

Background: Liver X receptor activation promotes formation of HDL-like particles at the blood-brain barrier (BBB).
Results: Cerebrovascular endothelial cells express phospholipid transfer protein (PLTP) that transfers phospholipids, remodels HDL, and supports cellular cholesterol efflux.
Conclusion: PLTP is involved in HDL genesis and remodeling at the BBB.
Significance: We demonstrate a direct role of PLTP in HDL metabolism at the blood-brain interface.
Keywords: Brain, Cholesterol, Endothelium, HDL, Nuclear Receptors, Aβ, Blood-Brain Barrier, Cholesterol Efflux, LXRs, Pre-β HDL
Abstract
Phospholipid transfer protein (PLTP) is a key protein involved in biogenesis and remodeling of plasma HDL. Several neuroprotective properties have been ascribed to HDL. We reported earlier that liver X receptor (LXR) activation promotes cellular cholesterol efflux and formation of HDL-like particles in an established in vitro model of the blood-brain barrier (BBB) consisting of primary porcine brain capillary endothelial cells (pBCEC). Here, we report PLTP synthesis, regulation, and its key role in HDL metabolism at the BBB. We demonstrate that PLTP is highly expressed and secreted by pBCEC. In a polarized in vitro model mimicking the BBB, pBCEC secreted phospholipid-transfer active PLTP preferentially to the basolateral (“brain parenchymal”) compartment. PLTP expression levels and phospholipid transfer activity were enhanced (up to 2.5-fold) by LXR activation using 24(S)-hydroxycholesterol (a cerebral cholesterol metabolite) or TO901317 (a synthetic LXR agonist). TO901317 administration elevated PLTP activity in BCEC from C57/BL6 mice. Preincubation of HDL3 with human plasma-derived active PLTP resulted in the formation of smaller and larger HDL particles and enhanced the capacity of the generated HDL particles to remove cholesterol from pBCEC by up to 3-fold. Pre-β-HDL, detected by two-dimensional crossed immunoelectrophoresis, was generated from HDL3 in pBCEC-derived supernatants, and their generation was markedly enhanced (1.9-fold) upon LXR activation. Furthermore, RNA interference-mediated PLTP silencing (up to 75%) reduced both apoA-I-dependent (67%) and HDL3-dependent (30%) cholesterol efflux from pBCEC. Based on these findings, we propose that PLTP is actively involved in lipid transfer, cholesterol efflux, HDL genesis, and remodeling at the BBB.
Introduction
Escalating evidence suggests that disparities in lipid metabolism, mainly associated with impaired cholesterol and lipoprotein homeostasis, play a role in the pathogenesis of several neurodegenerative diseases such as Alzheimer disease (AD)2 and Niemann-Pick disease type C (1, 2). Lipids represent approximately half of the brain's dry weight, and phospholipids and cholesterol are the main lipid constituents of mammalian brain. Compared with other organs, the brain is highly enriched in cholesterol (containing 25% of total unesterified body cholesterol) and covers its cholesterol requirement by de novo synthesis mainly by glial cells and oligodendrocytes (2). Cholesterol and phospholipid transport within the central nervous system (CNS) is mediated by high density lipoprotein (HDL)-like particles (3). Peripheral HDL protects against cardiovascular disease by promoting reverse cholesterol transport and thereby eliminating excess cholesterol and/or by antioxidant, anti-inflammatory, and anti-thrombotic properties ascribed to HDL (4). High plasma HDL-cholesterol and its main apolipoprotein (apo) A-I also protect against neurodegenerative disease; however, the underlying mechanisms are largely unexplored (5).
The presence of tight junctions between brain capillary endothelial cells (BCEC), constituting the blood-brain barrier (BBB), limits the exchange of circulating plasma lipoproteins with the brain. Nevertheless, cells forming the BBB (in particular BCEC) express several lipoprotein receptors, lipid transporters, and apolipoproteins important for both cholesterol turnover and HDL metabolism. We have shown that primary porcine brain capillary endothelial cells (pBCEC) are involved in the biogenesis of HDL-like particles at the “brain parenchymal side” of the BBB (6, 7). This process involves ATP-binding cassette transporter A1 (ABCA1)-mediated lipidation of apoA-I that is both expressed and secreted by pBCEC, induced by liver X receptor (LXR) activation (6, 7), and is able to transcytose the pBCEC monolayer in vitro (8).
Phospholipid transfer protein (PLTP) is a glycoprotein involved in lipid and lipoprotein metabolism. This 80-kDa, extensively N-glycosylated, protein belongs to the lipopolysaccharide (LPS)-binding protein family and facilitates the exchange of phospholipids, cholesterol, and vitamin E among various lipoproteins as well as between lipoproteins and cells (9, 10). Human plasma PLTP was shown to play a significant role in the conversion of circulating HDL into populations of larger (10.9 nm, HDL2) and smaller (7.8 nm pre-β) HDL particles (11). PLTP exists in a high and a low activity form in human plasma but only in active form in other body fluids (12, 13). Not much is known about the regulation of PLTP activity. Reportedly, PLTP activity increased following incubation in the presence of apolipoproteins such as apoA-I and apoE (14, 15). The gene expression of PLTP can be regulated by members of the nuclear receptor family of transcription factors as follows: LXRs, farnesoid X receptor, and peroxisome proliferator-activated receptor α (16–18).
Both pro- and anti-atherogenic roles of PLTP have been reported (19), and plasma PLTP activity is elevated in insulin resistance associated with obesity (20). In the CNS, PLTP is expressed in neurons and glial cells, and its phospholipid transfer-active form is present both in cerebrospinal fluid (CSF) and in brain tissue (21, 22). The potential roles of PLTP in the brain are still poorly understood. PLTP was proposed to play a role in the maintenance of functional and structural integrity of myelin and also to be an important regulator of signal transduction pathways in human neurons (13). PLTP deficiency significantly reduces brain vitamin E content and has been associated with increased anxiety in mice (23). PLTP was also found to be involved in regulating apoE (the major apolipoprotein in human brain) expression and secretion in primary human astrocytes (22). PLTP was further reported to be involved in the reduction of abnormal Tau phosphorylation in human neuronal cells (24). Interestingly, PLTP levels are altered in brain tissue and CSF of patients suffering from AD (21, 22), and PLTP deletion was recently demonstrated to increase amyloid-β (Aβ)-induced memory deficits in mice (25). These findings collectively suggest a significant role of PLTP in both physiological and pathophysiological processes in the brain.
The goal of this study was to investigate possible roles of PLTP at the BBB, the interface between the brain and the peripheral circulation. In particular, we investigated its expression, regulation, and potential functions in HDL metabolism. Furthermore, using an established in vitro model of the BBB, we assessed its role in lipid flux between the brain and the circulation.
EXPERIMENTAL PROCEDURES
Materials
Cell culture flasks, plates, and other plasticware were purchased from Greiner Bio-One (Kremsmünster, Austria). Transwell multiwell plates (polyester membrane inserts, 0.4 μm pore size) were obtained from Corning/Szabo-Scandic (Vienna, Austria). Medium M199, minimal essential medium, porcine serum, and dispase were obtained from Invitrogen, and bovine calf-skin collagen G was from Biochrom (Berlin, Germany). Culture media additives, trypsin/EDTA, and DMEM/Ham's F-12 medium were purchased from PAA (Pasching, Austria), and collagenase/dispase was from Roche Applied Science. Protease inhibitor mixture, Percoll, l-α-phosphatidylcholine (egg PC), butylated hydroxytoluene, hydrocortisone, and heparin were from Sigma. l-α-[3H]Dipalmitoylphosphatidylcholine (specific activity-1.550 TBq/mmol), [1,2-3H]cholesterol (specific activity, 1.772 TBq/mmol; [3H]cholesterol), and Ultima Gold scintillation mixture were purchased from PerkinElmer Life Sciences. 24(S)-Hydroxycholesterol was purchased from Medical Isotopes (Pelham, Canada) and TO901317 from Cayman Chemicals. RNeasy mini kit and small interfering RNA (siRNA) targeting for human PLTP were from Qiagen (Vienna, Austria). siRNA transfection reagents were obtained from Lonza and negative siRNA control from Dharmacon. PD-10 desalting columns and polyvinylidene fluoride (PVDF; 0.45 μm) transfer membranes were purchased from GE Healthcare. Centrifugal filter units (Amicon Ultra-15) for concentrating culture media from Merck. Antibodies were purchased from Abcam (PLTP, IgG; Cambridge, UK), Invitrogen (APP; Vienna, Austria), Millipore (A11 amyloid oligomer; Vienna, Austria) and Sigma (β-actin). PCR reagents were obtained from Bio-Rad and primers from Invitrogen. Anti-human apoA-I antibodies were raised in rabbits by immunizing the animals subcutaneously with 100 μg of apoA-I purified from human plasma HDL. All other solvents and chemicals were of reagent grade quality and purchased either from Sigma or Merck.
Immunohistochemical and Immunocytochemical Staining
Studies were carried out on 5-μm coronal cryosections of porcine brain samples. For immunocytochemistry, pBCEC were cultured on chamber slides. Tissue sections or cells were fixed in acetone for 5 min and air-dried 20 min prior to immunostaining using the UltraVision anti-polyvalent, LP HRP Detection System (Thermo Scientific, CA), according to the manufacturer's instructions. Sections were washed with 0.01 m PBS, pH 7.4, before primary antibodies were incubated for 30 min at room temperature. Antibody concentrations were 0.7 μg/ml for rabbit anti-human von Willebrand factor (Dako, CA), 1 μg/ml for rabbit anti-human PLTP (H-273; Santa Cruz Biotechnology, Santa Cruz, CA), and 1 μg/ml for normal rabbit immunoglobulin fraction (Dako). After rinsing for 5 min with PBS, slides were incubated with primary antibody enhancer (10 min) followed by horseradish peroxidase polymer (15 min). The slides were washed and immunolabeling was visualized by incubating with 3-amino-9-ethylcarbazole (Thermo Scientific) for 5 min. Slides were counterstained with Mayer's hematoxylin (Merck) and mounted with Kaiser's glycerol gelatin (Merck).
Isolation and Culture of Cells
pBCEC were isolated from freshly slaughtered pigs (about 6 months old) obtained from the local slaughterhouse as described by Franke et al. (26). After removal of the meninges and secretory areas of the porcine brain, pBCEC were isolated from the remaining cerebral cortex by sequential enzymatic digestion and centrifugation steps as described (26). pBCEC were plated onto collagen-coated (60 μg/ml) 75-cm2 culture flasks with M199 medium (containing 1% penicillin/streptomycin, 1% gentamycin, 1 mm l-glutamine, and 10% porcine serum). Cells were washed twice with PBS after 24 h to remove cell debris and nonadherent cells and cultured in fresh M199 (containing 1% penicillin/streptomycin, 1 mm l-glutamine, and 10% porcine serum) until confluent. After 3 days, the cells were trypsinized and plated onto collagen-coated (60 or 120 μg/ml) multiwell culture plates, flasks, or transwell filter plates and grown until confluent. For treatments, pBCEC monolayers were incubated in the absence or presence of the indicated concentrations of LXR agonists (24(S)-hydroxycholesterol or TO901317) for 24 h in serum-free medium. All cell culture incubations were performed at 37 °C in a humidified 5% CO2 incubator.
Transwell Studies
To establish polarized pBCEC cultures, cells were plated onto collagen-coated (120 μg/ml) transwell (6- or 12-well) culture dishes at a density of 40,000 cells/cm2. Cells were grown for 2–3 days depending on the transendothelial electrical resistance (TEER; ≥50 ohms/cm2). The tightness of the transwell culture was assessed by measuring the TEER using an EndOhm tissue resistance measurement chamber and EndOhm ohmmeter (World Precision Instruments, Florida). TEERs of collagen-coated, cell-free filters were used as blanks. Tight junction formation was induced (overnight) by adding DMEM/Ham's F-12 medium containing 550 nm hydrocortisone, 1% penicillin/streptomycin, and 0.7 mm l-glutamine along with the indicated concentrations of LXR agonists. Establishment of intact tight junctions was indicated by TEERs rising between 300 and 1000 ohms/cm2 in the in vitro BBB model system.
Immunoblot Analysis
pBCEC supernatants were collected and centrifuged (10,000 × g, 10 min, 4 °C), and secreted proteins were precipitated with 3% (v/v) trichloroacetic acid (TCA). Protein pellets were washed twice with acetone and resuspended in 1× sample buffer (Bio-Rad). For total cell proteins, pBCEC were lysed in protein lysis buffer (50 mm Tris, 10 mm EDTA, 1% Triton X-100, 0.5% v/v protease inhibitor mixture, pH 7.4), sonicated, and centrifuged (10,000 × g, 10 min, 4 °C). Cellular protein concentrations were quantified using the bicinchoninic acid (BCA) protein assay (Thermo Scientific, Vienna, Austria). Equal amounts of proteins (cellular and secreted) were loaded onto NuPAGE Novex 4–12% BisTris Midi gels (Invitrogen), and proteins were separated by SDS-PAGE in MOPS running buffer (Invitrogen) under denaturing conditions (200 V for 55 min). For immunoblotting, proteins were electrophoretically transferred to 0.45-μm PVDF membranes (50 V for 1 h). After blocking with 5% nonfat dry milk (Bio-Rad) for 90 min at room temperature, the membranes were probed using rabbit polyclonal anti-PLTP antibody (Abcam; 1:2000), rabbit polyclonal anti-APP antibody (Invitrogen; 1:1000), rabbit polyclonal A11 anti-amyloid oligomers antibody (Millipore; 1:10,000), or rabbit polyclonal anti-β-actin (Sigma; 1:5000) overnight at 4 °C. Membranes were washed and subsequently incubated with streptavidin-HRP (horseradish peroxidase; Bio-Rad)-labeled goat polyclonal antibody against rabbit IgG (Abcam; 1:5000) for 1 h at room temperature. Equal loading and transfer of proteins were confirmed by β-actin detection. Immunoreactive bands were detected on CL-XPosure films (Thermo Scientific) using enhanced chemiluminescence (Western C Immun-Star, Bio-Rad). Bands were quantified densitometrically using ImageJ version 1.42 software (National Institutes of Health).
PLTP mRNA Quantification
Total RNA was isolated from pBCEC using RNeasy mini kit according to the manufacturer's protocol, and RNA integrity was assessed by 1% agarose gel electrophoresis. RNA concentration was determined spectrophotometrically, and total RNA (1 μg) was reverse-transcribed using iScript cDNA synthesis kit (Bio-Rad) on a C1000 Thermal Cycler (Bio-Rad). Quantitative gene expression analysis of PLTP and reference genes HPRT1 (hypoxanthine phosphoribosyltransferase 1), ACTB (β-actin), GAPDH (glyceraldehyde-3-phosphate dehydrogenase), TBP (TATA box-binding protein), RPL4 (ribosomal protein L4), and HMBS (hydroxymethylbilane synthase) were performed on a CFX 96 Real Time System (Bio-Rad) using SYBR Green technology. In general, each reaction (10 μl) contained 1× iQ SYBR Green Supermix (Bio-Rad), 300 nm of each primer (Table 1), and 20 ng of cDNA template; PCR cycling conditions consisted of 40 cycles at 95 °C for 20 s, 60 °C for 40 s, and 72 °C for 40 s. All reactions were run in triplicate, and melting curve analyses were routinely performed to monitor the specificity of the PCR product. The relative gene expression ratio was determined using a standard curve method (27).
TABLE 1.
Primer sequences used for real time PCR
ss is Sus scrofa; hs is Homo sapiens; F is forward; and R is reverse.
| Gene | NCBI gene ID or Ref. | Primer sequence (5′–3′) | Amplicon length |
|---|---|---|---|
| bp | |||
| ssPLTP | 397527 | F, CCCTCTTCCTAGTGCTGCTG | 146 |
| R, CAGATCCGGAATGGTAATGG | |||
| hsPLTP | 26 | F, CAGGCGCACATGCAGAGTTC | 183 |
| R, GCAGCTCTGTGACCTTCACC | |||
| ssHPRT1 | 15452 | F, AGGACCTCTCGAAGTGTTGG | 247 |
| R, CAGATGGCCACAGGACTAGA | |||
| hsHPRT1 | 26 | F, GACCAGTCAACAGGGGACAT | 111 |
| R, CTGCATTGTTTTGCCAGTGT | |||
| ssACTB | 414396 | F, GGCCAGGTCATCACCATT | 140 |
| R, GGATGTCCACGTCACACTTC | |||
| hsACTB | 26 | F, TCCCTGGAGAAGAGCTACG | 131 |
| R, GTAGTTTCGTGGATGCCACA | |||
| ssGAPDH | 396823 | F, GCCGCGTCCCTGAGACAC | 262 |
| R, GGCGGGATCTCGCTCCT | |||
| hsGAPDH | 2597 | F, GAAGGTGAAGGTCGGAGTC | 226 |
| R, GAAGATGGTGATGGGATTTC | |||
| ssTBP | 27 | F, AACAGTTCAGTAGTTATGAGCCAGA | 153 |
| R,AGATGTTCTCAAACGCTTCG | |||
| hsTBP | 6908 | F, TGCACAGGAGCCAAGAGTGAA | 132 |
| R, CACATCACAGCTCCCCACCA | |||
| ssRPL4 | 27 | F, CAAGAGTAACTACAACCTTC | 122 |
| R, GAACTCTACGATGAATCTTC | |||
| hsRPL4 | 28 | F, GCTCTGGCCAGGGTGCTTTTG | 154 |
| R, ATGGCGTATCGTTTTTGGGTTGT | |||
| ssHMBS | 27 | F, AGGATGGGCAACTCTACCTG | 83 |
| R, GATGGTGGCCTGCATAGTCT | |||
| hsHMBS | 3145 | F, TGCTCGCATACAGACGGAC | 111 |
| R,GGTAAACAGGCTTTTCTCTCCAA | |||
| ssAPP | 53 | F, GTGAAGATGGATGCGGAGTT | 152 |
| R, GTGATGACAATCACGGTTGC | |||
| ssBACE1 | 100511707 | F, TGGACTGCCTCATGGTGTG | 155 |
| R, GTGACCAAAGTGAACCACCG |
Measurement of Phospholipid Transfer Activity
Phospholipid transfer activity of PLTP was assessed based on the transfer of l-α-[3H]dipalmitoylphosphatidylcholine from liposomes to HDL3 using an established radiometric assay (28). In brief, 129 μmol/μl egg PC, 1 nmol/μl butylated hydroxytoluene, and 1 μCi/μl l-α-[3H]dipalmitoylphosphatidylcholine were dried under nitrogen and resuspended in 1 ml of substrate buffer (10 mm Tris-HCl, 150 mm NaCl, and 1 mm EDTA, pH 7.4). To obtain clear liposomes, the above solution was sonicated and centrifuged (12,000 × g, 10 min, at room temperature). Radiolabeled liposomes (150 nmol) and plasma HDL3 (200 μg) were incubated with aliquots of pBCEC supernatants (300 μl) or cellular lysates (100 μl; in substrate buffer) for 1 h at 37 °C. The reaction was stopped, and liposomes were selectively precipitated by the addition of stop solution (536 mm NaCl, 363 mm MnCl2, and 52 units of heparin). After incubation for 30 min on ice, the mixture was centrifuged (12,000 × g for 10 min) at room temperature, and radioactivity transferred to HDL3 in the supernatant (500 μl) was determined on a Tri-Carb 2100 TR Liquid Scintillation Counter (Packard Bioscience Co.) after mixing with 5 ml of Ultima Gold scintillation mixture. The specificity of the PLTP activity assay in pBCEC lysates and supernatants was validated by antibody inhibition and heat inactivation control experiments, as described recently (29). To control for inter-assay variability, an aliquot (1 μl) of freshly thawed (−70 °C) human plasma was included in each assay. Cellular protein content was determined by BCA protein assay. PLTP activity was expressed in absolute activity as nanomoles of PL transferred per mg of total cell protein/ml medium or cell lysates and per h as measured under control conditions.
Isolation of Plasma HDL and ApoA-I
HDL3 (1.125–1.210 g/ml) was isolated fresh from normolipidemic human EDTA/plasma by density gradient ultracentrifugation (30). Briefly, density of plasma was adjusted with potassium bromide (KBr) to 1.24 g/ml, and density-adjusted plasma was then gently layered below an aqueous solution of KBr (1.063 g/ml). The samples were centrifuged at 694,000 × g for 4 h (10 °C) using an Optima l-90K Ultracentrifuge (Beckman Coulter, Vienna, Austria). HDL3 was carefully recovered, stored at 4 °C, and desalted before use with PD-10 columns containing Sephadex G-25 medium. Protein content of HDL3 was determined using Quanti-IT protein assay kit (Invitrogen). ApoA-I was purified after delipidation of HDL by size exclusion chromatography on a Sephacryl S-200 column (3 × 150 cm) as described (31).
Purification of Human Plasma PLTP
PLTP was purified from human plasma as described by Marques-Vidal et al. (32). Briefly, the human plasma fraction (d >1.22 g/ml) was applied sequentially to five different high performance chromatographic columns, and the active PLTP fraction was finally eluted at a sodium phosphate concentration of about 140 mm. Depending on the preparation, the activity of purified PLTP ranged between 5.0 and 9.0 μmol of PL transferred per ml/h. All of the preparations were free of cholesterol ester transfer protein, hepatic lipase, lecithin-cholesterol acyltransferase, and phospholipase activity.
HDL3 Conversion/Modification Assay
For HDL3 conversion assay, fresh human HDL3 (250 μg) was incubated with purified active human PLTP (250 nmol/h) at 37 °C for 24 h. Control incubations of HDL3 at 4 and 37 °C in the absence or presence of purified PLTP were also performed. HDL particle size was analyzed (10 μg of protein) by nondenaturing gradient gel electrophoresis, and PLTP-modified HDL particles were used as acceptors for cellular cholesterol efflux studies, as described below.
Nondenaturing Gradient Gel Electrophoresis (NDGGE) and Immunoblotting
Nondenaturing 4–15% (Tris-HCl gel, Bio-Rad) polyacrylamide gradient gel electrophoresis was used to determine HDL subclass distribution as described (33) with minor modifications. After pre-running the gels for 20 min at 140 V, samples were applied and electrophoresed for 30 min at 70 V, followed by another 6-h run at 140 V. Nascent HDL particle formation in pBCEC-conditioned media was determined by directly transferring the proteins after electrophoresis (4–20% Tris-HCl gel, Bio-Rad) to PVDF membranes (40 min at 100 V). All the above procedures were performed at 4 °C. Subsequent immunoblotting with polyclonal rabbit anti-human apoA-I antibody (1:2500, a kind gift from Dr. Ernst Malle, Medical University of Graz, Austria) was performed as described under “Immunoblot Analysis.” Gels were stained with Coomassie Brilliant Blue G-250 after fixing in 10% sulfosalicylic acid for 30 min. A high molecular weight protein marker (NativeMark, Invitrogen) was used as the size standard (7.1, 8.2, 11.0, 13.4, and 18.0 nm).
Pre-β-HDL Determination by Two-dimensional Crossed Immunoelectrophoresis
pBCEC monolayers grown in flasks (75 cm2) were incubated with fresh human HDL3 for 24 h at 37 °C. Cell-free control incubations of HDL3 in serum-free medium were performed at 4 and 37 °C. The next day, culture media were collected and concentrated using Amicon Ultra-15 Filter Devices. Pre-β-HDL formed in pBCEC conditioned media during incubation with HDL3 was quantified by two-dimensional crossed immunoelectrophoresis as described (34). In brief, in the first dimension, pre-β- and α-mobile lipoproteins present in the media (4 μg of protein) were resolved by 1% native agarose gel electrophoresis (4 °C, 2 h, 320 V). In the second dimension, separated lipoproteins were electrophoresed at right angles into a freshly applied layer of agarose gel (1%) containing 7.5% (v/v) rabbit anti-human apoA-I antiserum (4 °C for 20 h, 50 V). Gels were air-dried after staining with Coomassie Brilliant Blue R-250. The relative amount of pre-β-HDL was calculated as the percentage of the sum of α-HDL and pre-β-HDL peak areas.
Cellular Cholesterol Efflux Assay
Cellular cholesterol efflux was determined as described previously (6). In brief, pBCEC monolayers on 6- or 12-well plates were cholesterol-labeled with 0.5 μCi/ml [3H]cholesterol in M199 (containing 1% penicillin/streptomycin, 1 mm l-glutamine, and 10% porcine serum) for 24 h. Polarized pBCEC grown on 12-well transwell filter plates were labeled with 1.0 μCi/ml [3H]cholesterol added to the basolateral compartment (lower chamber, representing the brain parenchymal side) for 24 h. LXR agonist TO901317 was added to the apical compartment (upper chamber, representing the microvessel luminal side) during induction of tight junctions (16 h). Cells were washed and cellular cholesterol pools were equilibrated in serum-free medium for 2 h (transwell experiments) or 16 h (monolayer experiments). Subsequently, cells were washed twice with PBS, and cholesterol acceptors apoA-I (10 μg/ml), plasma HDL3 (50 μg/ml), or plasma HDL3 modified with purified human plasma PLTP (250 nmol/h, 37 °C, 24 h) were added in serum-free medium. For transwell experiments, plasma HDL3 (50 μg/ml) or plasma HDL3 modified with purified human plasma PLTP (250 nmol/h, 37 °C, 24 h) were added either to the apical or basolateral compartment. Aliquots of media were collected at the indicated time intervals, and radioactivity was determined with Ultima Gold scintillation mixture on a Tri-Carb 2100 TR Liquid Scintillation Counter (PerkinElmer Life Sciences). Cells were washed twice with ice-cold PBS and lysed in 0.3 n NaOH. Remaining [3H]cholesterol radioactivity in the cell lysates was determined, and total cellular protein concentration was quantified using the Qubit fluorometer (Quanti-IT protein assay kit, Invitrogen). Cholesterol efflux was calculated as the percentage of cpm/mg cell protein in the supernatants relative to the total counts in the supernatants plus cell lysates.
RNA Interference Mediated PLTP Silencing in pBCEC
A pool of four small interfering (si)RNAs targeting the human PLTP sequence was obtained from Qiagen. At 50–60% confluency, pBCEC were transfected with human PLTP siRNAs at a final concentration of 25 nm using PrimeFect siRNA transfection reagent (3 μl), as described by Stefulj et al. (35). Nontargeting control (NTC) with minimal unintended off-target effects was used as negative control. For cholesterol efflux assays, pBCEC were labeled with 0.5 μCi/ml [3H]cholesterol for 40 h, and simultaneously transfected with siRNA or NTC. Subsequently, cholesterol pools were equilibrated for 2 h and cholesterol efflux was determined as described under “Cellular Cholesterol Efflux Assay.” Silencing efficiency (relative to nontargeting siRNA) was monitored at the mRNA, protein, and phospholipid transfer activity level as described above.
Animal Experiments
Animal experiments were performed in accordance with the standards established by the Austrian Federal Ministry of Science and Research, Division of Genetic Engineering and Animal Experiments (Vienna, Austria). Male C57/BL6J mice 20 weeks of age (25–30g) were maintained under a 12-h light/12-h dark cycle in a temperature-controlled environment and had free access to chow diet (Ssniff®. Soest, Germany) and water. For 7 days the mice were fasted 4-h each day and dosed orally with vehicle (0.5% (w/v) carboxymethyl cellulose/H2O; 10 μl/g mouse) or synthetic LXR agonist T0901317 (50 mg/kg in 0.5% carboxymethyl cellulose). After 7 days of treatment, blood was drawn via retro-orbital puncture, and plasma was isolated for analysis. Mice were sacrificed by cervical dislocation. Brain and liver tissues were isolated, snap-frozen, and stored at −70 °C until analyzed. For phospholipid transfer activity assay, brain and liver samples were homogenized in 1 ml of substrate buffer containing protease inhibitor mixture at 4 °C using an Ultra-Turrax T25 homogenizer (IKA Labortechnic, Linz, Austria). Mouse brain capillary endothelial cells (mBCEC) were isolated from a pool of hemispheres (n = 7) essentially as described above for pBCEC. For PL transfer activity assay, mBCEC were homogenized in 1 ml of substrate buffer containing protease inhibitor mixture at 4 °C using Sonoplus HD 3080 (Bandelin, Berlin, Germany). After homogenization, the mixture was centrifuged (1000 × g, 5 min, 4 °C). PLTP activity (100 μl per assay) and total protein content were analyzed from the clear supernatant. PLTP activity was measured as described above and expressed as nanomoles of PL transferred per h/mg of protein.
Plasma PLTP and Lipid Analysis
Plasma (1 μl of undiluted mouse EDTA/plasma) phospholipid transfer activity was measured by radiometric assay as described above and expressed as nanomoles of PL transferred per ml/h. Plasma levels of total cholesterol, unesterified cholesterol, total phospholipids, and triglycerides were assayed using enzymatic kits according to the manufacturer's instructions (DiaSys Diagnostics, Holzheim, Germany).
Statistical Analysis
Results are reported as means ± S.E. unless stated otherwise. All experiments were performed at least two or more times in triplicate. Statistical significance (*, p < 0.05; **, p < 0.01; and ***, p < 0.001) was determined by two-tailed Student's t test or analysis of variance followed by Bonferroni's post hoc test using Prism software (Graphpad Version 5).
RESULTS
PLTP Is Synthesized in Porcine Cerebrovascular Endothelial Cells
Previous studies have identified the brain as one of the organs where PLTP is expressed (13). Immunohistochemistry has suggested that PLTP is present in neurons, astrocytes, and at the BBB (21). However, the contribution of BCEC to cerebral PLTP expression has not been evaluated. Hence, we first performed immunostaining on coronal sections of porcine midbrain using endothelial cell-specific marker von Willebrand factor (Fig. 1A) and PLTP antibody (Fig. 1B). Rabbit IgG was used as negative control (Fig. 1C). We detected distinct PLTP staining in cerebral vessels (Fig. 1A), more prominent than in brain parenchymal tissue, indicating that PLTP is highly expressed at the BBB. Immunocytochemical analysis of cultured primary pBCEC (Fig. 1, D–F) also showed distinct PLTP staining (Fig. 1E), confirming PLTP expression in the cerebral microvasculature. The presence of PLTP mRNA in isolated cerebral vessels confirmed that PLTP is of endogenous origin (Fig. 1G). Human liver expresses high levels of PLTP; hence, we used human liver RNA for comparison. PLTP mRNA expression levels (normalized to the geometric mean of four reference genes) were 6.8- and 1.8-fold higher in isolated porcine cerebral vessels as compared with whole porcine brain and liver, respectively (Fig. 1G). The enrichment of PLTP mRNA in isolated cerebral vessels as compared with total brain tissue strongly supports an important role of PLTP in BBB physiology.
FIGURE 1.

Cerebrovascular endothelial cells express PLTP. Immunocytochemical analysis of porcine brain endothelial cells is shown in situ on coronal sections of porcine midbrain (A–C) and in vitro in cultured pBCEC (D–F). Cerebral vessels and cultured endothelial cells express the endothelial marker von Willebrand factor (A and D) and PLTP (B and E). C and F, nonimmune rabbit IgG control. Cells were stained with Mayer's hematoxylin to visualize cell nuclei. Scale bar, 50 μm. G, total RNA was isolated from porcine brain tissue, porcine cerebral vessels, and human liver (positive control) and reverse-transcribed, and real time PCR was performed on a CFX 96 real time system (Bio-Rad) using SYBR Green technology. PLTP mRNA levels were normalized to four reference genes (HPRT1, GAPDH, RPL4, and HMBS). Data shown are means ± S.D. of triplicate analyses (***, p < 0.001 versus whole porcine brain).
PLTP Expression and Activity Are Enhanced by LXR Activation
It has been reported that LXR up-regulates expression of PLTP in HepG2 cells, macrophages, and murine liver and enhances PLTP activity in plasma (16, 17). Hence, we next investigated whether PLTP expression in pBCEC is modulated by LXR activation. The brain-specific natural LXR ligand, 24(S)-hydroxycholesterol (24OH-C) - under estimated physiological concentration (10 μm) - induced PLTP mRNA expression levels by 1.8-fold (Fig. 2A). Treatment with the synthetic LXR agonist TO901317 (5 μm) resulted in an even more pronounced 2.7-fold up-regulation of PLTP mRNA expression levels (Fig. 2A). PLTP protein was immunodetected both in cell lysates and in supernatants (Fig. 2B). LXR activation using both synthetic or natural ligands increased PLTP mass in pBCEC by 1.4–1.5-fold (Fig. 2B). We further investigated whether the PLTP secreted by pBCEC has the ability to transfer phospholipid, and thus we performed phospholipid transfer assays based on the rate of transport of radiolabeled PC from liposomes to human plasma HDL3. Both secreted and cellular PLTP efficiently transferred PC to HDL3 (Fig. 2C). Furthermore, LXR activation induced an increase in PLTP activity in both supernatants as well as in cell lysates, with somewhat less pronounced effects elicited by TO901317 (1.5- and 1.4-fold for supernatant and intracellular activity, respectively) as compared with the endogenous LXR ligand 24OH-C (2.0- and 1.6-fold, for supernatant and intracellular activity, respectively; Fig. 2C).
FIGURE 2.

LXR activation up-regulates PLTP expression and phospholipid transfer activity. Capillary endothelial cells were isolated from porcine brains and cultured on 6-well plates. Confluent pBCEC were incubated in the presence or absence of synthetic (5 μm TO901317, TO) or natural (10 μm 24OH-C) LXR ligands for 24 h in serum-free medium. Ctrl, control. A, total RNA was isolated and reverse-transcribed, and real time PCR was performed on a CFX 96 real time system (Bio-Rad) using SYBR Green technology. mRNA expression levels were normalized to HPRT1. Bars represent means ± S.E. of three independent experiments, each performed in triplicate (**, p < 0.01 versus controls). B, proteins were extracted from cells or TCA-precipitated from supernatants, separated by SDS-PAGE (4–12%), and blotted onto PVDF membranes. Secreted and intracellular PLTP was immunodetected using rabbit polyclonal anti-PLTP antibody. Image shown is a representative immunoblot of three independent experiments. (C, control; 24, 24(S)-hydroxycholesterol.) Band intensities were evaluated by densitometric scanning. Data shown are means ± S.E. of three experiments performed in triplicate (**, p < 0.01; ***, p < 0.001 versus controls). C, PLTP activity in the supernatants and cell lysates was determined based on the rate of transfer of [3H]phosphatidylcholine from liposomes to human HDL3. Values were normalized to total cellular protein contents and are expressed as means ± S.D. of one experiment representative of at least four independent experiments performed in triplicate (*, p < 0.05; **, p < 0.01 versus controls).
Polarized pBCEC Secrete PLTP Preferentially to the Brain Side of the in Vitro BBB Model
To characterize polarized expression/release of PLTP and to investigate how LXR agonists affect the polarized secretion pattern/activity of PLTP, pBCEC cells were grown on transwell filter plates, and tight junction formation was induced and assessed by measuring transendothelial electrical resistance. At 24 h, significant amounts of PLTP were detected that were secreted to both apical (mimicking the side facing the peripheral circulation) and basolateral (mimicking the side facing the brain parenchymal tissue) compartments (Fig. 3A). Notably, the amount of PLTP detected in the basolateral compartment was 1.8-fold higher as compared with the apical compartment (Fig. 3A). Furthermore, in line with results obtained with pBCEC monolayers, PLTP secretion was under control of LXR activation in both apical and basolateral compartments. PLTP secretion into the apical compartment was increased in response to 24OH-C (1.5-fold) and TO901317 (1.3-fold; Fig. 3A). PLTP protein levels secreted to the basolateral compartment within 24 h were 1.5-fold higher upon 24OH-C and 1.2-fold higher upon TO901317 treatment (as compared with basolateral levels detected under control conditions; Fig. 3A). We next investigated the phospholipid transfer activity of PLTP secreted to apical and basolateral compartments. In line with higher PLTP levels immunodetected in the basolateral compartments, the PL transfer activity was also enhanced (1.7-fold) in the basolateral supernatants (Fig. 3B). Furthermore, LXR activation induced an increase in PLTP activity in both apical as well as in basolateral compartments. The endogenous LXR ligand, 24OH-C elicited more pronounced effects on phospholipid transfer activity detected in both apical (1.6-fold relative control) and basolateral (1.7-fold relative to control) compartments as compared with TO901317 (1.4-fold PLTP activity in apical and 1.2-fold in basolateral supernatants) (Fig. 3B).
FIGURE 3.

Polarized release of active PLTP from pBCEC into apical (“blood compartment”) and basolateral (“brain parenchymal compartment”) direction is enhanced by LXR activation. pBCEC were cultured on 6-well transwell filter plates and incubated for 24 h in the presence or absence of synthetic (5 μm TO901317) or natural (10 μm 24OH-C) LXR ligands in serum-free medium. A, apical (1.5 ml/well) and basolateral (2.6 ml/well) media were collected, and proteins were TCA-precipitated; aliquots corresponding to 1 ml of supernatant and normalized to total cellular protein were separated by SDS-PAGE 4–12% and blotted onto PVDF membranes. Secreted PLTP (∼55 kDa) was immunodetected using rabbit polyclonal anti-PLTP antibody. Image shown is a representative immunoblot of four independent experiments. (C, control; 24, 24(S)-hydroxycholesterol.) Band intensities were evaluated by densitometric scanning. Data shown are means ± S.D. of one experiment representative of four independent experiments, each performed in triplicate (**, p < 0.01 versus controls). Ctrl, control; TO, TO901317; 24, 24(S)-hydroxycholesterol. B, PLTP activity in the supernatants was determined based on the transfer rate of [3H]phosphatidylcholine from liposomes to human HDL3. Values were normalized to total cellular protein contents and are expressed as PL transfer activity (per well) related to apical release under control conditions. Means ± S.D. of one experiment are representative of at least three independent experiments performed in triplicate (*, p < 0.05; **, p < 0.01; ***, p < 0.001 versus controls).
PLTP-modified HDL3 Elicits Enhanced Cholesterol Efflux from pBCEC
PLTP remodels HDL3, generating larger HDL particles with concomitant production of small apoA-I containing pre-β-HDL particles (11, 36). To investigate whether PLTP may alter the capacity of HDL particles to remove cellular cholesterol from pBCEC, we preincubated human plasma HDL3 with purified, active plasma PLTP (250 nmol/h) and used these HDL particles as acceptors. In accordance with previous reports (11, 36), particle size determination by NDGGE (Fig. 4A) confirmed that HDL3 incubation with PLTP resulted in the formation of two major distinct particle populations, one with larger (11 ± 0.5 nm) and the other with small mean diameter (7.4 ± 0.5 nm).
FIGURE 4.

PLTP-mediated remodeling increases the cholesterol removal capacity of HDL3. HDL3 (250 μg) was preincubated in the absence (PBS) or presence of plasma PLTP (250 nmol/h) at 37 °C for 24 h. A, subsequently, 10 μg of HDL protein was used for NDGGE analysis and stained with Coomassie Brilliant Blue G-250. PLTP-mediated particle remodeling is indicated by the arrows. Gel shown is representative of three independent experiments (STD, protein standards of known diameter). B–D, pBCEC were cultured on 12-well multiplates or transwell filter plates, labeled with [3H]cholesterol (0.5 or 1.0 μCi/ml, respectively) for 24 h in the absence (B) or presence of 24OH-C (10 μm; C) or TO901317 (5 μm; D). Cellular cholesterol pools were equilibrated, and time-dependent cellular [3H]cholesterol efflux to untreated (+ PBS) or PLTP-modified (+PLTP) HDL3 (50 μg/ml) was determined at the indicated time points. Aliquots of medium from apical (E) and basolateral (F) compartments were collected at the indicated time points, and time-dependent cellular [3H]cholesterol efflux to untreated (+PBS) or PLTP-modified (+PLTP) HDL3 (50 μg/ml) was determined. Data shown are means ± S.E. of three independent experiments performed in triplicate. δ, p < 0.0001 HDL3+ PLTP versus HDL3 + PBS; ψ, p < 0.0001 TO/24OH-C versus basal conditions in the presence of HDL3+ PLTP; ϕ, p < 0.0001 TO/24OH-C versus basal conditions in the presence of HDL3+ PBS (analysis of variance).
Strikingly, PLTP modification of HDL3 enhanced time-dependent cholesterol release from pBCEC, as compared with unmodified HDL3 particles, under basal (control) conditions (by 3.0- and 1.9-fold at 30 and 240 min, Fig. 4B) as well as in the presence of 24OH-C (from 1.5- to 1.8-fold at 240 and 30 min, respectively; Fig. 4C) or TO901317 (by 1.5- and 1.3-fold at 30 and 240 min, respectively; Fig. 4D). Cholesterol efflux to modified HDL3 particles was enhanced by 1.2- and 1.5-fold (at 30 min), respectively, upon 24OH-C and TO901317 treatment relative to control conditions, indicating additive effects of PLTP treatment and LXR activation (Fig. 4, B–D).
To further elucidate the effect of PLTP-mediated HDL3 modification on cholesterol mobilization, we added unmodified and PLTP-modified HDL particles to either apical or basolateral compartment of the polarized pBCEC cultures grown on transwell filters. Under basal conditions, PLTP modification of HDL3 increased cholesterol removal to the basolateral compartment by 2.0- and 1.6-fold at 30 and 240 min, respectively (Fig. 4E), but it did not have any significant effect on cellular cholesterol removal to the apical compartment (Fig. 4F). However, in the presence of TO901317, PLTP modification of HDL3 enhanced time-dependent cholesterol release from polarized pBCEC to both the basolateral (by 1.3- and 1.6-fold at 30 and 240 min, respectively) as well as to the apical (by 2.3- and 3.0-fold at 30 and 240 min, respectively) compartments (data not shown).
pBCEC Induce Remodeling of HDL3 and Pre-β-HDL Formation Is Augmented upon LXR Activation
Pre-β-HDL has been suggested to function among the most efficient HDL acceptors for cellular cholesterol (37). Previous studies have indicated that PLTP enhances the formation of pre-β-HDL particles from reconstituted HDL (38). To investigate whether PLTP secreted by pBCEC is capable of executing similar functions, we incubated the cells with human HDL3, and pre-β-HDL particles were quantified by two-dimensional crossed immunoelectrophoresis after 24 h. We immunodetected substantial amounts of pre-β-HDL in pBCEC-derived supernatants (Fig. 5). Moreover, the formation of pre-β-HDL particles was markedly increased (1.9-fold) in TO901317-treated pBCEC as compared with the untreated cells (Fig. 5, A, B and E), which is in line with (2.2-fold) increased PLTP activity detected in the supernatants (Fig. 5F). We were unable to immunodetect any pre-β-HDL particles in cell-free control incubations of HDL3 (Fig. 5, C and D).
FIGURE 5.

Pre-β-HDL and nascent HDL formation is amplified upon treatment of pBCEC with LXR agonists. pBCEC were cultured on 75-cm2 flasks and incubated in the absence or presence of TO901317 (TO) (5 μm) and/or HDL3 (50 μg/ml) for 24 h in serum-free medium. Supernatants were collected and concentrated (45-fold) using Amicon Ultra-10K centrifugal filters. Samples (5 μl) were analyzed by two-dimensional crossed immunoelectrophoresis with anti-human apoA-I antibody. A–D, representative patterns of α-HDL and/or pre-β-HDL generated under different conditions are shown. E, relative area of pre-β-HDL and α-HDL peaks was calculated by multiplication of peak height and peak width at half-height. The pre-β-HDL amount is expressed as percentage of the sum of α-HDL and pre-β-HDL peak areas. F, PLTP activity in supernatants was determined as described under “Experimental Procedures.” Means ± S.E. of three independent experiments performed in triplicate (**, p < 0.01 versus pBCEC + HDL3). G, LXR activation stimulates nascent HDL formation in pBCEC. Primary pBCEC were cultured on 75-cm2 flasks and incubated in the absence or presence of TO901317 (5 μm) or 24OH-C (10 μm) for 24 h in serum-free medium. Supernatants were collected and concentrated using Amicon Ultra-10K centrifugal filters. The presence of HDL-sized particles was assessed by NDGGE followed by immunoblotting using anti-human apoA-I antibody. Image shown is representative of three independent experiments. Ctrl, control.
Nascent HDL Formation Is Amplified upon LXR Activation in pBCEC
To evaluate the formation of apoA-I containing nascent particles in pBCEC, we examined supernatants of primary pBCEC cultured in serum-free medium for 24 h. We immunodetected apoA-I-containing nascent particles in pBCEC supernatants, and as expected, the amount of endogenous HDL particles formed was increased by LXR activation (Fig. 5G). Because of the heterogeneity in size of apoA-I containing particles, no distinct bands were visible. However, the size of the major fraction of nascent apoA-I containing particles produced by pBCEC varied between 12 and 18 nm (Fig. 5G).
PLTP Contributes to Both ApoA-I and HDL3-mediated Cholesterol Removal from pBCEC
To get further insights into the role of endogenous PLTP in apoA-I- and HDL3-mediated cholesterol removal from pBCEC, we silenced the gene using RNA interference (RNAi) technology (Fig. 6). Real time PCR and immunoblotting analyses confirmed that transfection with siRNA (25 nm) targeting human PLTP sequence resulted in 75% (Fig. 6A) and 60% (Fig. 6B) reduction in PLTP mRNA and protein levels, respectively. Furthermore, PLTP activity assays confirmed a significant reduction (73%) in PL transfer activity in PLTP-silenced cells (Fig. 6C). PLTP silencing had no effect on mRNA expression levels of ABCA1 (data not shown). Silencing of PLTP resulted in an up to 67% (at 24 h) reduction in time-dependent cholesterol release to apoA-I (Fig. 6D). We also evaluated HDL3-mediated cholesterol release in PLTP-silenced cells and observed a reduction by up to 30% in cholesterol removal capacity from PLTP-silenced cells as compared with the nonsilenced cells (Fig. 6E), whereby the PLTP-silencing effects became more evident at the later time points (4 and 24 h) investigated (Fig. 6E).
FIGURE 6.

Both apoA-I-and HDL3-mediated cholesterol removal from pBCEC are diminished upon PLTP silencing. pBCEC were cultured on 12-well plates. Cells were labeled with [3H]cholesterol (0.5 μCi/ml) and simultaneously transfected with human PLTP siRNA (25 nm) for 40 h. NTC siRNA was used as the negative control. A, RNA was isolated and reverse-transcribed, and real time PCR was performed using SYBR Green technology. mRNA expression levels were normalized to HPRT1 (means ± S.E. of four experiments performed in triplicate; **, p < 0.01 versus NTC). B, proteins were TCA-precipitated from supernatants; cells were lysed, and immunoblotting was performed using rabbit polyclonal anti-PLTP and β-actin antibodies as described under “Experimental Procedures.” Image shown is a representative immunoblot. Band intensities were evaluated by densitometric scanning. Data shown are means ± S.E. of three experiments performed in duplicates (**, p < 0.01; ***, p < 0.001 versus controls). C, phospholipid transfer activity in the supernatants was determined after PLTP silencing as described under “Experimental Procedures.” (Means ± S.D. of one experiment are representative of at least two independent experiments performed in triplicate. ***, p < 0.001 versus NTC.) D and E, cellular cholesterol pools were equilibrated for 2 h, and time-dependent cellular [3H]cholesterol release to apoA-I (D, 10 μg/ml) or HDL3 (E, 50 μg/ml) was measured. (Means ± S.D. are of one experiment representative of at least four independent experiments performed in triplicate, *, p < 0.05; **, p < 0.01, versus NTC).
PLTP Modulates Amyloid β (Aβ) Oligomerization in pBCEC
To investigate whether PLTP might have a direct or indirect role in Aβ metabolism, we treated pBCEC with HDL3, PLTP-modified HDL particles, or with increasing amounts of active plasma PLTP. Analyses of intracellular Aβ oligomers by immunoblotting revealed a significant reduction of Aβ octamers by 63% upon HDL3, by 56% upon 1000 nmol/ml active PLTP, and an albeit nonsignificant reduction by 47% upon 50 nmol/ml active PLTP and 30% upon PLTP-modified HDL treatments (Fig. 7A). Although not significant, we observed a trend toward decreased Aβ tetramer levels with the above-mentioned treatments (Fig. 7B), although we were unable to detect any changes in intracellular APP protein levels (Fig. 7C).
FIGURE 7.

Decreased Aβ oligomerization in pBCEC treated with HDL3 and active PLTP and increased expression of BACE1 mRNA in PLTP-silenced cells. A–C, confluent pBCEC were incubated for 24 h in serum-free medium containing HDL3, PLTP-modified HDL particles increasing amounts of active plasma PLTP, or vehicle only (Ctrl). Intracellular proteins were extracted from cells, separated by SDS-PAGE (4–12%), and blotted onto PVDF membranes. Intracellular Aβ oligomers (A and B) and APP (C) were immunodetected using appropriate rabbit polyclonal antibodies. Image shown is a representative immunoblot of three independent experiments. Band intensities were evaluated by densitometric scanning. Ctrl, control. Data shown are means ± S.E. of three experiments performed in triplicate (*, p < 0.05 versus controls; 1, 2, 3, 4, and 5 represents control, HDL3 + PLTP, HDL3 + PBS, 50 nmol/ml PLTP, and 1000 nmol/ml PLTP, respectively). D and E, pBCEC were transfected with human PLTP siRNA (25 nm) for 40 h, and NTC siRNA was used as the negative control. RNA was isolated and reverse-transcribed, and real time PCR was performed using SYBR Green technology. BACE1 (D) and APP (E) mRNA expression levels were normalized to HPRT1 (Means ± S.E. are of two experiments performed in triplicate. *, p < 0.05 versus NTC.)
In parallel, we further analyzed the mRNA levels of β-secretase (β-site APP cleavage enzyme 1, BACE1) and APP in PLTP-silenced pBCEC. Interestingly, there was a significant increase (1.8-fold) in β-secretase mRNA levels in PLTP-silenced cells as compared with the nonsilenced cells (Fig. 7D), although the APP mRNA levels remained unchanged (Fig. 7E).
LXR Activation Up-regulates PLTP Activity in Murine BCEC in Vivo
It has been previously reported that PLTP expression in mouse liver is up-regulated by LXR activation in vivo (16, 17). To explore whether PLTP activity in BCEC could be regulated by LXR activation in vivo, we administered C57BL6 mice with synthetic LXR agonist TO901317 for 7 days and assayed the PLTP activity in plasma, mBCEC, whole brain, and liver. Plasma lipid analysis revealed a significant increase in total cholesterol (1.4-fold) and phospholipid (1.9-fold) content in TO901317-treated animals as compared with vehicle control (Table 2). Plasma PL transfer activity in TO901317-fed mice was increased to 1.7-fold compared with vehicle control (Fig. 8A). Whole brain (1.3-fold) and liver (1.9-fold) PLTP activity was also increased in TO901317-fed mice (Fig. 8B). More importantly, isolated mBCEC were highly enriched in PL transfer activity and displayed a 2.5-fold increase in PLTP activity in TO901317-treated animals compared with vehicle control (Fig. 8B).
TABLE 2.
Plasma lipid profile in C57/BL6 mice treated with LXR agonist TO901317 or vehicle
Data are expressed as means ± S.D.; n = 7 per group.
| Lipid profile | Vehicle | TO901317 (50 mg/kg) |
|---|---|---|
| Triglycerides (mg/dl) | 86.8 ± 24 | 77.3 ± 19.1 |
| Total cholesterol (mg/dl) | 46.2 ± 14 | 64.3 ± 7.9a |
| Free cholesterol (mg/dl) | 23.4 ± 5.3 | 28.7 ± 5.9 |
| Phospholipids (mmol/liter) | 1.6 ± 0.5 | 3.0 ± 0.9b* |
a p < 0.05.
b p < 0.01 versus vehicle.
FIGURE 8.

PLTP activity in BCEC is regulated by LXR agonist administration in vivo. C57/BL6 mice were administered orally with either vehicle or TO901317 (TO) (50 mg/kg/day) for 1 week. PLTP activity in plasma (A), isolated mBCEC, whole brain, and liver (B) was measured based on the transfer rate of [3H]phosphatidylcholine from liposomes to human HDL3. Data represent mean values ± S.D. (n = 7). *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus vehicle.
DISCUSSION
The protective role of HDL against Alzheimer disease and/or dementia is under extensive investigation because, strikingly, prospective studies point toward an inverse relationship between plasma HDL levels and the risk of cognitive decline or AD development (39, 40). Cholesterol metabolism in the brain appears to be completely separated from that in the periphery due to the presence of tight junctions established by brain microvascular endothelial cells. However, these cells are also actively involved in HDL metabolism (6, 7) at the blood-brain interface. PLTP is an LXR target gene involved in the regulation of HDL biogenesis in the periphery and is also expressed in neurons and glial cells (12, 21). These findings, collectively, prompted us to investigate the potential role of PLTP in HDL metabolism at the BBB using an in vitro model consisting of primary pBCEC.
The results presented here demonstrate for the first time that PLTP is synthesized by BCEC of the BBB (Figs. 1 and 2). One of the most interesting findings is that polarized pBCEC secrete substantial amounts of PLTP into both apical and basolateral compartments but more favorably toward the basolateral (i.e. parenchymal or brain) side (Fig. 3). PLTP secreted to both compartments is active in transferring phospholipids, strongly supporting a role of BCEC-derived PLTP in lipid transport and metabolism at the blood-brain interface and possibly also in deeper regions of the brain. Intriguingly, and unlike reported recently in human fetoplacental endothelial cells (29, 41), PLTP protein and activity were detectable also in pBCEC lysates, indicating that pBCEC retain a fraction of the produced protein in intracellular pool(s), whereas human fetoplacental endothelial cells release the entire amount of synthesized PLTP into the culture medium. Differentiated intracellular levels of PLTP have also been detected in neurons, and active PLTP has been found (trans)located in the nucleus; however, the possible function(s) of intracellular PLTP are thus far unknown (42).
Furthermore, we established that expression and activity of PLTP in pBCEC are under the control of LXRs (Figs. 2 and 3). In line with this, a direct regulatory influence of LXRs on PLTP has been reported in HepG2 cells, macrophages, and in mouse liver (16, 17); however, PLTP mRNA levels were not elevated in HUVEC upon treatment with LXR agonists (43). The PLTP promoter region contains a high affinity LXR response element (DR-4A and DR-4B) that binds LXR/retinoid X receptor heterodimers, and the regulatory effects of LXR ligands on PLTP expression/activity were lost in animals or cells lacking LXRs (16, 17). Our present findings are of physiological relevance because under in vivo conditions BCEC are in contact with 24(S)OH-cholesterol, the major brain cholesterol metabolite that can readily cross the BBB to be transported by HDL and LDL to the liver for metabolism/elimination (2). Hence, the current results suggest that LXR agonists may regulate lipid metabolism at the blood-brain interface through modulating the expression levels of PLTP.
It has been well described that PLTP modulates HDL size and composition thereby enhancing the ability of HDL to remove (excess) cholesterol and phospholipid from cells (11, 12). The smaller HDL particles, termed pre-β-HDL, are formed as a result of PLTP-mediated HDL remodeling and serve as the ideal initial acceptors for cellular cholesterol (37, 44). The role of PLTP in HDL-mediated reverse cholesterol transport has been established (45). We previously reported an auto-regulatory reverse sterol transport mechanism operating in pBCEC at the BBB (6). In this study we established a firm role of pBCEC-derived PLTP in this process. When using PLTP-modified HDL as compared with native HDL3 as exogenous cholesterol acceptors, pBCEC responded with enhanced cholesterol release, and LXR activation augmented the effect further (Fig. 4). It is interesting to note that the addition of PLTP-modified HDL3 to the basolateral compartment of polarized pBCEC resulted in an enhanced removal/transfer of cholesterol to the brain parenchymal side as compared with native HDL3 acceptor particles (Fig. 4F). Neurons require high amounts of cholesterol, cannot efficiently produce all cholesterol required, and thus rely on external sources such as glial cells (46). Based on our findings, we speculate that BCEC may form an additional source of external cholesterol. pBCEC efficiently release cholesterol into the basolateral direction, and PLTP present at the brain parenchymal side remodels HDL particles thereby enhancing the transfer of cholesterol from the BBB to the brain. In addition, we immunodetected pre-β-HDL formation in pBCEC supernatants incubated with native HDL3, and the relative amount of pre-β-HDL was also augmented by LXR activation (Fig. 5). These results together strongly suggest that PLTP, of either exogenous (i.e. plasma) or endogenous (i.e. BCEC) origin, mediates HDL remodeling at the BBB and thereby markedly enhances cholesterol release from pBCEC.
pBCEC express ABCA1, scavenger receptor class B, type I (SR-BI), and apoA-I, all involved in sterol transport at the BBB (6, 7). The LXR/ABCA1/apoA-I cholesterol efflux pathway facilitates the assembly of particles possessing a density corresponding to that of HDL particles preferentially at the basolateral side (brain side) of the BBB (6). PLTP is known to interact with and stabilize ABCA1 in peripheral cells, thus enhancing cholesterol efflux to apoA-I (47, 48). Because PLTP, apoA-I, and ABCA1 are all more abundant at the basolateral side, it is reasonable to hypothesize that PLTP expressed in BCEC could similarly interact with and/or stabilize ABCA1 and in concert with apoA-I promote HDL genesis at the brain side of the BBB.
Assembly of cellular phospholipids and cholesterol with apoA-I form a crucial step in HDL formation (37). Our RNA interference studies to knock down PLTP in pBCEC resulted in a 67% reduction (Fig. 6D) of cholesterol release to apoA-I, confirming a key role of PLTP in the process of HDL genesis. Characterization of apoA-I-containing particles in pBCEC-conditioned media by NDGGE/immunoblotting analyses also clearly pointed toward the formation of nascent HDL particles at the BBB, which was amplified upon LXR activation (Fig. 5G). In previous studies nascent apoA-I-containing particles with diameters varying from 8 to 20 nm have been detected in various cell lines (49). The nascent HDL particles detected in pBCEC supernatants have a similar size as that reported for human CSF lipoproteins (13–18 nm) that are also mainly enriched in apoA-I (50). Because previous studies employed different separation methods, further characterization of nascent HDL particles formed by pBCEC is required to draw firm conclusions on their composition. However, based on our current findings, it is reasonable to assume that 24(S)OH-cholesterol, although crossing the BBB, can induce PLTP expression and activity together with ABCA1 and apoA-I to enhance lipid efflux and HDL formation. Hence, PLTP expressed in pBCEC together with apoA-I may serve to integrate plasma and cerebrovascular lipid metabolism. Efflux of cellular cholesterol is also mediated by HDL3 representing a ligand for SR-BI and ABCG1 (6, 51). Present PLTP gene-silencing experiments in pBCEC suggest an association of PLTP also with HDL3-mediated cholesterol efflux, accounting for ∼30% (Fig. 6E). Our findings, collectively, underline a crucial role for PLTP in HDL metabolism at the BBB.
It has been documented that HDL particles mediate clearance of Aβ from cells (52, 53). Because we here revealed a critical role for PLTP in HDL biogenesis at the BBB, we assumed that it might be also involved in the regulation of Aβ homeostasis. Indeed, incubation of pBCEC in the presence of active plasma PLTP significantly reduced intracellular levels of Aβ oligomers (Fig. 7, A and B). Furthermore, PLTP-modified HDL3 particles reduced intracellular levels of Aβ oligomers, although to a lesser extent than unmodified HDL3. Although here we have not addressed the underlying mechanism, one potential explanation is that HDL binds and removes Aβ thereby reducing intracellular Aβ/oligomers. According to our results, Aβ would bind more efficiently to unmodified than to PLTP-modified HDL3. The mechanism on how PLTP itself reduces Aβ oligomers in pBCEC is presently unknown. Interestingly, we found increased levels of BACE1 mRNA in PLTP-silenced pBCEC (Fig. 7D). This is intriguing as increased levels of BACE may enhance generation of Aβ by redirecting the APP processing toward the amyloidogenic pathway (54). A recent study in PLTP-deficient aged mice also reported an increased expression and activity of β-secretase in various brain regions (55). Further investigation is required to explore the mechanism(s) behind these findings.
Our animal studies confirm that LXR activation modulates PLTP activity in vivo and for the first time show that this effect applies also to BCEC (Fig. 8). Following LXR activation, the PLTP activity was up-regulated in plasma as well as in all the tissue/cell samples analyzed, i.e. liver, whole brain, and isolated mBCEC. It is important to note that PLTP activity detected in mBCEC was higher than in whole brain, confirming a significant role of PLTP at the BBB (Fig. 8B). Because LXR agonist modulated PLTP activity in BCEC in vivo, it is very tempting to state that PLTP regulates lipid metabolism at the blood-brain interface.
Apolipoproteins documented in the CSF and CNS are mainly apoE, apoA-I, apoA-IV, apoD, apoH, and apoJ, and the lipoproteins detected have a density resembling HDL in plasma (56, 57). Continuous supplies of lipids are required for neurons mainly for membrane synthesis, and they depend greatly on exogenous sources. Lipoprotein-mediated clearance mechanisms to eliminate excess lipids are also functioning in neurons (58). Previous studies undoubtedly established that brain HDL has a crucial role in cholesterol turnover in the CNS (59). In recent years, several neuroprotective properties of HDL have been reported. HDL can decrease oxidative stress and reduce neuroinflammation (59). HDL is able to reduce production of amyloid β (Aβ), the major component of senile amyloid plaques, by activating reverse cholesterol transport with the help of ABC transporters (60). HDL can directly bind Aβ and inhibit its assembly to fibrils thereby attenuating its neurotoxic effects (51). HDL transports Aβ in both CSF and plasma, and thus it might remove excess Aβ piled up in the vessels (52), although the mechanisms have not yet been described. Furthermore, plasma HDL cholesterol levels correlate with the maintenance of BBB integrity in patients with mild and moderate AD (61). We recently reported that cerebrovascular endothelial cells actively contribute to cerebral APP/Aβ synthesis. Strikingly, LXR activation redirected APP synthesis and processing by pBCEC toward the beneficial nonamyloidogenic pathway (54). We hypothesize that HDL and mechanisms of reverse cholesterol transport operating at the BBB trigger these beneficial LXR agonist-mediated effects on APP processing. Results of this study strongly suggest that PLTP produced by BCEC forms an additional source of PLTP to the brain pool. As an LXR target and key determinant of HDL metabolism at the BBB, PLTP might be involved in regulating APP/Aβ homeostasis. A model of the potential roles of PLTP and HDL at the BBB is outlined in Fig. 9.
FIGURE 9.
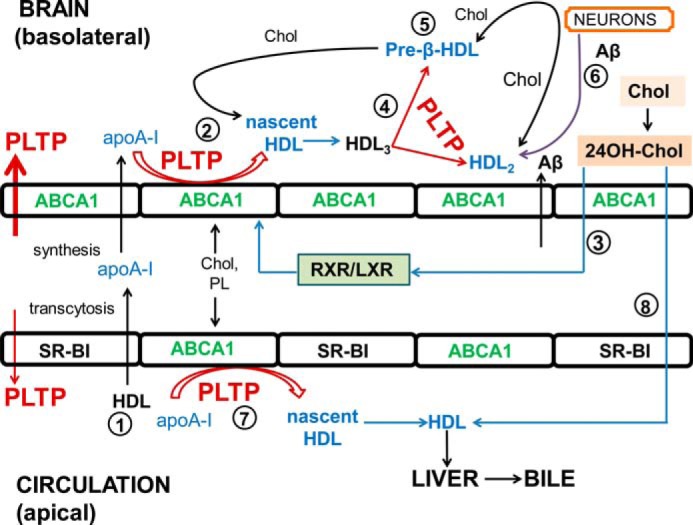
Model of proposed PLTP functions in HDL metabolism and HDL functions at the BBB. 1, small fraction of apoA-I from circulating HDL can transcytose across cerebrovascular endothelial cells (8). In addition, apoA-I is expressed by BCEC and released bidirectionally but mainly to the basolateral compartment (6). 2, active PLTP is also secreted bidirectionally but mainly to the basolateral compartment. Nascent HDL particles are formed at the brain side with the aid of ABCA1 and PLTP. 3, LXR activation by 24OH-cholesterol (or synthetic agonists) promotes the formation of nascent apoA-I containing particles. 4, nascent HDL particles mature to form spherical HDL3, and PLTP further remodels HDL3 into HDL2 and pre-β-HDL particles. 5, pre-β-HDL (and also HDL2) accepts excess cellular cholesterol. 6, HDL may directly bind excess Aβ (from neurons and from BCEC); both PLTP and HDL may reduce intracellular Aβ oligomers, and HDL may facilitate the elimination of excess Aβ into the circulation (56, 59). 7, HDL particles are also formed and remodeled at the apical side, through endogenous and exogenous (plasma) PLTP. 8, HDL in the circulation with the help of SR-BI accepts 24(S)-hydroxycholesterol (6), which will be finally removed through bile. (HDL, high density lipoproteins; ABCA1, ATP-binding cassette transporter A1; RXR, retinoid X receptor; PL, phospholipid; 24OH-Chol, 24(S)-hydroxycholesterol; Chol, cholesterol; SR-BI, scavenger receptor, class B, type I).
In summary, this study identified for the first time that phospholipid transfer protein is expressed in cerebrovascular endothelial cells, regulated by LXRs in BCEC, and actively involved in cholesterol efflux, HDL genesis, and remodeling at the blood-brain barrier in vitro. Further investigation is needed to fully disclose the prospective roles of PLTP at the BBB, in particular under conditions of lipid-related neurodegenerative diseases, like AD.
Acknowledgments
We acknowledge the assistance by Kerstin Hingerl (Institute of Cell Biology, Histology, and Embryology) and Carmen Tam-Amersdorfer (Institute of Pathophysiology and Immunology), Medical University of Graz, Austria, during immuno-histo/cytochemical analyses.
This work was supported by the Austrian Science Fund Grants DK-MCD W1226-B18 (to U. P. and D. K.), P24783 B19 (to U. P.), and SFB LIPOTOX F30 and P38223P (to D. K.), by the Medical University of Graz Doctoral College of Metabolic and Cardiovascular Diseases, Franz Lanyar Foundation 365 (to U. P.).
- AD
- Alzheimer disease
- 24OH-C
- 24(S)-hydroxycholesterol
- ABCA1
- ATP binding cassette transporter A1
- apo A-I
- apolipoprotein A-I
- BBB
- blood-brain barrier
- CSF
- cerebrospinal fluid
- HPRT1
- hypoxanthine phosphoribosyltransferase 1
- LXR(s)
- liver X receptor(s)
- NDGGE
- nondenaturing gradient gel electrophoresis
- PC
- phosphatidyl choline
- PL
- phospholipid
- PLTP
- phospholipid transfer protein
- pBCEC
- porcine brain capillary endothelial cell
- TEER
- transendothelial electrical resistance
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- Aβ
- amyloid β
- mBCEC
- mouse brain capillary endothelial cell
- APP
- amyloid precursor protein
- NTC
- nontargeting control
- SR-BI
- scavenger receptor class B, type I.
REFERENCES
- 1. Vaya J., Schipper H. M. (2007) Oxysterols, cholesterol homeostasis, and Alzheimer disease. J. Neurochem. 102, 1727–1737 [DOI] [PubMed] [Google Scholar]
- 2. Björkhem I., Meaney S. (2004) Brain cholesterol: long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 24, 806–815 [DOI] [PubMed] [Google Scholar]
- 3. Ladu M. J., Reardon C., Van Eldik L., Fagan A. M., Bu G., Holtzman D., Getz G. S. (2000) Lipoproteins in the central nervous system. Ann. N.Y. Acad. Sci. 903, 167–175 [DOI] [PubMed] [Google Scholar]
- 4. Rye K. A., Bursill C. A., Lambert G., Tabet F., Barter P. J. (2009) The metabolism and anti-atherogenic properties of HDL. J. Lipid Res. 50, S195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Merched A., Xia Y., Visvikis S., Serot J. M., Siest G. (2000) Decreased high density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer's disease. Neurobiol. Aging 21, 27–30 [DOI] [PubMed] [Google Scholar]
- 6. Panzenboeck U., Balazs Z., Sovic A., Hrzenjak A., Levak-Frank S., Wintersperger A., Malle E., Sattler W. (2002) ABCA1 and scavenger receptor class B, type I, are modulators of reverse sterol transport at an in vitro blood-brain barrier constituted of porcine brain capillary endothelial cells. J. Biol. Chem. 277, 42781–42789 [DOI] [PubMed] [Google Scholar]
- 7. Panzenboeck U., Kratzer I., Sovic A., Wintersperger A., Bernhart E., Hammer A., Malle E., Sattler W. (2006) Regulatory effects of synthetic liver X receptor- and peroxisome-proliferator activated receptor agonists on sterol transport pathways in polarized cerebrovascular endothelial cells. Int. J. Biochem. Cell Biol. 38, 1314–1329 [DOI] [PubMed] [Google Scholar]
- 8. Balazs Z., Panzenboeck U., Hammer A., Sovic A., Quehenberger O., Malle E., Sattler W. (2004) Uptake and transport of high density lipoprotein (HDL) and HDL-associated α-tocopherol by an in vitro blood-brain barrier model. J. Neurochem. 89, 939–950 [DOI] [PubMed] [Google Scholar]
- 9. Albers J. J., Tu A. Y., Paigen B., Chen H., Cheung M. C., Marcovina S. M. (1996) Transgenic mice expressing human phospholipid transfer protein have increased HDL/non-HDL cholesterol ratio. Int. J. Clin. Lab. Res. 26, 262–267 [DOI] [PubMed] [Google Scholar]
- 10. Huuskonen J., Jauhiainen M., Ehnholm C., Olkkonen V. M. (1998) Biosynthesis and secretion of human plasma phospholipid transfer protein. J. Lipid Res. 39, 2021–2030 [PubMed] [Google Scholar]
- 11. Jauhiainen M., Metso J., Pahlman R., Blomqvist S., van Tol A., Ehnholm C. (1993) Human plasma phospholipid transfer protein causes high density lipoprotein conversion. J. Biol. Chem. 268, 4032–4036 [PubMed] [Google Scholar]
- 12. Huuskonen J., Olkkonen V. M., Jauhiainen M., Ehnholm C. (2001) The impact of phospholipid transfer protein (PLTP) on HDL metabolism. Atherosclerosis 155, 269–281 [DOI] [PubMed] [Google Scholar]
- 13. Albers J. J., Vuletic S., Cheung M. C. (2012) Role of plasma phospholipid transfer protein in lipid and lipoprotein metabolism. Biochim. Biophys. Acta 1821, 345–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jänis M. T., Metso J., Lankinen H., Strandin T., Olkkonen V. M., Rye K. A., Jauhiainen M., Ehnholm C. (2005) Apolipoprotein E activates the low-activity form of human phospholipid transfer protein. Biochem. Biophys. Res. Commun. 331, 333–340 [DOI] [PubMed] [Google Scholar]
- 15. Robciuc M. R., Metso J., Sima A., Ehnholm C., Jauhiainen M. (2010) Human apoA-I increases macrophage foam cell derived PLTP activity without affecting the PLTP mass. Lipids Health Dis. 9, 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Laffitte B. A., Joseph S. B., Chen M., Castrillo A., Repa J., Wilpitz D., Mangelsdorf D., Tontonoz P. (2003) The phospholipid transfer protein gene is a liver X receptor target expressed by macrophages in atherosclerotic lesions. Mol. Cell. Biol. 23, 2182–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cao G., Beyer T. P., Yang X. P., Schmidt R. J., Zhang Y., Bensch W. R., Kauffman R. F., Gao H., Ryan T. P., Liang Y., Eacho P. I., Jiang X. C. (2002) Phospholipid transfer protein is regulated by liver X receptors in vivo. J. Biol. Chem. 277, 39561–39565 [DOI] [PubMed] [Google Scholar]
- 18. Tu A. Y., Albers J. J. (2001) Glucose regulates the transcription of human genes relevant to HDL metabolism: responsive elements for peroxisome proliferator-activated receptor are involved in the regulation of phospholipid transfer protein. Diabetes 50, 1851–1856 [DOI] [PubMed] [Google Scholar]
- 19. Tzotzas T., Desrumaux C., Lagrost L. (2009) Plasma phospholipid transfer protein (PLTP): review of an emerging cardiometabolic risk factor. Obes. Rev. 10, 403–411 [DOI] [PubMed] [Google Scholar]
- 20. Riemens S. C., van Tol A., Sluiter W. J., Dullaart R. P. (1998) Plasma phospholipid transfer protein activity is related to insulin resistance: impaired acute lowering by insulin in obese type II diabetic patients. Diabetologia 41, 929–934 [DOI] [PubMed] [Google Scholar]
- 21. Vuletic S., Jin L. W., Marcovina S. M., Peskind E. R., Moller T., Albers J. J. (2003) Widespread distribution of PLTP in human CNS: evidence for PLTP synthesis by glia and neurons, and increased levels in Alzheimer's disease. J. Lipid Res. 44, 1113–1123 [DOI] [PubMed] [Google Scholar]
- 22. Vuletic S., Peskind E. R., Marcovina S. M., Quinn J. F., Cheung M. C., Kennedy H., Kaye J. A., Jin L. W., Albers J. J. (2005) Reduced CSF PLTP activity in Alzheimer's disease and other neurologic diseases; PLTP induces ApoE secretion in primary human astrocytes in vitro. J. Neurosci. Res. 80, 406–413 [DOI] [PubMed] [Google Scholar]
- 23. Desrumaux C., Risold P. Y., Schroeder H., Deckert V., Masson D., Athias A., Laplanche H., Le Guern N., Blache D., Jiang X. C., Tall A. R., Desor D., Lagrost L. (2005) Phospholipid transfer protein (PLTP) deficiency reduces brain vitamin E content and increases anxiety in mice. FASEB J. 19, 296–307 [DOI] [PubMed] [Google Scholar]
- 24. Dong W., Albers J. J., Vuletic S. (2009) Phospholipid transfer protein reduces phosphorylation of tau in human neuronal cells. J. Neurosci. Res. 87, 3176–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Desrumaux C., Pisoni A., Meunier J., Deckert V., Athias A., Perrier V., Villard V., Lagrost L., Verdier J. M., Maurice T. (2013) Increased amyloid-β peptide-induced memory deficits in phospholipid transfer protein (PLTP) gene knockout mice. Neuropsychopharmacology 38, 817–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Franke H., Galla H., Beuckmann C. T. (2000) Primary cultures of brain microvessel endothelial cells: a valid and flexible model to study drug transport through the blood-brain barrier in vitro. Brain Res. Brain Res. Protoc. 5, 248–256 [DOI] [PubMed] [Google Scholar]
- 27. Larionov A., Krause A., Miller W. (2005) A standard curve based method for relative real time PCR data processing. BMC Bioinformatics 6, 62–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jauhiainen M., Ehnholm C. (2005) Determination of human plasma phospholipid transfer protein mass and activity. Methods 36, 97–101 [DOI] [PubMed] [Google Scholar]
- 29. Scholler M., Wadsack C., Lang I., Etschmaier K., Schweinzer C., Marsche G., Dieber-Rotheneder M., Desoye G., Panzenboeck U. (2012) Phospholipid transfer protein in the placental endothelium is affected by gestational diabetes mellitus. J. Clin. Endocrinol. Metab. 97, 437–445 [DOI] [PubMed] [Google Scholar]
- 30. Sattler W., Mohr D., Stocker R. (1994) Rapid isolation of lipoproteins and assessment of their peroxidation by high performance liquid chromatography postcolumn chemiluminescence. Methods Enzymol. 233, 469–489 [DOI] [PubMed] [Google Scholar]
- 31. Kratzer I., Wernig K., Panzenboeck U., Bernhart E., Reicher H., Wronski R., Windisch M., Hammer A., Malle E., Zimmer A., Sattler W. (2007) Apolipoprotein A-I coating of protamine-oligonucleotide nanoparticles increases particle uptake and transcytosis in an in vitro model of the blood-brain barrier. J. Control. Release 117, 301–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marques-Vidal P., Jauhiainen M., Metso J., Ehnholm C. (1997) Transformation of high density lipoprotein 2 particles by hepatic lipase and phospholipid transfer protein. Atherosclerosis 133, 87–95 [DOI] [PubMed] [Google Scholar]
- 33. Ashida Y., Takagi A., Ikeda Y. (1999) Improved method for non-denaturing polyacrylamide gradient gel electrophoresis for detection of small-sized LDL produced during postprandial hypertriglyceridaemia. Scand. J. Clin. Lab. Invest. 59, 663–670 [DOI] [PubMed] [Google Scholar]
- 34. van Haperen R., van Tol A., Vermeulen P., Jauhiainen M., van Gent T., van den Berg P., Ehnholm S., Grosveld F., van der Kamp A., de Crom R. (2000) Human plasma phospholipid transfer protein increases the antiatherogenic potential of high density lipoproteins in transgenic mice. Arterioscler. Thromb. Vasc. Biol. 20, 1082–1088 [DOI] [PubMed] [Google Scholar]
- 35. Stefulj J., Panzenboeck U., Becker T., Hirschmugl B., Schweinzer C., Lang I., Marsche G., Sadjak A., Lang U., Desoye G., Wadsack C. (2009) Human endothelial cells of the placental barrier efficiently deliver cholesterol to the fetal circulation via ABCA1 and ABCG1. Circ. Res. 104, 600–608 [DOI] [PubMed] [Google Scholar]
- 36. Tu A. Y., Nishida H. I., Nishida T. (1993) High density lipoprotein conversion mediated by human plasma phospholipid transfer protein. J. Biol. Chem. 268, 23098–23105 [PubMed] [Google Scholar]
- 37. Rye K. A., Barter P. J. (2004) Formation and metabolism of preβ-migrating, lipid-poor apolipoprotein A-I. Arterioscler. Thromb. Vasc. Biol. 24, 421–428 [DOI] [PubMed] [Google Scholar]
- 38. Settasatian N., Duong M., Curtiss L. K., Ehnholm C., Jauhiainen M., Huuskonen J., Rye K. A. (2001) The mechanism of the remodeling of high density lipoproteins by phospholipid transfer protein. J. Biol. Chem. 276, 26898–26905 [DOI] [PubMed] [Google Scholar]
- 39. Singh-Manoux A., Gimeno D., Kivimaki M., Brunner E., Marmot M. G. (2008) Low HDL cholesterol is a risk factor for deficit and decline in memory in midlife: the Whitehall II study. Arterioscler. Thromb. Vasc. Biol. 28, 1556–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reitz C., Tang M. X., Schupf N., Manly J. J., Mayeux R., Luchsinger J. A. (2010) Association of higher levels of high density lipoprotein cholesterol in elderly individuals and lower risk of late-onset Alzheimer disease. Arch. Neurol. 67, 1491–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scholler M., Wadsack C., Metso J., Chirackal Manavalan A. P., Sreckovic I., Schweinzer C., Hiden U., Jauhiainen M., Desoye G., Panzenboeck U. (2012) Phospholipid transfer protein is differentially expressed in human arterial and venous placental endothelial cells and enhances cholesterol efflux to fetal HDL. J. Clin. Endocr. Metab. 97, 2466–2474 [DOI] [PubMed] [Google Scholar]
- 42. Vuletic S., Dong W., Wolfbauer G., Day J. R., Albers J. J. (2009) PLTP is present in the nucleus, and its nuclear export is CRM1-dependent. Biochim. Biophys. Acta 1793, 584–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Norata G. D., Ongari M., Uboldi P., Pellegatta F., Catapano A. L. (2005) Liver X receptor and retinoic X receptor agonists modulate the expression of genes involved in lipid metabolism in human endothelial cells. Int. J. Mol. Med. 16, 717–722 [PubMed] [Google Scholar]
- 44. Vikstedt R., Metso J., Hakala J., Olkkonen V. M., Ehnholm C., Jauhiainen M. (2007) Cholesterol efflux from macrophage foam cells is enhanced by active phospholipid transfer protein through generation of two types of acceptor particles. Biochemistry 46, 11979–11986 [DOI] [PubMed] [Google Scholar]
- 45. Yazdanyar A., Yeang C., Jiang X. C. (2011) Role of phospholipid transfer protein in high density lipoprotein-mediated reverse cholesterol transport. Curr. Atheroscler. Rep. 13, 242–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kang J., Rivest S. (2012) Lipid metabolism and neuroinflammation in Alzheimer's disease: A role for liver X receptors. Endocr. Rev. 33, 715–746 [DOI] [PubMed] [Google Scholar]
- 47. Oram J. F., Wolfbauer G., Vaughan A. M., Tang C., Albers J. J. (2003) Phospholipid transfer protein interacts with and stabilizes ATP-binding cassette transporter A1 and enhances cholesterol efflux from cells. J. Biol. Chem. 278, 52379–52385 [DOI] [PubMed] [Google Scholar]
- 48. Oram J. F., Wolfbauer G., Tang C., Davidson W. S., Albers J. J. (2008) An amphipathic helical region of the N-terminal barrel of phospholipid transfer protein is critical for ABCA1-dependent cholesterol efflux. J. Biol. Chem. 283, 11541–11549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Krimbou L., Hajj Hassan H., Blain S., Rashid S., Denis M., Marcil M., Genest J. (2005) Biogenesis and speciation of nascent apoA-I-containing particles in various cell lines. J. Lipid Res. 46, 1668–1677 [DOI] [PubMed] [Google Scholar]
- 50. Koch S., Donarski N., Goetze K., Kreckel M., Stuerenburg H. J., Buhmann C., Beisiegel U. (2001) Characterization of four lipoprotein classes in human cerebrospinal fluid. J. Lipid Res. 42, 1143–1151 [PubMed] [Google Scholar]
- 51. Kennedy M. A., Barrera G. C., Nakamura K., Baldan A., Tarr P., Fishbein M. C., Frank J., Francone O. L., Edwards P. A. (2005) ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 1, 21–31 [DOI] [PubMed] [Google Scholar]
- 52. Olesen O. F., Dagø L. (2000) High density lipoprotein inhibits assembly of amyloid β-peptides into fibrils. Biochem. Biophys. Res. Commun. 270, 62–66 [DOI] [PubMed] [Google Scholar]
- 53. Kontush A., Chapman M. J. (2008) HDL: close to our memories? Arterioscler. Thromb. Vasc. Biol. 28, 1418–1420 [DOI] [PubMed] [Google Scholar]
- 54. Schweinzer C., Kober A., Lang I., Etschmaier K., Scholler M., Kresse A., Sattler W., Panzenboeck U. (2011) Processing of endogenous AβPP in blood-brain barrier endothelial cells is modulated by liver X receptor agonists and altered cellular cholesterol homeostasis. J. Alzheimers Dis. 27, 341–360 [DOI] [PubMed] [Google Scholar]
- 55. Wang H., Yu Y., Chen W., Cui Y., Luo T., Ma J., Jiang XC., Qin S. (2013) PLTP deficiency impairs learning and memory capabilities partially due to alteration of amyloid-β metabolism in old mice. J. Alzheimers Dis. 10.3233/JAD-130812 [DOI] [PubMed] [Google Scholar]
- 56. Elliott D. A., Weickert C. S., Garner B. (2010) Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clin. Lipidol. 51, 555–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yu C., Youmans K. L., LaDu M. J. (2010) Proposed mechanism for lipoprotein remodelling in the brain. Biochim. Biophys. Acta 1801, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Beffert U., Danik M., Krzywkowski P., Ramassamy C., Berrada F., Poirier J. (1998) The neurobiology of apolipoproteins and their receptors in the CNS and Alzheimer's disease. Brain Res. Rev. 27, 119–142 [DOI] [PubMed] [Google Scholar]
- 59. Wahrle S. E., Jiang H., Parsadanian M., Kim J., Li A., Knoten A., Jain S., Hirsch-Reinshagen V., Wellington C. L., Bales K. R., Paul S. M., Holtzman D. M. (2008) Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Invest. 118, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kontush A. (2004) Apolipoprotein Aβ: black sheep in a good family. Brain Pathol. 14, 433–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bowman G. L., Kaye J. A., Quinn J. F. (2012) Dyslipidemia and blood-brain barrier integrity in Alzheimer's disease. Curr. Gerontol. Geriatr. Res. 2012:184042. [DOI] [PMC free article] [PubMed] [Google Scholar]