

Background: This work explores proteins participating in trophoblastic cell fusion.
Results: Gln-deamidated GAPDH accumulates at the plasma membrane and promotes cell fusion.
Conclusion: Transglutaminase-2 regulates trophoblastic cell fusion through GAPDH deamidation.
Significance: GAPDH and transglutaminase-2 have a novel function in cell fusion.
Keywords: Cell Differentiation, Cytoskeleton, Protein Deamidation, Transglutaminases, Trophoblast, Cell Fusion, Glyceraldehyde-3-phosphate Dehydrogenase
Abstract
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a multifunctional protein as well as a classic glycolytic enzyme, and its pleiotropic functions are achieved by various post-translational modifications and the resulting translocations to intracellular compartments. In the present study, GAPDH in the plasma membrane of BeWo choriocarcinoma cells displayed a striking acidic shift in two-dimensional electrophoresis after cell-cell fusion induction by forskolin. This post-translational modification was deamidation of multiple glutaminyl residues, as determined by molecular mass measurement and tandem mass spectrometry of acidic GAPDH isoforms. Transglutaminase (TG) inhibitors prevented this acidic shift and reduced cell fusion. Knockdown of the TG2 gene by short hairpin RNA reproduced these effects of TG inhibitors. Various GAPDH mutants with replacement of different numbers (one to seven) of Gln by Glu were expressed in BeWo cells. These deamidated mutants reversed the suppressive effect of wild-type GAPDH overexpression on cell fusion. Interestingly, the mutants accumulated in the plasma membrane, and this accumulation was increased according to the number of Gln/Glu substitutions. Considering that GAPDH binds F-actin via an electrostatic interaction and that the cytoskeleton is rearranged in trophoblastic cell fusion, TG2-dependent GAPDH deamidation was suggested to participate in actin cytoskeletal remodeling.
Introduction
The placenta is a vital organ for feto-maternal exchange of gases, nutrients, and waste products. These functions are carried out mainly by the trophoblasts covering chorionic villi, which are exposed to maternal blood flow into the intervillous space. Trophoblasts are composed of syncytiotrophoblasts and cytotrophoblasts. The former is a terminally differentiated multinucleated cell without generative potency. It is formed by fusion of a cytotrophoblast, which is present between a syncytiotrophoblast and its basement membrane. Syncytiotrophoblasts are continuously supplied with cellular materials, such as enzymes, organelles, and nucleic acids derived from fusing with a cytotrophoblast. The cell-cell fusion or the syncytial formation of trophoblasts is regulated by numerous factors, such as growth factors, membrane proteins, proteases, physicochemical factors, and membrane architecture. One of the major players in the downstream signaling for cell fusion is protein kinase A (PKA), and this pathway has been largely delineated in vitro using the choriocarcinoma cell line BeWo. Treatment with cyclic AMP (cAMP) or agents such as forskolin (1) induces BeWo cell fusion. Forskolin increases intracellular cAMP levels by activating adenylyl cyclase and activates PKA. In turn, PKA activates transcription factors such as GCMα (glial cell missing α) (2–4), and the target genes of GCMα include syncytin-1 and -2 (5, 6). Syncytin is a fusogenic membrane glycoprotein of human endogenous retroviral origin and is essential for trophoblast cell differentiation and syncytiotrophoblast morphogenesis during placental development (7–9). In addition to the cAMP/PKA pathway, two mitogen-activated protein kinase (MAPK) family members, ERK1/2 and p38, are suggested to mediate trophoblast cell fusion and differentiation downstream from epidermal growth factor receptor activation. Induction of these MAPKs activates the PPARγ/RXRα signal directly regulating syncytin-1 for cell fusion (10). Although syncytin is a key factor mediating cell fusion of cytotrophoblasts, many other proteins and signaling pathways, including those involved in cytoskeletal remodeling and degradation of adhesion proteins, also participate in trophoblast fusion, and the whole picture of the syncytialization process is not yet completely understood.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH; EC 1.2.1.12) is a multifunctional protein with diverse activities. Besides its classic function in glycolysis, this enzyme is directly involved in gene regulation, vesicular transport, cell signaling, chromatin structure, DNA repair, autophagy, and apoptosis (for a review, see Ref. 11). To exert these functions, GAPDH undergoes dynamic changes in subcellular localization and post-translational modification as well as in its interaction with other proteins. For example, upon exposure to oxidative stress, GAPDH is S-nitrosylated at its active site, cysteine, and this S-nitrosylation increases binding of GAPDH to an E3 ubiquitin ligase, Siah1 (12). The GAPDH-Siah1 complex stabilizes Siah1, whose nuclear localization signal mediates translocation of GAPDH, facilitating the ubiquitin-mediated degradation of nuclear proteins and thereby apoptotic cell death. Furthermore, the GAPDH-Siah1 complex binds to p300/CBP to form a second nuclear protein complex and triggers a pleiotropic cascade involving p53, Bax, and p21 (13).
In the present study, GAPDH was deamidated at multiple glutaminyl residues during BeWo cell fusion. This post-translational modification was catalyzed by transglutaminase 2 (TG22 or tissue transglutaminase; EC 2.3.2.13). The deamidated GAPDH accumulates at the plasma membrane and probably participates in the cytoskeletal rearrangement necessary for cell fusion. This is a novel function of GAPDH, not previously reported.
MATERIALS AND METHODS
Cell Culture
The BeWo human choriocarcinoma-derived cell line was cultured in Ham's F-12 medium (Wako, Osaka, Japan) supplemented with 10% heat-inactivated fetal calf serum (Nichirei, Tokyo, Japan) and penicillin/streptomycin (Invitrogen). To induce differentiation into a multinuclear syncytium, 25 μm forskolin (Calbiochem) was added to the medium, and the cells were incubated on type 1 collagen (BD Biosciences)-coated plates for 3 days. To inhibit TG activity, 250 μm cystamine dihydrochloride (Tokyo Chemical Industry Co., Ltd., Tokyo, Japan) or 200 μm monodansylcadaverine (Sigma-Aldrich) was added 24 h after the stimulation with forskolin.
Isolation of Apical-Plasma Membrane (A-PM) Fraction
The A-PM fraction was obtained using the method of Chaney and Jacobson (14) with minor modification. Briefly, the BeWo cells were washed once with a plasma membrane coating (PMC) buffer (20 mm MES, 135 mm NaCl, 0.5 mm CaCl2, 1 mm MgCl2, pH 5.3) and incubated with 5% (w/v) cationic colloidal silica (Sigma-Aldrich) in PMC buffer on ice for 2 min for surface coating. Subsequently, the cells were incubated with 35% (w/v) polyacrylic acid (Sigma-Aldrich) in the PMC buffer on ice for 2 min for cross-linking and neutralization of the silica surface. The cells were then lysed with 2.5 mm imidazole, and the lysate was fractionated by density gradient centrifugation using Nicodenz (Axis-Shield PoC, Oslo, Norway).
Two-dimensional Electrophoresis (2-DE)
The isolated A-PM proteins in 1% SDS were dialyzed against the 2-DE buffer, containing 5 m urea, 1.4 m thiourea, and 1 mm dithiothreitol (DTT) and were subjected to first dimension separation by isoelectric focusing using an immobilized pH gradient (IPG) dry strip, pH 3–10 (GE Healthcare). After electrofocusing, the strip was equilibrated in an SDS-equilibrating buffer (50 mm Tris-HCl, 6 m urea, 30% glycerol, 2% SDS, 0.002% bromphenol blue, pH 8.8) and subsequently reduced and alkylated with 10 mg/ml DTT and 25 mg/ml iodoacetamide, respectively, for 15 min each at room temperature. The strip was then inserted into a 10% SDS-polyacrylamide gel for the second dimension separation. The electrophoresed gel was stained with SYPRO Ruby (Invitrogen) to visualize the proteins.
In-gel Digestion and Peptide Mass Fingerprinting
In-gel digestion was carried out according to the method of Shevchenko et al. (15). Briefly, the protein spots were cut out of the 2-DE gel, and the proteins in the gel slices were then rinsed with acetonitrile. The dehydrated gels were incubated with a mixture of trypsin (modified trypsin from bovine pancrease; Promega) and lysylendopeptidase (Wako) in 50 μl of 100 mm ammonium hydrogen carbonate on ice for 45 min, and the solution was then replaced by a new ammonium hydrogen carbonate solution without enzymes, followed by incubation overnight at 37 °C. The peptides were extracted from the gel employing a 5% formic acid and 50% acetonitrile solution at room temperature for 15 min and then dried with a SpeedVac concentrator (Tomy, Tokyo, Japan). The peptide samples were desalted employing a Zip-Tip (Millipore), and mixed with 20 mm 2,5-dihydroxybenzoic acid (Wako) solution on a matrix-assisted laser desorption/ionization (MALDI) sample plate. Mass spectrometry (MS) was carried out with a Voyager DE-Pro time-of-flight mass spectrometer (AB Sciex), and the protein database search was performed using the MASCOT search engine (available on the Matrix Science Web site).
Isolation of BeWo Cell Surface Proteins
The cell surface proteins were biotinylated and isolated employing streptavidin, as follows. The BeWo cells on culture plates were washed twice with ice-cold phosphate-buffered saline (PBS) at pH 7.4 and then incubated with Biotin-Sulfo-OSu (Dojindo, Kumamoto, Japan) dissolved in PBS under gentle rotation at 4 °C for 30 min. After removal of the excess reagent by washing twice with an ice-cold buffer of 50 mm Tris-HCl (pH 8.0), 0.1 mm EDTA, and 150 mm NaCl at pH 8.0, the cells were recovered by scraping and incubated in a lysis buffer of 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1% Triton X-100, 1 μg/ml aprotinin, and 1 mm phenylmethanesulfonyl fluoride (PMSF) on ice for 30 min. The cell lysate was centrifuged at 17,400 × g at 4 °C for 20 min, and the supernatant was collected. After protein concentration measurement by the Bradford method (Bio-Rad), the supernatant was incubated with streptavidin-coupled agarose beads (Pierce) under rotation at 4 °C overnight. The beads were collected by centrifugation and washed five times with the lysis buffer. Subsequently, the biotinylated proteins were eluted by boiling the beads with the SDS-PAGE sample buffer for 5 min and then subjected to 10% SDS-PAGE.
Western Blotting
The total cell lysate was dissolved in a buffer containing 20 mm Tris-HCl (pH7.2), 150 mm NaCl, 0.1% Nonidet P-40, 0.3% Triton X-100, 5 mm EDTA, 1 μg/ml aprotinin, 1 mm PMSF, 1 mm sodium pyrophosphate, 1 mm sodium fluoride, and 1 mm sodium vanadate and then centrifuged to remove cell debris. The proteins in the supernatant were separated by SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Millipore). The membrane was then incubated with a blocking buffer (2.5% skim milk, 0.1% Tween 20 in PBS). After washing with PBS-T (0.1% Tween 20 in PBS) three times, each for 10 min, the membrane was incubated with IgG antibodies in the blocking buffer at 4 °C overnight. The antibodies were mouse monoclonal anti-GAPDH (Fitzgerald), mouse monoclonal anti-E-cadherin (BD Biosciences), goat polyclonal anti-enolase (Toyobo, Osaka, Japan), and mouse monoclonal anti-TG2 (Thermo Fisher). After washing with PBS-T three times, each for 10 min, the membrane was incubated with horseradish peroxidase-conjugated antibodies against mouse IgG (Dako), goat IgG (Dako), or rabbit IgG (Promega) in the blocking buffer at room temperature for 1 h, followed by washing with PBS-T three times. Immunoreactive bands were developed with Western Lightning Plus-ECL (PerkinElmer Life Sciences) according to the manufacturer's instructions.
OFFGEL Fractionation
The A-PM proteins were separated according to their isoelectric point (pI) by using an OFFGEL fractionator (Agilent), as follows. The solvent (1% SDS) of the A-PM fraction was replaced by a solution containing 5 m urea, 1.4 m thiourea, and 0.1 mm DTT using a cellulose ester dialysis membrane (molecular weight cut-off 3,500–5,000; Spectrum Labs), and the protein concentration was adjusted to 1 mg/1.8 ml by dilution with a Protein OFFGEL stock solution containing 6.7 m urea, 1.9 m thiourea, 62.2 mm DTT, glycerol, and OFFGEL Ampholyte pH3–10 (Agilent). An equal volume (150 μl) of protein solution was loaded over the IPG gel (pH 3–10) in each of 12 wells, and the apparatus was run under a programmed method consisting of 50 μA, 4,500 V, and 200 milliwatts at maximum and up to 20 kV-h.
LC-Nanoelectrospray Ionization (Nano-ESI) MS
LC-nano-ESI/MS was carried out by using reversed phase Inertsil 300C4 (GL Science, Tokyo, Japan) trap (0.3 × 2 mm) and separation (0.3 × 150 mm) columns directly coupled with an LTQ-XL ion trap mass spectrometer (Thermo Fisher). Each OFFGEL fraction (∼20 μl) was mixed with 1 μl of 1% formic acid, injected into the trap column, and washed with 0.1% formic acid. The proteins were eluted by a gradient of 0–90% acetonitrile in 0.1% formic acid at a flow rate of 5 μl/min and infused into the mass spectrometer through a Monospray tip (GL Science).
Purification of GAPDH with Different pI
Immunoaffinity gel was prepared by coupling mouse monoclonal anti-GAPDH antibody with Protein G-Sepharose (Amersham Biosciences) using a dimethyl pimelimidate imidoester cross-linker (Pierce) according to the manufacturer's instructions. Each OFFGEL fraction after isoelectric separation was dialyzed against the lysis buffer and then applied to an immunoaffinity gel column (200-μl bed volume), as described above. The column was washed with PBS three times, equilibrated with the lysis buffer, and incubated at 4 °C for 5 h. The column was then washed with PBS, and the proteins were eluted employing 0.1 m glycine-HCl, pH 2.5.
Fluorescent Microscopy
BeWo cells were fixed with 4% paraformaldehyde in PBS at 4 °C for 20 min. After the cells had been incubated with PBS containing 0.2% Triton X-100 for 3 min, they were treated with 2.5% (w/v) bovine serum albumin in PBS at room temperature for 1 h and finally incubated with mouse monoclonal anti-E-cadherin IgG at 4 °C overnight. The samples were washed three times, for 5 min each, with PBS at room temperature and then exposed to Alexa488-labeled anti-mouse IgG (Molecular Probes) or Alexa633-labeled anti-phalloidin (Molecular Probes). Nuclear DNA was fluorescently stained with 0.2 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) (Dojindo). The cells were observed using an Olympus IX51 fluorescent microscope or a Leica TCS SP8 confocal fluorescent microscope.
Gene Knockdown
For TG2 knockdown, the shRNA-encoding TG2 genes were created employing a pBAsi-hU6 Pur DNA vector (Takara, Shiga, Japan) according to the manufacturer's protocol. The sequences for the coding strand of shRNA were as follows: GATCCGTCGACTGTGGATTGGCATCTGTGAAGCCACAGATGGGATGCCAATCCACAGTCGACTTTTTTA (5′-3′) and AGCTTAAAAAAGTCGACTGTGGATTGGCATCCCATCTGTGGCTTCACAGATGCCAATCCACAGTCGACG (5′-3′) for the controls, GATCCGCAGTGACTTTGACGTCTTTGCTGTGAAGCCACAGATGGGCAAAGACGTCAAAGTCACTGCTTTTTTA (5′-3′) and AGCTTAAAAAAGCAGTGACTTTGACGTCTTTGCCCATCTGTGGCTTCACAGCAAAGACGTCAAAGTCACTGCG for human TG2 shRNA-1; and GATCCAGTGTGGCACCAAGTACCTGCTCAACTGTGAAGCCACAGATGGGTTGAGCAGGTACTTGGTGCCACACTTTTTTTA (5′-3′) and AGCTTAAAAAA AGTGTGGCACCAAGTACCTGCTCAACCCATCTGTGGCTTCACAGTTGAGCAGGTACTTGGTGCCACACTG for human TG2 shRNA-2.
For GAPDH gene knockdown, the shRNA-encoding GAPDH vectors were created employing a pBAsi-hU6 Pur DNA vector according to the manufacturer's protocol. The sequences for the coding strand of shRNA were GATCCGGTCGGAGTCAACGGATTTCTGTGAAGCCACAGATGGGAAATCCGTTGACTCCGACCTTTTTTA (5′-3′) and AGCTTAAAAAAGGTCGGAGTCAACGGATTTCCCATCTGTGGCTTCACAGAAATCCGTTGACTCCGACCG (5′-3′) for controls and GATCCGTCGACTGTGGATTGGCATCTGTGAAGCCACAGATGGGATGCCAATCCACAGTCGACTTTTTTA (5′-3′) and AGCTTAAAAAAGTCGACTGTGGATTGGCATCCCATCTGTGGCTTCACAGATGCCAATCCACAGTCGACG (5′-3′) for human GAPDH shRNA.
These constructs were introduced into BeWo cells using an Effectene Transfection Reagent (Qiagen) according to the manufacturer's instructions. The cells stably expressing shRNA were selected employing puromycin, and down-regulation of the protein was assessed by Western blotting.
The pEGFP-TG2 plasmid encoding the wild-type construct, which was kindly provided by Dr. Marc Antonyak (Cornell University, Ithaca, NY), was subcloned into a CSII-RfA-CMV-EGFP vector, which was kindly provided by Dr. Hiroyuki Miyoshi (RIKEN BRC, Ibaraki, Japan), using an L-R clonase reaction (Invitrogen) according to the manufacturer's protocol.
Gln/Glu-mutated GAPDH
Human GAPDH cDNA was amplified from BeWo cells using random primers and was inserted into the XhoI/BamHI site located at the 3′-end of a human U6 promoter of the pENTER4 (Life Technologies) vector. The cDNAs in pENTER4 were then subcloned into a CSII-RfA-CMV-HA or a CSII-RfA-CMV-EYFP vector using the L-R clonase reaction. Site-directed mutagenesis to generate the mutants with replacement of Gln by Glu was performed using a KOD-Plus mutagenesis kit (Toyobo, Osaka, Japan) according to the manufacturer's protocol.
RESULTS
Post-translational Modification of GAPDH upon Syncytial Formation
Forskolin induces differentiation of BeWo cells via activation of cAMP-sensitive pathways. The cells undergo fusion to form a syncytium, as demonstrated by the disappearance of E-cadherin at the lateral cell boundaries 72 h after activation (Fig. 1A) (16). To identify the molecules involved in the fusion process occurring at the cell membrane, the A-PM was isolated from the cells, in the presence or absence of forskolin, by using colloidal silica (14, 17) and subjected to 2-DE differential display followed by peptide mass fingerprinting. We identified several proteins displaying quantitative or qualitative change by forskolin treatment: cytokeratin 18 (0.1), galectin-3 (0.7), keratin 19 (0.5), desmolase/cytochrome P450 (18.8), ER-60 protease/protein-disulfide isomerase (1.6) and heat shock 70kDa protein 5 (2.0), where -fold change is shown in parentheses. In addition, GAPDH displayed a remarkable change or “hypershift” in its isoelectric point (pI), indicating post-translational modification (Fig. 1B). This change was unrelated to the use of colloidal silica for isolation, because it was also found to affect the GAPDH in the membrane fraction prepared by sucrose density gradient ultracentrifugation without using colloidal silica (data not shown).
FIGURE 1.

Post-translational modification of GAPDH upon syncytial formation. A, immunofluorescence microscopy for E-cadherin (E-cad) (Alexa488) and nuclear DNA (DAPI) in BeWo cells. BeWo cells were cultured in the absence (NT; a, c, and e) or presence (For; b, d, and f) of forskolin (25 μm) for 72 h. Disappearance of E-cadherin at cell-cell contact indicates syncytium formation (a and b). Merged images for E-cadherin and DAPI (c and d) and the corresponding high magnification images (e and f) are shown. The dotted line indicates the margin of syncytial cells. Scale bars, 50 μm (a–d), 100 μm (e and f). B, 2-DE images of A-PM fraction before and after forskolin treatment. The gels were stained with SYPRO Ruby. Protein spots indicated by arrows or arrowheads in the inset were subjected to in-gel digestion followed by peptide mass fingerprinting, and GAPDH was identified. C, Western blotting of GAPDH in total cell lysate (TCL) and the A-PM fraction. The amounts of protein loaded onto the lane are indicated, and the SYPRO Ruby protein stain images of the SDS-polyacrylamide gel are presented in the bottom panel.
Next, endogenous levels of GAPDH before and after forskolin treatment were determined by Western blotting. The amount of GAPDH recovered from the A-PM fraction was increased after forskolin treatment, whereas the level of total GAPDH in the cell was unchanged (Fig. 1C). The labeling of cell surface proteins revealed that GAPDH was not exposed to the extracellular milieu, suggesting localization beneath the plasma membrane (data not shown).
Separation and Characterization of the Modified GAPDH
To characterize the post-translational modification leading to the acidic hypershift, the acidic isoforms in the A-PM were separated according to their pI values using an OFFGEL fractionator. The SDS-PAGE indicated that the isoforms had been successfully separated into several fractions (Fig. 2, A and B). These fractions were subjected to LC-nano-ESI/MS for molecular mass measurement. Although the mass spectra of multiply charged ions displayed no gross changes, the deconvolution to singly charged species suggested an increase in the molecular mass by one or a few Da according to the acidic shift (Fig. 2C). This feature of acidic GAPDH isoforms allowed us to speculate that deamidation or citrullination had occurred, both accompanying an increase by 1 Da of the molecule.
FIGURE 2.

Separation and characterization of modified GAPDH. A, SDS-PAGE of OFFGEL fractions. The fractions separated according to isoelectric points by the OFFGEL fractionator were analyzed by 10% SDS-PAGE and silver-stained. B, Western blot for GAPDH in the SDS-polyacrylamide gels. Only the areas corresponding to those indicated by the rectangle in A are presented. C, calculated masses of the GAPDH in different OFFGEL fractions. The mass of each ion observed in the nano-ESI mass spectrum was transformed into the molecular mass, and the mean and S.E. values were calculated. D, product ion mass spectrum from a protonated peptide (residues 67–84) derived from the GAPDH in fraction 9. The isotopic distribution for y10 ions indicated the presence of a +1-Da species, suggesting partial deamidation of Gln-78. E, the isotopic distribution of y12 product ions from the protonated peptide (sequence 67–84). The observed spectrum (open profile) was compared with the theoretical distribution (closed profile).
Subsequently, GAPDH was immunopurified from each fraction and digested with trypsin, and the resulting peptides were analyzed by tandem MS. The isotopic distribution of the product ions from protonated peptide LVINGNPITIFQERDPSK (residues 67–84) indicated that residue 78 (Gln) but not 80 (Arg) was largely responsible for the +1 Da increase, suggesting partial deamidation of the glutaminyl residue (Fig. 2D). The same change was observed in 204 (Gln) and suggested in other glutaminyl residues (data not shown). In addition, the isotopic profile of the product ions indicated that the deamidation level was higher in peptides derived from the GAPDH isoforms with lower pI values (Fig. 2E).
Effects of TG Inhibitors on GAPDH Modification and Cell Fusion
Non-enzymatic deamidation of at least seven asparaginyl residues has been demonstrated for GAPDH (UniProtKB/Swiss-Prot; P04406, G3P_HUMAN) and was actually present in the GAPDH samples analyzed herein (data not shown). On the other hand, deamidation of glutaminyl residues can occur enzymatically (i.e. with TG, which binds the side chain amide of γ-glutaminyl residues of proteins and then forms an isopeptide bond with an amine donor or hydrolyzes it to free acid (glutamic acid) when a suitable amine donor is absent). Dansylcadaverine is a competitive inhibitor substrate, and cystamine blocks the active site of TG (18). In a study by Robinson et al. (19), these TG inhibitors down-regulated fusion and differentiation of (cyto)trophoblasts in primary culture. To study the role of TG in BeWo cell fusion, we examined the effects of these inhibitors on the modification of GAPDH. The cells were treated with forskolin, and then either 0.2 m dansylcadaverine or 0.25 m cystamine was added to the medium. After 48 h of incubation, A-PM was obtained from the cells and subjected to 2-DE followed by Western blotting. As shown in Fig. 3 (A and B), these inhibitors prevented the hypershift, and the number of GAPDH spots identified in the TG-inhibited cells was comparable with that of the cells without forskolin and the inhibitors. In this experiment, GAPDH expression was unchanged by either dansylcadaverine or cystamine (data not shown).
FIGURE 3.
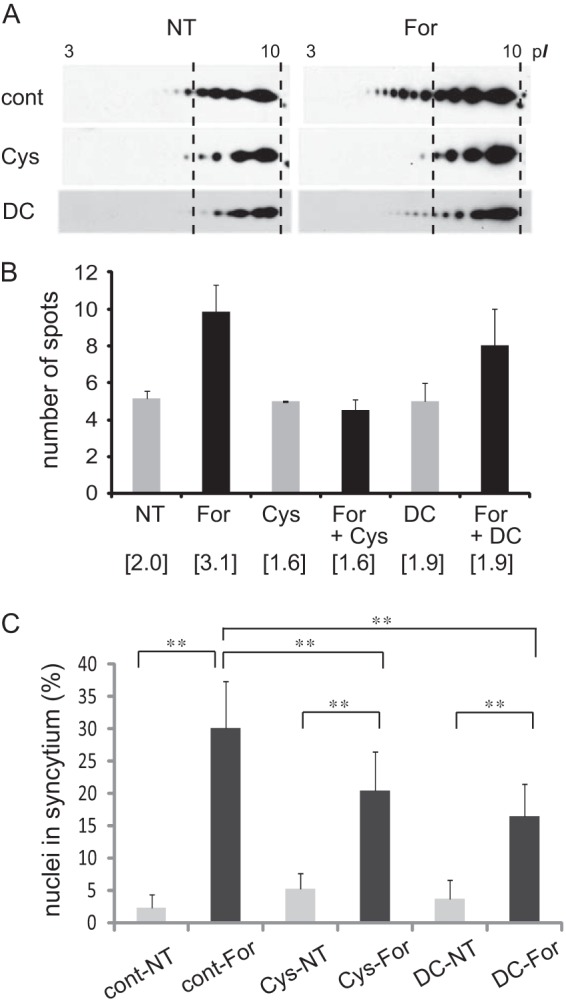
Effects of TG inhibitors on GAPDH modification and cell fusion. A, 2-DE Western blot images of GAPDH in the A-PM fraction of BeWo cell cultures with 250 μm cystamine (Cys) or 200 μm dansylcadaverine (DC) for 72 h. NT, no treatment. For, forskolin. B, number of observed spots for GAPDH when separated by 2-DE. Results of three independent experiments are presented as means ± S.D. (n = 3–5). The spots of GAPDH isoforms were numbered from the high pI side, and the density was quantified by densitometry. The weighted average of the density was calculated and is shown in parentheses. C, percentage of syncytial cell nuclei among total nuclei. More than 100 nuclei in each microscopic field (×100 magnification) were counted (n = 15). Error bars, S.D. **, p < 0.01 (Tukey-Kramer statistical test).
Next, the effects of TG inhibitors on BeWo cell fusion were investigated by counting the nuclei in the cells whose nuclei and cell borders were labeled with DAPI and E-cadherin, respectively, and the percentage of multinucleated cell nuclei among total nuclei was calculated. As shown in Fig. 3C, BeWo cell fusion was significantly suppressed by these inhibitors. These findings suggested that TG is responsible for the glutaminyl deamidation of GAPDH and is involved in the cell fusion mechanism.
TG2 Involvement in GAPDH Modification and BeWo Cell Fusion
At least eight TGs encoded by different genes are distributed in the human body (for a review, see Ref. 20). To identify the TG specifically catalyzing GAPDH deamidation, we first analyzed the expressions of TG genes and proteins in BeWo cells. TG2 (tissue TG) but not TG1 protein was detected by Western blotting, whereas TG1 and TG2 mRNAs were expressed in BeWo cells (data not shown). These results were consistent with TG1 being expressed in skin, whereas TG2 is ubiquitous, and suggested that TG2 is responsible for deamidation of GAPDH during trophoblastic cell fusion. To specify the nature of the involvement of TG2 in GAPDH deamidation and cell fusion, TG2 gene manipulation was conducted. We generated two clones of shRNA with different constructs completely suppressing TG2 expression (Fig. 4A). In the TG2-deficient cells, the amount of TG2 protein in the A-PM fraction was low, and interestingly, the hypershift was not observed (Fig. 4B). On the other hand, overexpression of TG2 did not cause substantial change in the modification of GAPDH or cell fusion.
FIGURE 4.

Effects of TG2 knockdown on GAPDH deamidation and cell fusion. A, Western blot of TG2 and enolase in the total cell lysate of BeWo cells expressing scrambled RNA (mock) or shRNA of TG2 (sh-TG2). B, BeWo cells expressing scrambled RNA (mock), sh-TG2, or GFP-TG2 (overexpression) were cultured in the presence or absence of 25 μm forskolin for 72 h (For). The A-PM fraction was separated by 2-DE and analyzed by Western blotting. NT, no treatment. C, representative images of BeWo cells expressing mock (left panels) or sh-TG2 (middle and right panels) in the presence (a–f) or absence (g–i) of forskolin. The dotted line indicates the syncytium outline (d–f). Scale bars, 100 μm. D, percentage of syncytial cell nuclei among total nuclei. More than 100 nuclei in each microscopic field (×100 magnification) were counted (n = 10). Error bars, S.D. **, p < 0.01 (Tukey-Kramer statistical test).
Subsequently, the effect of TG2 knockdown on cell fusion was analyzed, employing microscopic observations and statistical evaluation. As shown in Fig. 4 (C and D), the TG2 knockdown impaired cell fusion, which is consistent with the effects of the TG inhibitors described above. All of these findings indicated that TG2 is involved in GAPDH deamidation and cell fusion.
Effects of Gln-deamidated GAPDH on Cell Fusion
Human GAPDH contains seven Gln residues at positions 48, 78, 113, 185, 204, 264, and 280, six of which are known to be conserved among four mammalian species (i.e. humans, pigs, mice, and rabbits). To more directly demonstrate that the deamidation of glutaminyl residues of GAPDH is responsible for cell fusion, we generated four types of HA-tagged GAPDH mutants, in which one (Q78E or Q204E), two (Q78E/Q204E), or all (7QE) glutaminyl residues are replaced by glutamic acid and expressed these mutants in BeWo cells using a lentivirus vector. As shown in Fig. 5A, GAPDH accumulated in the A-PM fraction in response to forskolin treatment, whereas the total GAPDH level was unchanged. Furthermore, accumulation in the A-PM was prominent for the 7QE mutant. This property of the 7QE GAPDH was reproduced by EYFP-tagged mutants, as shown in Fig. 5B. In the EYFP-tagged experiment, the mutant with replacement of three (Q204E/Q264E/Q280E; 3QE) or four (Q48E/Q78E/Q113E/Q185E; 4QE) Gln residues was included in addition to the single (Q78E) and 7QE mutants, and, interestingly, localization to the A-PM was increased as the number of substitutions increased (Fig. 5B).
FIGURE 5.

Effects of Gln/Glu-substituted GAPDH mutants on cell fusion. A, Western blots of wild-type and HA-tagged mutant GAPDH in total cell lysate (TCL) and the A-PM. HA-tagged wild-type, Q78E, Q204E, Q78E/Q204E, or 7QE mutated GAPDH was expressed in BeWo cells using a lentivirus vector. The cells were cultured in the presence or absence of 25 μm forskolin for 72 h. All of the Gln residues were replaced by Glu in the 7QE mutant. For A-PM samples, the SYPRO Ruby protein stain images of the SDS-polyacrylamide gel are presented in the bottom panel. The endogenous GAPDH and HA-tagged mutant were not discriminated from each other in the GAPDH band. B, Western blots of wild-type and EYFP-tagged GAPDH mutants. EYFP-tagged wild-type, Q78E, Q204E/Q264E/Q280E (3QE), Q48E/Q78E/Q113E/Q185E (4QE), or 7QE GAPDH was expressed in BeWo cells using a lentivirus vector. The cells were cultured in the presence or absence of 25 μm forskolin for 72 h. For A-PM samples, the SYPRO Ruby protein stain images of the SDS-polyacrylamide gel are presented in the bottom panel. The GAPDH band does not include EYFP-tagged fusion GAPDH. C, percentage of syncytial cell nuclei among total nuclei in the HA-tagged experiment. More than 100 nuclei in each microscopic field (×100 magnification) were counted (n = 5). Error bars, S.D. **, p < 0.01 (Tukey-Kramer statistical test). D, effects of GAPDH knockdown on cell fusion. Western blot of GAPDH in the total cell lysate of BeWo cells expressing scrambled shRNA (sh-control) or shRNA of GAPDH (sh-GAPDH) (top). The cells were cultured in the presence or absence of 25 μm forskolin for 72 h. Bottom, numbers of nuclei in the fused cells 72 and 96 h after forskolin treatment. Two different cells harboring either red fluorescent protein in the cytoplasm (DsRed cells) or enhanced cyan fluorescent protein with a nuclear localization signal (CFP-Nuc cells) were co-cultured, and nuclei were counted in the double positive cells (n > 300). E, localization of the YFP-GAPDH-7QE mutant. The mutant GAPDH co-localizes with F-actin at the plasma membrane. Scale bars, 25 μm (left) and 10 μm (right). The fluorescent image was obtained with a Leica TCS SP8 confocal fluorescent microscope.
Finally, the effect of GAPDH deamidation on BeWo cell differentiation was examined by microscopic observation. As summarized in Fig. 5C, syncytium formation in response to forskolin treatment was suppressed by overexpression of wild-type GAPDH, and this suppression was reversed by the Gln/Glu-mutated GAPDH. Overexpression of wild-type or mutant GAPDH did not change syncytin expression (data not shown). The knockdown of GAPDH promoted BeWo cell fusion (Fig. 5D), suggesting that the basal GAPDH property inhibiting cell fusion is lost with TG2-dependent deamidation. Under microscopic observation, the 7QE mutant showed accumulation at the plasma membrane (Fig. 5E), supporting the accumulation of deamidated mutants at the A-PM in Fig. 5 (A and B). Taking these observations together, we can reasonably speculate that the Gln deamidation of GAPDH promotes cell fusion at the plasma membranes of trophoblastic cells.
DISCUSSION
Syncytiotrophoblasts are terminally differentiated cells lacking proliferative capacity and are maintained by fusion with underlying cytotrophoblasts. The present results allow us to propose a novel GAPDH function in trophoblast cell fusion through deamidation of glutaminyl residues. GAPDH is a multifunctional protein acting in a number of fundamental cell pathways, and its functional diversity is controlled by dynamic changes in subcellular distributions and post-translational modifications, including phosphorylation of tyrosine or serine, S-nitrosylation or poly-ADP-ribosylation of cysteine, acetylation of lysine, and O-GlcNAcylation of threonine (for a review, see Ref. 21). Herein, GAPDH was found to be deamidated by TG2, and the deamidated GAPDH accumulated at the plasma membrane and facilitated trophoblast cell fusion. The biological role of Gln deamidation has not been studied in sufficient detail, as compared with that of cross-linking, although both mechanisms are catalyzed by TG. Exceptionally, the potential pathophysiological consequences of TG2-dependent deamidation have been reported in βB2- and βB3-crystallins (20–25-kDa proteins containing multiple Gln residues) (22), β-amyloid peptide (23), and substance P (24). Deamidation influences macromolecular assembly or induces aggregation of β-crystallins and β-amyloid (total of 40 amino acid residues, including one Gln), and deamidated substance P (total of 11 residues, including one Gln) shows increased agonist potency toward the receptor. In these cases, the physicochemical properties of proteins and peptides are altered by deamidation.
It is well known that the glycolytic enzymes, including GAPDH, are associated with F-actin at physiological ionic strengths (25). This interaction is suggested to be one of the mechanisms underlying the compartmentalization of glycolytic enzymes, which allows them to function efficiently in the cytoplasm, and is electrostatic in nature. In addition, associations of GAPDH with cytoskeletal proteins, such as actin and those comprising microtubules, have been reported in studies on vesicular trafficking or microtubule-membrane interactions (26–28). Considering that rearrangement of the actin cytoskeleton is required for trophoblastic cells to become competent for fusion (17), it is conceivable that deamidated GAPDH is involved in the cytoskeletal remodeling process. GAPDH binds acidic residues of F-actin, and residues of GAPDH affecting GAPDH/F-actin binding are widely distributed throughout the sequence (25). In addition, in the present study, the number of Gln/Glu mutations correlated with the accumulation of GAPDH at the A-PM. Furthermore, multiple deamidations occurred in GAPDH. It is thus conceivable that a change in the overall charge due to multiple Gln deamidations but not deamidation at a specific Gln residue(s) of GAPDH might play a role in cytoskeletal dynamics during trophoblastic cell fusion.
TG2 is ubiquitously expressed in many tissues and organs and is involved in a variety of cellular processes, including adhesion, migration, growth, survival, apoptosis, differentiation, and extracellular matrix organization, and the wide variety of these TG2 functions is substantiated by the diverse localization of TG2 in cellular compartments, such as in the cytoplasm, beneath the plasma membrane, in the nucleus, in mitochondria, in early/late/recycling endosomes and lysosomes, on the cell surface, in the extracellular matrix, and in extracellular microvesicles (for a review, see Ref. 29). In the human placenta, strong activity was identified at the syncytial microvillous membrane (30), where TG2 is seen in both syncytio- and cytotrophoblasts in association with the cytoskeletal structure, including peripheral actin bundles and stress fibers (19). Furthermore, TG2 might mediate polymerization of cytoskeletal proteins by cross-linking with, and thereby stabilizing, particle materials shed from placental villi (19). The association of TG2 with cytoskeletal proteins in the microvillous membrane supports our notion that deamidated GAPDH accumulated at the membrane as part of the cytoskeletal rearrangement process. Indeed, GAPDH was included among the proteins associated with TG2 (19).
Expression of TG2 is regulated on many levels from epigenetics to degradation (31). In the present study, TG2 transcription was up-regulated by forskolin treatment. However, the intracellular pool of TG2 is in a catalytically inactive state, and its activity is regulated by physiological factors, such as Ca2+ ions, guanine nucleotides, and the redox potential. Most notably, binding of Ca2+ ions is essential for TG2 catalytic activity (32). Forskolin resensitizes cell receptors by activating adenylyl cyclase and increases the intracellular cAMP level. It would be intriguing to reconcile cAMP and TG2 up-regulation in the BeWo cell fusion observed in our experiment. Among the multiple effectors activated by cAMP, Epac (exchange protein directly activated by cAMP) mediates cAMP signaling in many cellular activities. Epac proteins (Epac1 and Epac2) are cAMP-binding proteins directly activating small GTPases, Rap1 and Rap2 (33, 34). In a recent report, cAMP-dependent activation of Epac1 and Rap1 but not PKA was shown to have the capacity to activate CaMK1 and to mediate Ser-47 phosphorylation of GCM1, eventually stimulating cell fusion (35). This cAMP/Epac1/CaMK1 signaling cascade could lead to Ca2+-mediated activation of TG2.
The involvement of TG2 in trophoblast cell fusion may have clinical implications. Celiac disease is caused by intolerance to dietary gluten, resulting in immunologically mediated inflammatory damage of the small intestinal mucosa, malabsorption, and nutritional deficiency, and TG2 is the major autoantigen found in celiac disease patients (36). A number of cohort studies have suggested an association of celiac disease with poor pregnancy outcomes, including recurrent miscarriage, low birth weight babies, stillbirth, and fetal growth restriction, in untreated women (37). Although one plausible explanation for this link is obviously nutrient deficiencies, recent observational and in vitro studies have suggested that anti-TG2 autoantibody may impair placental growth and function and thereby cause fetal growth restriction (37–39). It will be intriguing to analyze GAPDH deamidation in the trophoblasts during pregnancy of celiac disease women and to study a possible mechanism underlying the placental damage by anti-TG2 autoantibodies in view of the physiological role of TG2 in trophoblastic cell fusion.
Acknowledgments
We thank Dr. Hiroyuki Miyoshi (RIKEN BRC, Ibaraki, Japan) and Dr. Marc Antonyak (Cornell University, Ithaca, NY) for providing recombinant lentiviruses and a TG2 plasmid, respectively, and Machiko Kadoya for technical assistance.
This work was supported in part by Grants-in-aid for Scientific Research B (23390081) and C (24591618) and for JSPS Fellows (22-3627) from the Japan Society for the Promotion of Science.
- TG
- transglutaminase
- A-PM
- apical plasma membrane
- ESI
- electrospray ionization
- EYFP
- enhanced yellow fluorescent protein
- IPG
- immobilized pH gradient
- PMC
- plasma membrane coating
- 2-DE
- two-dimensional electrophoresis
- 7QE
- GAPDH mutant in which all Gln residues are replaced with Glu (Q48E/Q78E/Q113E/Q185E/Q204E/Q264E/Q280E).
REFERENCES
- 1. Wice B., Menton D., Geuze H., Schwartz A. L. (1990) Modulators of cyclic AMP metabolism induce syncytiotrophoblast formation in vitro. Exp. Cell Res. 186, 306–316 [DOI] [PubMed] [Google Scholar]
- 2. Schreiber J., Riethmacher-Sonnenberg E., Riethmacher D., Tuerk E. E., Enderich J., Bösl M. R., Wegner M. (2000) Placental failure in mice lacking the mammalian homolog of glial cells missing, GCMa. Mol. Cell Biol. 20, 2466–2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anson-Cartwright L., Dawson K., Holmyard D., Fisher S. J., Lazzarini R. A., Cross J. C. (2000) The glial cells missing-1 protein is essential for branching morphogenesis in the chorioallantoic placenta. Nat. Genet. 25, 311–314 [DOI] [PubMed] [Google Scholar]
- 4. Knerr I., Schubert S. W., Wich C., Amann K., Aigner T., Vogler T., Jung R., Dötsch J., Rascher W., Hashemolhosseini S. (2005) Stimulation of GCMa and syncytin via cAMP mediated PKA signaling in human trophoblastic cells under normoxic and hypoxic conditions. FEBS Lett. 579, 3991–3998 [DOI] [PubMed] [Google Scholar]
- 5. Yu C., Shen K., Lin M., Chen P., Lin C., Chang G. D., Chen H. (2002) GCMa regulates the syncytin-mediated trophoblastic fusion. J. Biol. Chem. 277, 50062–50068 [DOI] [PubMed] [Google Scholar]
- 6. Liang C. Y., Wang L. J., Chen C. P., Chen L. F., Chen Y. H., Chen H. (2010) GCM1 regulation of the expression of syncytin 2 and its cognate receptor MFSD2A in human placenta. Biol. Reprod. 83, 387–395 [DOI] [PubMed] [Google Scholar]
- 7. Mi S., Lee X., Li X., Veldman G. M., Finnerty H., Racie L., LaVallie E., Tang X. Y., Edouard P., Howes S., Keith J. C., Jr., McCoy J. M. (2000) Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403, 785–789 [DOI] [PubMed] [Google Scholar]
- 8. Dupressoir A., Vernochet C., Bawa O., Harper F., Pierron G., Opolon P., Heidmann T. (2009) Syncytin-A knockout mice demonstrate the critical role in placentation of a fusogenic, endogenous retrovirus-derived, envelope gene. Proc. Natl. Acad. Sci. U.S.A. 106, 12127–12132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vargas A., Moreau J., Landry S., LeBellego F., Toufaily C., Rassart E., Lafond J., Barbeau B. (2009) Syncytin-2 plays an important role in the fusion of human trophoblast cells. J. Mol. Biol. 392, 301–318 [DOI] [PubMed] [Google Scholar]
- 10. Ruebner M., Langbein M., Strissel P. L., Henke C., Schmidt D., Goecke T. W., Faschingbauer F., Schild R. L., Beckmann M. W., Strick R. (2012) Regulation of the human endogenous retroviral Syncytin-1 and cell-cell fusion by the nuclear hormone receptors PPARγ/RXRα in placentogenesis. J. Cell Biochem. 113, 2383–2396 [DOI] [PubMed] [Google Scholar]
- 11. Sirover M. A. (2011) On the functional diversity of glyceraldehyde-3-phosphate dehydrogenase. Biochemical mechanisms and regulatory control. Biochim. Biophys. Acta 1810, 741–751 [DOI] [PubMed] [Google Scholar]
- 12. Hara M. R., Agrawal N., Kim S. F., Cascio M. B., Fujimuro M., Ozeki Y., Takahashi M., Cheah J. H., Tankou S. K., Hester L. D., Ferris C. D., Hayward S. D., Snyder S. H., Sawa A. (2005) S-Nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 7, 665–674 [DOI] [PubMed] [Google Scholar]
- 13. Sen N., Hara M. R., Kornberg M. D., Cascio M. B., Bae B. I., Shahani N., Thomas B., Dawson T. M., Dawson V. L., Snyder S. H., Sawa A. (2008) Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat. Cell Biol. 10, 866–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chaney L. K., Jacobson B. S. (1983) Coating cells with colloidal silica for high yield isolation of plasma membrane sheets and identification of transmembrane proteins. J. Biol. Chem. 258, 10062–10072 [PubMed] [Google Scholar]
- 15. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 16. Coutifaris C., Kao L. C., Sehdev H. M., Chin U., Babalola G. O., Blaschuk O. W., Strauss J. F., 3rd (1991) E-cadherin expression during the differentiation of human trophoblasts. Development 113, 767–777 [DOI] [PubMed] [Google Scholar]
- 17. Shibukawa Y., Yamazaki N., Kumasawa K., Daimon E., Tajiri M., Okada Y., Ikawa M., Wada Y. (2010) Calponin 3 regulates actin cytoskeleton rearrangement in trophoblastic cell fusion. Mol. Biol. Cell 21, 3973–3984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Birckbichler P. J., Orr G. R., Patterson M. K., Jr., Conway E., Carter H. A. (1981) Increase in proliferative markers after inhibition of transglutaminase. Proc. Natl. Acad. Sci. U.S.A. 78, 5005–5008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robinson N. J., Baker P. N., Jones C. J., Aplin J. D. (2007) A role for tissue transglutaminase in stabilization of membrane-cytoskeletal particles shed from the human placenta. Biol. Reprod. 77, 648–657 [DOI] [PubMed] [Google Scholar]
- 20. Griffin M., Casadio R., Bergamini C. M. (2002) Transglutaminases. Nature's biological glues. Biochem. J. 368, 377–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sirover M. A. (2012) Subcellular dynamics of multifunctional protein regulation. Mechanisms of GAPDH intracellular translocation. J. Cell Biochem. 113, 2193–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boros S., Wilmarth P. A., Kamps B., de Jong W. W., Bloemendal H., Lampi K., Boelens W. C. (2008) Tissue transglutaminase catalyzes the deamidation of glutamines in lens βB(2)- and βB(3)-crystallins. Exp. Eye Res. 86, 383–393 [DOI] [PubMed] [Google Scholar]
- 23. Schmid A. W., Condemi E., Tuchscherer G., Chiappe D., Mutter M., Vogel H., Moniatte M., Tsybin Y. O. (2011) Tissue transglutaminase-mediated glutamine deamidation of β-amyloid peptide increases peptide solubility, whereas enzymatic cross-linking and peptide fragmentation may serve as molecular triggers for rapid peptide aggregation. J. Biol. Chem. 286, 12172–12188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fornelli L., Schmid A. W., Grasso L., Vogel H., Tsybin Y. O. (2011) Deamidation and transamidation of substance P by tissue transglutaminase revealed by electron-capture dissociation Fourier transform mass spectrometry. Chemistry 17, 486–497 [DOI] [PubMed] [Google Scholar]
- 25. Forlemu N. Y., Njabon E. N., Carlson K. L., Schmidt E. S., Waingeh V. F., Thomasson K. A. (2011) Ionic strength dependence of F-actin and glycolytic enzyme associations. A Brownian dynamics simulations approach. Proteins 79, 2813–2827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kumagai H., Sakai H. (1983) A porcine brain protein (35 K protein) which bundles microtubules and its identification as glyceraldehyde 3-phosphate dehydrogenase. J. Biochem. 93, 1259–1269 [DOI] [PubMed] [Google Scholar]
- 27. Reiss N., Oplatka A., Hermon J., Naor Z. (1996) Phosphatidylserine directs differential phosphorylation of actin and glyceraldehyde-3-phosphate dehydrogenase by protein kinase C. Possible implications for regulation of actin polymerization. Biochem. Mol. Biol. Int. 40, 1191–1200 [DOI] [PubMed] [Google Scholar]
- 28. Andrade J., Pearce S. T., Zhao H., Barroso M. (2004) Interactions among p22, glyceraldehyde-3-phosphate dehydrogenase and microtubules. Biochem. J. 384, 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nurminskaya M. V., Belkin A. M. (2012) Cellular functions of tissue transglutaminase. Int. Rev. Cell Mol. Biol. 294, 1–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hager H., Gliemann J., Hamilton-Dutoit S., Ebbesen P., Koppelhus U., Jensen P. H. (1997) Developmental regulation of tissue transglutaminase during human placentation and expression in neoplastic trophoblast. J. Pathol. 181, 106–110 [DOI] [PubMed] [Google Scholar]
- 31. Del Bello A., Congy N., Sallusto F., Cardeau-Desangles I., Fort M., Esposito L., Guitard J., Cointault O., Lavayssière L., Nogier M. B., Game X., Blancher A., Rostaing L., Kamar N. (2012) Anti-human leukocyte antigen immunization after early allograft nephrectomy. Transplantation 93, 936–941 [DOI] [PubMed] [Google Scholar]
- 32. Király R., Demény M., Fésüs L. (2011) Protein transamidation by transglutaminase 2 in cells. A disputed Ca2+-dependent action of a multifunctional protein. FEBS J. 278, 4717–4739 [DOI] [PubMed] [Google Scholar]
- 33. Kawasaki H., Springett G. M., Mochizuki N., Toki S., Nakaya M., Matsuda M., Housman D. E., Graybiel A. M. (1998) A family of cAMP-binding proteins that directly activate Rap1. Science 282, 2275–2279 [DOI] [PubMed] [Google Scholar]
- 34. Bos J. L. (2006) Epac proteins. Multi-purpose cAMP targets. Trends Biochem. Sci. 31, 680–686 [DOI] [PubMed] [Google Scholar]
- 35. Chang C. W., Chang G. D., Chen H. (2011) A novel cyclic AMP/Epac1/CaMKI signaling cascade promotes GCM1 desumoylation and placental cell fusion. Mol. Cell Biol. 31, 3820–3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Green P. H., Cellier C. (2007) Celiac disease. N. Engl. J. Med. 357, 1731–1743 [DOI] [PubMed] [Google Scholar]
- 37. Kiefte-de Jong J. C., Jaddoe V. W., Uitterlinden A. G., Steegers E. A., Willemsen S. P., Hofman A., Hooijkaas H., Moll H. A. (2013) Levels of antibodies against tissue transglutaminase during pregnancy are associated with reduced fetal weight and birth weight. Gastroenterology 144, 726–735.e2 [DOI] [PubMed] [Google Scholar]
- 38. Anjum N., Baker P. N., Robinson N. J., Aplin J. D. (2009) Maternal celiac disease autoantibodies bind directly to syncytiotrophoblast and inhibit placental tissue transglutaminase activity. Reprod. Biol. Endocrinol. 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Di Simone N., Silano M., Castellani R., Di Nicuolo F., D'Alessio M. C., Franceschi F., Tritarelli A., Leone A. M., Tersigni C., Gasbarrini G., Silveri N. G., Caruso A., Gasbarrini A. (2010) Anti-tissue transglutaminase antibodies from celiac patients are responsible for trophoblast damage via apoptosis in vitro. Am. J. Gastroenterol. 105, 2254–2261 [DOI] [PubMed] [Google Scholar]