

Background: Initial cleavage by chymotrypsin C regulates degradation of human cationic trypsin.
Results: Cleavage is reversible and favors calcium-dependent bond formation in trypsin, but not in trypsinogen.
Conclusion: Trypsin resistance to degradation derives from the regulated thermodynamic stability of a specific peptide bond that is responsive to physiological environment.
Significance: This new paradigm explains the robustness of trypsin functioning in the protease-rich intestinal milieu.
Keywords: Calcium-binding Proteins, Pancreas, Protein Degradation, Proteolytic Enzymes, Serine Protease, Chymotrypsin C, Proteolytic Stability, Trypsin, Trypsinogen, Zymogen Activation
Abstract
Human cationic trypsinogen, precursor of the digestive enzyme trypsin, can be rapidly degraded to protect the pancreas when pathological conditions threaten, while trypsin itself is impressively resistant to degradation. For either form, degradation is controlled by two necessary initial proteolytic events: cleavage of the Leu81–Glu82 peptide bond by chymotrypsin C (CTRC) and cleavage of the Arg122–Val123 peptide bond by trypsin. Here we demonstrate that the Leu81–Glu82 peptide bond of human cationic trypsin, but not trypsinogen, is thermodynamically stable, such that cleavage by CTRC leads to an equilibrium mixture containing 10% cleaved and 90% uncleaved trypsin. When cleaved trypsin was incubated with CTRC, the Leu81–Glu82 peptide bond was re-synthesized to establish the same equilibrium. The thermodynamic stability of the scissile peptide bond was not dependent on CTRC or Leu-81, as re-synthesis was also accomplished by other proteases acting on mutated cationic trypsin. The Leu81–Glu82 peptide bond is located within a calcium binding loop, and thermodynamic stability of the bond was strictly dependent on calcium and on the calcium-coordinated residue Glu-85. Trypsinolytic cleavage of the Arg122–Val123 site was also delayed in trypsin relative to trypsinogen in a calcium-dependent manner, but for this bond cleavage was modulated by kinetic rather than thermodynamic control. Our results reveal that the trypsinogen to trypsin conformational switch modulates cleavage susceptibility of nick sites by altering both the thermodynamics and kinetics of cleavage to protect human cationic trypsin from premature degradation.
Introduction
Proteins are the versatile molecular workhorses of biology; they catalyze chemical reactions, perform complex mechanical tasks, and also comprise much of the structural scaffolding of living organisms. In the animal kingdom, essential amino acid building blocks for protein synthesis are derived from digestion of protein-rich foods, or through the salvage and degradation of cellular and secreted proteins that have passed their useful life span (1). Paradoxically, the proteolytic enzymes evolved to break proteins down into their component amino acids are themselves proteins. How does such a protein, residing in a compartment filled with other digestive proteases, remain intact, and functionally active for long enough to carry out its function?
Trypsin, an archetypal example of a digestive enzyme, must function in the digestive juice of the small intestine, comprised of a mixture of active digestive proteases at concentrations that may exceed one milligram per milliliter (2, 3). It retains its structure and activity for long enough to completely digest dietary protein in the duodenum, being only very slowly inactivated during its intestinal transit. In the terminal ileum trypsin retains about 10–20% of the activity measured in the duodenum; this gradual degradation is likely to serve the physiological purpose of mitigating loss of digestive enzyme protein through the stool (2, 4, 5). In the case of human cationic trypsin, the slow degradation in the digestive tract is ultimately initiated by sequential cleavage at two nick sites: Arg122–Val1234 is cleaved by trypsin itself and Leu81–Glu82 by the regulatory protease chymotrypsin C (CTRC)5 (6).
Trypsin is produced in the pancreas as trypsinogen, an inactive precursor possessing a short N-terminal activation peptide. Trypsinogen is normally activated in the duodenum upon cleavage by enteropeptidase, but under pathological conditions can be susceptible to autoactivation in the pancreas, where in turn it will activate other digestive zymogens including CTRC (7). In this event, degradation of remaining trypsinogen in the pancreas serves as an important protective mechanism to limit the scope of the damage. Trypsinogen degradation is initiated by CTRC and trypsin cleavage at the same two nick sites, but here will proceed much more rapidly to completion.
In this study, we explored the basis for control of trypsin/trypsinogen proteolytic stability, and identified a novel mechanism by which the subtle trypsinogen to trypsin conformational switch modulates nick site cleavage susceptibility. Efficient digestion of a protein by a protease is generally understood to require (a) colocalization of protein and protease, (b) an amino acid sequence motif within the protein matching the specificity requirement of the protease, and (c) physical accessibility and sufficient flexibility of the cleavage site (8–10). Remarkably, we find that alterations in protein flexibility can modulate not only the accessibility of the nick site or the kinetics of proteolysis, but also the fundamental thermodynamic stability of the nick site peptide bond. Whereas in trypsinogen the cleavage of Leu81–Glu82 by CTRC proceeds rapidly to completion, in trypsin the enhanced thermodynamic stability of this same peptide bond results in a cleavage/re-ligation equilibrium in which the intact form predominates by a factor of 10. This mechanism, in which trypsin activity is temporarily prolonged in the presence of degrading enzymes by thermodynamic stability of a peptide bond, may be a widespread contributor to protein stability in vivo that has previously remained unrecognized.
MATERIALS AND METHODS
Plasmid Construction and Mutagenesis
The pTrapT7 expression plasmid harboring the coding sequence for human cationic trypsinogen (PRSS1) and the pcDNA3.1(−) expression plasmids containing the human chymotrypsinogen C (CTRC) and proelastase 3B (CELA3B) coding sequences with a C-terminal 10His tag were previously described (6, 11–13). Mutations in trypsinogen were generated by overlap extension PCR mutagenesis and cloned into the pTrapT7 plasmid. With the exception of the K23Q mutant, all trypsinogen constructs harbored the S200A-inactivating mutation.
Expression, Refolding, Purification, and Activation of Human Cationic Trypsinogen
Wild-type and mutant trypsinogens were expressed in Escherichia coli BL21(DE3) as cytosolic inclusion bodies. In vitro refolding and purification with ecotin affinity chromatography were carried out as described previously (11, 12, 14, 15). Trypsinogen concentrations were estimated from ultraviolet absorbance at 280 nm using the extinction coefficient 37,525 m−1cm−1. Trypsinogen (8–10 μm) was activated with 0.4 μg/ml final concentration of recombinant human enteropeptidase (R&D Systems, Minneapolis, MN) in 0.1 m Tris-HCl (pH 8.0) at 37 °C for 45 min.
Expression and Purification of Chymotrypsinogen C and Proelastase 3B
Zymogens for human CTRC and CELA3B were expressed with transient transfection of HEK 293T cells. The C-terminally His-tagged proenzymes were purified from 200 ml of conditioned medium using Ni-NTA superflow cartridge (Qiagen, Valencia, CA) (13). The proenzymes were activated using immobilized bovine trypsin (Pierce/Thermo Fisher Scientific) in 0.1 m Tris-HCl, pH 8.0, 0.05% Tween-20, and the trypsin beads were removed with centrifugation at 2500 × g, 22 °C, for 5 min. The concentration of active proteases was determined by active site titration against ecotin (16).
Cleavage and Re-synthesis Assays
Cleavage reactions were carried out using 2 μm human cationic trypsin or trypsinogen containing given mutations and 100 nm cleaving protease (CTRC, cationic trypsin or CELA3B), as indicated. Incubations were performed at 37 °C in 0.1 m Tris-HCl (pH 8.0) and 15 μm, 100 μm or 1 mm CaCl2, as indicated. To study peptide bond re-synthesis in trypsin, given mutants were first pre-treated with the cleaving protease at 37 °C in 0.1 m Tris-HCl, pH 8.0 and 2 mm K-EDTA to achieve nearly complete cleavage of the scissile peptide bond. For the re-synthesis of the Leu81–Glu82 peptide bond, S200A-trypsin was pre-cleaved with 25 nm CTRC for 1 h and re-synthesis was then initiated by adding 3 mm CaCl2 and 100 nm CTRC. For the re-synthesis of the Arg81–Glu82 peptide bond, the L81R,R122A,S200A trypsin mutant was pre-treated with 125 nm cationic trypsin for 3 h and re-synthesis was then initiated by adding 3 mm CaCl2. For the re-synthesis of the Ala81–Glu82 peptide bond, the L81A,N84A,S200A trypsin mutant was pre-treated with 125 nm CELA3B for 1 h and re-synthesis was initiated by adding 3 mm CaCl2. To study re-synthesis of the Leu81–Glu82 peptide bond in S200A-trypsinogen, samples were pre-cleaved at 37 °C in 0.1 m Tris-HCl (pH 8.0) with 25 nm CTRC for 15 min and re-synthesis was initiated by adding 1 mm CaCl2 and 100 nm CTRC.
Gel Electrophoresis and Densitometry
Samples (75 μl) were precipitated with trichloroacetic acid (10% final concentration), the precipitate was recovered by centrifugation, dissolved in 15 μl of Laemmli sample buffer containing 100 mm dithiothreitol (final concentration), and heat-denatured at 95 °C for 5 min. Electrophoretic separation was performed on 15% SDS-PAGE mini gels in standard Tris-glycine buffer. Gels were stained with Coomassie Brilliant Blue R. Quantitation of bands was carried out with the Quantity One 4.6.9 (Bio-Rad) software. Rectangles were drawn around each band of interest, and an identical rectangle was used in each lane for background subtraction.
When cleavage and re-synthesis of the Leu81–Glu82 peptide bond was studied, the intensity of the uncleaved, intact trypsin band (24 kDa, migrating at ∼30 kDa) was expressed as percent of the total intensity of the cleaved and uncleaved trypsin bands. In this calculation only the larger, C-terminal cleavage fragment (18 kDa, migrating at ∼20 kDa), was considered and the small N-terminal fragment (6.5 kDa, migrating at ∼9 kDa) was ignored because its staining intensity was highly variable. This approach was validated by running samples under non-reducing conditions, when the cleaved trypsin formed a single band on gels migrating slower than intact, uncleaved trypsin. Results of the densitometric evaluation of the cleavage reactions were essentially identical when samples were analyzed under reducing or non-reducing conditions. Cleavage of the Arg122–Val123 peptide bond in trypsinogen resulted in two fragments (12 and 13 kDa) that co-migrated on reducing gels as a single band at ∼15 kDa. Densitometry was performed by measuring the intensity of the uncleaved, intact trypsinogen band (25 kDa, migrating at ∼31 kDa) as percent of the total intensity of the cleaved and uncleaved bands. In trypsin, cleavage of the Arg122–Val123 peptide bond generated two cleavage fragments (11 and 13 kDa, migrating at ∼13 and ∼15 kDa on reducing gels, respectively). The intensity of the uncleaved, intact trypsin band was determined by densitometry as percent of the total intensity of the intact band plus the two cleavage product bands.
Structure Figures
Human cationic trypsin structures were rendered with PYMOL 1.3 software using the coordinates of Protein Data Bank file 2RA3 (17). The mutated His-122 found in this structure was replaced with the native Arg in the model.
RESULTS
Cleavage and Re-synthesis of the Leu81–Glu82 Peptide Bond in Human Cationic Trypsin by Chymotrypsin C
Previously we discovered that degradation of human cationic trypsin is regulated by CTRC cleavage at the Leu81–Glu82 bond (Fig. 1A), which is necessary for subsequent autolytic breakdown (6). To selectively study the kinetics of this initial cleavage event without interference from autolysis, the catalytic serine residue in human cationic trypsin was mutated to alanine and constructs were made in this inactive S200A background. Incubation of cationic trypsin with a catalytic amount of CTRC in 1 mm calcium initially resulted in the appearance of products formed by cleavage at the Leu81–Glu82 peptide bond, but surprisingly the reaction did not proceed to completion; minimal (<10%) cleavage occurred within 10 min but no further progress was observed over the course of one hour (Fig. 1, B and D, left panel). We hypothesized that a cleavage equilibrium might be established with about 90% intact and 10% cleaved trypsin forms present, corresponding to a Khyd = [cleaved]/[intact] of ∼0.1.
FIGURE 1.

Cleavage and re-synthesis of the Leu81–Glu82 peptide bond by CTRC. A, ribbon diagram of human cationic trypsin (Protein Data Bank ID: 2RA3 (17)) showing the position of the Leu81–Glu82 cleavage site (red arrow) in the calcium binding loop (green) relative to the active site catalytic triad of Ser-200 (here mutated to Ala), His-63, and Asp-107 (red) and the Arg-122 loop (blue). The active site and substrate binding site are shaped in part by the activation domain (yellow), comprised of the N terminus and three surface loops that are unstructured in trypsinogen; N-terminal Ile-24 forms an H-bond (dotted line) with Asp-199 upon activation. Structure was rendered with PyMOL 1.3. B-C, cleavage and re-synthesis of human cationic trypsin (Tr) and trypsinogen (Tg) containing the S200A mutation were performed in 1 mm CaCl2, as described in “Materials and Methods.” At the indicated time points, samples were precipitated with trichloroacetic acid and analyzed by SDS-PAGE and Coomassie blue staining, as detailed in “Materials and Methods.” The asterisk indicates a secondary cleavage product of trypsinogen. Representative gels of two to four experiments are shown. D, densitometric quantitation of the intact trypsin and trypsinogen bands. Error bars were omitted for clarity; the error was within 8% of the mean.
To test our hypothesis that this was a true thermodynamic equilibrium, we asked whether CTRC could catalyze re-synthesis of the Leu81–Glu82 peptide bond. We generated two-chain cationic trypsin almost fully cleaved at Leu-81 by taking advantage of the empirical observation that trypsin is more readily cleaved by CTRC in the absence of calcium and the presence of 2 mm EDTA. When Leu-81-cleaved two-chain trypsin was supplemented with 3 mm calcium (i.e. 1 mm free calcium after chelating EDTA) and incubated with CTRC, intact, full-length trypsin accumulated as a function of time indicating re-synthesis of the Leu81–Glu82 peptide bond (Fig. 1, B and D, right panel). Importantly, the cleavage equilibrium (Khyd ∼0.1) was identical to that obtained in the forward cleavage reaction. Inhibition of CTRC by a high-affinity peptide inhibitor prevented re-formation of intact trypsin, confirming that re-synthesis of the Leu81–Glu82 peptide bond was catalyzed by CTRC (Fig. 2).
FIGURE 2.
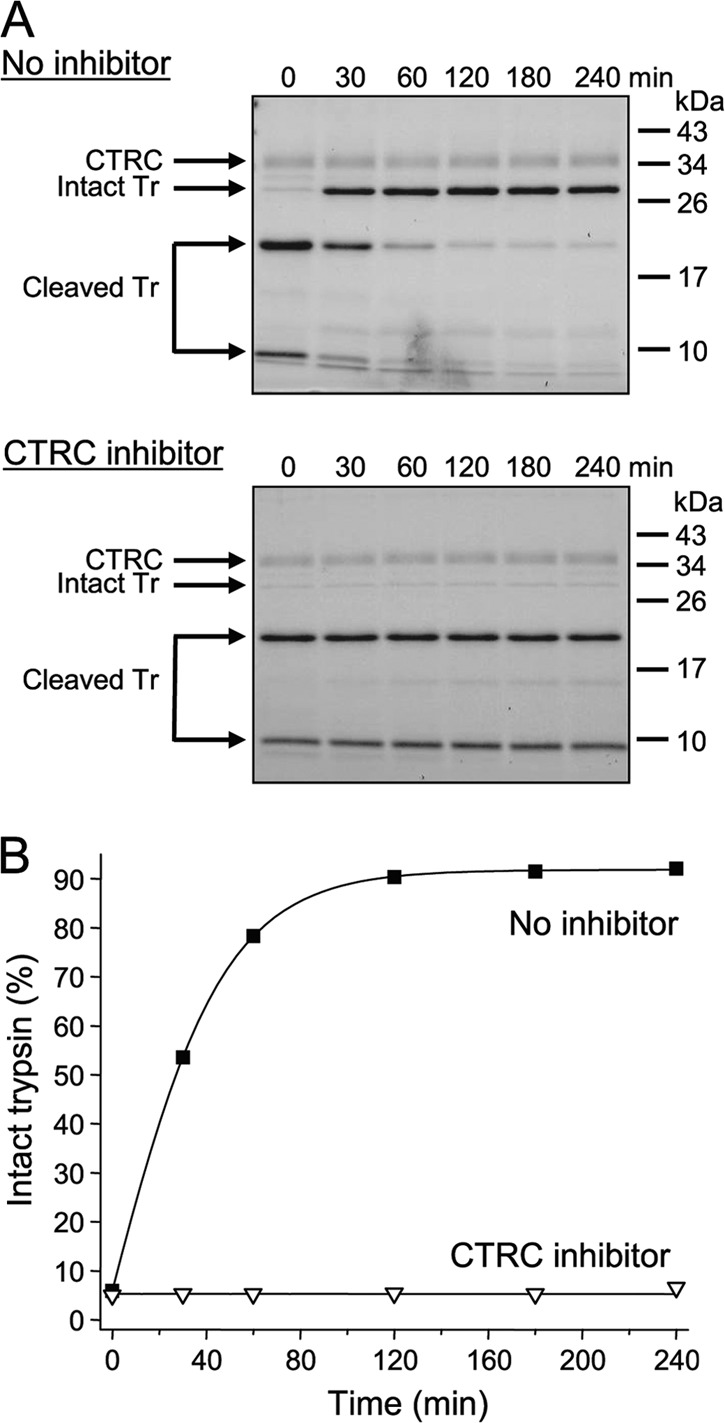
Effect of a CTRC inhibitor on the re-synthesis of the Leu81–Glu82 peptide bond by human CTRC. The re-synthesis experiment was performed with S200A-trypsin (Tr) as described in “Materials and Methods,” in the absence or presence of 500 nm CTRC inhibitor C4. This is a phage-display selected variant of the Schistocerca gregaria proteinase inhibitor-2 (SGPI-2), which inhibits human CTRC with a Ki of 20 pm. See Ref. (16) for details. A, representative gels of two experiments are shown. B, densitometric quantitation of the intact trypsin band. Error bars were omitted for clarity; the error was within 5% of the mean.
Activation of trypsin from its precursor trypsinogen involves formation of an intramolecular salt bridge by the new N terminus, which results in structural stabilization of several loops that are disordered in trypsinogen, termed the “activation domain” (Fig. 1A). Although the Leu81–Glu82 bond lies within the calcium binding loop and is not part of the activation domain, we reasoned that its positioning adjacent to the activation domain might impact the thermodynamic stability of the bond. To determine whether CTRC cleavage of trypsinogen is characterized by a thermodynamic equilibrium similar to that of trypsin, we repeated our proteolysis experiments using cationic trypsinogen as the substrate. By contrast with trypsin, the Leu81–Glu82 peptide bond in trypsinogen was fully cleaved by CTRC within 40 min and the primary cleavage products were further degraded. No re-synthesis of the Leu81–Glu82 peptide bond was detectable with Leu-81-cleaved two-chain cationic trypsinogen, which was also rapidly degraded by CTRC (Fig. 1, C and D). This observation reveals a novel mechanism of biological regulation that to our knowledge is unprecedented: the fundamental thermodynamic stability of a peptide bond is altered by the trypsinogen to trypsin transition, such that thermodynamic control favors cleavage and subsequent degradation for the zymogen versus stability for the active enzyme.
The Cleavage Equilibrium of the Leu81–Glu82 Peptide Bond Is Independent of Leu-81 and Chymotrypsin C
Like other true catalysts, enzymes do not alter the relative thermodynamic stability of their substrates and products, and so do not shift chemical equilibria, but simply accelerate a chemical interconversion. We therefore anticipated that the high thermodynamic stability of the Leu81–Glu82 peptide bond in trypsin should be determined by the structure of the protein in the vicinity of the calcium binding loop and not by the cleaving protease (i.e. CTRC). Because the side-chain of Leu-81 points toward the solvent and does not participate in stabilizing interactions with neighboring amino acid residues, Leu-81 per se was predicted to be relatively unimportant for determining the cleavage equilibrium. To test these predictions, we mutated Leu-81 to Arg (L81R) in the inactive S200A trypsin background and carried out proteolysis experiments using a catalytic amount of active trypsin as the cleaving protease (Fig. 3A). To prevent tryptic cleavage at the Arg122–Val123 peptide bond, the construct also carried the R122A mutation (L81R,R122A,S200A). Although trypsin-mediated cleavage and re-synthesis of the Arg81–Glu82 peptide bond proceeded at a much slower rate than the analogous reactions with CTRC and a true equilibrium was not reached, the extrapolated hydrolytic equilibrium (Khyd ∼0.18) was comparable to that observed with CTRC acting on the Leu81–Glu82 peptide bond.
FIGURE 3.

Cleavage and re-synthesis of Leu-81-mutated trypsins by trypsin or elastase. A, cleavage and re-synthesis of the Arg81–Glu82 peptide bond by human cationic trypsin in the L81R,R122A,S200A trypsin (Tr) mutant. B, cleavage and re-synthesis of the Ala81–Glu82 peptide bond by human elastase 3B (CELA3B) in the L81A,N84A,S200A trypsin (Tr) mutant. See text for details. Reactions and densitometric evaluation were performed as given in “Materials and Methods.” Error bars were omitted for clarity; the error was within 7% of the mean. Insets show representative gels of three experiments.
Similar results were obtained when the L81A trypsin mutant was incubated with human elastase 3B (CELA3B). This construct also carried mutation N84A, as previously we found that the L81A,N84A double mutant was cleaved by CELA3B at the Ala81–Glu82 peptide bond much more efficiently than the L81A mutant (18). As shown in Fig. 3B, the Ala81–Glu82 peptide bond in the L81A,N84A,S200A mutant cationic trypsin was cleaved and re-synthesized by CELA3B, although neither reaction reached equilibrium during the time course studied and a Khyd value could not be assigned. More extended time courses resulted in degradation due to secondary cleavages. Taken together, properties of the L81R and L81A trypsin mutants clearly demonstrate that the high thermodynamic stability of the Leu81–Glu82 peptide bond is an inherent property of the protein not determined by Leu-81 or dependent on CTRC as the cleaving protease. The small changes in Khyd observed for the Leu-81 trypsin mutants are likely attributable to small adjustments in the local conformation of the calcium binding loop and an associated impact on stability.
Calcium Binding and a Hydrogen Bond Network Are Essential for the Thermodynamic Stability of the Leu81–Glu82 Peptide Bond
The experiments presented above were performed in 1 mm calcium, as this concentration approximates physiological levels in the pancreatic juice; at this concentration, the calcium site is expected to be fully occupied (6). Examination of the cationic trypsin structure reveals that calcium is coordinated by the carbonyl oxygens of Asn-77 and Val-80 and by the side chain carboxylates of Glu-75 and Glu-85 (Fig. 4A). Binding of calcium to trypsin is expected to limit the backbone conformational flexibility of the calcium binding loop, which may contribute to the high thermodynamic stability of the Leu81–Glu82 peptide bond. Indeed, when the forward cleavage reaction with CTRC was performed in 100 μm and 15 μm calcium concentrations, the Khyd values increased from 0.1 to 0.7 and 2.3, respectively (Fig. 4, B and C), indicating that calcium dissociation dramatically decreases the thermodynamic stability of the Leu81–Glu82 peptide bond, shifting the hydrolysis equilibrium.
FIGURE 4.
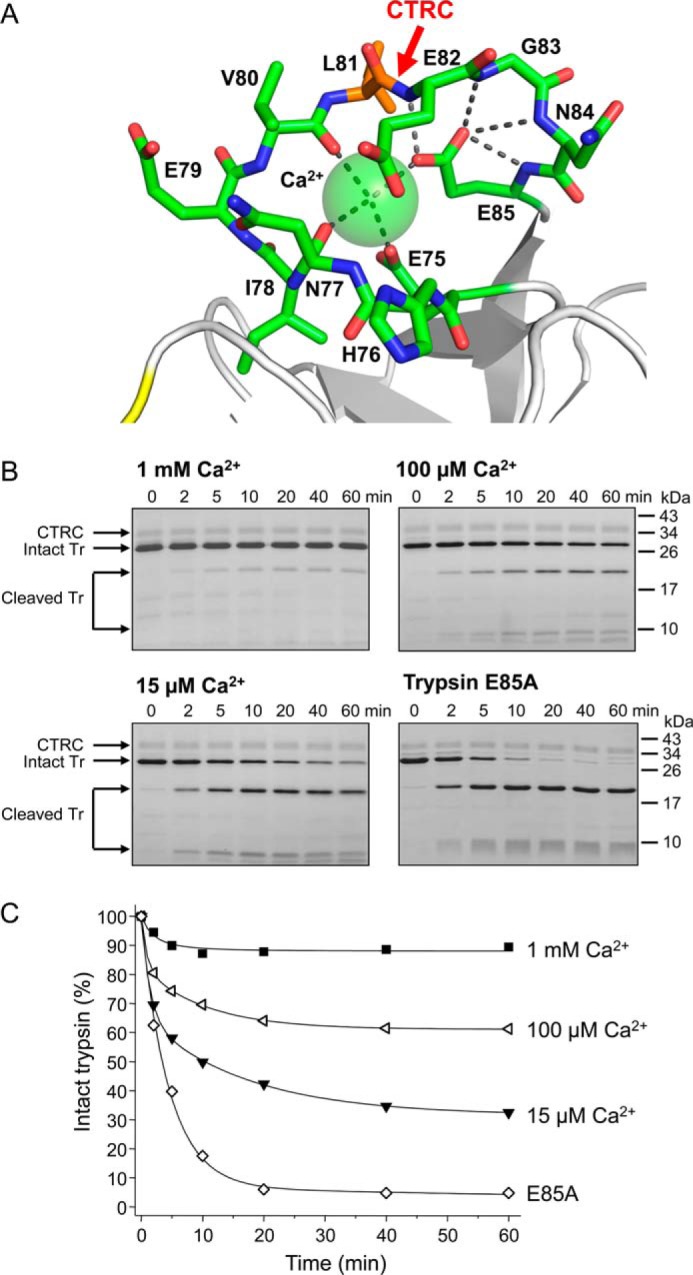
Effect of calcium and mutation E85A on the hydrolysis equilibrium of the Leu81–Glu82 peptide bond. A, structure of the human cationic trypsin calcium-binding loop, with carbon atoms shown in green and nitrogen and oxygen atoms displayed in blue and red, respectively. The bound calcium ion is represented as a sphere. Metal coordination bonds and hydrogen bonds are shown as gray dashed lines. The CTRC cleavage site is indicated (red arrow) between Leu-81 (orange) and Glu-82. B, cleavage of S200A-trypsin (Tr) with human CTRC was performed in 1 mm, 100 μm or 15 μm CaCl2, as described in “Materials and Methods.” Trypsin mutant E85A,S200A was incubated with CTRC in 1 mm CaCl2. At the indicated time points, aliquots were withdrawn, precipitated with trichloroacetic acid, and analyzed by SDS-PAGE, and Coomassie blue staining; representative gels of three experiments are shown. C, cleavage reactions were quantitated with densitometry and plotted. Error bars were omitted for clarity; the error was within 8% of the mean.
A likely explanation for these data is that by stabilizing the loop conformation, the bound calcium limits mobility of the newly formed termini after CTRC cleavage. Cleavage events that occur in more flexible regions of proteins proceed to completion, driven in part by the entropy gain realized due to increased rotational freedom of the newly formed termini (19–21). Importantly, by restricting the mobility of the newly formed termini, the bound calcium ion will limit the gain in entropy upon cleavage, shifting the equilibrium toward the intact form. The structure suggests that calcium binding can directly dampen mobility for the residues of the loop that are N-terminal to Leu-81, by its coordination to Glu-75, Asn-77, and Val-80 (Fig. 4A). By contrast, the immobilization of C-terminal residues Glu-82, Gly-83, and Asn-84 appear to be dependent on bridging interactions of the Glu-85 carboxylate, which in addition to coordinating calcium lies within hydrogen bonding distance of the backbone amide nitrogens of Glu-82, Gly-83, Asn-84, and Glu-85 itself (Fig. 4A). To test the critical nature of these bridging interactions, we mutated Glu-85 to alanine. The Leu81–Glu82 peptide bond in the E85A,S200A mutant trypsin was almost completely cleaved by CTRC, yielding a Khyd value of ∼20 (Fig. 4, B and C).
Kinetic Rather Than Thermodynamic Properties of the Arg122–Val123 Trypsin Nick Site Are Primarily Affected by Calcium Binding
Trypsin and trypsinogen degradation require dual initial proteolytic events at CTRC nick site Leu81–Glu82 and at trypsin nick site Arg122–Val123. The trypsin nick site lies in a loop adjacent to the calcium-binding loop (Fig. 1A), and is conformationally stabilized by a hydrogen bond network (Fig. 5A). We previously found that the Arg122–Val123 peptide bond in human cationic trypsinogen also exhibits appreciable thermodynamic stability (22), although the significance of this phenomenon has remained unknown. Trypsinolytic cleavage of the Arg122–Val123 bond in the physiologically relevant calcium concentration range (0.5–5 mm) led to a cleavage/re-synthesis equilibrium with Khyd ∼0.7 (22). Although not characterized quantitatively, the high thermodynamic stability of the Arg122–Val123 peptide bond was also observed in trypsin (22).
FIGURE 5.
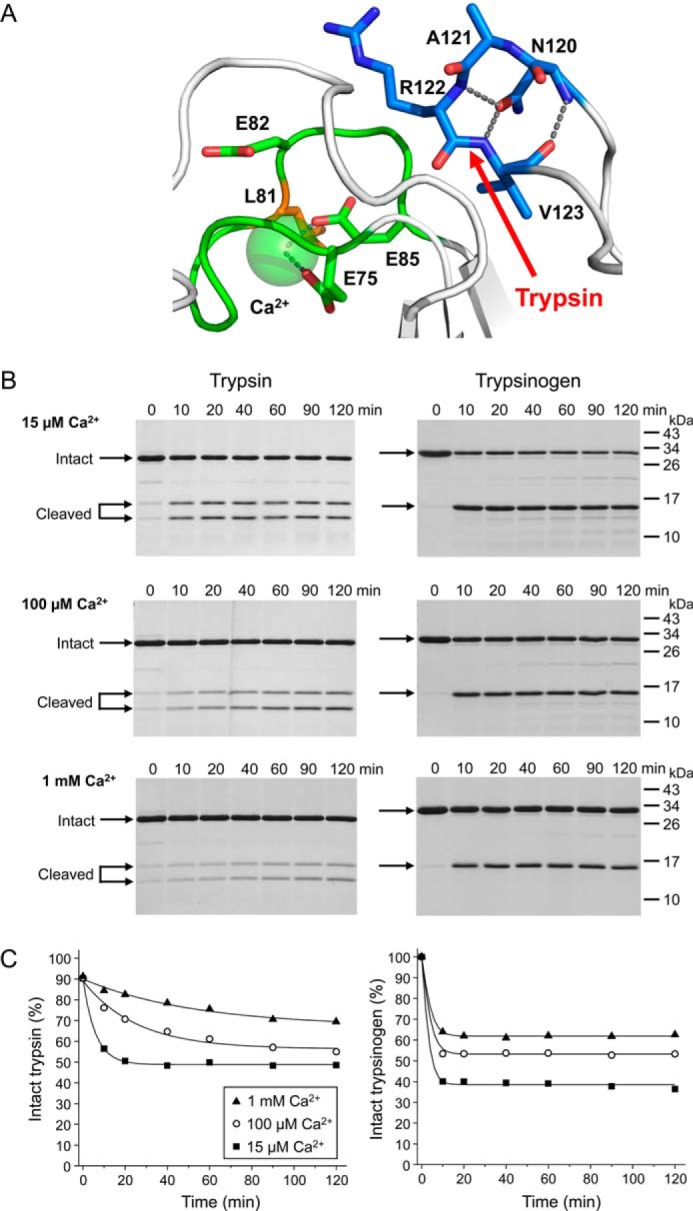
Effect of calcium on the hydrolysis equilibrium of the Arg122–Val123 peptide bond in human cationic trypsin and trypsinogen. A, structure of the human cationic trypsin Arg-122 loop. Carbon atoms of the Arg-122 loop are shown in blue with nitrogen and oxygen atoms displayed in darker blue and red, respectively. The adjacent calcium binding loop is shown in green. Hydrogen bonds and metal coordination bonds are shown as gray dashed lines. The Arg122–Val123 trypsin cleavage site is indicated (red arrow). B, cleavage of S200A-trypsin or K23Q-trypsinogen with human cationic trypsin was performed in 15 μm, 100 μm or 1 mm CaCl2, as described in “Materials and Methods.” At the indicated time points, aliquots were withdrawn, precipitated with trichloroacetic acid and analyzed by SDS-PAGE and Coomassie blue staining. Representative gels from three experiments performed at each CaCl2 concentration are shown. C, densitometric quantitation of the cleavage reactions. Error bars were omitted for clarity; the error was within 5% of the mean.
To determine (a) whether calcium is important for the thermodynamic stability of the Arg122–Val123 peptide bond (as it clearly is for the Leu81–Glu82 peptide bond), and (b) whether the transition of trypsinogen to trypsin modulates the thermodynamic stability of the Arg122–Val123 peptide bond (as it does for the Leu81-Glu82 peptide bond), we measured Khyd values at different calcium concentrations in trypsin and trypsinogen. In these experiments the inactive S200A trypsin mutant and the non-activable K23Q trypsinogen mutant were cleaved with a catalytic quantity of cationic trypsin. The Arg122–Val123 peptide bond was cleaved in trypsin slowly and a true equilibrium was only reached at 15 μm calcium (Khyd 1.0) within the time course studied (Fig. 5, B and C, left panel). The extrapolated hydrolysis equilibrium in 100 μm calcium seemed to approach the same Khyd value, whereas in 1 mm calcium the rate of cleavage was so slow that a cleavage equilibrium could not be estimated reliably. Nonetheless, it appeared that calcium has only a small effect, if any, on the Khyd of the Arg122–Val123 peptide bond in trypsin. Consistent with previous observations (22), calcium had a clear but modest effect on the hydrolysis equilibrium of the Arg122–Val123 peptide bond in trypsinogen; the Khyd values were 1.5, 0.8, and 0.6 in 15 μm, 100 μm, and 1 mm calcium, respectively (Fig. 5, B and C, right panel). Notably, while the rate of cleavage after Arg-122 by trypsin was much faster in trypsinogen than in trypsin, the hydrolysis equilibrium was affected very little. We conclude that unlike the regulatory cleavage at the Leu81–Glu82 peptide bond, which operates under thermodynamic control, the cleavage of the Arg122–Val123 peptide bond is subject to kinetic control. Nevertheless, these divergent regulatory mechanisms have the same functional consequence, in that each enables the relatively facile cleavage and degradation of the zymogen while supporting the stability of the active enzyme.
DISCUSSION
In the present study we demonstrated that the CTRC-sensitive Leu81–Glu82 peptide bond in the calcium binding loop of human cationic trypsin is thermodynamically stable with a hydrolysis equilibrium of 0.1, which is at least 2–4 orders magnitude lower than typical hydrolysis equilibria of proteolytically cleaved peptide bonds in proteins or cyclic peptides (19, 23, 24). The trypsin-sensitive Arg122–Val123 peptide bond also exhibits unusually high thermodynamic stability (22), as well as a very slow rate of proteolysis in the presence of physiological calcium levels. Since cleavage of these two peptide bonds is required for proteolytic inactivation of trypsin, degradation will be slow; productive cleavage at the two peptide bonds at the same time is a rare occurrence, as only 1 in 10 encounters with the Leu81–Glu82 peptide bond and about 1 in 2 encounters with the Arg122–Val123 peptide bond will result in hydrolysis. The proteolytic stability of these peptide bonds represents a defense mechanism of the enzyme, increasing its robustness and functionality in the presence of the high protease concentrations found in the digestive juices, while still allowing its eventual breakdown and recovery of amino acid content.
Importantly, the behavior of both nick sites is sensitive to calcium, albeit through different mechanisms; calcium binding is required for the thermodynamic stability of the Leu81–Glu82 peptide bond, while the kinetics of Arg122–Val123 cleavage are altered when calcium is bound. Thus, higher calcium concentrations can most effectively decrease the rate of trypsin degradation. Free calcium concentrations in the small intestine are estimated to be millimolar, with potentially higher concentrations in the duodenum and proximal intestine where digestive processes are the most active and trypsin stabilization would be the most beneficial (25, 26).
In contrast to the slow intestinal degradation of trypsin, a rapid mechanism for trypsinogen degradation is required in the pancreas. Trypsinogen is normally activated following secretion to the duodenum through cleavage by enteropeptidase, a serine protease localized to the brush-border of enterocytes (27), but trypsinogen can also undergo pathological intra-pancreatic autoactivation, leading to pancreatitis (7). In the pancreas, CTRC-dependent trypsinogen degradation serves a protective function, and mutations in either trypsinogen or CTRC that interfere with this function increase the risk for chronic pancreatitis (13, 28). Thus, it is functionally advantageous for trypsinogen to be vulnerable to CTRC-dependent degradation while trypsin is more resistant. Remarkably, the proteolytic stability of Leu81–Glu82 to CTRC and of Arg122–Val123 to trypsin are both controlled by the trypsinogen to trypsin conformational switch, but via different mechanisms; whereas Arg122–Val123 hydrolysis is limited by kinetic control, the Leu81–Glu82 peptide bond undergoes alteration of its fundamental thermodynamic stability.
It is not immediately obvious exactly how the zymogen to enzyme transition modulates thermodynamic stability of the Leu81–Glu82 peptide bond, but structural analyses and earlier biochemical reports may offer insights. Crystal structures of bovine trypsinogen, which serves as a structural model for understanding the trypsinogen to trypsin transition, show that the boundaries between the activation domain and the structured regions of trypsinogen are sharply delimited (29). Flexibility begins abruptly at the interface between “hinge” residues, most of which are Gly, and adjacent “anchor” residues which maintain the trypsinogen structure. Comparisons of trypsinogen crystal structures with those of trypsins reveal no evidence of significant differences in the conformation or apparent flexibility of the calcium binding loop. Furthermore, for the bovine enzyme it has been shown convincingly that the calcium binding site has very similar affinity for calcium in trypsin and trypsinogen (30). These studies would seem to predict a conformationally stabilized and protease resistant calcium binding loop in trypsinogen as well as trypsin; however, comparable structural and calcium binding data are lacking for the two-chain trypsin and trypsinogen that have been cleaved at Leu81–Glu82, so here we are left to speculate.
In human cationic trypsin, the residues anchoring the 145–155 loop of the activation domain are Trp-144 and Asp-156 (Fig. 6A). These anchor residues form H-bonds with backbone atoms of His-76 and Ile-78, residues that lie within the calcium binding loop and flank the calcium-coordinating Asn-77 (Fig. 6A). It seems plausible that allosteric communication between the activation domain and the calcium binding loop may be mediated through these anchor residues. We speculate that when trypsin is cleaved at Leu81–Glu82, interactions between the calcium binding loop and the anchor residues, which are stabilized by the structured activation domain, may favor the retention of a native-like structure and enhance the thermodynamic stability of the Leu81–Glu82 bond, in turn favoring re-ligation. By contrast, when trypsinogen is cleaved at Leu81-Glu82, the anchor residues are destabilized from both above and below, allowing motions of the activation domain to be propagated into the cleaved calcium binding loop; the result may be loss of structure in the loop and loss of bound calcium, disfavoring re-ligation. A scheme outlining a proposed model is shown in Fig. 6B. In this model, both trypsin and trypsinogen bind to and are stabilized by calcium; in the absence of calcium, both are more susceptible to limited proteolysis. Whereas the cleaved form of trypsin remains competent to bind calcium and forms a calcium-dependent cleavage/re-synthesis equilibrium of the Leu81–Glu82 bond, the cleaved form of trypsinogen cannot efficiently stabilize the protein structure in the vicinity of this bond, and secondary cleavage events outpace re-ligation.
FIGURE 6.
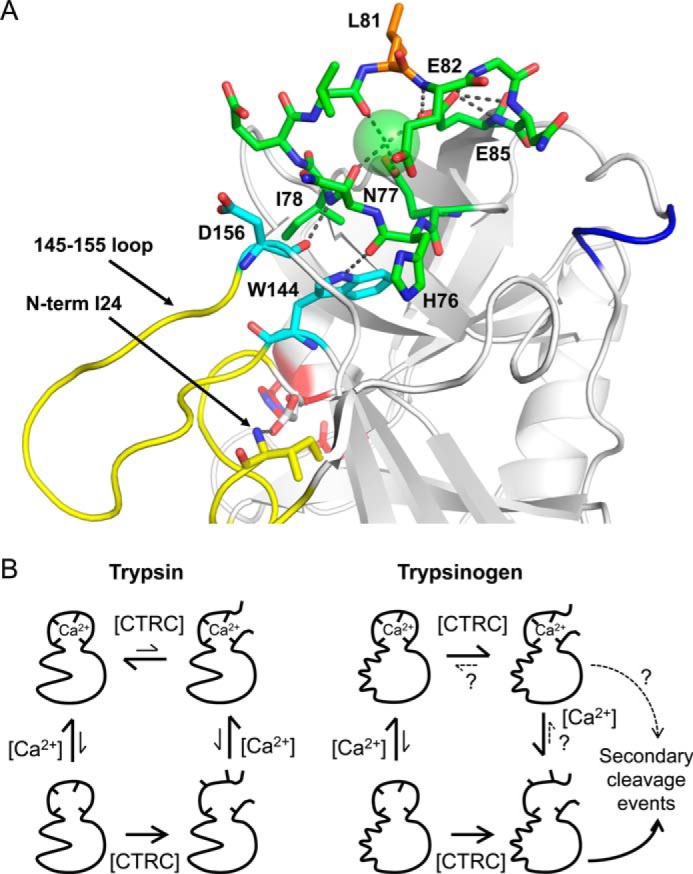
Model for effects of calcium and activation domain structure on Leu81–Glu82 cleavage/re-synthesis equilibrium. A, ribbon diagram of human cationic trypsin showing anchor residues (cyan) linking the activation domain (yellow) to the calcium binding loop (green). The Arg-122 loop is colored blue as in Fig. 1A. The Trp-144 side chain is H-bonded to the His-76 backbone carbonyl, and the Asp-156 backbone carbonyl is H-bonded to the Ile-78 amide nitrogen; in trypsinogen these contacts may allow allosteric transmission of motion from the unstructured zymogen activation domain to the destabilized calcium binding loop following cleavage of Leu81–Glu82 by CTRC (see text). B, proposed scheme consistent with biochemical and structural data. Trypsin and trypsinogen have similar affinities for calcium, binding of which lends a measure of protection from CTRC proteolysis; in the absence of calcium, both zymogen and enzyme become fully cleaved at Leu81–Glu82. Trypsin maintains affinity for calcium in its cleaved form, and forms a calcium-dependent cleavage/re-synthesis equilibrium that thermodynamically favors the intact form. The cleaved form of trypsinogen loses calcium binding affinity and fails to efficiently re-ligate the Leu81–Glu82 bond; competing secondary cleavage events lead to further degradation.
The ability of a thermodynamically stable peptide bond to prolong trypsin activity in the intestinal environment has obvious benefits, raising the question of whether this mechanism has been exploited to advantage in other biological settings. Additional examples may be found in the canonical protease inhibitors. These inhibitors, which follow a “standard mechanism” described by Laskowski (31), possess a thermodynamically stable bond; when bound to a target protease, this “reactive site” bond is ideally positioned for proteolytic attack by the inhibited enzyme (32). Structures of cleaved canonical inhibitors bound to target serine or metalloproteases show that the cleaved termini are structurally stabilized in an orientation favorable to re-ligation (33, 34), as we speculate is also the case for the cleaved Leu81–Glu82 bond of trypsin. Trypsin and the canonical inhibitors represent but a few of the proteins required to function in protease-rich environments, and the tantalizing possibility exists that the proteomes of digestive compartments and organelles may contain many more examples yet to be uncovered. Peptide bond thermodynamic stability may represent a previously unappreciated, evolutionarily tunable parameter for optimizing the kinetics of protein turnover in vivo.
In summary, we demonstrated a unique mechanism of protection from proteolytic degradation, in which a requirement for independent cleavages at two thermodynamically stable peptide bonds enables cationic trypsin to function effectively for an extended time frame in the presence of very high protease concentrations. Remarkably, the fundamental thermodynamic stability of the Leu81–Glu82 peptide bond is modulated both by calcium and by the trypsinogen to trypsin transition, enabling more facile degradation of trypsinogen in the pancreas as a failsafe measure to stem a pathological cascade of premature activation. Our observations also suggest the possibility that thermodynamic versus kinetic control of regulatory proteolytic events may be a more widespread phenomenon in biological systems than appreciated so far.
This work was supported, in whole or in part, by National Institutes of Health Grants R01DK058088, R01DK082412, and R01DK095753 (to M. S.-T.) and R01CA154387 (to E. S. R.).
The human cationic trypsinogen residue numbering used here is based on sequential numbering of the trypsinogen precursor.
- CTRC
- chymotrypsin C
- CELA3B
- elastase 3B
- H-bond
- hydrogen bond.
REFERENCES
- 1. Freeman H. J., Kim Y. S. (1978) Digestion and absorption of protein. Annu. Rev. Med. 29, 99–116 [DOI] [PubMed] [Google Scholar]
- 2. Borgstrom B., Dahlqvist A., Lundh G., Sjovall J. (1957) Studies of intestinal digestion and absorption in the human. J. Clin. Invest. 36, 1521–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tarlow M. J., Hadorn B., Arthurton M. W., Lloyd J. K. (1970) Intestinal enterokinase deficiency. A newly-recognized disorder of protein digestion. Arch. Dis. Child. 45, 651–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bohe M., Borgström A., Genell S., Ohlsson K. (1986) Metabolism of 131I-labelled human pancreatic cationic trypsin after intraduodenal administration. Digestion 34, 127–135 [DOI] [PubMed] [Google Scholar]
- 5. Layer P., Go V. L., DiMagno E. P. (1986) Fate of pancreatic enzymes during small intestinal aboral transit in humans. Am. J. Physiol. 251, G475–G480 [DOI] [PubMed] [Google Scholar]
- 6. Szmola R., Sahin-Tóth M. (2007) Chymotrypsin C (caldecrin) promotes degradation of human cationic trypsin: identity with Rinderknecht's enzyme Y. Proc. Natl. Acad. Sci. U.S.A. 104, 11227–11232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sahin-Tóth M. (2006) Biochemical models of hereditary pancreatitis. Endocrinol. Metab. Clin. North Am. 35, 303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hubbard S. J. (1998) The structural aspects of limited proteolysis of native proteins. Biochim. Biophys. Acta 1382, 191–206 [DOI] [PubMed] [Google Scholar]
- 9. Kazanov M. D., Igarashi Y., Eroshkin A. M., Cieplak P., Ratnikov B., Zhang Y., Li Z., Godzik A., Osterman A. L., Smith J. W. (2011) Structural determinants of limited proteolysis. J. Proteome Res. 10, 3642–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Timmer J. C., Zhu W., Pop C., Regan T., Snipas S. J., Eroshkin A. M., Riedl S. J., Salvesen G. S. (2009) Structural and kinetic determinants of protease substrates. Nat. Struct. Mol. Biol. 16, 1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sahin-Tóth M. (2000) Human cationic trypsinogen. Role of Asn-21 in zymogen activation and implications in hereditary pancreatitis. J. Biol. Chem. 275, 22750–22755 [DOI] [PubMed] [Google Scholar]
- 12. Sahin-Tóth M., Tóth M. (2000) Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem. Biophys. Res. Commun. 278, 286–289 [DOI] [PubMed] [Google Scholar]
- 13. Szabó A., Sahin-Tóth M. (2012) Increased activation of hereditary pancreatitis-associated human cationic trypsinogen mutants in presence of chymotrypsin C. J. Biol. Chem. 287, 20701–20710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lengyel Z., Pál G., Sahin-Tóth M. (1998) Affinity purification of recombinant trypsinogen using immobilized ecotin. Protein Expr. Purif. 12, 291–294 [DOI] [PubMed] [Google Scholar]
- 15. Király O., Guan L., Sahin-Tóth M. (2011) Expression of recombinant proteins with uniform N-termini. Methods Mol. Biol. 705, 175–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szabó A., Héja D., Szakács D., Zboray K., Kékesi K. A., Radisky E. S., Sahin-Tóth M., Pál G. (2011) High affinity small protein inhibitors of human chymotrypsin C (CTRC) selected by phage display reveal unusual preference for P4′ acidic residues. J. Biol. Chem. 286, 22535–22545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Salameh M. A., Soares A. S., Hockla A., Radisky E. S. (2008) Structural basis for accelerated cleavage of bovine pancreatic trypsin inhibitor (BPTI) by human mesotrypsin. J. Biol. Chem. 283, 4115–4123 [DOI] [PubMed] [Google Scholar]
- 18. Szabó A., Sahin-Tóth M. (2012) Determinants of chymotrypsin C cleavage specificity in the calcium-binding loop of human cationic trypsinogen. FEBS J. 279, 4283–4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Niekamp C. W., Hixson H. F., Jr., Laskowski M., Jr. (1969) Peptide-bond hydrolysis equilibria in native proteins. Conversion of virgin into modified soybean trypsin inhibitor. Biochemistry 8, 16–22 [DOI] [PubMed] [Google Scholar]
- 20. Sealock R. W., Laskowski M., Jr. (1973) Thermodynamics and kinetics of the reactive site peptide-bond hydrolysis in bovine pancreatic secretory trypsin inhibitor (Kazal). Biochemistry 12, 3139–3146 [DOI] [PubMed] [Google Scholar]
- 21. Finkenstadt W. R., Hamid M. A., Mattis J. A., Schrode J., Sealock R. W., Wang D., Laskowski M., Jr. (1974) Kinetics and thermodynamics of the interaction of proteinases with protein inhibitors in Proteinase Inhibitors (Fritz H., Tschesche H., Greene L., Truscheit E., eds), pp. 389–411, Springer Berlin; Heidelberg [Google Scholar]
- 22. Kukor Z., Tóth M., Pál G., Sahin-Tóth M. (2002) Human cationic trypsinogen. Arg117 is the reactive site of an inhibitory surface loop that controls spontaneous zymogen activation. J. Biol. Chem. 277, 6111–6117 [DOI] [PubMed] [Google Scholar]
- 23. Homandberg G. A., Mattis J. A., Laskowski M., Jr. (1978) Synthesis of peptide bonds by proteinases. Addition of organic cosolvents shifts peptide bond equilibria toward synthesis. Biochemistry 17, 5220–5227 [DOI] [PubMed] [Google Scholar]
- 24. Homandberg G. A., Laskowski M., Jr. (1979) Enzymatic resynthesis of the hydrolyzed peptide bond(s) in ribonuclease S. Biochemistry 18, 586–592 [DOI] [PubMed] [Google Scholar]
- 25. Bronner F. (2003) Mechanisms of intestinal calcium absorption. J. Cell. Biochem. 88, 387–393 [DOI] [PubMed] [Google Scholar]
- 26. Duflos C., Bellaton C., Pansu D., Bronner F. (1995) Calcium solubility, intestinal sojourn time and paracellular permeability codetermine passive calcium absorption in rats. J. Nutr. 125, 2348–2355 [DOI] [PubMed] [Google Scholar]
- 27. Rinderknecht H. (1986) Activation of pancreatic zymogens. Normal activation, premature intrapancreatic activation, protective mechanisms against inappropriate activation. Dig. Dis. Sci. 31, 314–321 [DOI] [PubMed] [Google Scholar]
- 28. Rosendahl J., Witt H., Szmola R., Bhatia E., Ozsvári B., Landt O., Schulz H. U., Gress T. M., Pfützer R., Löhr M., Kovacs P., Blüher M., Stumvoll M., Choudhuri G., Hegyi P., te Morsche R. H., Drenth J. P., Truninger K., Macek M., Jr., Puhl G., Witt U., Schmidt H., Büning C., Ockenga J., Kage A., Groneberg D. A., Nickel R., Berg T., Wiedenmann B., Bödeker H., Keim V., Mössner J., Teich N., Sahin-Tóth M. (2008) Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat. Genet. 40, 78–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huber R., Bode W. (1978) Structural basis of the activation and action of trypsin. Acc. Chem. Res. 11, 114–122 [Google Scholar]
- 30. Delaage M., Lazdunski M. (1967) The binding of Ca2+ to trypsinogen and its relation to the mechanism of activation. Biochem. Biophys. Res. Commun. 28, 390–394 [DOI] [PubMed] [Google Scholar]
- 31. Laskowski M., Jr., Kato I. (1980) Protein inhibitors of proteinases. Annu. Rev. Biochem. 49, 593–626 [DOI] [PubMed] [Google Scholar]
- 32. Radisky E. S., Koshland D. E., Jr. (2002) A clogged gutter mechanism for protease inhibitors. Proc. Natl. Acad. Sci. U.S.A. 99, 10316–10321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zakharova E., Horvath M. P., Goldenberg D. P. (2009) Structure of a serine protease poised to resynthesize a peptide bond. Proc. Natl. Acad. Sci. U.S.A. 106, 11034–11039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arolas J. L., Botelho T. O., Vilcinskas A., Gomis-Rüth F. X. (2011) Structural evidence for standard-mechanism inhibition in metallopeptidases from a complex poised to resynthesize a peptide bond. Angew. Chem. Int. Ed. Engl. 50, 10357–10560 [DOI] [PubMed] [Google Scholar]