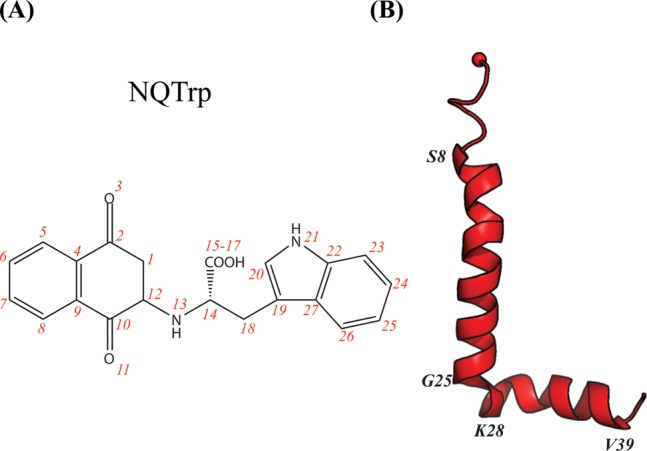
Structures of the NQTrp molecule and Aβ monomer. (A) Chemical structure of the NQTrp molecule. The red numbers label all the heavy atoms. (B) Initial Aβ conformation. The red sphere represents the Cα atom of Asp1.
To this end, we performed all-atom simulations of Aβ1–42 dimer with two NQTrp molecules. To overcome the sampling issue, we used REMD simulation in explicit solvent with 64 replicas between 315 and 450 K. Our previous REMD study of Aβ1–42 dimer in the absence or presence of 10 EGCG molecules demonstrated it is an efficient way to study Aβ42 dimer–inhibitor interactions.30 Our aim in this study is to provide an atomic picture of the modes of action of NQTrp on Aβ1–42 dimer to improve our understanding of its inhibitory mechanism on Aβ oligomerization and toxicity.
Results
Convergence
The simulation convergence was assessed by several criteria. As shown in Figure 2A, one representative replica visits a wide range of temperatures over 250 ns, indicating an efficient walk in temperature space. The conformational entropy at the lowest temperature, 315 K, shown in Figure 2B, remains relatively constant after 150 ns. The coil, turn, α-helix, and β-strand contents of the peptides as a function of temperature are shown in panels C–F, respectively, of Figure 2, using three different time intervals, 100–150, 150–200, and 200–250 ns. The superposition of the curves for 150–200 and 200–250 ns indicates that the propensities for the four secondary structures have converged. The high degree of similarity of the α-helical propensity profiles among the three regimes clearly demonstrates that the memory of the originally assumed structure does not play any role. The coil propensity as a function of the amino acids for the three time intervals is shown in Figure 3A. The percentage of coil for each amino acid remains constant as the simulation progresses from 100 to 250 ns. The fraction of exposed side chain surface area for each amino acid, shown in Figure 3B, does not vary from one time interval to another. The inter-center-of-mass distance between the two two Aβ peptides at 315 K is monitored in Figure 4A. During the first 100 ns, the Aβ peptides approach gradually and then remain at a distance of 1.2 nm in the following 150 ns. Figure 4B shows the time evolution of the total number of heavy atom contacts between the two Aβ peptides at 315 K. The number of contacts increases during the first 100 ns and then fluctuates around 1000 during the 100–250 ns period. Taken together, all these results indicate that the conformational ensemble has reasonably converged within 250 ns. In what follows, analysis is based on the ensemble trajectory at 315 K from 100 to 250 ns.
Figure 2.

Simulation convergence. (A) Time evolution of one replica in temperature space. (B) Time evolution of the conformational entropy as estimated by the quasi-harmonic approach based on the ensemble trajectory at 315 K. (C) Propensity of coil as a function of temperature in the time intervals of 100–150, 150–200, and 200–250 ns. (D) Propensity of turn as a function of temperature in the three time intervals. (E and F) Propensities of α-helix and β-strand, respectively, as a function of temperature in the three time intervals.
Figure 3.
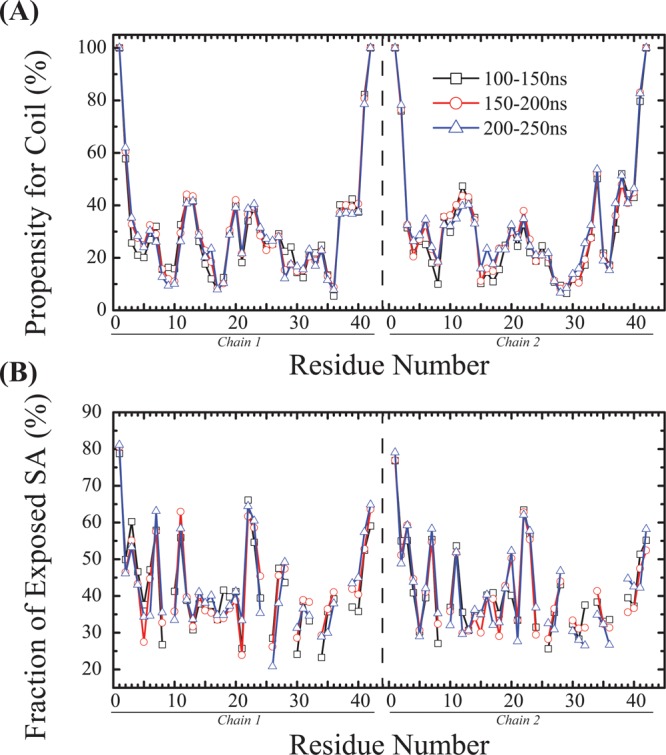
Per-residue convergence assessment. (A) Coil propensity of each Aβ residue at 315 K and (B) fraction of exposed side chain surface area at 315 K using the three time windows. Note the curves are discontinuous at glycine.
Figure 4.
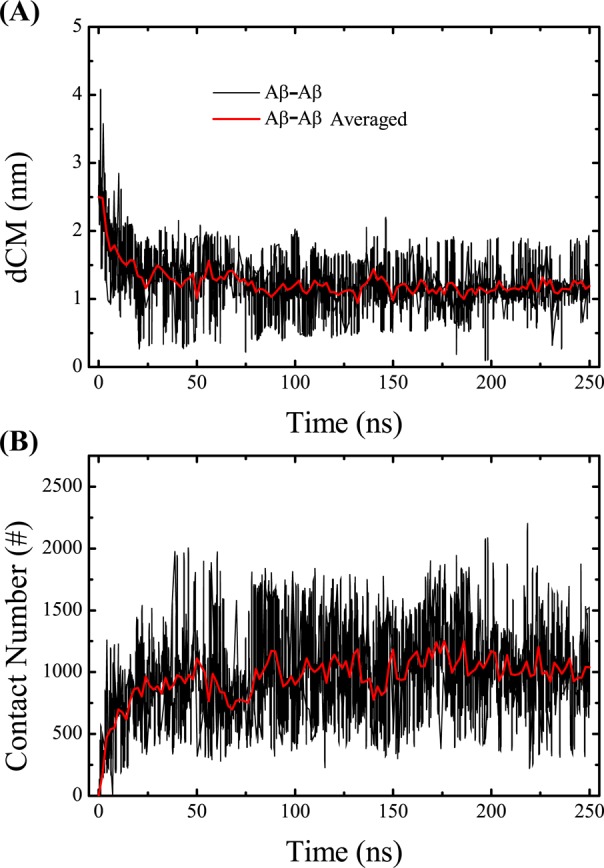
Time evolution of two global properties of Aβ dimer. (A) Time evolution of the inter-center-of-mass distance, dCM, between the two Aβ peptides (gray) and the averaged value (red). (B) Time evolution of the total number of heavy atom contacts between the Aβ peptides (gray) and the averaged value (red).
Two-Dimensional Structures and Contact Maps of Aβ Dimer
Averaged over the two peptides, the secondary structure propensities at 315 K show that coil is the most populated (32.0 ± 1.0%) and β-strand is the least populated (6.8 ± 0.9%). Bend, turn, and α-helix have populations of 17.5 ± 0.4, 24.2 ± 1.5, and 19.5 ± 0.7%, respectively. Via examination of the per-residue secondary structure propensity, in Figure 5, the β-strand content is mainly observed in the CT region (12%) and then in the CHC (8%) and NT (3.8%) regions. The α-helix content is observed at residues 12–18 (32%), residues 30–35 of the CT region (22%), and the FL region (19%).
Figure 5.
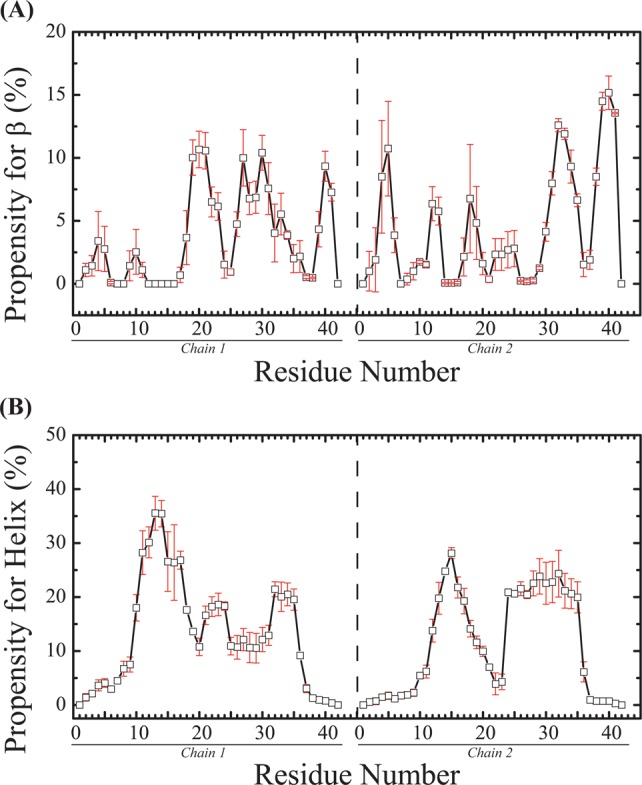
Secondary structure propensities of each amino acid at 315 K: (A) β-strand and (B) α-helix. The vertical dashed line separates the two chains. The error bars represent the standard errors estimated by block averaging.
The Aβ dimer interface is assessed by the weak intermolecular side chain–side chain contact map, as seen in Figure 6. The interface is characterized by CT–CT interactions among residues 30–35 with a lifetime probability of 8.4 ± 0.2%, and the strongest interaction between residues Ile31 and Leu34 has a lifetime of 27%. The CT–CHC interactions have a lifetime probability of 5.1 ± 0.7% using residues 30–35 for CT, and the strongest interaction between Phe19 and Ile38 has a lifetime of 18%. The CHC–CHC interactions have a lifetime probability of 3.3 ± 0.3%, and the strongest interaction between Phe19 and Phe20 has a lifetime of 12%. In contrast, the intra- and intermolecular interactions between NT and the rest of the protein are almost negligible.
Figure 6.
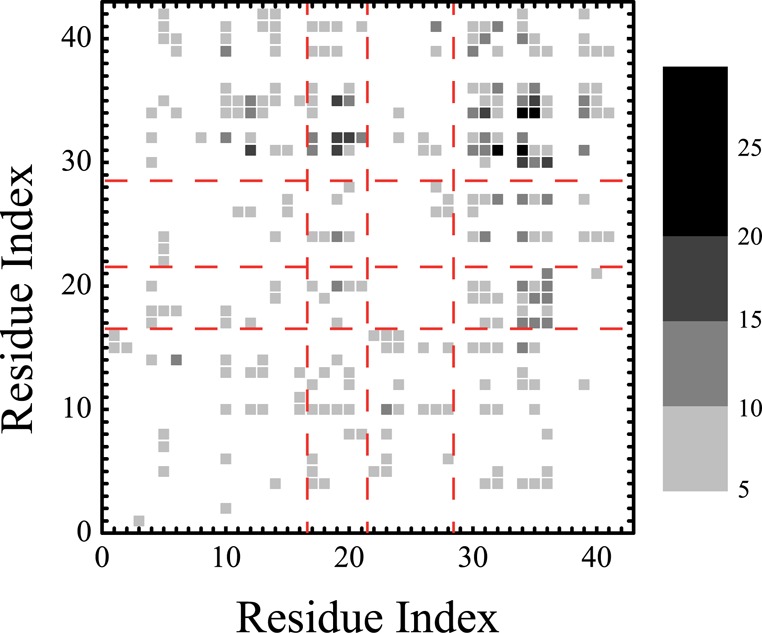
Interpeptide side chain–side chain contact maps. The color scale defines the contact probability. The red dashed lines define the four Aβ regions: NT, CHC, FL, and CT.
Binding of NQTrp to Aβ Dimer
The distribution of NQTrp molecules around the Aβ dimer is assessed by the radial distribution function (RDF) shown in Figure 7. The RDF and the height of the peak at 0.36 nm from Aβ1–42 dimer surface indicate direct binding of NQTrp to Aβ peptides. For a system containing two Aβ peptides and two NQTrp molecules, six different states for characterizing the binding exist. These states can be defined as P1 (all peptides and NQTrp are separated), P2 (one Aβ interacts with one NQTrp), P3 (one Aβ interacts with two NQTrp molecules), P4 (the two peptides interact with each other, but not with NQTrp), P5 (the two peptides interact with each other and with one NQTrp, whereas the second NQTrp is devoid of any intermolecular interactions), and P6 (the two peptides interact with each other and with the two NQTrp molecules). Using all conformations within the time frame of 100–250 ns at 315 K, the population of the P1–P3 states is 0%, indicating the absence of free Aβ monomers. The population of the P4 state is also 0%, indicating no dimers free of NQTrp. The populations of the P5 and P6 states are 1.7 ± 0.2 and 98.3 ± 0.2%, respectively, indicating that most of the conformational ensemble is characterized by two Aβ peptides forming a dimer and interacting with both NQTrp molecules. Via examination of the P6 heterotetramer interactions, the most dominant binding mode (59.2 ± 0.2%) is described by the two NQTrp molecules interacting and intercalated between both peptides. The second state (33.5 ± 0.1%) has one NQTrp interacting with both peptides, whereas the second NQTrp interacts with only one Aβ; finally, the third state (7.3 ± 0.1%) is such that one NQTrp interacts with one single Aβ.
Figure 7.
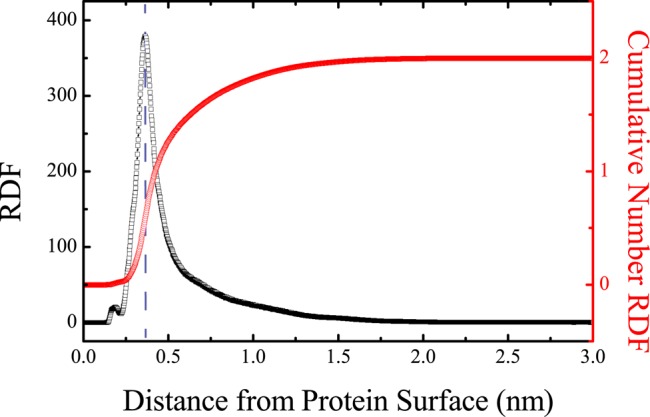
Radial distribution functions of NQTrp from Aβ dimer. The RDF is shown with black squares. The cumulative number RDF is shown with red circles. The vertical blue dashed line labels the position of the highest peak in the RDF curve.
The binding of NQTrp to Aβ1–42 dimer can be further examined by the contact maps between the heavy NQTrp atoms and the heavy Aβ main chain and side chain atoms shown in panels A and B of Figure 8, respectively. The carboxyl group (atoms 15–17) of NQTrp displays a larger number of contacts with the main chain atoms of Aβ peptides than the other atoms (Figure 8A). The same carboxyl group and the outward atoms of the two aromatic moieties of NQTrp (atoms 3 and 5–8 from naphtoquinone and atoms 25 and 26 from indole) also form roughly 25% more contacts with the side chain atoms of Aβ than the other atoms. The main chain atoms of residues Met35/Leu34, Gly37/Gly38, Gly9/Tyr10, Gly29, Ser26, Lys16, and Arg5 interact strongly with NQTrp (Figure 8A), while the side chain atoms of residues Phe4/Arg5, Tyr10, His13, Lys16, Phe19/Phe20, Ile31/Ile32, and Leu34/Met35 interact preferentially with NQTrp (Figure 8B). The H-bond propensity between the Aβ amino acids and NQTrp, shown in Figure 9A, reveals that residues Glu3, Arg7, Ser8, Glu11, Glu22, Asp23, Gly29, and Ala30 have a probability of forming H-bonds with NQTrp of 6–8%. The other residues except residues Asp1, Ala2, Glu11, Ala21, Val24, Ile31/32, Val36, Gly38, and Ile41 have a H-bond probability of 2–6%. As expected, residues Gly29 and Ala30 form backbone H-bonds with NQTrp, while residues Glu3, Arg7, Ser8, Glu11, Glu22, and Asp23 form H-bonds with NQTrp mainly by their side chains. Figure 9B–F also shows the H-bond propensity between the CO (NH) atoms of NQTrp and the main chain and side chains of Aβ, revealing the heterogeneity of the interactions, even if CO atoms 10 and 11 of naphtoquinone show a much lower H-bond propensity than NH atoms 13 and 21 and COOH atoms 15–17.
Figure 8.

Contact maps between the heavy atoms of NQTrp and Aβ1–42 residues. (A) Interactions with the backbone Aβ atoms. (B) Interactions with the side chain Aβ atoms. The labeling of the NQTrp heavy atoms is shown in Figure 1A. The color scale reflects the number of contacts. The cumulative number of contacts for each heavy atom of NQTrp and Aβ is shown in the top panels and right panels, respectively. Amino acids with a large number of contacts are labeled. The red dashed lines label the naphthoquinone, peptide bond, and indole groups of NQTrp from left to right on the x-axis, and the four Aβ regions (NT, CHC, FL, and CT) on the y-axis from bottom to top, respectively.
Figure 9.

H-Bond propensities between Aβ amino acids and NQTrp. (A) Propensities between NQTrp and the main chain and side chain atoms of Aβ peptides are shown with white and black bars, respectively. (B–F) Propensities between NQTrp functional groups and the main chain and side chain atoms of Aβ peptides are shown as white and black bars, respectively.
The strength of the interaction between NQTrp and each Aβ residue can be estimated by the molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) method. The total interaction free energies between NQTrp and each amino acid, as well as the contributions from van der Waals interactions, electrostatic interactions, and solvation, are shown in Figure 10. As seen in Figure 10A, all Aβ residues display a favorable interaction (<0 kcal/mol), but 10 residues, Tyr10, Leu34/Met35, Arg5, Lys16, Asp7, His13, Lys28, and Phe20/Phe19, are the most favorable binding partners. The favorable interaction free energies between NQTrp and residues Tyr10, Leu34, Met35, and Phe20/Phe19 result mainly from van der Waals interactions and solvation (Figure 10B,D). The favorable interaction free energy between NQTrp and Arg5 comes mostly from van der Waals interactions, while the favorable interaction free energy between NQTrp and Lys16 and Asp7 results from electrostatic interactions (Figure 10C).
Figure 10.

Interaction free energies between NQTrp and each Aβ amino acid. (A) Total interaction free energies. (B) Contribution of van der Waals interactions. (C) Contribution of electrostatic interactions. (D) Contribution of solvation.
Finally, to predict the binding pockets of NQTrp on Aβ1–42 dimer, the Aβ1–42 configurations were clustered, and the representative structures of the first 10 most populated clusters are shown in Figure 11. The population of each cluster is shown in Table 1. The first cluster displays two α-helices spanning the CHC or part of CT (residues 30–35) region in each chain. The second cluster is essentially random coil with two short helices of residues 3–6 in one chain and residues 24–28 in the other chain. Cluster 3 shows two α-helices for residues 13–17 in both chains, while cluster 4 is characterized by four short helices spanning the CHC and FL regions in both chains. Cluster 5 displays two long α-helices spanning residues 24–36 in one chain and residues 13–21 in the second chain, as well a short helix at residues 26–28 in the second chain. Cluster 6 is similar to cluster 5, but with only one long helix spanning residues 6–20 in the second chain. Cluster 7 shows one long α-helix spanning residues 13–25 and one short helix spanning residues 31–34 in one chain, and two short helices spanning residues 17–24 and 32–35 in the second chain. Cluster 8 features a β-hairpin formed by residues 32–34 and 37–40 of one chain packed against a third strand formed by residues 39–41 of the other chain. Cluster 8 also displays four short helices spanning the flanking CHC amino acids in both chains. Cluster 9 is similar to clusters 5 and 6 and is characterized by a long helix spanning residues 24–36 in one chain and two short helices spanning residues 13–15 and 26–32 in the second chain. Finally, cluster 10 displays a long α-helix spanning residues 11–24 in one chain packed against a random coil second chain. The binding residues to NQTrp in the first 10 Aβ1–42 clusters are listed in Table 1. The key residues involved in the fifth, sixth, and ninth binding sites are rather similar, differing by several residues, while the other sites are different. Table S1 of the Supporting Information gives the binding residues in the 11–20 Aβ1–42 clusters. We find there are eight different binding pockets in the first 10 clusters and 15 in the first 20 clusters, which include 29.2 and 46.3% of all Aβ1–42 configurations, respectively. Overall, 555 clusters are found for Aβ1–42 dimer, with the first 50 and 100 clusters including 69.8 and 82.6% of the configurations, respectively, and the number of binding pockets is on the same order of magnitude.
Figure 11.

First 10 clusters of Aβ and the NQTrp binding sites. Chains A and B are colored red and blue, respectively. The red and blue spheres represent the Cα atoms of Asp1. The center of mass of NQTrp is shown as green spheres, and we show NQTrp if it forms a <4 Å contact with any heavy atom of Aβ. Residues colored yellow assist in the reading. Panels A–J represent the 1st to 10th largest clusters, respectively.
Table 1. The binding residues to NQTrp from the first ten Aβ1-42 clusters. These are shown in Figure 11.
| amino
acids at binding pockets |
||||
|---|---|---|---|---|
| cluster | population (%) | chain A | chain B | averaged area of binding interface (Å2) |
| 1 | 4.29 | Ser26, Lys28, Ile31–Ile40 | Glu3–Arg5, Ser8–Gln15, Val18, Phe20, Ala21, Gly29, Ile32–Val39 | 253 ± 4.3 |
| 2 | 3.84 | Phe4–Gln15, Phe19, Met35 | Ala2, Leu17, Val18, Val24, Ala30–Gly37, Val39, Ile41 | 272 ± 4.4 |
| 3 | 3.66 | His13, Phe19, Ile32–Met35, Gly37–Val39 | Asp1–Asp7, Gly9, His14–Val18, Ala30–Ala42 | 304 ± 9.6 |
| 4 | 3.39 | Ala30–Val39 | Glu3–His6, Val12–Lys16, Phe19, Phe20, Gly25, Lys28–Val36, Val40 | 188 ± 5.0 |
| 5 | 2.91 | Val12, Phe19, Ser26, Asn27, Ile31, Leu34–Gly37 | His6, Val12, Leu17–Val36 | 248 ± 5.5 |
| 6 | 2.59 | Val12, Phe19, Gly25, Ala30, Leu34–Val36, Val40 | Arg5, His6, Val12, Leu17–Val36 | 250 ± 4.0 |
| 7 | 2.40 | Val24–Ser26, Gly29, Ile32, Gly33, Val36–Ile41 | Arg5–His14, Leu17, Phe20, Ala21, Val24, Asn27–Ile41 | 266 ± 7.4 |
| 8 | 2.09 | Leu17–Phe19, Glu22–Gly38 | Asp1, Val17, Phe20–Lys28, Ala30–Gly33, Gly38–Ile41 | 274 ± 4.5 |
| 9 | 2.04 | Val12, Phe19, Phe20, Asn27, Ile31, Leu34–Gly38 | His6, Val12, Leu17–Gly37 | 278 ± 6.6 |
| 10 | 2.02 | Arg5, Leu17, Val24, Lys28–Val36 | Tyr10, Glu11, Phe20–Val24, Ser25–Ile31, Leu34–Val40 | 225 ± 6.7 |
Discussion
Experimentally, NQTrp has been reported to reduce the level of aggregation and toxicity of Aβ1–42.40 In the NMR study of the Aβ12–28 monomer with various NQTrp:Aβ molar ratios reported strong amide proton chemical shift changes at Phe20, Ala21, and Glu22 and smaller variations at Val18 and Val24. All-atom MD in implicit solvent showed three dominant binding sites between NQTrp and the Aβ18–21 region.34 Following MD simulations in explicit solvent of Aβ12–28 monomer with NQTrp38 and Aβ14–20, Aβ16–22, or Aβ18–24 trimers with one single NQTrp39 revealed that the most frequently observed intermolecular contacts involve mainly the region of residues 13–20 with the side chains of Phe19 and Phe20 constituting the site of highest interaction probability and providing therefore the most favorable van der Waals energy. Our total interaction free energies and van der Waals energies using the MM/PBSA method show that the most favorable binding partners are residues Tyr10, Leu34/Met35, Arg5, Lys16, Asp7, Phe20/Phe19, Lys28, and His13, providing therefore a binding picture different from that from the studies with short fragments. After the addition of the information about the highest H-bond interaction probability to our analysis, the preferred binding of NQTrp to Aβ is in the region of residues 3–35, not only the region of residues 13–20. At the molecular level, our analysis indicates that the dominant binding mechanism with a population of 59% at 315 K involves the two Aβ peptides interacting together and simultaneously with the two NQTrp molecules. Although there are multiple binding modes of action, the analysis of intermolecular contact maps shows the NQTrp molecule binds strongly to the CHC and FL regions, reducing therefore the strength of the interpeptide interactions among residues 16–35, interactions that are seen in many simulations of Aβ1–40 and Aβ1–42 dimers in the absence of inhibitors.17,25−32,45 Our result is also in agreement with a recent hydrogen–deuterium exchange coupled with mass spectrometry study showing that the region of residues 20–35 is the first to aggregate, followed by the region of residues 36–42 and then the region of residues 1–19 during Aβ1–42 oligomerization.46
At the residual and atomic level, we have determined that the NQTrp molecules bind mostly to the side chains of residues Arg5, Asp7, Tyr10, His13, Lys16, Phe19/Phe20, Lys28, and Leu34/Met35. All the residues that we identify are known to play very important roles in both Aβ1–42 aggregation and toxicity.
Triple substitution of Arg5, Lys16, and Lys28 with Ala results in a significant loss of Aβ1–40 fibril toxicity in human embryonic kidney cells.47 Using CD and fluorescence spectroscopy, transmission electron microscopy, a reactive oxygen species (ROS) fluorescent assay, and neuronal cell viability, mouse Aβ1–42 as a three-site mutant (Arg5Gly, Tyr10Phe, and His13Arg) of human Aβ1–42 alters the metal copper and zinc binding sites, reduces the likelihood of forming β-sheet structures and aggregated fibrils, alleviates the generation of ROS, and decreases toxicity.48 Binding of NQTrp to Arg5, Tyr10, and His13 should, therefore, reduce the level of metal binding and toxicity.
The Asp7Asn mutation alters the self-assembly and increases the toxicity of Aβ1–40 and Aβ1–42 peptides.49 Binding of NQTrp to Asp7 should therefore reduce both the level of assembly and toxicity.
The Lys16Ala mutation impacts Aβ self-assembly and dramatically reduces the toxicity of Aβ1–40 and Aβ1–42 peptides.50 A novel familial Aβ Lys16Asn peptide has been discovered. The peptide itself is not harmful to neuronal cells, but toxicity is observed for a mixture of Aβ K16N and its wild-type (WT) counterpart.51 All-atom MD simulations of both Aβ1–40 and Aβ1–42 WT and D7N dimers found an interplay between the Asp1–Arg5 and Asp1–Lys16 salt bridges upon introduction of the D7N mutation.24 Other studies suggested Lys16 is exposed to solvent and interacting with other monomers in Aβ aggregation.52−54 We find that the side chain of Lys16 is 47% of the time exposed to the solvent, thereby limiting partially interactions with free monomers during Aβ oligomerization (Table S2 of the Supporting Information). The formation of the Asp23–Lys28 salt bridge is also known to accelerate fibril formation. We find that Lys28 is 63% of the time accessible to solvent and the intramolecular Glu22–Lys28 and Asp23–Lys28 salt bridges are formed 7.0 ± 2.3% and 9.4 ± 1.2% of the time, respectively (Table S3 of the Supporting Information).
Using mutagenesis, Leu34 was found to be a key residue for Aβ42 aggregation, the Leu34Cys mutation leading to the disruption of hexamer and tetramer formation.55 In addition, Leu34 is found to be packing with Phe19 in both oligomer and fibril models.56 We find that the side chain of Leu34 is exposed only 39% of the time to solvent, and the binding of NQTrp prevents Leu34 from interacting with Phe19. Finally, the substitution of Met35 with Val was found to accelerate the kinetics of aggregation of Aβ42, and the presence of Met35 is not important for toxicity.57 We find that the side chain of Met35 is exposed to solvent 42% of the time.
Taken together, our results indicate that the reduced level of Aβ1–42 oligomerization and toxicity induced by NQTrp binding results from a multifactorial mechanism. It is also of interest to determine the extent to which our results compare with three recent simulations of Aβ1–42 with other small compounds.
In the REMD simulation of Aβ1–42 dimer interacting with 10 EGCG molecules, EGCG was found buried in the interface between the Aβ peptides and bound to the hydrophobic side chain atoms of residues Phe4, Phe19/Phe20, Tyr10, Ile31/Ile32, Met35/Val36, Val39, and Ile41, and the hydrophilic N-terminal amino acids Asp1, Glu3, Arg5, Asp7, and Glu11 by H-bonds.30 Via comparison with our pattern of interactions with NQTrp, the hot residues Arg5, Asp7, Tyr10, Phe19/Phe20, and Met35 are recovered, but overall, the sites with the highest interaction probability are clearly different.
Zhu et al. determined the most populated clusters of Aβ1–42 monomer from 100 ns REMD in solvent. Then, the structures were subjected to fragment-based calculations, and they identified 35 clusters displaying binding pockets made essentially of the CHC region and residues Phe4, Tyr10, Ile31, and Met35, though hydrophilic residues can contribute, as well. The binding of curcumin and Congo red revealed that in some of the complexes, the ligands remain bound during the 80 ns MD at 300 K.58 Though Tyr10, Met35, and the CHC region are identified by our study, it is not surprising to observe differences because it is well-known that the configurations of Aβ1–42 monomer and dimer are different.17,23,59
Finally, we can compare our results with implicit solvent MD simulations of Aβ1–42 monomer interacting with carnosine, a dipeptide naturally occurring in the brain and rescuing cells from Aβ-induced toxicity.60 Using a total MD trajectory of 3 μs, carnosine was found to interact transiently by salt bridges with charged residues Arg5, Lys16, and Lys28, and the CHC and flanked residues. Though their binding residues differ from those identified our study, it is interesting that Arg5, Lys16, Lys28, and the CHC region are found in both studies.
Conclusion
We have performed atomistic REMD simulation of the Aβ1–42 dimer with two NQTrp molecules at a salt concentration of 150 mM. Though there are multiple binding sites, hydrophobic residues Phe19/Phe20 and Leu34/Met35 and hydrophilic residues Arg5, Tyr10, Lys16, Asp7, and Lys18 are identified as hot spots for NQTrp binding, providing therefore a different structural and dynamical picture compared to that from Aβ fragments with NQTrp. Interestingly, because all these residues are known to play a critical role in both Aβ self-assembly and toxicity, our atomistic study therefore explains the beneficial effect of the NQTrp molecule.
Using simulations, NQTrp has clearly a better binding affinity for Aβ42 than carnosine and EGCG. At equilibrium, the population of free Aβ42 monomers is 0% in the presence of NQTrp, while in the presence of 10 EGCG molecules, there is a population of 5.2% of free Aβ42 monomers that would be able to associate with larger toxic and nontoxic aggregates.30 Carnosine was found to be in contact with Aβ1–42 monomer for only 20% of the simulation time at 300 K and at a concentration of 5 mM versus a concentration of 11 mM in our study.
Despite the increased affinity, the number of Aβ42 clusters is still very high in the presence of NQTrp, reaching a total of 555, and the number of binding sites is estimated to be on the order of 102. This result provides a picture different from that provided by standard protein–drug interaction.61 While, on average, the contact surface between a small-molecule ligand and its protein receptor is 300–1000 Å2,62 we find that the average contact surface of NQTrp with Aβ42, calculated using all conformations, is only 259 ± 7.4 Å2 (with a minimum of 68 Å2 and a maximum of 396 Å2). Taken together, our results indicate that there is room to design more efficient drugs targeting Aβ42 dimer against AD.
Methods
Simulation
The Aβ1–42 sequence is DAEFRHDSGY10 EVHHQKLVFF20 AEDVGSNKGA30 IIGLMVGGVV40 IA. The initial coordinates of Aβ1–42 monomer were taken from model 1 of PDB entry 1IYT.63 This conformation observed by NMR in an apolar environment is characterized by two α-helices spanning residues 8–25 and 28–39 (Figure 1B). The monomer with the N- and C-termini treated as NH3+ and COO– was then replicated and translated to obtain the initial dimer structures in a parallel orientation with no interchain atomic distances of <10 Å. The recently developed AMBER99sb*-ILDN force field was employed to parametrize Aβ1–42.64,65 We chose the AMBER99sb*-ILDN force field rather than older versions of AMBER, and the OPLS-AA or GROMOS force field,66 because it provides a better description of the structural and dynamical properties of well-structured proteins and allows folding of diverse proteins into their NMR structures.64 The parameters for NQTrp, obtained from quantum calculations, were taken from our previous study.31
Aβ dimer was initially put in the center of a dodecahedron box with periodic boundary conditions. The initial volume of the box is ∼283.4 nm3. Then, two NQTrp molecules were randomly added. This molar ratio of NQTrp to Aβ matches that used experimentally.40 Then, 8661 TIP3P water molecules65 were added to the box. Finally, 32 Na+ and 26 Cl– ions were added by randomly replacing water molecules to keep the system neutral at a physiological salt concentration of 150 mM. The pH was set to 7 with the Arg and Lys residues positively charged, the Glu and Asp residues negatively charged, and the His residues neutral with a hydrogen on the ε-nitrogen site.
GROMACS (version 4.5.5)66 was used. The LINCS protocol67 was used to constrain the bonds involving hydrogen atoms, allowing an integration time step of 2 fs. The particle mesh Ewald method68 with a cutoff of 0.9 nm was used to treat the electrostatic interactions. A cutoff of 1.2 nm was used for the van der Waals interactions. The nonbonded pair lists were updated every 0.010 ps. The coordinates were saved every 2 ps. The REMD simulation69 was conducted with 64 replicas with temperature ranging from 315 to 450 K by using the method developed by Patriksson and van der Spoel.70 Temperatures were controlled by the Bussi–Donadio–Parrinello velocity rescaling thermostat found to sample the canonical ensemble.71 Exchanges between neighboring replicas were attempted every 2 ps, leading to an average acceptance ratio of 39.4%. The REMD simulation ran for 250 ns.
Analysis
The conformational entropy was calculated using the quasi-harmonic approximation.72 The secondary structures of Aβ were calculated using the DSSP algorithm.73 The fraction of exposed side chain surface area was calculated using the reference data presented by Miller et al.74 A contact was defined if the interheavy atom distance was <5 Å. A hydrogen bond was considered formed when the acceptor–donor distance was not more than 3.5 Å and the acceptor–donor–hydrogen angle was not more than 30°. The root-mean-square deviation-based clustering method described by Daura et al.75 with a cutoff of 3.5 Å for backbone heavy atoms was used to extract the representative Aβ structures. Then, for each Aβ cluster, the binding sites were obtained by analyzing all bound NQTrp molecules. The binding interfacial area was calculated as being half of the buried surface areas in the Aβ–NQTrp complex. The interaction free energy of the Aβ–NQTrp complex was estimated using the MM/PBSA method.76 The data set containing 1500 snapshots was extracted from the last 150 ns at 315 K with a time interval of 100 ps. The interaction free energies between NQTrp and Aβ residues were calculated using the pairwise per-residue decomposition, with all parameters and, notably, the solvation energy taken from our previous work.30 Statistical errors were estimated by block (time interval) averaging. To facilitate discussion, the N-terminal region (NT) covers residues 1–16 and the fibril-loop region (FL) spans residues 22–28.
Supporting Information Available
Binding sites of Aβ1–42 clusters 11–20, lifetimes of the side chain exposed to solvent for each residue, and lifetimes of salt bridges. This material is available free of charge via the Internet at http://pubs.acs.org.
We are grateful for the support of the French-Singapore MERLION Ph.D. 2010 program (Project 5.08.10). This work was partially supported by IDA Cloud Computing Call 4, NTU Tier 1 Grant RG 23/11, and the A*STAR Computational Resource Centre (http://www.acrc.a-star.edu.sg) through the use of its high-performance computing facilities. P.D. also acknowledges financial support from ANR GRAL SIMI 12-BS07-0017, IUF, and the Pierre de Gilles Foundation.
The authors declare no competing financial interest.
Supplementary Material
References
- Selkoe D. J. (2003) Folding proteins in fatal ways. Nature 426, 900–904. [DOI] [PubMed] [Google Scholar]
- Dobson C. M. (2003) Protein folding and misfolding. Nature 426, 884–890. [DOI] [PubMed] [Google Scholar]
- Walsh D. M.; Klyubin I.; Fadeeva J. V.; Cullen W. K.; Anwyl R.; Wolfe M. S.; Rowan M. J.; Selkoe D. J. (2002) Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- Thompson L. K. (2003) Unraveling the secrets of Alzheimer’s β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 100, 383–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eanes E. D.; Glenner G. G. (1968) X-ray diffraction studies on amyloid filaments. J. Histochem. Cytochem. 16, 673–677. [DOI] [PubMed] [Google Scholar]
- Kirschner D. A.; Abraham C.; Selkoe D. J. (1986) X-ray diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimer disease indicates cross-β conformation. Proc. Natl. Acad. Sci. U.S.A. 83, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova A. T.; Ishii Y.; Balbach J. J.; Antzutkin O. N.; Leapman R. D.; Delaglio F.; Tycko R. (2002) A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A. 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lührs T.; Ritter C.; Adrian M.; Riek-Loher D.; Bohrmann B.; Döbeli H.; Schubert D.; Riek R. (2005) 3D structure of Alzheimer’s amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. U.S.A. 102, 17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkitadze M. D.; Bitan G.; Teplow D. B. (2002) Paradigm shifts in Alzheimer’s disease and other neurodegenerative disorders: The emerging role of oligomeric assemblies. J. Neurosci. Res. 69, 567–577. [DOI] [PubMed] [Google Scholar]
- Klein W. L.; Stine W. B. Jr.; Teplow D. B. (2004) Small assemblies of unmodified amyloid β-protein are the proximate neurotoxin in Alzheimer’s disease. Neurobiol. Aging 25, 569–580. [DOI] [PubMed] [Google Scholar]
- Shankar G. M.; Li S.; Mehta T. H.; Garcia-Munoz A.; Shepardson N. E.; Smith I.; Brett F. M.; Farrell M. A.; Rowan M. J.; Lemere C. A.; Regan C. M.; Walsh D. M.; Sabatini B. L.; Selkoe D. J. (2008) Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M.; Shepardson N.; Yang T.; Chen G.; Walsh D.; Selkoe D. J. (2011) Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 5819–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein S. L.; Dupuis N. F.; Lazo N. D.; Wyttenbach T.; Condron M. M.; Bitan G.; Teplow D. B.; Shea J. E.; Ruotolo B. T.; Robinson C. V.; Bowers M. T. (2009) Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem. 1, 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkitadze M. D.; Bitan G.; Teplow D. B. (2002) Paradigm shifts in Alzheimer’s disease and other neuro degenerative disorders: The emerging role of oligomeric assemblies. J. Neurosci. Res. 69, 567–577. [DOI] [PubMed] [Google Scholar]
- Kirkitadze M. D.; Condron M. M.; Teplow D. B. (2001) Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J. Mol. Biol. 312, 1103–1119. [DOI] [PubMed] [Google Scholar]
- Teplow D. B. (2006) Preparation of Amyloid β-Protein for Structural and Functional Studies. In Methods in Enzymology (Indu K., and Ronald W., Eds.) pp 20–33, Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- Cote S.; Laghaei R.; Derreumaux P.; Mousseau N. (2012) Distinct dimerization for various alloforms of the amyloid-β protein: Aβ(1–40), Aβ(1–42), and Aβ(1–40)(D23N). J. Phys. Chem. B 116, 4043–4055. [DOI] [PubMed] [Google Scholar]
- Rosenman D. J.; Connors C. R.; Chen W.; Wang C.; Garcia A. E. (2013) Aβ monomers transiently sample oligomer and fibril-like configurations: Ensemble characterization using a combined MD/NMR approach. J. Mol. Biol. 425, 3338–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball K. A.; Phillips A. H.; Wemmer D. E.; Head-Gordon T. (2013) Differences in β-strand populations of monomeric Aβ40 and Aβ42. Biophys. J. 104, 2714–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. S.; Bowman G. R.; Beauchamp K. A.; Pande V. S. (2012) Investigating how peptide length and a pathogenic mutation modify the structural ensemble of amyloid β monomer. Biophys. J. 102, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chebaro Y.; Derreumaux P. (2009) Targeting the early steps of Aβ16–22 protofibril disassembly by N-methylated inhibitors: A numerical study. Proteins: Struct., Funct., Bioinf. 75, 442–452. [DOI] [PubMed] [Google Scholar]
- Scherzer-Attali R.; Convertino M.; Pellarin R.; Gazit E.; Segal D.; Caflisch A. (2013) Methylations of tryptophan-modified naphthoquinone affect its inhibitory potential toward Aβ aggregation. J. Phys. Chem. B 117, 1780–1789. [DOI] [PubMed] [Google Scholar]
- Melquiond A.; Dong X.; Mousseau N.; Derreumaux P. (2008) Role of the region 23–28 in Aβ fibril formation: Insights from simulations of the monomers and dimers of Alzheimer’s peptides Aβ40 and Aβ42. Curr. Alzheimer Res. 5, 244–250. [DOI] [PubMed] [Google Scholar]
- Viet M. H.; Nguyen P. H.; Ngo S. T.; Li M. S.; Derreumaux P. (2013) Effect of the Tottori Familial Disease Mutation (D7N) on the Monomers and Dimers of Aβ40 and Aβ42. ACS Chem. Neurosci. 4(11), 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitternacht S.; Staneva I.; Hard T.; Irback A. (2010) Comparing the folding free-energy landscapes of Aβ42 variants with different aggregation properties. Proteins: Struct., Funct., Bioinf. 78, 2600–2608. [DOI] [PubMed] [Google Scholar]
- Mitternacht S.; Staneva I.; Hard T.; Irback A. (2011) Monte Carlo study of the formation and conformational properties of dimers of Aβ42 variants. J. Mol. Biol. 410, 357–367. [DOI] [PubMed] [Google Scholar]
- Chong S. H.; Ham S. (2012) Atomic-level investigations on the amyloid-β dimerization process and its driving forces in water. Phys. Chem. Chem. Phys. 14, 1573–1575. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Bora R. P.; Barman A.; Singh R.; Prabhakar R. (2012) Dimerization of the full-length Alzheimer amyloid β-peptide (Aβ42) in explicit aqueous solution: A molecular dynamics study. J. Phys. Chem. B 116, 4405–4416. [DOI] [PubMed] [Google Scholar]
- Barz B.; Urbanc B. (2012) Dimer Formation Enhances Structural Differences between Amyloid β-Protein (1–40) and (1–42): An Explicit-Solvent Molecular Dynamics Study. PLoS One 7, e34345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T.; Zhang J.; Derreumaux P.; Mu Y. (2013) Molecular Mechanism of the Inhibition of EGCG on the Alzheimer Aβ1–42 Dimer. J. Phys. Chem. B 117, 3993–4002. [DOI] [PubMed] [Google Scholar]
- Chebaro Y.; Jiang P.; Zang T.; Mu Y.; Nguyen P. H.; Mousseau N.; Derreumaux P. (2012) Structures of Aβ17–42 Trimers in Isolation and with Five Small-Molecule Drugs Using a Hierarchical Computational Procedure. J. Phys. Chem. B 116, 8412–8422. [DOI] [PubMed] [Google Scholar]
- Meral D.; Urbanc B. (2013) Discrete Molecular Dynamics Study of Oligomer Formation by N-Terminally Truncated Amyloid β-Protein. J. Mol. Biol. 425, 2260–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharoar M. G.; Shahnawaz M.; Islam M. I.; Ramasamy V. S.; Shin S. Y.; Park I. S. (2013) The inhibitory effects of Escherichia coli maltose binding protein on β-amyloid aggregation and cytotoxicity. Arch. Biochem. Biophys. 538, 41–48. [DOI] [PubMed] [Google Scholar]
- Ghanta J.; Shen C. L.; Kiessling L. L.; Murphy R. M. (1996) A strategy for designing inhibitors of β-amyloid toxicity. J. Biol. Chem. 271, 29525–29528. [DOI] [PubMed] [Google Scholar]
- Soto C.; Kindy M. S.; Baumann M.; Frangione B. (1996) Inhibition of Alzheimer’s amyloidosis by peptides that prevent β-sheet conformation. Biochem. Biophys. Res. Commun. 226, 672–680. [DOI] [PubMed] [Google Scholar]
- Tjernberg L. O.; Naslund J.; Lindqvist F.; Johansson J.; Karlstrom A. R.; Thyberg J.; Terenius L.; Nordstedt C. (1996) Arrest of β-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 271, 8545–8548. [DOI] [PubMed] [Google Scholar]
- Fradinger E. A.; Monien B. H.; Urbanc B.; Lomakin A.; Tan M.; Li H.; Spring S. M.; Condron M. M.; Cruz L.; Xie C. W.; Benedek G. B.; Bitan G. (2008) C-terminal peptides coassemble into Aβ42 oligomers and protect neurons against Aβ42-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 14175–14180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrnhoefer D. E.; Bieschke J.; Boeddrich A.; Herbst M.; Masino L.; Lurz R.; Engemann S.; Pastore A.; Wanker E. E. (2008) EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 15, 558–566. [DOI] [PubMed] [Google Scholar]
- Frydman-Marom A.; Levin A.; Farfara D.; Benromano T.; Scherzer-Attali R.; Peled S.; Vassar R.; Segal D.; Gazit E.; Frenkel D.; Ovadia M. (2011) Orally administrated cinnamon extract reduces β-amyloid oligomerization and corrects cognitive impairment in Alzheimer’s disease animal models. PLoS One 6, e16564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzer-Attali R.; Pellarin R.; Convertino M.; Frydman-Marom A.; Egoz-Matia N.; Peled S.; Levy-Sakin M.; Shalev D. E.; Caflisch A.; Gazit E.; Segal D. (2010) Complete Phenotypic Recovery of an Alzheimer’s Disease Model by a Quinone-Tryptophan Hybrid Aggregation Inhibitor. PLoS One 5, e11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necula M.; Kayed R.; Milton S.; Glabe C. G. (2007) Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 282, 10311–10324. [DOI] [PubMed] [Google Scholar]
- Pawar A. P.; Dubay K. F.; Zurdo J.; Chiti F.; Vendruscolo M.; Dobson C. M. (2005) Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 350, 379–392. [DOI] [PubMed] [Google Scholar]
- Scherzer-Attali R.; Shaltiel-Karyo R.; Adalist Y. H.; Segal D.; Gazit E. (2012) Generic inhibition of amyloidogenic proteins by two naphthoquinone–tryptophan hybrid molecules. Proteins: Struct., Funct., Bioinf. 80, 1962–1973. [DOI] [PubMed] [Google Scholar]
- Convertino M.; Vitalis A.; Caflisch A. (2011) Disordered binding of small molecules to Aβ(12–28). J. Biol. Chem. 286, 41578–41588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand P.; Nandel F. S.; Hansmann U. H. E. (2008) The Alzheimer β-amyloid (Aβ1–39) dimer in an implicit solvent. J. Chem. Phys. 129, 195102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Rempel D. L.; Zhang J.; Sharma A. K.; Mirica L. M.; Gross M. L. (2013) Pulsed hydrogen-deuterium exchange mass spectrometry probes conformational changes in amyloid β (Aβ) peptide aggregation. Proc. Natl. Acad. Sci. U.S.A. 110, 14604–14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiike Y.; Akagi T.; Takashima A. (2007) Surface structure of amyloid-β fibrils contributes to cytotoxicity. Biochemistry 46, 9805–9812. [DOI] [PubMed] [Google Scholar]
- Lv X.; Li W.; Luo Y.; Wang D.; Zhu C.; Huang Z. X.; Tan X. (2013) Exploring the differences between mouse mAβ(1–42) and human hAβ(1–42) for Alzheimer’s disease related properties and neuronal cytotoxicity. Chem. Commun. 49, 5865–5867. [DOI] [PubMed] [Google Scholar]
- Ono K.; Condron M. M.; Teplow D. B. (2010) Effects of the English (H6R) and Tottori (D7N) familial Alzheimer disease mutations on amyloid β-protein assembly and toxicity. J. Biol. Chem. 285, 23186–23197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S.; Lopes D. H. J.; Bitan G. (2012) A Key Role for Lysine Residues in Amyloid β-Protein Folding, Assembly, and Toxicity. ACS Chem. Neurosci. 3, 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaden D.; Harmeier A.; Weise C.; Munter L. M.; Althoff V.; Rost B. R.; Hildebrand P. W.; Schmitz D.; Schaefer M.; Lurz R.; Skodda S.; Yamamoto R.; Arlt S.; Finckh U.; Multhaup G. (2012) Novel APP/Aβ mutation K16N produces highly toxic heteromeric Aβ oligomers. EMBO Mol. Med. 4, 647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Casey N.; Lee J. P. (1998) Residual structure in the Alzheimer’s disease peptide: Probing the origin of a central hydrophobic cluster. Folding Des. 3, 413–422. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Iwata K.; Lachenmann M. J.; Peng J. W.; Li S.; Stimson E. R.; Lu Y.; Felix A. M.; Maggio J. E.; Lee J. P. (2000) The Alzheimer’s peptide Aβ adopts a collapsed coil structure in water. J. Struct. Biol. 130, 130–141. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Krause G.; Reif B. (2005) Structure and orientation of peptide inhibitors bound to β-amyloid fibrils. J. Mol. Biol. 354, 760–776. [DOI] [PubMed] [Google Scholar]
- Ngo S.; Guo Z. (2011) Key residues for the oligomerization of Aβ42 protein in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 414, 512–516. [DOI] [PubMed] [Google Scholar]
- Ahmed M.; Davis J.; Aucoin D.; Sato T.; Ahuja S.; Aimoto S.; Elliott J. I.; Van Nostrand W. E.; Smith S. O. (2010) Structural conversion of neurotoxic amyloid-β(1–42) oligomers to fibrils. Nat. Struct. Mol. Biol. 17, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitan G.; Tarus B.; Vollers S. S.; Lashuel H. A.; Condron M. M.; Straub J. E.; Teplow D. B. (2003) A Molecular Switch in Amyloid Assembly: Met35 and Amyloid β-Protein Oligomerization. J. Am. Chem. Soc. 125, 15359–15365. [DOI] [PubMed] [Google Scholar]
- Zhu M.; De Simone A.; Schenk D.; Toth G.; Dobson C. M.; Vendruscolo M. (2013) Identification of small-molecule binding pockets in the soluble monomeric form of the Aβ42 peptide. J. Chem. Phys. 139, 035101–035110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K.; Condron M. M.; Teplow D. B. (2009) Structure-neurotoxicity relationships of amyloid β-protein oligomers. Proc. Natl. Acad. Sci. U.S.A. 106, 14745–14750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attanasio F.; Convertino M.; Magno A.; Caflisch A.; Corazza A.; Haridas H.; Esposito G.; Cataldo S.; Pignataro B.; Milardi D.; Rizzarelli E. (2013) Carnosine inhibits Aβ(42) aggregation by perturbing the H-bond network in and around the central hydrophobic cluster. ChemBioChem 14, 583–592. [DOI] [PubMed] [Google Scholar]
- Shan Y.; Kim E. T.; Eastwood M. P.; Dror R. O.; Seeliger M. A.; Shaw D. E. (2011) How does a drug molecule find its target binding site?. J. Am. Chem. Soc. 133, 9181–9183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. D.; Hu L.; Falkner J. A.; Benson M. L.; Nerothin J. P.; Carlson H. A. (2006) Exploring protein-ligand recognition with Binding MOAD. J. Mol. Graphics Modell. 24, 414–425. [DOI] [PubMed] [Google Scholar]
- Crescenzi O.; Tomaselli S.; Guerrini R.; Salvadori S.; D’Ursi A. M.; Temussi P. A.; Picone D. (2002) Solution structure of the Alzheimer amyloid β-peptide (1–42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur. J. Biochem. 269, 5642–5648. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Maragakis P.; Piana S.; Eastwood M. P.; Dror R. O.; Shaw D. E. (2012) Systematic Validation of Protein Force Fields against Experimental Data. PLoS One 7, e32131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. [Google Scholar]
- Van der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. C. (2005) GROMACS: Fast, flexible, and free. J. Comput. Chem. 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. (1997) LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472. [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593. [Google Scholar]
- Sugita Y.; Okamoto Y. (1999) Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314, 141–151. [Google Scholar]
- Patriksson A.; van der Spoel D. (2008) A temperature predictor for parallel tempering simulations. Phys. Chem. Chem. Phys. 10, 2073–2077. [DOI] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101–014107. [DOI] [PubMed] [Google Scholar]
- Schlitter J. (1993) Estimation of absolute and relative entropies of macromolecules using the covariance matrix. Chem. Phys. Lett. 215, 617–621. [Google Scholar]
- Kabsch W.; Sander C. (1983) Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Miller S.; Janin J.; Lesk A. M.; Chothia C. (1987) Interior and surface of monomeric proteins. J. Mol. Biol. 196, 641–656. [DOI] [PubMed] [Google Scholar]
- Daura X.; Gademann K.; Jaun B.; Seebach D.; van Gunsteren W. F.; Mark A. E. (1999) Peptide Folding: When Simulation Meets Experiment. Angew. Chem., Int. Ed. 38, 236–240. [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheatham T. E. III (2000) Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.