

Background: Intracellular GPR37 accumulation is neurotoxic and associated with parkinsonism, whereas plasma membrane association is protective.
Results: The endogenous GPR37 ligand prosaposin promotes GPR37 surface density and association with GM1-enriched lipid rafts.
Conclusion: GPR37, prosaposin, and GM1 constitute a pathway that improves cell viability through GPR37 trafficking to the plasma membrane.
Significance: Targeting this pathway could reduce toxic intracellular GPR37 accumulation observed in parkinsonism.
Keywords: Fluorescence Correlation Spectroscopy, G Protein-coupled Receptors (GPCR), Ganglioside, Lipid Raft, Parkinson Disease, Pael-R
Abstract
The subcellular distribution of the G protein-coupled receptor GPR37 affects cell viability and is implicated in the pathogenesis of parkinsonism. Intracellular accumulation and aggregation of GPR37 cause cell death, whereas GPR37 located in the plasma membrane provides cell protection. We define here a pathway through which the recently identified natural ligand, prosaposin, promotes plasma membrane association of GPR37. Immunoabsorption of extracellular prosaposin reduced GPR37tGFP surface density and decreased cell viability in catecholaminergic N2a cells. We found that GPR37tGFP partitioned in GM1 ganglioside-containing lipid rafts in the plasma membrane of live cells. This partitioning required extracellular prosaposin and was disrupted by lipid raft perturbation using methyl-β-cyclodextrin or cholesterol oxidase. Moreover, complex formation between GPR37tGFP and the GM1 marker cholera toxin was observed in the plasma membrane. These data show functional association between GPR37, prosaposin, and GM1 in the plasma membrane. These results thus tie together the three previously defined components of the cellular response to insult. Our findings identify a mechanism through which the receptor's natural ligand and GM1 may protect against toxic intracellular GPR37 aggregates observed in parkinsonism.
Introduction
GPR37 (or Pael-R) is a G protein-coupled receptor (GPCR)2 that is enriched in the brain (1–4) and that has been implicated in the pathogenesis of Parkinson disease (PD). It is a substrate of the E3 ubiquitin ligase parkin, and aggregates in the substantia nigra of patients with autosomal recessive juvenile PD caused by parkin mutations (3), as well as in patients with sporadic PD (5). Indeed, when highly overexpressed, or under conditions of reduced parkin activity, GPR37 has a propensity to aggregate and cause cell death via endoplasmic reticulum stress that is unique among GPCRs (3, 6–8). Conversely, inhibition of GPR37 accumulation in the endoplasmic reticulum and potentiation of membrane trafficking improve cell viability in GPR37-overexpressing cells (9). Moreover, GPR37 receptors at the plasma membrane are protective against 1-methyl-4-phenylpyridinium-, rotenone-, and 6-hydroxydopamine-induced toxicity in a neuron-like catecholaminergic cell line (10).
Prosaposin (PSAP, sulfated glycoprotein-1, or sphingolipid activator protein-1) is a dual-function protein that either is cleaved in the lysosome to produce the biologically active peptides, saposins A, B, C, and D, or released extracellularly as the full-length protein acting as a neuroprotective, neurotrophic, and gliotrophic factor (11–14). The neurotrophic sequence of full-length PSAP is a 12-amino acid motif within the saposin C region (15–17). It has long been known that PSAP and prosaptides, which are short synthetic peptides derived from the neurotrophic sequence of PSAP, protect neurons against various cellular insults, including MPTP, oxidative stress, and ischemia (15, 18, 19). PSAP and prosaptides exert their neurotrophic actions via an unknown Gi/o-coupled GPCR at the plasma membrane. Very recently, extracellular PSAP along with prosaptides were identified as agonists at GPR37 (and its homologue GPR37L1) (20). GPR37 and GPR37L1, but not other GPCRs tested, were pulled down with prosaptide in biotinylation assays, and PSAP and prosaptides stimulated Gi/o and ERK1/2 signaling via GPR37. Moreover, GPR37 was shown to mediate PSAP-induced protection against H2O2 toxicity in primary astrocytes.
It is also known that PSAP localizes to lipid rafts containing GM1 and GM3 gangliosides at plasma membranes (21, 22) and that ganglioside-containing lipid rafts are required for PSAP-activated GPCR signaling and neurotrophic effects (22). This study evaluated the hypothesis that PSAP, prosaptides, and gangliosides may regulate the plasma membrane levels of GPR37 suggested to be crucial for neuroprotective actions of GPR37. For this purpose, catecholaminergic N2a cells stably expressing GPR37 genetically fused with the turbo green fluorescent protein (GPR37tGFP) were studied using various approaches, most notably confocal microscopy and fluorescence correlation spectroscopy (FCS) in live cells. The FCS technique is unique in its ability to quantitatively characterize molecular diffusion and complex formation in live cells.
EXPERIMENTAL PROCEDURES
Cell Culture
Reagents for cell culture and transfection were from Invitrogen. A stable cell line overexpressing GPR37-turboGFP (GPR37tGFP) was generated as described previously (10). The GPR37tGFP construct was bought from Origene (Rockville, MD). Cells were maintained in culture medium containing 50 μg ml−1 geneticin at 37 °C, 5% CO2, and split 1:4 every 3–4 days. The cells were seeded in differentiating medium (phenol red- and serum-free Opti-MEM supplemented with 100 units ml−1 penicillin, 100 μg ml−1 streptomycin, and 1 mm dibutyryl cyclic AMP (Sigma)) and subjected to various treatments 3–4 days thereafter. For confocal laser scanning microscopy (CLSM) and fluorescence correlation/cross-correlation spectroscopy (FCS/FCCS) measurements, the cells were cultured in 8-well chambers on a 1.0 borosilicate coverglass (Nunc Lab-Tek, Thermo Scientific, Stockholm, Sweden).
Immunoblotting
Samples were subjected to SDS-PAGE on 10% acrylamide gels and then transferred to Immobilon-P membranes (Millipore, Billerica, MA). Membranes were blocked in 5% milk (Tris-buffered saline, 5% fat-free milk powder, 0.12% Tween 20) for 1 h and then incubated with primary antibodies against prosaposin (ab68466, Abcam, Cambridge, UK) or GPR37 (14820-1-AP, Proteintech, Manchester, UK) diluted in 5% milk. Membranes were then washed and incubated with HRP-linked or fluorescently labeled secondary antibodies for film or fluorescence detection, respectively, followed by a second washing. For film detection, membranes were incubated with ECL Plus substrate (PerkinElmer Life Sciences), and the signal was detected on BioMax XAR film (Eastman Kodak). For fluorescence detection, the signal was read on a LI-COR Odyssey CLx (LI-COR Biosciences, Cambridge, UK).
MTT Cell Viability Assays
Cells were seeded in 96-well plates at a density of 1 × 104 cells per well and incubated at 37 °C, 5% CO2 for 3 days before addition of prosaposin (PSAP)-specific antibody anti-PSAP (sc-32876, Santa Cruz Biotechnology, Dallas, TX) at 40 μg ml−1 or anti-PS769 (generated as described previously (21)) at 8.5 μg ml−1. Cytotoxicity was assayed 8, 16, and 24 h later or in a separate experiment 48 h later. MTT (0.5 mg ml−1) was added to the medium, and cells were incubated for 1 h at 37 °C, 5% CO2. The medium was removed, and cells were lysed in isopropyl alcohol containing 0.04 m HCl. Absorbance was measured at 550 nm using an Anthos 2020 microplate reader (Biochrom, Berlin, Germany). Average values for untreated controls within each cell type and time point were set to 100%. Values measured in treatment groups were expressed as a percentage of control within cell type and time point. Data were pooled from three independent experiments.
Immunoprecipitation of PSAP
Cells were seeded in 8-well chambers at a density of 2 × 104 cells per well and incubated at 37 °C, 5% CO2 for 3 days before addition of PSAP-specific antibody anti-PSAP (sc-32876, Santa Cruz Biotechnology, 40 μg ml−1) or normal IgG (12-370, Millipore, 40 μg ml−1) as a negative isotype control. After incubation for 2.5 h at room temperature, medium was removed from the cells and incubated with protein A-coated magnetic beads to pull down the antibodies. Beads and media were separated, and the medium was subjected to Novex SDS-PAGE on 4–12% BisTris gels (Invitrogen) followed by transfer to Immobilon-P membranes (Millipore). Membranes were subjected to immunoblotting as described above using a PSAP-specific primary antibody (1:200, sc-32876, Santa Cruz Biotechnology) and conformation-specific HRP-labeled anti-rabbit secondary antibody (1:4000, 5127S, Cell Signaling Technology).
Fluorescent Peptides
The PSAP-derivative prosaptide TX14(A) (PS-TX14(A)TAMRA), and β-endorphin (β-EndTAMRA) peptides fluorescently labeled at the C terminus with carboxytetramethylrhodamine (TAMRA), were purchased from Biomatik (Wilmington, DE). The primary sequences of the peptides were T(d-Ala)LIDNNATEEILY and YGGFMTSEKSQTPLVTLFKNAIIKNAYKKGQK for PS-TX14(A) and β-endorphin. Both peptides were purified by the producer to a purity level of >98% using HPLC with gradient elution of water/acetonitrile and 0.1% trifluoroacetic acid. The molecular masses of the peptides were 2.1 and 4 kDa for PS-TX14(A)TAMRA and β-EndTAMRA, respectively, as determined by electrospray ionization-mass spectrometry. Peptides were stored in powder at −20 °C and were used without any further purification. Aqueous solutions of the peptides were prepared just before cell treatment. 5–15-μl aliquots of the peptide solution were added to the cell culture medium (350 μl) and gently mixed. In experiments with PS-TX14(A)TAMRA and β-EndTAMRA, the medium was replaced twice with a 45-min interval before addition of the peptide. This was done to reduce the amount of endogenous PSAP in the medium and its potential interference with fluorescently labeled peptides binding to GPR37.
Cholesterol Sequestration
Membrane cholesterol was sequestered by treatment with 4 mm methyl-β-cyclodextrin (Sigma) for 45 min or 4 units ml−1 cholesterol oxidase (Sigma) for 1 h. Both treatments were performed at room temperature.
CLSM and FCS/FCCS
CLSM imaging, FCS/FCCS, and fluorescence recovery after photobleaching (FRAP) measurements were performed using the LSM510 ConfoCor3 instrument (Carl Zeiss, Jena, Germany) individually modified to enable fluorescence imaging with silicone avalanche photodiodes (SPCM-AQR-1X; PerkinElmer Life Sciences) (23). Background on FCS/FCCS measurements and data analysis are described below and in Figs. 1 and 2. Image adjustments only included brightness and/or contrast adjustments applied to the entire image and to corresponding controls. Nonlinear image processing and filtering were not applied.
FIGURE 1.
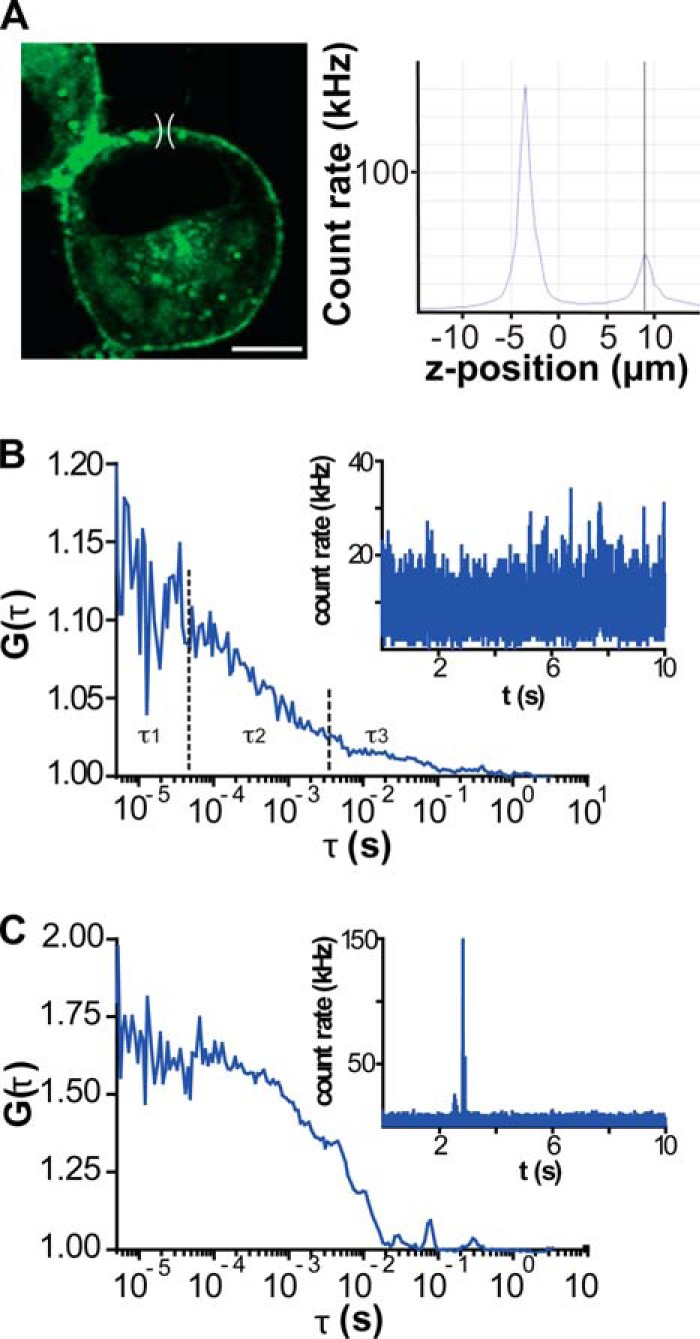
FCS at the apical plasma membrane of N2a cells expressing GPR37tGFP. A, left panel, confocal image of an N2a cell expressing GPR37tGFP. For FCS analysis, the observation volume element (drawn to scale) was positioned above the cell nucleus, which has the lowest level of autofluorescence. Scale bar, 5 μm. Right panel, axial fluorescence intensity scan (z-scan) was used to localize the position of the apical plasma membrane. The first maximum reflects the position of the basal membrane, and the second one reflects the position of the apical membrane. B, typical autocorrelation curve obtained after a single 10-s measurement of fluorescence intensity fluctuations at the apical plasma membrane of an N2a cell expressing GPR37tGFP. The corresponding fluorescence intensity fluctuations are shown in the inset. C, autocorrelation curve reflecting a sporadic observation of a very bright intracellular vesicle traveling through the observation volume element. The corresponding fluorescence intensity fluctuations are shown in the inset. Such individual recordings were excluded from further analysis and did not contribute to the average autocorrelation curves.
FIGURE 2.

Fluorescence intensity fluctuations recorded at the apical plasma membrane of N2a cells expressing GPR37tGFP are generated by molecular movement. A, autocorrelation curves recorded at varying observation volume sizes show that the amplitudes of the GPR37tGFP autocorrelation curves decrease as the pinhole size in front of the detector is increased, reflecting an increase in the number of molecules in the observation volume element. B, for varying sizes of the observation volume element, a linear relationship was observed between the number of observed molecules (N) and the (pinhole size). C, autocorrelation curves at two different pinhole sizes show that characteristic times increase with increasing the observation volume size. D, linear relationship was observed between the first characteristic time (τD1) and the (pinhole size). The positive intercept suggests that partitioning between domains may occur. E, linear relationship was observed between the second characteristic time (τD2) and the (pinhole size). The negative intercept suggests that some confinement by the cytoskeletal protein meshwork may also occur. F, linear relationship between the number of molecules characterized by slow (N2) and fast (N1) diffusion was observed for different surface densities of GPR37tGFP.
Optical Setting
tGFP fluorescence was excited using the 488-nm line of the Ar/ArKr laser, and the 543-nm He/Ne laser was used for Alexa Fluor 594 and TAMRA. Emitted light was separated from the incident light using the main dichroic beam splitter HFT 488/543 and split using the secondary dichroic beam splitter NFT 545. Further spectral separation of the emitted light was achieved using specialized filters in front of the detectors. For imaging, emitted light was collected in the multitrack acquisition mode using the bandpass filter BP 505–530 for tGFP and the bandpass filter BP 615–680 for Alexa Fluor 594 and TAMRA. Images were acquired without averaging, using a scanning speed of 51.2 μs/pixel and a 512 × 512 pixels frame. For FCS/FCCS, emitted light was collected using the bandpass filter BP 505–530 for tGFP and the long pass filter LP 580 or bandpass filter BP 615–680 for Alexa Fluor 594 and TAMRA. All FCS/FCCS measurements were performed on the plasma membrane that is at the apical end of the cell, above the fraction of the plasma membrane that is adhering to the coverslip and the virtually transparent nucleus. The apical plasma membrane was identified by a fluorescence intensity scan across the vertical axis of the cell (Fig. 1A). Fluorescence intensity fluctuations were recorded in arrays of 10–50 consecutive measurements, each measurement lasting 10–20 s. Autocorrelation curves (Fig. 1, B and C) were individually inspected to exclude traces where the passage of bright transporting intracellular vesicles interfered with the FCS measurements (Fig. 1C). Thus, the generated average autocorrelation curves (Fig. 2A) were analyzed using the dedicated ConfoCor3 Zeiss software (Carl Zeiss, Jena, Germany).
Temperature and Atmosphere Control
CLSM imaging, FCS/FCCS, and FRAP were performed under controlled temperature (37 °C) and atmospheric conditions (humid air with 5% CO2) and at room temperature (20 °C) and atmospheric conditions. Temperature and atmosphere regulations were achieved using a heating insert at the microscope stage (Heat Insert P, PeCon GMBH), temperature regulating unit (TempControl 37-2 digital, PeCon GMBH), and an atmosphere controlling unit (CTI-Controller 3700 digital, PeCon GMBH) with an IR absorption-based CO2 sensor for continuous monitoring of CO2 concentration in the heated circulating air stream. Differential effects of temperature/atmosphere on GPR37 trafficking were not observed. All measurements reported here were performed at room temperature.
Background on FCS
FCS is a quantitative method with single molecule sensitivity that uses statistical approaches in the form of temporal autocorrelation and/or photon-counting histogram analysis of fluorescence intensity fluctuations recorded over time to obtain information about molecular numbers and their transporting properties (temporal autocorrelation) or molecular brightness distribution (photon-counting histogram) (24). In this study, fluorescence intensity fluctuations recorded over time (Fig. 1B, inset) were analyzed using temporal autocorrelation to extract information about the average number of molecules in the observation volume element, i.e. the concentration, and the diffusion times. For this purpose, the normalized autocorrelation function G(τ) was first derived as shown in Equation 1,
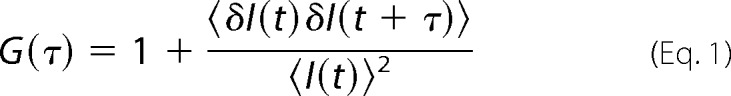 |
G(τ) gives the correlation between the deviation of fluorescence intensity measured at a certain time point t, δI(t) = I(t) − 〈I(t)〉, which is given as the difference in fluorescence intensity I(t) and the mean fluorescence intensity over the recorded time series (〈I(t)〉), and its intensity measured at a later time, δI(t + τ) = I(t + τ) − 〈I(t)〉. G(τ) is then plotted as a function of different lag times τ, yielding the experimental autocorrelation curve (see for example Fig. 1B).
To extract the molecular numbers (N) and diffusion times (τD), the experimental autocorrelation curves were fitted using autocorrelation functions derived for appropriate model systems (25, 26). For measurements performed in the cell culturing medium, FCS curves were fitted using a model for free three-dimensional diffusion with triplet contribution as shown in Equation 2,
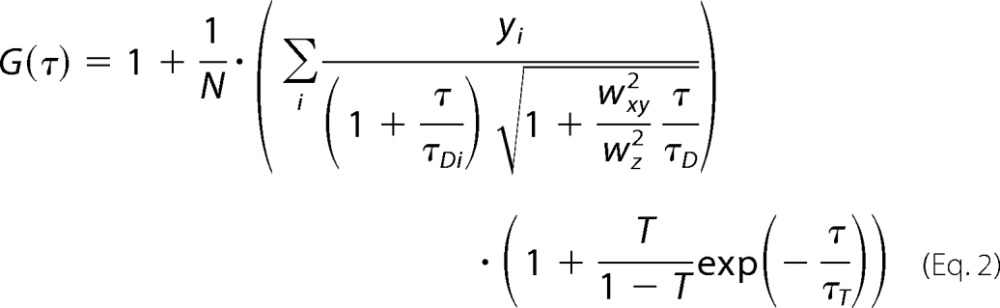 |
For measurements performed at the plasma membrane, a model for free two-dimensional diffusion with triplet contribution was used as shown in Equation 3,
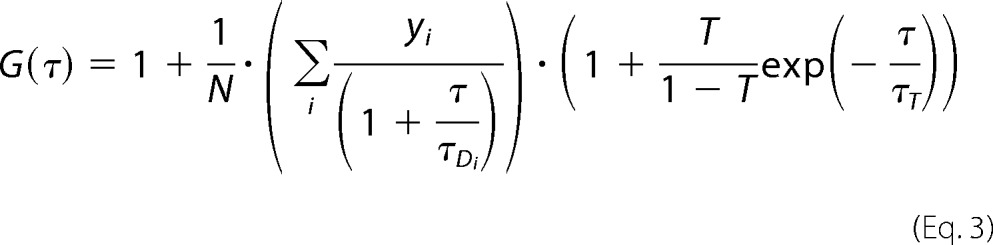 |
In Equations 2 and 3, T is the average equilibrium fraction of molecules in the triplet state; τT is the triplet state relaxation time; i is the number of components; τDi is the diffusion time of the ith component, and yi is its relative amplitude (Σyi = 1). In Equation 2, wxy and wz are the distances from the center of the laser beam focus in the radial and axial directions, respectively, where the collected fluorescence intensity has dropped by a factor of e2 compared with its peak value for the Gaussian beam profile. The parameter (wxy/wz)2 is determined by instrument calibration using standard aqueous solutions of rhodamine 6G for λex = 488 nm and TAMRA for λex = 543 nm. Data fitting using Equation 2 for a single component (i = 1) yielded diffusion times τD, Rh6G = 27 ± 3 μs and τD, TAMRA = 33 ± 3 μs and structural parameters 7 for λex = 488 nm and 8 for λex = 543 nm.
For fitting the experimental autocorrelation curves, the simplest model, i.e. the model with the lowest number of components, was always used. For a first hand characterization of two-dimensional diffusion, the instrument running software was used (5, 9).
Analysis of Autocorrelation Curves for GPR37tGFP and Interpretation of Autocorrelation Decay Times
Autocorrelation curves for GPR37tGFP recorded at the apical plasma membrane show several characteristic decay times (Fig. 1B). To identify the most appropriate model to evaluate the experimental autocorrelation curves, we needed to first identify the underlying processes that lead to fluorescence intensity fluctuations. In general, fluorescence intensity fluctuations can be generated by molecular diffusion, chemical interactions, and/or photo-physical processes, such as intersystem crossing (singlet to triplet state transition) (27). To identify the contribution of these processes in our system, we varied the size of the observation volume element and investigated the effect of the observation volume size on the observed decay times (28, 29). In this assay, the size of the observation volume element from which the photons are collected is changed by changing the size of the pinhole in front of the detector. If fluorescence intensity fluctuations are generated by molecular diffusion, the characteristic decay time of the autocorrelation function should increase as the size of the observation volume element increases. In contrast, if fluorescence intensity fluctuations arise as a consequence of photo-physical or chemical transformations, such as transition to the triplet state or a chemical reaction where fluorescence is gained/lost, the characteristic decay time of the autocorrelation function would not be affected (27).
We observed that the autocorrelation curves recorded at the apical plasma membrane shift to longer decay times when the size of the observation element is increased (Fig. 2, C–E), indicating that fluorescence intensity fluctuations characterized with decay times τ >100 μs are generated by molecular movement. Accordingly, a two-component diffusion model with triplet contribution (Equation 2, i = 2) was used for fitting the experimental autocorrelation curves. The two-component diffusion model yielded diffusion times τD1 = (3.5 ± 1.5) × 10−4 s and τD2 = (2.5 ± 1.5) × 10−2 s. For all cells investigated (n = 25), the relative contribution of the second component was y2 = 0.17 ± 0.08 (Fig. 2F). The indicated standard deviation reflects differences between cells and not between different measurements on the same cell.
The pinhole size scan assay also showed that both diffusion times τD1 and τD2 increase as the size of the observation volume element is increased, but they follow different diffusion laws (30). Thus, a positive intercept was observed for τD1 = f (ph2) (Fig. 2D), whereas a small negative intercept was observed for τD2 = f (ph2) (Fig. 2E). This indicates that GPR37tGFP lateral organization and dynamics in the plasma membrane is complex, involving partitioning between microdomains and possibly some confinement by the cytoskeletal protein meshwork (30, 31). It is also possible that the faster component originates from an intracellular pool of GPR37tGFP, as was suggested for serotonin 5-HT1A (31). However, one would expect in this case that the fraction of molecules characterized by fast or slow diffusion would be independent from one another, showing different relative contributions for different cells. The fact that a linear relationship between the number of molecules characterized by slow (N2) and fast (N1) diffusion was observed for different surface densities of GPR37tGFP (Fig. 2F) supports the interpretation that a dynamic sorting mechanism is observed at the plasma membrane. In addition, unpublished studies from our laboratory3 on other GPCRs show that the relative contribution of one fraction with respect to the other is different for different GPCRs. Taken together, these observations strengthen the interpretation that both diffusion times reflect processes at the plasma membrane.
This interpretation is further supported by our observation that a fraction of GPR37tGFP interacts with GM1 and resides in GM1-enriched domains (see under “Results”). If GPR37tGFP sorting between the lipid bilayer and GM1-enriched domains is slow compared with the later diffusion time of GPR37tGFP in the lipid bilayer, two components will be detected by FCS, with the fast one reflecting GPR37tGFP diffusion in the lipid bilayer and the slow one reflecting the diffusion of the GM1-enriched domains. (A detailed explanation for chemical reactions in three dimensions can be found in Ref. 27.) Recent studies on model membranes verified that peptides/protein can spontaneously partition between liquid disordered and solid ordered raft-like microdomains (32). This study also showed that the diffusion coefficient of the investigated peptides/protein in the fluid phase is in the order of a few μm2 s−1, whereas the diffusion coefficient of the microdomains is about 1 × 10−1 μm2 s−1 (32). The difference between the experimentally measured diffusion times for GPR37tGFP, τD1 = (3.5 ± 1.5) × 10−4 s, and τD2 = (2.5 ± 1.5) × 10−2 s, falls in the same range. However, we cannot unequivocally determine the diffusion coefficients based on the lateral diffusion times, because GPR37tGFP is excluded from some domains in the plasma membrane, and the actual area that is available for GPR37tGFP diffusion is smaller than the FCS observation area (see “Results”).
Analysis of Autocorrelation Curves for PS-TX14(A)TAMRA and Interpretation of Autocorrelation Decay Times
Using wild type N2a cells expressing low levels of GPR37 and GPR37tGFP-overexpressing cells (Fig. 3), we studied PS-TX14(A)TAMRA interactions with GPR37tGFP (Figs. 4 and 5). The autocorrelation curve for PS-TX14(A)TAMRA in the bulk medium could be fitted with a single component model for free three-dimensional diffusion and triplet formation (Equation 3, i = 1, yi = 1), with the characteristic diffusion time τD = 70 ± 5 μs. In contrast, the single color autocorrelation curve for PS-TX14(A)TAMRA recorded at the plasma membrane of N2a cells expressing GPR37tGFP showed an additional component with a significantly longer diffusion time τD2 = 15 ± 10 ms (Fig. 4B).
FIGURE 3.

WT and GPR37tGFP-expressing N2a cells both express low levels of endogenous GPR37 and secrete PSAP but respond differently to inhibition of extracellular PSAP. A, Western blot for GPR37 on lysates from WT and GPR37tGFP-expressing cells showing a weak band at the size corresponding to untagged GPR37 in both cell types and an additional band at the size corresponding to GPR37tGFP in the GPR37tGFP-expressing cells. B, Western blot for extracellular PSAP using medium from WT and GPR37tGFP cells shows that both cell types secrete equal amounts of PSAP (n = 9, p = 0.99). C, estimate of PSAP concentrations in the conditioned medium of differentiated GPR37tGFP cells based on Western blot analysis. The calibration curve was generated using a standard series of PSAP solutions that were analyzed on the same gel as the conditioned medium. D, immunoprecipitation of PSAP from conditioned cell medium using anti-PSAP (well 1) or normal rabbit IgG (well 2) as a negative isotype control. 40 μg ml−1 anti-PSAP precipitates 83–96% of all PSAP compared with beads alone. Western blot films show three separate immunoprecipitation experiments. E, quantification of GPR37 density at the plasma membrane versus cytoplasm before and after inhibition of extracellular PSAP using anti-PSAP (n = 15 visual fields). Representative images of live cells show distribution of GPR37 with or without anti-PSAP. Data from Western blots and GPR37 density were analyzed by Student's t test. Data from MTT assays were analyzed by two-way analysis of variance followed by t test for pairwise comparisons, ***, p < 0.001 compared with WT. F, MTT assay of WT and GPR37-overexpressing cells untreated or treated with the PSAP-specific antibodies anti-PSAP (antibody: F(2,138) = 3.6, p < 0.05; cell type: F(1,138) = 5.0, p < 0.05; interaction: F(2,138) = 0.9, p = 0.41) or anti-PS769 (antibody: F(1,60) = 42.4, p < 0.001; cell type: F(1,60) = 9.2, p < 0.01; interaction: F(1,60) = 9.6, p < 0.01) (n = 24 in each group). ###, p < 0.001 compared with WT anti-PS769; *, p < 0.05 compared with WT anti-PSAP.
FIGURE 4.
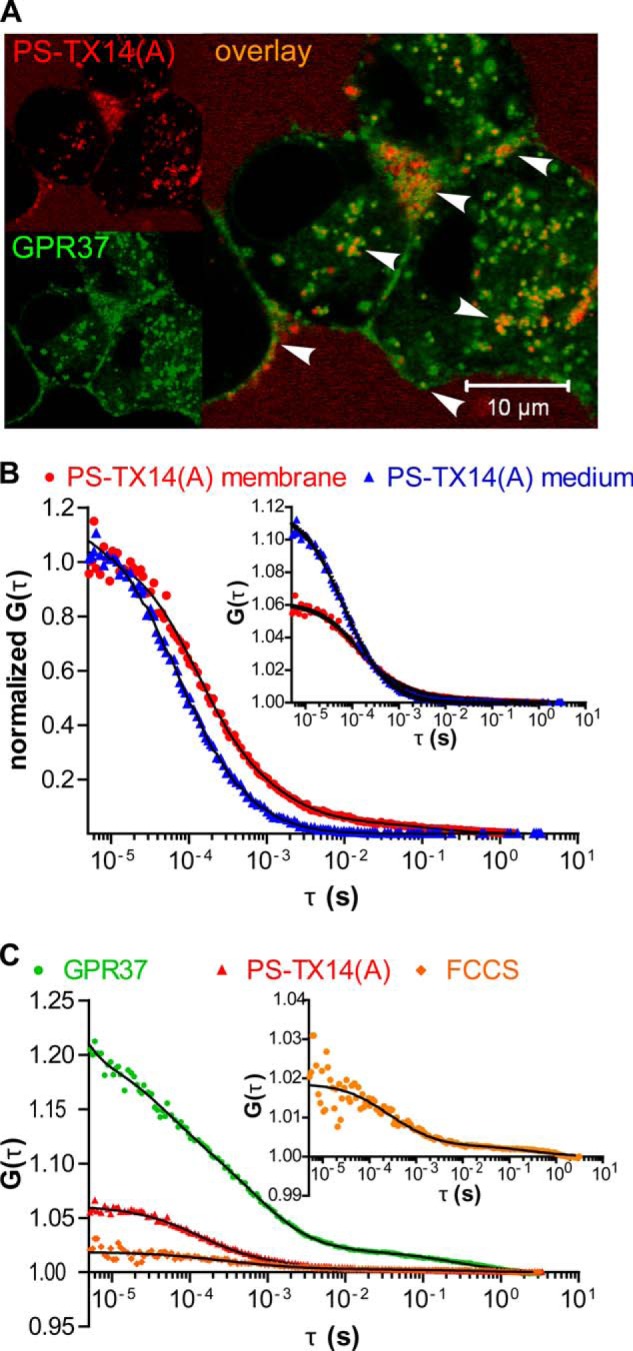
GPR37 interacts with PSAP-derived PS-TX14(A). A, confocal microscopy shows partial co-localization of GPR37tGFP (green) with PS-TX14(A)TAMRA (red) in live cells. PSAP co-localizes with GPR37 at the plasma membrane and in intracellular endosomes/organelles. B, FCS for PS-TX14(A)TAMRA at the plasma membrane compared with the bulk medium 200 μm above the cell. In the bulk medium, the diffusion time of PS-TX14(A)TAMRA was τD = 73 μs. At the plasma membrane, 93% of the molecules showed fast diffusion, τD1 = 130 μs, and 7% diffused at τD2 = 14 ms. C, FCCS for GPR37tGFP and PS-TX14(A)TAMRA at the plasma membrane (same cell as in B) shows a significant cross-correlation with 90% of the complexes diffusing at τD1 = 410 μs and 10% at τD2 = 43 ms.
FIGURE 5.
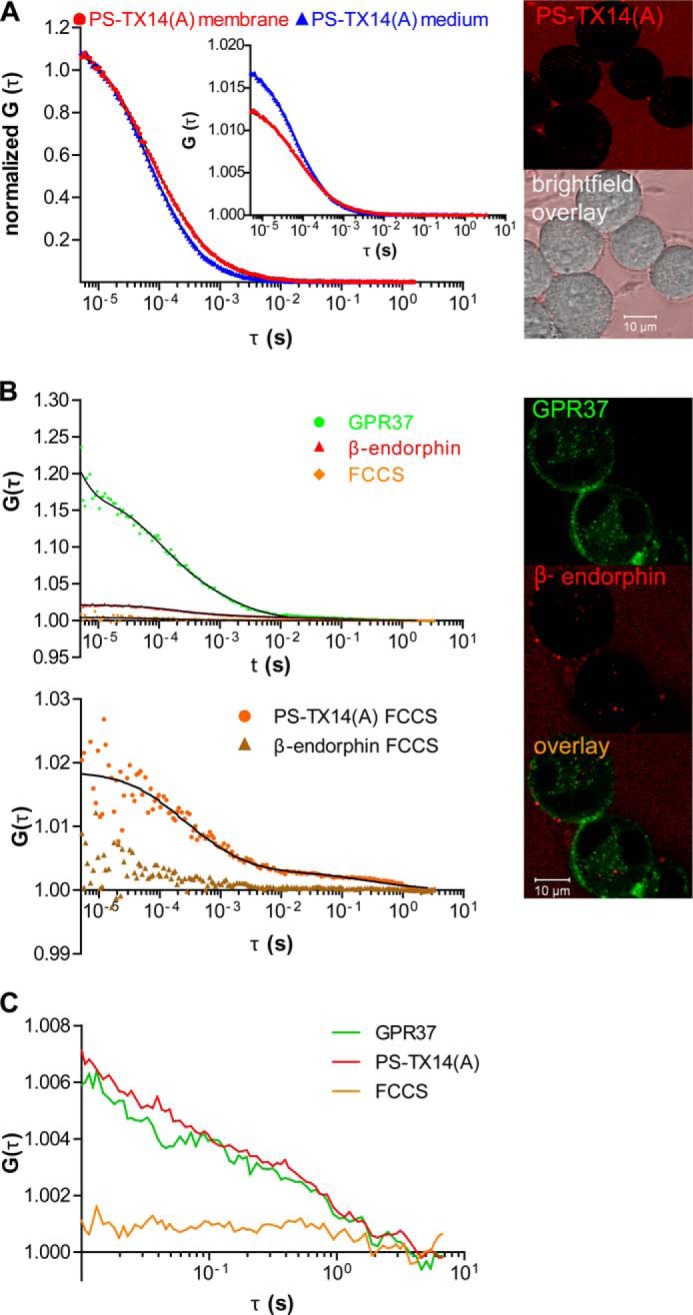
Interactions of PS-TX14(A)TAMRA with GPR37tGFP are specific. A, autocorrelation curves (left panel) recorded in the bulk medium (blue) and on the surface of WT N2a cells (red) show that neither the diffusion nor the concentration of PS-TX14(A)TAMRA are significantly altered at the plasma membrane of WT N2a cells, indicating that PS-TX14(A)TAMRA does not bind nonspecifically to the plasma membrane of WT N2a cells. Representative images (right panel) show limited binding of PS-TX14(A)TAMRA to membranes of WT N2a cells with low expression levels of GPR37. B, top left panel, FCCS measurements showed no cross-correlation between GPR37tGFP and β-EndTAMRA. Bottom left panel, cross-correlation curves confirming interactions between PS-TX14(A)TAMRA and GPR37tGFP reflect specific binding (orange), as compared with lack of cross-correlation between β-EndTAMRA and GPR37tGFP signal (brown). Right panel, representative images show absence of co-localization between β-EndTAMRA and GPR37tGFP. C, autocorrelation and cross-correlation curves for GPR37tGFP and PS-TX14(A)TAMRA using the sequential illumination mode at a switching rate of 100 μs.
Analysis of Cross-correlation Curves and Interpretation of Cross-correlation Decay Times
Dual-color FCCS relies on the usage of spectrally distinct fluorescent markers and the co-variance of their signals to establish complex formation (24, 33, 34). The cross-correlation curves showed two characteristic decay times and were fitted using a two-component diffusion model without triplet contribution. The apparent dissociation constant of the GPR37tGFP·PS-TX14(A)TAMRA complexes (KDapp) was determined using the uncorrected form of the cross-correlation function to derive the number of free versus bound GPR37tGFP and PS-TX14(A)TAMRA molecules (35, 36) as shown in Equations 4 and 5,
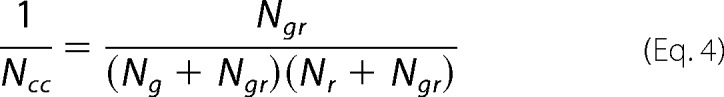 |
 |
where Ng is the number of GPR37tGFP molecules in the observation volume element; Nr is the number of PS-TX14(A)TAMRA molecules in the observation volume element, and Ngr is the number of their complexes in the observation volume element. In the equivalent Equation 5, ACC, Ag, and Ar are the amplitudes of the cross-correlation curve, the amplitude of the autocorrelation curve recorded in the “green” channel, reflecting the contributions from GPR37tGFP and GPR37tGFP·PS-TX14(A)TAMRA complexes, and the amplitude of the autocorrelation curve recorded in the “red” channel, reflecting the contributions from PS-TX14(A)TAMRA and GPR37tGFP·PS-TX14(A)TAMRA complexes, respectively, at τ = 1·10−5 s.
The apparent value of the dissociation constant of GPR37tGFP·PS-TX14(A)TAMRA complexes (KDapp) obtained using the uncorrected cross-correlation function in Equations 4 or 5 should be regarded as an approximate value that may significantly differ from the actual dissociation constant KD. On the technical side, the following factors can affect the amplitudes of auto- and cross-correlation curves, and hence the experimentally determined KD value; the observation volume elements for the spectrally distinct fluorophores have different volumes and can be displaced due to chromatic aberrations and misaligned excitation lasers, cross-talk between the channels, background fluorescence, and resonance energy transfer between the fluorophores (Förster resonance energy transfer (FRET)). These processes have been recognized and meticulously analyzed in several publications, from the first report by Földes-Papp et al. (37) to the recent work by Wohland and co-workers (38) and references therein. The main conclusion of these studies is that technical limitations do not contribute significantly (<25%) (37), except under conditions when FRET between the fluorophores is very strong (33, 35, 37, 38), which seems not to be the case in our studies. For the purpose of our studies, it is most important to ascertain that cross-talk between the channels does not yield a false-positive result. To verify this, we performed control experiments using the μ-opioid receptor selective peptide β-endorphin fluorescently labeled with TAMRA (β-EndTAMRA). Cross-correlation curves recorded for GPR37tGFP/β-EndTAMRA (Fig. 5B, lower left panel, brown curve) and GPR37tGFP·PS-TX14(A)TAMRA (Fig. 5B, lower left panel, orange curve) clearly show that cross-talk between channels is minimal and cannot account for the cross-correlation observed for GPR37tGFP·PS-TX14(A)TAMRA.
The formation of complexes with long characteristic times was further confirmed by sequential FCCS (Fig. 5C). Switching between channels was performed at 100 μs to enable acquisition of sufficient number of photons. Unfortunately, this resulted in losing the information for lag times shorter than 1 ms.
In addition to the technical challenges discussed above, one should also note that endogenous nonfluorescent molecules, protein constructs with fluorophores residing in dark states, or protein constructs with photobleached fluorophores may compete in bimolecular interactions, shifting the apparent equilibrium to higher values of KDapp (38).
FRAP
FRAP measurements were performed on the same instrument using the same optical settings as for tGFP confocal imaging (described above). Circular regions of interest (ROIs) 2 μm in diameter were defined for bleaching. To measure the fluorescence intensity before bleaching, 15 measurements were first performed using low laser intensity (2% transmission), followed by bleaching with the 488 nm line of the Ar/ArKr laser at 100% transmission to achieve 60–90% bleaching. Fluorescence was then measured at 3-s intervals for 5 min, using the same laser transmission at acquisition as before bleaching. Fluorescence intensities were normalized so that the average intensity before bleaching was 1 and after bleaching was 0 (39).
To investigate the effect of cholesterol depletion on GPR37 dynamics, nearby regions of the same cell were subjected to FRAP before and 45 min after addition of mβCD or 1 h after addition of cholesterol oxidase. We first ascertained in control experiments that this procedure is valid and that the first and second FRAP measurement yielded the same results when no treatment was applied. Fitting of the FRAP curves was not performed.
Quantification of Fluorescence Intensity and Co-localization
For fluorescence intensity quantification at the plasma membrane versus the cytoplasm, five visual fields per well from three separate experiments were imaged before and 2.5–3 h after addition of the PSAP antibody. This yielded 349 and 346 cells, respectively, for analysis in the untreated and treated group. The membrane was identified in overlaid images of the transmission channel and the tGFP channel. Image analysis was performed using the Carl Zeiss LSM image browser software (Carl Zeiss, Germany). The average fluorescence intensities (I) in defined ROI comprising the plasma membrane and the cytoplasm, respectively, were determined for each cell. The average of all Imembrane values in a given image was divided by the average of all Icytoplasm values in the same image, and the ratios of averages for all images were plotted.
In co-localization studies with Alexa 594-tagged cholera toxin β-subunit (CTxB), cells were treated with CTxB alone or pretreated with PSAP antibody for 2.5 h followed by addition of CTxB. For quantification of co-localization, 10 visual fields per well from three separate experiments yielded a minimum of 69 cells per group. The plasma membrane was identified as described above, and ROIs comprising the plasma membrane were defined. The co-localization function in the Zeiss LSM image browser was then used to derive the Pearson's correlation (P) and Manders' coefficient of green overlapping red (c2) for each ROI. From these values, the average P and c2 within each group was calculated.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software, San Diego). Data sets with two grouping variables were analyzed by two-way analysis of variance, and the post hoc test was performed using Student's t test for pairwise comparisons. Data sets with one variable and two groups were analyzed by unpaired two-tailed Student's t test. Values are reported as mean ± S.E.M., and effects were considered significant at p < 0.05. Numbers of cells analyzed in each graph are given in the respective figure legends.
RESULTS
N2a Cells Secrete PSAP
Overexpression of GPR37 improves cell survival after toxin exposure (10), which suggests constitutive receptor activity and/or the presence of agonist in the cell culture medium. Here, we studied wild type (WT) N2a cells, expressing low levels of GPR37 and nondetectable levels of GPR37-L1, and GPR37tGFP-overexpressing cells (Fig. 3A). Because PSAP was recently reported to act as an agonist at GPR37 (20), conditioned medium from WT and GPR37tGFP-expressing cells was tested for the presence of PSAP. Western blot indicated similar concentrations of PSAP in the conditioned medium from WT and GPR37tGFP cells (Fig. 3B). The conditioned medium, collected from differentiated cells 3 days after seeding at a density of 5 × 104 cells ml−1, was estimated to contain 0.1–0.4 μm PSAP (Fig. 3C).
Immunoabsorption of Extracellular PSAP Causes Decreased Density of GPR37 at the Plasma Membrane
Differentiation of N2a cells leads to increased density of stably transfected GPR37tGFP at the plasma membrane (10). Preliminary observations suggested that replacement of the conditioned medium from differentiated cells with new medium temporarily reverses this process. This led us to hypothesize that the density of GPR37 at the plasma membrane depends on secreted PSAP. The extracellular pool of PSAP was therefore inhibited by immunoabsorption with an antibody raised against amino acids 301–385 comprising the neurotrophic saposin C region of PSAP (anti-PSAP). To confirm that most of extracellular PSAP was indeed bound by the amount of antibody added, immunoprecipitation was performed using protein A-coupled magnetic beads. In three separate experiments, 83–96% of extracellular PSAP was precipitated with the antibody compared with magnetic beads alone (Fig. 3D). Quantification of GPR37tGFP at the plasma membrane compared with the cytoplasm revealed a decrease of membrane density 2.5–3 h after anti-PSAP addition (Fig. 3E).
Immunoabsorption of Extracellular PSAP Decreases Cell Viability in N2a Cells with a Delayed Time Course in GPR37-overexpressing Cells
Protective effects of GPR37 overexpression against dopaminergic toxins in N2a cells correlate with increased receptor levels at the plasma membrane (10). To test for neurotrophic effects of PSAP in differentiated N2a cells and the possible involvement of GPR37 in such effects, we performed MTT cell viability assays after inhibiting extracellular PSAP using anti-PSAP. Cell viability was determined after 8, 16, and 24 h of treatment. Depletion of PSAP decreased cell viability in both WT and GPR37tGFP cells, but with a delayed time course in the latter cells (Fig. 3F). In WT cells, the maximum effect was reached at 16 h, whereas it was reached at 24 h in GPR37tGFP cells (Fig. 3F, blue and green). In a follow-up experiment, WT and GPR37tGFP cells were incubated with another anti-PSAP antibody raised against the neurotrophic 22-mer prosaptide 769 (anti-PS769) (21) for 16 h (Fig. 3F, purple and brown). Based on titration studies, this antibody binds PSAP at a 1:2 antibody/target ratio (data not shown), and the antibody concentration added in these experiments should thus bind 100 nm PSAP in solution. Also in these experiments, GPR37tGFP cells showed less reduction in cell viability as compared with WT cells (Fig. 3F). We also confirmed that the reduction in cell viability at 24 h was in fact the maximum effect for both cell types by carrying out an additional MTT assay after 48 h of PSAP depletion. After 48 h, cell viability was reduced by 33 ± 5 and 33 ± 4% in WT and GPR37-overexpressing cells, respectively (n = 24 for each group).
GPR37 Co-localizes, Co-migrates, and Forms Complexes with the PSAP-derived Peptide Prosaptide TX14(A)
To study interaction in live cells of GPR37tGFP and the synthetic PSAP derivative prosaptide-TX14(A) fluorescently labeled with carboxytetramethylrhodamine (PS-TX14(A)TAMRA), we first used CLSM imaging, which revealed co-localization of GPR37tGFP with PS-TX14(A)TAMRA at the plasma membrane (Fig. 4A). Both GPR37tGFP and PS-TX14(A)TAMRA fluorescence was discrete, forming punctate patterns at the plasma membrane, indicating compartmentalization in membrane microdomains. Confocal imaging of PS-TX14(A)TAMRA interactions with WT N2a cells showed very limited interaction of PS-TX14(A)TAMRA with the plasma membrane, confirming that PS-TX14(A)TAMRA binding to membranes of GPR37tGFP-overexpressing cells is specific for GPR37 (Fig. 5A). CLSM also demonstrated co-localization of GPR37tGFP with PS-TX14(A)TAMRA in intracellular vesicles of live cells (Fig. 4A). Based on this observation, we stained the cells with MitoTracker® or LysoTracker® to investigate whether GPR37tGFP co-localized with either mitochondria or lysosomes. Vesicles containing GPR37tGFP were found to largely co-localize with LysoTracker staining but not with Mitotracker staining (Fig. 6).
FIGURE 6.

GPR37 co-localizes with lysosomes but not with mitochondria. A, representative images of GPR37 and LysoTracker® showing co-localization in a majority of GPR37-containing intracellular vesicles. B, representative images of GPR37 and MitoTracker® showing no co-localization.
GPR37tGFP interactions with PS-TX14(A)TAMRA at the plasma membrane were further characterized using FCS and dual-color FCCS. FCCS can detect complex formation in living cells in a manner that is independent of distance between the fluorophores by measuring the co-variance of fluorescence intensity fluctuations of the dually labeled complexes (24, 33, 34).
Single color FCS experiments showed a longer diffusion time of PS-TX14(A)TAMRA at the plasma membrane of GPR37GFP cells when compared with the extracellular medium, as evident from the shift of the autocorrelation curve toward longer decay times (Fig. 4B, red autocorrelation curve versus blue curve). These longer decay times indicate that PS-TX14(A)TAMRA interacted with components of the plasma membrane. This was also confirmed by the appearance of an additional component in the autocorrelation curve derived from measurements at the plasma membrane, compared with the autocorrelation curve that was measured in the extracellular medium (Fig. 4B). In addition, FCS analysis showed that PS-TX14(A)TAMRA accumulates in the plasma membrane, as evident from the decrease in the amplitude of the autocorrelation curve (Fig. 4B, inset). When WT cells were treated with PS-TX14(A)TAMRA, the autocorrelation curve of PS-TX14(A)TAMRA was not significantly altered, showing that interaction of PS-TX14(A)TAMRA with the plasma membrane of N2a cells required significant density of GPR37 (Fig. 5A).
FCCS analysis at the plasma membrane showed that addition of PS-TX14(A)TAMRA (100 nm, as determined by FCS in the bulk medium (Fig. 4B)) to GPR37tGFP-expressing cells yielded cross-correlation between fluorescence intensity fluctuations of the two spectrally distinct reporters TAMRA and tGFP, demonstrating complex formation and suggesting ligand-receptor interaction (Fig. 4C). Control measurements with the μ-opioid receptor-selective peptide ligand β-endorphinTAMRA and GPR37tGFP did not produce any cross-correlation, showing that the cross-correlation did not arise from spectral bleed through between the channels or unspecific binding of peptides to GPR37tGFP (Fig. 5B). The apparent dissociation constant (KDapp) of the PS-TX14(A)TAMRA·GPR37tGFP complexes was determined to be KDapp = 550 ± 350 nm. It should, however, be noted that the conditioned medium contained high concentrations of endogenously secreted PSAP and that a large fraction of the receptors may thus already have been occupied. The KD values determined in our experiments may therefore be considerably higher than KD values in a system lacking extracellular PSAP.
In the Presence of PSAP, GPR37 Accumulates in and Internalizes with GM1-containing Lipid Rafts at the Plasma Membrane
As mentioned above, both GPR37tGFP and PS-TX14(A)TAMRA fluorescence was discrete, forming punctate patterns at the plasma membrane, indicating compartmentalization in membrane microdomains. To test whether GPR37 compartmentalizes into lipid rafts at the plasma membrane, cells were stained with Alexa 594-labeled CTxB. CTxB binds to GM1 gangliosides and hence labels lipid raft structures with a high density of GM1, as well as endosomes originating from stained plasma membrane microdomains (40). GPR37tGFP was found to co-localize with CTxB and thus to accumulate in GM1-dense lipid rafts of the plasma membrane (Fig. 7A). This was confirmed by quantification of fluorescent signals using Manders' coefficient of pixel overlap and Pearson's correlation of pixel intensity (Fig. 7C). Indeed, the average Manders' coefficient for all analyzed images showed that a substantial fraction of GPR37tGFP co-localized with CTxB. The average Pearson's correlation confirmed that fluorescence intensity in the green channel (GPR37tGFP) positively correlated with fluorescence intensity in the red channel (CTxBAlexa594).
FIGURE 7.

Inhibition of extracellular PSAP decreases membrane localization of GPR37 to GM1-dense lipid rafts. Representative images are shown for co-localization of GPR37 and CTxB in live cells at baseline (A) or after inhibition of extracellular PSAP (B). C, average Pearson's correlation of pixel intensity, as well as Manders' coefficient for fraction of green pixels overlapping red, was calculated. For each group, calculations were made from ROIs comprising the plasma membrane of each cell in 30 randomly selected fields of vision, nuntreated = 69 cells and nanti-PSAP = 70 cells. A marked reduction of the Pearson's correlation confirms reduced accumulation of GPR37 in GM1-dense membrane microdomains after inhibition of extracellular PSAP.
To investigate whether accumulation of GPR37 in GM1-dense lipid rafts is dependent on PSAP binding to the receptor, we used the anti-PSAP antibody to inhibit interactions between the extracellular PSAP and GPR37tGFP. Confocal fluorescence microscopy revealed that accumulation of GPR37tGFP in strongly CTxB-stained microdomains was reduced 2.5 h after antibody addition (Fig. 7B). This was reflected by an 11% reduction of the average Manders' coefficient and a 43% reduction of the average Pearson's correlation of pixel intensity (Fig. 7C). Importantly, FCCS analysis showed that GPR37tGFP not only co-localizes but also forms complexes with GM1 gangliosides (Fig. 8A, red cross-correlation curve).
FIGURE 8.

Lipid rafts disruption with mβCD alters distribution and trafficking of GPR37, PS-TX14(A)TAMRA, and the complex between GPR37tGFP and PS-TX14(A)TAMRA at the plasma membrane. A, comparison of FCCS curves for GPR37tGFP and CTxB from untreated, mβCD-treated, or cholesterol oxidase-treated cells shows a significant cross-correlation (i.e. complex formation) between GPR37tGFP and CTxB in control cells with 85% of the complexes diffusing at τD1 = 0.5 ms and 15% at τD2 = 0.2 s. This cross-correlation was largely disrupted by mβCD and cholesterol oxidase. B, FRAP measurements for GPR37tGFP were performed on live cells (n = 23 cells for each treatment). Measurements were made on the same cell before and after treatment. Either mβCD or cholesterol oxidase increased GPR37tGFP fluorescence recovery compared with untreated cells, and the average amplitude of the FRAP curves at t = 350 s was significantly higher after mβCD or cholesterol oxidase treatment than before (**, p < 0.01; ***, p < 0.001, Student's t test). C, comparison of FCCS curves for GPR37tGFP and PS-TX14(A)TAMRA averaged from three separate untreated, mβCD-treated, or cholesterol oxidase-treated cells shows that cholesterol depletion shifts the curve toward shorter diffusion times. In untreated cells, 75 ± 15% of the complexes diffused at τD1 = 1.2 ± 0.6 ms and 25 ± 15% at τD2 = 330 ± 200 ms. In mβCD-treated cells, 80 ± 12% of the complexes diffused at τD1 = 550 ± 250 μs and 20 ± 12% at τD = 29 ± 15 ms. In cholesterol oxidase-treated cells, 89 ± 5% of the complexes diffused at τD1 = 170 ± 40 μs and 11 ± 5% at τD2 = 15 ± 9 ms.
Lipid Raft Disruption Alters Trafficking of GPR37 and Complexes between PS-TX14(A) and GPR37 at the Plasma Membrane
Methyl-β-cyclodextrin (mβCD) and cholesterol oxidase are commonly used to sequester membrane cholesterol and to disrupt lipid rafts (41, 42). To study whether trafficking of GPR37 is dependent upon the integrity of lipid raft microdomains, FCCS, CLSM, and FRAP analyses were performed before and after mβCD or cholesterol oxidase treatment and compared (Fig. 8). FCCS experiments at the plasma membrane showed that both treatments largely disrupted GPR37 interactions with GM1, as shown by the loss of cross-correlation between GPR37tGFP and CTxBAlexa 594 (Fig. 8A, blue curves).
Baseline FRAP measurements at the plasma membrane revealed low recovery of GPR37tGFP fluorescence (14 ± 4 and 7 ± 2% over 5 min for Fig. 8B, left and right panels, respectively), further indicating compartmentalization and scaffolding of the receptors (Fig. 8B, black curves). After mβCD treatment, fluorescence recovery increased to 35 ± 5% over 5 min (Fig. 8B, left, blue curve), and after cholesterol oxidase treatment, fluorescence recovery increased to 40 ± 5% over 5 min (Fig. 8B, right, blue curve).
FCCS measurements with GPR37tGFP and PS-TX14(A)TAMRA at the plasma membrane were also performed at baseline and after mβCD or cholesterol oxidase treatment (Fig. 8C). Comparison of FCCS curves with or without mβCD (Fig. 8C, left) or cholesterol oxidase (Fig. 8C, right) shows that the FCCS curves shifted toward shorter diffusion times after disruption of lipid rafts. This is consistent with the interpretation that GPR37·PS-TX14(A) complexes are located in and migrate with lipid rafts, which are disrupted after cholesterol sequestration.
DISCUSSION
Emerging evidence points toward a signaling pathway in which GPR37 located at the plasma membrane reduces effects of cellular insult (9, 10, 20). This signaling pathway appears to be relatively susceptible to perturbation and once compromised may instead exacerbate cellular stress through intracellular accumulation of GPR37 aggregates (3, 6–8). PSAP and gangliosides also regulate neuronal survival and improve neuronal resistance to various insults via mechanisms that are not yet defined (12, 15, 43, 44). This study provides detailed live cell imaging and fluorescence spectroscopy-based data strongly supporting the notion of functional PSAP-GPR37-ganglioside interactions at the plasma membrane. These interactions reduce intracellular GPR37 accumulation and may both regulate neuroprotective actions and attenuate cytotoxic aggregation of GPR37.
PSAP has long been known to exert neurotrophic and neuroprotective effects via GPCR-dependent signaling at the plasma membrane (45). The PSAP receptor couples to Gi/o and activates the MAPK and PI3K signaling pathways as well as modulates the activity of voltage-gated calcium channels (45–47). Very recently, GPR37 and GPR37L1 were identified as receptors for PSAP and PS-TX14(A), an active synthetic peptide derived from the neurotrophic sequence of PSAP. In our present experiments, we extend these observations by demonstrating binding in live cells of PS-TX14(A) to GPR37.
Importantly, our study demonstrates that inhibition of extracellular PSAP reduces plasma membrane levels of GPR37. Here, we used an anti-PSAP antibody added to the extracellular medium instead of siRNA to specifically inhibit the function of extracellular PSAP without depleting the cells of all intracellular saposins, which are critical for lysosomal function and cell survival (14). Our observations of PSAP-induced effects on GPR37 trafficking support the idea of functional coupling between extracellular PSAP and GPR37. With inhibition of extracellular PSAP, membrane levels of GPR37 decline. This suggests a pathway for intracellular build up of GPR37, which eventually may reach the critical concentration for aggregation. Given that GPR37 is known to cause degeneration of dopaminergic cells via intracellular aggregation and endoplasmic reticulum stress (3, 5–7), PSAP or its active fragments may via this mechanism protect against the progress of PD, as was indicated in previous experimental studies (15, 48).
A mechanism where the GPR37 agonist PSAP promotes GPR37 trafficking to the cell surface may seem at odds with the traditional view on GPCR activation, which in most cases leads to receptor internalization. However, there are now a number of examples of both antagonist-induced GPCR internalization and agonist-induced GPCR insertion into the plasma membrane (49–55). One example is the δ-opioid receptor (DOR), which similarly to GPR37 has a low surface density in the absence of agonist (49, 56). Agonist binding to DORs at the plasma membrane leads to receptor internalization but also triggers intracellular signaling cascades that promote slow and sustained insertion of DORs from the intracellular pool into the plasma membrane (49). The net effect of these two processes is a positive feedback mechanism leading to a sustained increase of the DOR surface density as long as both extracellular agonist and intracellular receptors are available. It is possible that a similar mechanism applies to GPR37. Such a mechanism would be consistent with the slow kinetics for the anti-PSAP-induced internalization of GPR37tGFP as well as with the PS-TX14(A)-induced GPR37 internalization observed by Meyer et al. (20), and with the co-localization of PS-TX14(A)TAMRA and GPR37tGFP in intracellular vesicles observed in this study.
Mounting evidence indicates that lipids are critical for GPCR folding and that lipid rafts regulate GPCR trafficking and signaling (57). Moreover, beneficial effects of GM1 gangliosides against PD symptoms and progression have been shown both in animal models and in clinical studies (44, 58). We therefore characterized the lipid environment of GPR37tGFP at the plasma membrane in the presence or absence of PSAP. PSAP localizes to ganglioside-containing membrane microdomains (21, 22), and ganglioside-containing lipid rafts are required for PSAP-induced neurotrophic GPCR signaling (22). The mechanism for how lipid rafts mediate PSAP function, however, has been elusive. The present data suggest a model of how lipid rafts mediate PSAP function, as co-localization studies revealed that GPR37 accumulates in GM1-containing lipid rafts in the presence, but not upon inhibition, of extracellular PSAP. Moreover, a significant cross-correlation between GPR37 and CTxB showed that GPR37 not only co-localizes but also forms complexes with GM1 gangliosides in the presence of PSAP. The results from these experiments collectively suggest that PSAP induces trafficking of GPR37 to ganglioside-containing lipid rafts and consequently complex formation of the receptor with GM1 gangliosides. Notably lipid raft disruption using mβCD or cholesterol oxidase altered trafficking of both GPR37tGFP and the GPR37tGFP·prosaptide complex at the plasma membrane. This suggests that not only GPR37, but also GPR37·prosaptide complexes, are located in lipid rafts. Lipid rafts thus provide a scaffold in which GPR37, prosaptide, and GM1 may interact to promote cell viability.
We also found that inhibition of extracellular PSAP reduced cell viability in GPR37tGFP cells with a delayed time course as compared with WT cells. This correlates well with the study of Meyer et al. (20) showing that GPR37 mediates PSAP-induced protection against H2O2-induced toxicity and our previous study showing that overexpression of GPR37 in N2a cells leads to substantial protection against toxicity when the receptor is located at the plasma membrane (10). Because N2a cells endogenously express low levels of GPR37, the GPR37tGFP cell line used in our studies can be considered a gain-of-function, rather than a heterologous, system. The accumulated effect of more strongly activated intracellular signaling cascades is likely to cause the delayed response to anti-PSAP in GPR37tGFP cells. The observed effects of PSAP depletion are therefore consistent with neurotrophic PSAP signaling via GPR37 and suggest that GPR37 at least in part mediates the neurotrophic effects of PSAP in these cells.
In conclusion, our studies show that constitutively secreted PSAP promotes trafficking of GPR37tGFP to the cell surface, where it forms complexes with GM1 gangliosides and associates with GM1-enriched lipid rafts. Inhibiting extracellular PSAP disrupts GPR37tGFP interaction with GM1-enriched microdomains, promoting its intracellular accumulation. Thus, our data provide evidence for the functional association between GPR37, PSAP, and GM1 gangliosides in the plasma membrane, which conveys protection against intracellular accumulation of GPR37. These findings define a mechanistic frame wherein PSAP, and its derivative prosaptides, and GM1 may protect against the progress of PD by stabilizing GPR37 in the plasma membrane.
This work was supported by the Swedish Research Council and Karolinska Institutet.
E. G. Lundius, V. Vukojević, and P. Svenningsson, unpublished data.
- GPCR
- G protein-coupled receptor
- PD
- Parkinson disease
- FCS
- fluorescence correlation spectroscopy
- FCCS
- fluorescence cross-correlation spectroscopy
- PSAP
- prosaposin
- CLSM
- confocal laser scanning microscopy
- FRAP
- fluorescence recovery after photobleaching
- ROI
- region of interest
- TAMRA
- carboxytetramethylrhodamine
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- CTxB
- cholera toxin β
- mβCD
- methyl-β-cyclodextrin
- DOR
- δ-opioid receptor.
REFERENCES
- 1. Marazziti D., Gallo A., Golini E., Matteoni R., Tocchini-Valentini G. P. (1998) Molecular cloning and chromosomal localization of the mouse Gpr37 gene encoding an orphan G-protein-coupled peptide receptor expressed in brain and testis. Genomics 53, 315–324 [DOI] [PubMed] [Google Scholar]
- 2. Zeng Z., Su K., Kyaw H., Li Y. (1997) A novel endothelin receptor type-B-like gene enriched in the brain. Biochem. Biophys. Res. Commun. 233, 559–567 [DOI] [PubMed] [Google Scholar]
- 3. Imai Y., Soda M., Inoue H., Hattori N., Mizuno Y., Takahashi R. (2001) An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105, 891–902 [DOI] [PubMed] [Google Scholar]
- 4. Leng N., Gu G., Simerly R. B., Spindel E. R. (1999) Molecular cloning and characterization of two putative G protein-coupled receptors which are highly expressed in the central nervous system. Brain Res. Mol. Brain Res. 69, 73–83 [DOI] [PubMed] [Google Scholar]
- 5. Murakami T., Shoji M., Imai Y., Inoue H., Kawarabayashi T., Matsubara E., Harigaya Y., Sasaki A., Takahashi R., Abe K. (2004) Pael-R is accumulated in Lewy bodies of Parkinson's disease. Ann. Neurol. 55, 439–442 [DOI] [PubMed] [Google Scholar]
- 6. Wang H. Q., Imai Y., Inoue H., Kataoka A., Iita S., Nukina N., Takahashi R. (2008) Pael-R transgenic mice crossed with parkin-deficient mice displayed progressive and selective catecholaminergic neuronal loss. J. Neurochem. 107, 171–185 [DOI] [PubMed] [Google Scholar]
- 7. Kitao Y., Imai Y., Ozawa K., Kataoka A., Ikeda T., Soda M., Nakimawa K., Kiyama H., Stern D. M., Hori O., Wakamatsu K., Ito S., Itohara S., Takahashi R., Ogawa S. (2007) Pael receptor induces death of dopaminergic neurons in the substantia nigra via endoplasmic reticulum stress and dopamine toxicity, which is enhanced under condition of parkin inactivation. Hum. Mol. Genet. 16, 50–60 [DOI] [PubMed] [Google Scholar]
- 8. Dusonchet J., Bensadoun J. C., Schneider B. L., Aebischer P. (2009) Targeted overexpression of the parkin substrate Pael-R in the nigrostriatal system of adult rats to model Parkinson's disease. Neurobiol. Dis. 35, 32–41 [DOI] [PubMed] [Google Scholar]
- 9. Mimori S., Okuma Y., Kaneko M., Kawada K., Hosoi T., Ozawa K., Nomura Y., Hamana H. (2012) Protective effects of 4-phenylbutyrate derivatives on the neuronal cell death and endoplasmic reticulum stress. Biol. Pharm. Bull. 35, 84–90 [DOI] [PubMed] [Google Scholar]
- 10. Lundius E. G., Stroth N., Vukojević V., Terenius L., Svenningsson P. (2013) Functional GPR37 trafficking protects against toxicity induced by 6-OHDA, MPP+, or rotenone in a catecholaminergic cell line. J. Neurochem. 124, 410–417 [DOI] [PubMed] [Google Scholar]
- 11. Campana W. M., Mohiuddin L., Misasi R., O'Brien J. S., Calcutt N. A. (2000) Prosaposin-derived peptides enhanced sprouting of sensory neurons in vitro and induced sprouting at motor endplates in vivo. J. Peripher. Nerv. Syst. 5, 126–130 [DOI] [PubMed] [Google Scholar]
- 12. Hiraiwa M., Campana W. M., Mizisin A. P., Mohiuddin L., O'Brien J. S. (1999) Prosaposin: a myelinotrophic protein that promotes expression of myelin constituents and is secreted after nerve injury. Glia 26, 353–360 [PubMed] [Google Scholar]
- 13. O'Brien J. S., Carson G. S., Seo H. C., Hiraiwa M., Kishimoto Y. (1994) Identification of prosaposin as a neurotrophic factor. Proc. Natl. Acad. Sci. U.S.A. 91, 9593–9596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kishimoto Y., Hiraiwa M., O'Brien J. S. (1992) Saposins: structure, function, distribution, and molecular genetics. J. Lipid Res. 33, 1255–1267 [PubMed] [Google Scholar]
- 15. Liu J., Wang C. Y., O'Brien J. S. (2001) Prosaptide D5, a retro-inverso 11-mer peptidomimetic, rescued dopaminergic neurons in a model of Parkinson's disease. FASEB J. 15, 1080–1082 [DOI] [PubMed] [Google Scholar]
- 16. O'Brien J. S., Carson G. S., Seo H. C., Hiraiwa M., Weiler S., Tomich J. M., Barranger J. A., Kahn M., Azuma N., Kishimoto Y. (1995) Identification of the neurotrophic factor sequence of prosaposin. FASEB J. 9, 681–685 [DOI] [PubMed] [Google Scholar]
- 17. Qi X., Kondoh K., Krusling D., Kelso G. J., Leonova T., Grabowski G. A. (1999) Conformational and amino acid residue requirements for the saposin C neuritogenic effect. Biochemistry 38, 6284–6291 [DOI] [PubMed] [Google Scholar]
- 18. Morita F., Wen T. C., Tanaka J., Hata R., Desaki J., Sato K., Nakata K., Ma Y. J., Sakanaka M. (2001) Protective effect of a prosaposin-derived, 18-mer peptide on slowly progressive neuronal degeneration after brief ischemia. J. Cereb. Blood Flow Metab. 21, 1295–1302 [DOI] [PubMed] [Google Scholar]
- 19. Ochiai T., Takenaka Y., Kuramoto Y., Kasuya M., Fukuda K., Kimura M., Shimeno H., Misasi R., Hiraiwa M., Soeda S. (2008) Molecular mechanism for neuro-protective effect of prosaposin against oxidative stress: its regulation of dimeric transcription factor formation. Biochim. Biophys. Acta 1780, 1441–1447 [DOI] [PubMed] [Google Scholar]
- 20. Meyer R. C., Giddens M. M., Schaefer S. A., Hall R. A. (2013) GPR37 and GPR37L1 are receptors for the neuroprotective and glioprotective factors prosaptide and prosaposin. Proc. Natl. Acad. Sci. U.S.A. 110, 9529–9534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Misasi R., Sorice M., Garofalo T., Griggi T., Campana W. M., Giammatteo M., Pavan A., Hiraiwa M., Pontieri G. M., O'Brien J. S. (1998) Colocalization and complex formation between prosaposin and monosialoganglioside GM3 in neural cells. J. Neurochem. 71, 2313–2321 [DOI] [PubMed] [Google Scholar]
- 22. Sorice M., Molinari S., Di Marzio L., Mattei V., Tasciotti V., Ciarlo L., Hiraiwa M., Garofalo T., Misasi R. (2008) Neurotrophic signalling pathway triggered by prosaposin in PC12 cells occurs through lipid rafts. FEBS J. 275, 4903–4912 [DOI] [PubMed] [Google Scholar]
- 23. Vukojevic V., Heidkamp M., Ming Y., Johansson B., Terenius L., Rigler R. (2008) Quantitative single-molecule imaging by confocal laser scanning microscopy. Proc. Natl. Acad. Sci. U.S.A. 105, 18176–18181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Elson E. L. (2013) Brief introduction to fluorescence correlation spectroscopy. Methods Enzymol. 518, 11–41 [DOI] [PubMed] [Google Scholar]
- 25. Weisshart K., Jüngel V., Briddon S. J. (2004) The LSM 510 META-ConfoCor 2 system: an integrated imaging and spectroscopic platform for single-molecule detection. Curr. Pharm. Biotechnol. 5, 135–154 [DOI] [PubMed] [Google Scholar]
- 26. Vukojević V., Pramanik A., Yakovleva T., Rigler R., Terenius L., Bakalkin G. (2005) Study of molecular events in cells by fluorescence correlation spectroscopy. Cell. Mol. Life Sci. 62, 535–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Elson E. L. (2001) Fluorescence correlation spectroscopy measures molecular transport in cells. Traffic 2, 789–796 [DOI] [PubMed] [Google Scholar]
- 28. Vukojevic V., Papadopoulos D. K., Terenius L., Gehring W. J., Rigler R. (2010) Quantitative study of synthetic Hox transcription factor-DNA interactions in live cells. Proc. Natl. Acad. Sci. U.S.A. 107, 4093–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hendrix J., Gijsbers R., De Rijck J., Voet A., Hotta J., McNeely M., Hofkens J., Debyser Z., Engelborghs Y. (2011) The transcriptional co-activator LEDGF/p75 displays a dynamic scan-and-lock mechanism for chromatin tethering. Nucleic Acids Res. 39, 1310–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lenne P. F., Wawrezinieck L., Conchonaud F., Wurtz O., Boned A., Guo X. J., Rigneault H., He H. T., Marguet D. (2006) Dynamic molecular confinement in the plasma membrane by microdomains and the cytoskeleton meshwork. EMBO J. 25, 3245–3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ganguly S., Chattopadhyay A. (2010) Cholesterol depletion mimics the effect of cytoskeletal destabilization on membrane dynamics of the serotonin1A receptor: A zFCS study. Biophys. J. 99, 1397–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weise K., Triola G., Janosch S., Waldmann H., Winter R. (2010) Visualizing association of lipidated signaling proteins in heterogeneous membranes–partitioning into subdomains, lipid sorting, interfacial adsorption, and protein association. Biochim. Biophys. Acta 1798, 1409–1417 [DOI] [PubMed] [Google Scholar]
- 33. Weidemann T., Schwille P. (2013) Dual-color fluorescence cross-correlation spectroscopy with continuous laser excitation in a confocal setup. Methods Enzymol. 518, 43–70 [DOI] [PubMed] [Google Scholar]
- 34. Bleicken S., Otsuki M., Garcia-Saez A. J. (2011) Quantification of protein-protein interactions within membranes by fluorescence correlation spectroscopy. Curr. Protein Pept. Sci. 12, 691–698 [DOI] [PubMed] [Google Scholar]
- 35. Földes-Papp Z., Rigler R. (2001) Quantitative two-color fluorescence cross-correlation spectroscopy in the analysis of polymerase chain reaction. Biol. Chem. 382, 473–478 [DOI] [PubMed] [Google Scholar]
- 36. Rigler R., Földes-Papp Z., Meyer-Almes F. J., Sammet C., Völcker M., Schnetz A. (1998) Fluorescence cross-correlation: a new concept for polymerase chain reaction. J. Biotechnol. 63, 97–109 [DOI] [PubMed] [Google Scholar]
- 37. Földes-Papp Z., Angerer B., Ankenbauer W., Rigler R. (2001) Fluorescent high-density labeling of DNA: error-free substitution for a normal nucleotide. J. Biotechnol. 86, 237–253 [DOI] [PubMed] [Google Scholar]
- 38. Foo Y. H., Naredi-Rainer N., Lamb D. C., Ahmed S., Wohland T. (2012) Factors affecting the quantification of biomolecular interactions by fluorescence cross-correlation spectroscopy. Biophys. J. 102, 1174–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ulrich M., Kappel C., Beaudouin J., Hezel S., Ulrich J., Eils R. (2006) Tropical–parameter estimation and simulation of reaction-diffusion models based on spatio-temporal microscopy images. Bioinformatics 22, 2709–2710 [DOI] [PubMed] [Google Scholar]
- 40. Radhakrishnan A., Anderson T. G., McConnell H. M. (2000) Condensed complexes, rafts, and the chemical activity of cholesterol in membranes. Proc. Natl. Acad. Sci. U.S.A. 97, 12422–12427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sjögren B., Hamblin M. W., Svenningsson P. (2006) Cholesterol depletion reduces serotonin binding and signaling via human 5-HT(7(a)) receptors. Eur. J. Pharmacol. 552, 1–10 [DOI] [PubMed] [Google Scholar]
- 42. Castro B. M., Torreno-Piña J. A., van Zanten T. S., Gracia-Parajo M. F. (2013) Biochemical and imaging methods to study receptor membrane organization and association with lipid rafts. Methods Cell Biol. 117, 105–122 [DOI] [PubMed] [Google Scholar]
- 43. Merlo Pich E., Grimaldi R., Zoli M., Biagini G., Solfrini V., Toffano G., Fuxe K., Agnati L. F. (1992) Siagoside selectively attenuates morphological and functional striatal impairments induced by transient forebrain ischemia in rats. Stroke 23, 234–241 [DOI] [PubMed] [Google Scholar]
- 44. Schneider J. S., Pope A., Simpson K., Taggart J., Smith M. G., DiStefano L. (1992) Recovery from experimental parkinsonism in primates with GM1 ganglioside treatment. Science 256, 843–846 [DOI] [PubMed] [Google Scholar]
- 45. Hiraiwa M., Campana W. M., Martin B. M., O'Brien J. S. (1997) Prosaposin receptor: evidence for a G-protein-associated receptor. Biochem. Biophys. Res. Commun. 240, 415–418 [DOI] [PubMed] [Google Scholar]
- 46. Yan L., Otero D. A., Hiraiwa M., O'Brien J. S. (2000) Prosaptide D5 reverses hyperalgesia: inhibition of calcium channels through a pertussis toxin-sensitive G-protein mechanism in the rat. Neurosci. Lett. 278, 120–122 [DOI] [PubMed] [Google Scholar]
- 47. Campana W. M., Hiraiwa M., O'Brien J. S. (1998) Prosaptide activates the MAPK pathway by a G-protein-dependent mechanism essential for enhanced sulfatide synthesis by Schwann cells. FASEB J. 12, 307–314 [DOI] [PubMed] [Google Scholar]
- 48. Gao H. L., Li C., Nabeka H., Shimokawa T., Saito S., Wang Z. Y., Cao Y. M., Matsuda S. (2013) Attenuation of MPTP/MPP(+) toxicity in vivo and in vitro by an 18-mer peptide derived from prosaposin. Neuroscience 236, 373–393 [DOI] [PubMed] [Google Scholar]
- 49. Bao L., Jin S. X., Zhang C., Wang L. H., Xu Z. Z., Zhang F. X., Wang L. C., Ning F. S., Cai H. J., Guan J. S., Xiao H. S., Xu Z. Q., He C., Hökfelt T., Zhou Z., Zhang X. (2003) Activation of δ opioid receptors induces receptor insertion and neuropeptide secretion. Neuron 37, 121–133 [DOI] [PubMed] [Google Scholar]
- 50. Coleman S. K., Möykkynen T., Jouppila A., Koskelainen S., Rivera C., Korpi E. R., Keinänen K. (2009) Agonist occupancy is essential for forward trafficking of AMPA receptors. J. Neurosci. 29, 303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Roettger B. F., Ghanekar D., Rao R., Toledo C., Yingling J., Pinon D., Miller L. J. (1997) Antagonist-stimulated internalization of the G protein-coupled cholecystokinin receptor. Mol. Pharmacol. 51, 357–362 [PubMed] [Google Scholar]
- 52. Pfeiffer R., Kirsch J., Fahrenholz F. (1998) Agonist and antagonist-dependent internalization of the human vasopressin V2 receptor. Exp. Cell Res. 244, 327–339 [DOI] [PubMed] [Google Scholar]
- 53. Gray J. A., Roth B. L. (2001) Paradoxical trafficking and regulation of 5-HT(2A) receptors by agonists and antagonists. Brain Res. Bull. 56, 441–451 [DOI] [PubMed] [Google Scholar]
- 54. Navarro A., Zapata R., Canela E. I., Mallol J., Lluis C., Franco R. (1999) Epidermal growth factor (EGF)-induced up-regulation and agonist- and antagonist-induced desensitization and internalization of A1 adenosine receptors in a pituitary-derived cell line. Brain Res. 816, 47–57 [DOI] [PubMed] [Google Scholar]
- 55. Achour L., Labbé-Jullié C., Scott M. G., Marullo S. (2008) An escort for GPCRs: implications for regulation of receptor density at the cell surface. Trends Pharmacol. Sci. 29, 528–535 [DOI] [PubMed] [Google Scholar]
- 56. Cahill C. M., McClellan K. A., Morinville A., Hoffert C., Hubatsch D., O'Donnell D., Beaudet A. (2001) Immunohistochemical distribution of δ opioid receptors in the rat central nervous system: evidence for somatodendritic labeling and antigen-specific cellular compartmentalization. J. Comp. Neurol. 440, 65–84 [DOI] [PubMed] [Google Scholar]
- 57. Björk K., Svenningsson P. (2011) Modulation of monoamine receptors by adaptor proteins and lipid rafts: role in some effects of centrally acting drugs and therapeutic agents. Annu. Rev. Pharmacol. Toxicol. 51, 211–242 [DOI] [PubMed] [Google Scholar]
- 58. Schneider J. S., Gollomp S. M., Sendek S., Colcher A., Cambi F., Du W. (2013) A randomized, controlled, delayed start trial of GM1 ganglioside in treated Parkinson's disease patients. J. Neurol. Sci. 324, 140–148 [DOI] [PMC free article] [PubMed] [Google Scholar]