

Background: Dok proteins are negative regulators of immunoreceptor signaling and, potentially, integrin adhesion receptors.
Results: Deficiency of Dok-2 results in enhanced shear-dependent integrin adhesion in platelets, leading to accelerated platelet thrombus growth.
Conclusion: Dok-2 is a shear-specific negative regulator of blood clot formation.
Significance: Dok-2 regulates biomechanical platelet adhesion, and targeting this molecule may provide new avenues to regulate thrombosis.
Keywords: Adaptor Proteins, Integrins, Mechanotransduction, Platelets, Shear Stress
Abstract
The Dok proteins are a family of adaptor molecules that have a well defined role in regulating cellular migration, immune responses, and tumor progression. Previous studies have demonstrated that Doks-1 to 3 are expressed in platelets and that Dok-2 is tyrosine-phosphorylated downstream of integrin αIIbβ3, raising the possibility that it participates in integrin αIIbβ3 outside-in signaling. We demonstrate that Dok-2 in platelets is primarily phosphorylated by Lyn kinase. Moreover, deficiency of Dok-2 leads to dysregulated integrin αIIbβ3-dependent cytosolic calcium flux and phosphatidylinositol(3,4)P2 accumulation. Although agonist-induced integrin αIIbβ3 affinity regulation was unaltered in Dok-2−/− platelets, Dok-2 deficiency was associated with a shear-dependent increase in integrin αIIbβ3 adhesive function, resulting in enhanced platelet-fibrinogen and platelet-platelet adhesive interactions under flow. This increase in adhesion was restricted to discoid platelets and involved the shear-dependent regulation of membrane tethers. Dok-2 deficiency was associated with an increased rate of platelet aggregate formation on thrombogenic surfaces, leading to accelerated thrombus growth in vivo. Overall, this study defines an important role for Dok-2 in regulating biomechanical adhesive function of discoid platelets. Moreover, they define a previously unrecognized prothrombotic mechanism that is not detected by conventional platelet function assays.
Introduction
The excessive accumulation of platelets at sites of vascular injury is of central importance to the development of arterial thrombosis and is the primary pathogenic mechanism underlying acute coronary syndromes and ischemic stroke (1). Arterial thrombotic diseases are the leading cause of morbidity and mortality in industrialized societies (1–3), and, as a consequence, the platelet represents a key therapeutic target in the management of cardiovascular diseases (2–4). The molecular mechanisms regulating platelet aggregation have been well defined and are critically dependent on the activation of the major platelet integrin αIIbβ3. Activated integrin αIIbβ3 engages various adhesive ligands, including von Willebrand factor, fibrinogen, and fibronectin, which promote platelet aggregation and the development of the primary hemostatic plug. The importance of integrin αIIbβ3 in primary hemostasis is well established and is underscored by the severe bleeding tendency associated with individuals with qualitative or quantitative defects in integrin αIIbβ3 (Glanzmann thrombasthenia) (5).
An exaggerated level of integrin αIIbβ3 activation is a key factor promoting thrombus development. Increased integrin αIIbβ3 activation in hyperactive platelets is primarily a manifestation of enhanced “inside-out” signaling in which signals generated from within the cell up-regulate the affinity status of integrins by inducing specific conformational changes within the intracellular and extracellular domains of the receptors (6). When activated and cross-linked by adhesive ligands, integrin αIIbβ3 transduces specific “outside-in” signals that modulate a subset of platelet functional responses, including irreversible platelet aggregation, spreading, and clot retraction (6). Abnormalities in integrin αIIbβ3 outside-in signaling have been associated with an increased bleeding tendency (7). Whether dysregulation of integrin αIIbβ3 outside-in signaling processes can enhance platelet adhesive function, leading to a prothrombotic phenotype, remains unknown.
One potential family of proteins that may be involved in integrin αIIbβ3 outside-in signaling is the downstream of tyrosine kinase (Dok) family of adaptor proteins. Dok proteins consist of an amino-terminal pleckstrin homology domain, a phosphotyrosine binding domain, a Dok homology domain, and a proline- and tyrosine-rich carboxyl-terminal region (8–13). These domains support the interactions of Dok proteins with SH22 and SH3 domain-containing proteins as well as integrins bearing NPXY- or NPXY-like motifs (8, 14). Dok-1 to 3 are primarily hematopoietic in origin (8, 9) and have been shown to be present in platelets (15–17), whereas other members, Dok-4 to 7, show diverse expression patterns (10, 12, 13, 18). Dok proteins have been shown to be involved in the negative regulation of immune responses and/or tumor progression in various cellular systems (8, 19–25). In platelets, Dok-2 is primarily phosphorylated downstream of integrin αIIbβ3 through a process dependent on calcium flux and Src kinases, leading to the physical association of Dok-2 with activated integrin αIIbβ3 (16).
In this study we investigated the functional importance of Dok-2 in platelets. We demonstrate here that Dok-2 deficiency is associated with a shear-dependent increase in integrin αIIbβ3 adhesive function, leading to accelerated platelet aggregation and thrombus development under flow. Significantly, this increase in adhesive function was restricted to discoid platelets and was not associated with any other detectable changes in integrin αIIbβ3 adhesive function following agonist stimulation of platelets. Mechanistically, the increased adhesion of Dok-2−/− platelets was due to enhanced integrin αIIbβ3 bond stability and more stable discoid platelet aggregation that was coincident with an exaggerated cytosolic calcium response. This study defines a role for Dok-2 in regulating the biomechanical adhesive function of platelets linked to thrombus development.
EXPERIMENTAL PROCEDURES
Additional detailed materials and methods used in this study are described in the supplemental Experimental Procedures.
Materials
A detailed description of specific materials used in this study can be found in the supplemental Materials. All other reagents are described in sources published previously (16, 26–28).
Mouse Strains
Dok-2−/− mice backcrossed for eight generations to a C57BL/6 background were imported from the Department of Cell Regulation, Medical Research Institute, Tokyo Medical and Dental University (Tokyo, Japan) (24). C57Bl Lyn+/+ and Lyn−/− mice were generated at the Ludwig Institute for Cancer Research (Melbourne, Australia) and were a gift from Dr. Margaret Hibbs (29). All procedures involving the use of mice were approved by the Alfred Medical Research and Education Precinct Animal Ethics Committee (Melbourne, Australia) under project numbers E/0492/2006/M, E/0677/2008/M, E/0734E/2008/M, E/0865/2009/M, and E/0889/2009/M.
Whole Blood and Platelet Preparation
All procedures involving the collection of mouse and human blood were performed in accordance with the Alfred Medical Research and Education Precinct Animal Ethics Committee (SOP19, Collection of Whole Blood from Mice) and the Standing Committee on Ethics in Research Involving Humans (project number CF07/0141-2007000025), respectively. Mouse whole blood was collected in hirudin (0.5 μg/μl, Refludan, Pharmion, Calgene, Summit, NJ). Mouse platelets were isolated according to Maxwell et al. (30). In some studies, mouse platelets were reconstituted with washed human RBCs, isolated as described (26).
In Vitro Perfusion Studies
In vitro perfusion assays were performed according to the modifications of Maxwell et al. (26) and Goncalves et al. (31). For a detailed description of the methods used for in vitro perfusion studies, refer to the supplemental Experimental Procedures.
Scanning Electron Microscopy (SEM)
Platelets were perfused across hexamethyldisilazane-derived fibrinogen-coated microslides or spread platelet monolayers, fixed (4% paraformaldehyde, 1 h), and prepared for SEM essentially as described (32). Samples were imaged using a Hitachi S570 scanning electron microscope (Tokyo, Japan) at 15 kV of accelerating voltage.
Analysis of Calcium Flux
Calcium flux in isolated platelets under static and shear conditions was quantified as described previously (31). In some experiments, platelets were stimulated with thrombin (0.1–1.0 units/ml), collagen-related peptide (1–10 μg/ml), or ADP (2–25 μm) in the presence or absence of EDTA (1 mm), EGTA (1 mm), or 2-APB (10 μm), either alone or in combination, prior to measurement of calcium concentrations. In other studies, murine platelets were treated with EDTA (1 mm), EGTA (1 mm), or 2-APB (10 μm), either alone or in combination, for 15 min at 37 °C prior to measurement of resting calcium concentrations.
Statistical Analysis
Statistical significance was determined using one-way analysis of variance or Student's t test (unpaired, two-way t test) calculated using GraphPAD Prism software (Prism Software, GraphPAD Software for Science, San Diego, CA). The p values are as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Data are presented as the mean ± S.E., and n equals the number of independent experiments performed.
RESULTS
Increased Platelet Thrombus Formation in Dok-2−/− Mice
In preliminary studies, we confirmed that platelet counts were normal in Dok-2−/− mice and that the platelets had normal surface expression of the major platelet adhesion receptors GPIbα; integrin αIIbβ3; and GPVI; as well as integrins β1, α2, and α5, both in resting and activated platelets,3 and normal intracellular levels of Dok-1 (supplemental Fig. S1). Furthermore, Dok-2−/− mice had no spontaneous bleeding or increase in tail bleeding time following surgical transection.3 However, the rate and extent of platelet thrombus formation when anticoagulated whole blood was perfused over an immobilized collagen substrate (1800 s−1) was increased in Dok-2−/− mice relative to WT controls (Fig. 1). This difference was consistent over a broad range of collagen-coating concentrations (5–100 μg/ml),3 resulting in an approximately 3-fold increase in thrombus size (Fig. 1, A–C). Notably, we could not detect any differences in the rate or extent of platelet aggregation between Dok-2−/− platelets and matched controls following ADP, thrombin, or collagen-related peptide stimulation even when stimulated with threshold concentrations of agonists (supplemental Fig. S2A). Similarly, there was no difference in thrombin- or collagen-related peptide-induced α-granule release (measured by P-selectin surface expression) in Dok-2−/− platelets (supplemental Fig. S2B). These studies suggest that Dok-2 deficiency leads to an exaggerated platelet thrombotic response that is unrelated to increased platelet sensitivity to soluble agonist stimulation.
FIGURE 1.
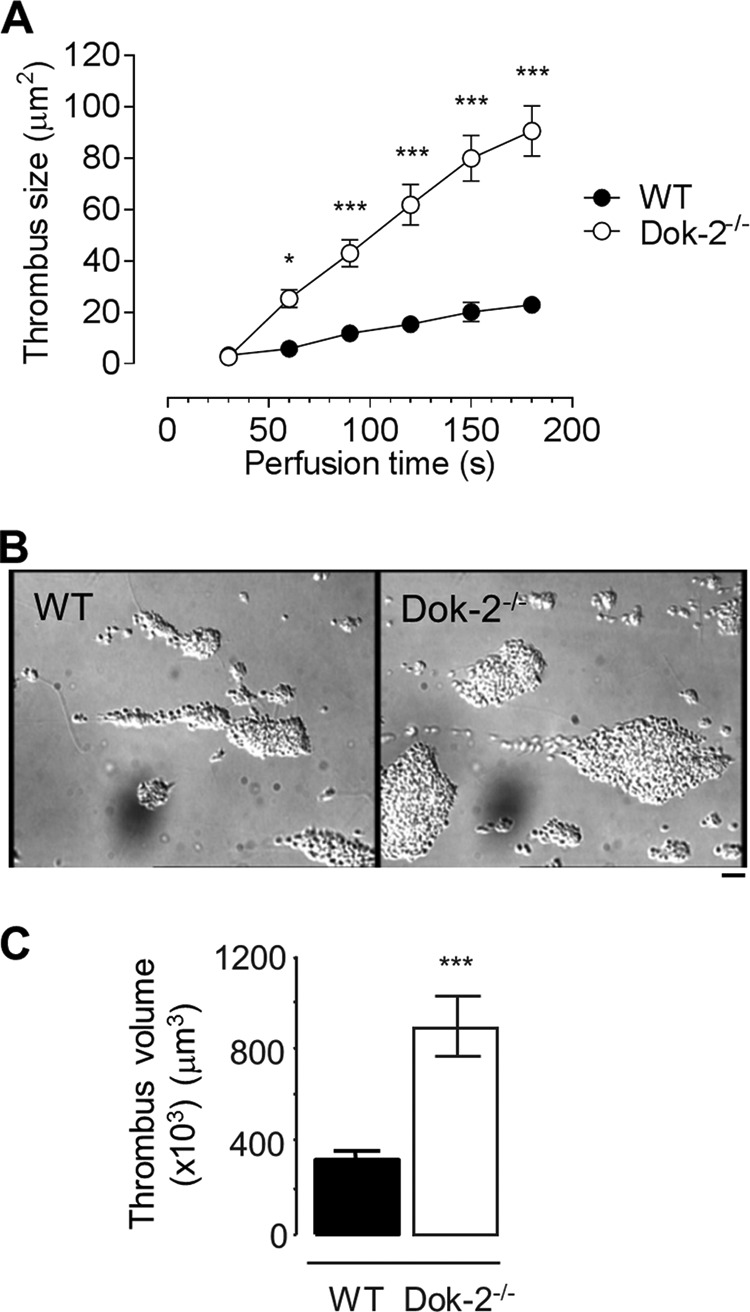
Dok-2−/− platelets form larger thrombi in vitro. Anticoagulated whole blood from WT or Dok-2−/− mice was perfused through type I collagen (10 μg/ml)-coated microslides at 1800 s−1 for 3 min. A, total surface area (in square micrometers) of thrombi forming over time (s) was quantified as described under “Experimental Procedures.” B, differential interference contrast images taken from paired, representative flows. Scale bar = 10 μm. C, following 3 min of perfusion, microslides were fixed and labeled with DiOC6 prior to confocal sectioning (1 μm) to determine total thrombus volume. Data are mean ± S.E. (n = 3). ***, p < 0.001.
Dok-2 Deficiency Results in Enhanced Platelet-Platelet Interactions under Flow
To investigate whether Dok-2−/− platelets had enhanced reactivity to collagen under flow, primary platelet adhesion studies were performed on an immobilized collagen matrix under experimental conditions preventing platelet aggregation (Fig. 2). Analysis of the number of platelets recruited to the collagen substrate revealed no difference in platelet adhesion between Dok-2−/− platelets and matched controls (Fig. 2A). In contrast, when anticoagulated whole blood from Dok-2−/− mice and WT controls were perfused over the surface of preformed thrombi, platelet recruitment to the thrombus surface was enhanced significantly in Dok-2−/− platelets (Fig. 2B), as reflected by the increased lifetime of adhesive interactions, raising the possibility that Dok-2 plays a role in regulating the shear-dependent adhesive interactions between aggregating platelets.
FIGURE 2.
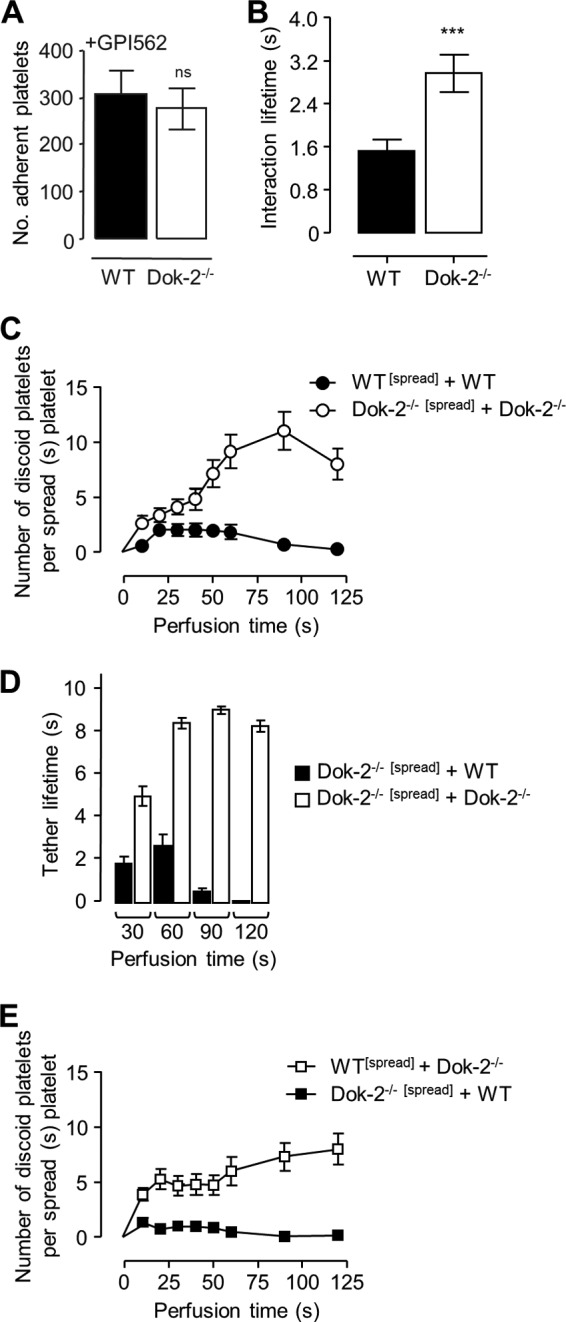
Dok-2−/− platelets exhibit increased platelet-platelet interactions through an increased αIIbβ3-dependent tether lifetime. A, whole blood from WT or Dok-2−/− mice was pretreated with the integrin αIIbβ3 inhibitor GPI562 (10 μm) prior to perfusion through type I collagen (50 μg/ml)-coated microslides at 1,800 s−1 for 3 min, and the number of adherent platelets (per 25% of field) was determined as described under “Experimental Procedures.” ns, not significant. B, WT whole blood was perfused over immobilized collagen (50 μg/ml) for 1 min to allow thrombi to form. Non-adherent platelets were removed, and WT or Dok-2−/− whole blood was perfused over the preformed thrombi. The interaction lifetime (s) of adherent platelets was determined by quantifying the number of frames for which a platelet remained attached to preformed thrombi (25 frames = 1 s). ***, p < 0.001. C–E, whole blood from WT or Dok-2 mice was perfused across spread platelets of the same genotype (●, WT[spread]+WT; ○, Dok-2−/−[spread] + Dok-2−/−) or alternative genotypes (■, Dok-2−/−[spread]+WT; □, WT[spread] + Dok-2−/−). C and E, the number of incoming platelets attached to a spread ([spread]) platelet of the indicated genotype. D, the tether lifetime of adherent platelets (s), which was determined by quantifying the number of frames for which a platelet remained attached to a spread platelet (25 frames = 1 s). These results represent six independent experiments (mean ± S.E.).
To further investigate the impact of Dok-2 deficiency on platelet-platelet interactions under flow, experiments were carried out using a two-stage perfusion assay in which an initial population of firmly adherent spread platelets was established (indicated as “[spread]”), followed by perfusion of a second platelet population over the stably adherent population (26). Perfusion of WT platelets over the surface of WT spread platelets resulted in the formation of predominantly transient adhesive interactions between adhering platelets (Fig. 2C, ●, and supplemental Fig. S3). In contrast, perfusion of Dok-2−/− platelets over Dok-2−/− spread platelet monolayers was associated with a marked increase in the number of platelets forming sustained adhesion contacts with the spread platelet surface (supplemental Fig. S3), with a tendency for these platelets to form small aggregates (Fig. 2C, ○). The enhanced adhesion response was primarily due to the increased adhesion lifetimes of perfused Dok-2−/− platelets (Fig. 2D) rather than an alteration in the reactivity of spread platelets because WT platelets perfused over a Dok-2−/− platelet monolayer formed transient adhesive interactions (Fig. 2E). In further control studies, we confirmed that GPIb-dependent tethering of WT and Dok-2−/− platelets to spread platelet monolayers was similar (supplemental data S3, B and C), whereas subsequent integrin αIIbβ3-dependent stabilization of platelet adhesion contacts was specifically increased with Dok-2−/− platelets (supplemental data S3, B and C).
Enhanced Shear-dependent Adhesion of Dok-2−/− Platelets to Immobilized Fibrinogen
To investigate more directly the impact of Dok-2 deficiency on integrin αIIbβ3 adhesive function under flow, we performed perfusion studies on a purified fibrinogen matrix. Immobilized fibrinogen selectively binds integrin αIIbβ3 and induces conformational changes in the receptor that initiates outside-in signaling events to stimulate platelet activation (33). As demonstrated in Fig. 3A, Dok-2 deficiency was associated with a marked up-regulation in shear-dependent platelet adhesion to fibrinogen at each of the time points examined (Fig. 3A and supplemental Movie 1). This increase in adhesion was observed over a broad range of fibrinogen-coating concentrations (5–100 μg/ml) and was associated with enhanced platelet spreading (Fig. 3, B and C, and supplemental Fig. S4) and an increased propensity to form platelet aggregates, particularly at higher matrix densities (100 μg/ml).3 The increased adhesion of Dok-2−/− platelets required platelet activation because it was inhibited by pretreating platelets with the activation inhibitors PGE1 and theophylline.3 In further control studies, we confirmed that the activation state of integrin αIIbβ3, as assessed by Oregon green fibrinogen (supplemental Fig. S2, C–E) or JON/A3 binding was no different on the surface of resting or activated platelets relative to matched controls. Furthermore, under static conditions, there was no significant difference in the level of adhesion or spreading of Dok-2−/− platelets relative to WT controls (supplemental Fig. S4),3 indicating that Dok-2 deficiency leads to a shear-selective increase in platelet adhesion.
FIGURE 3.
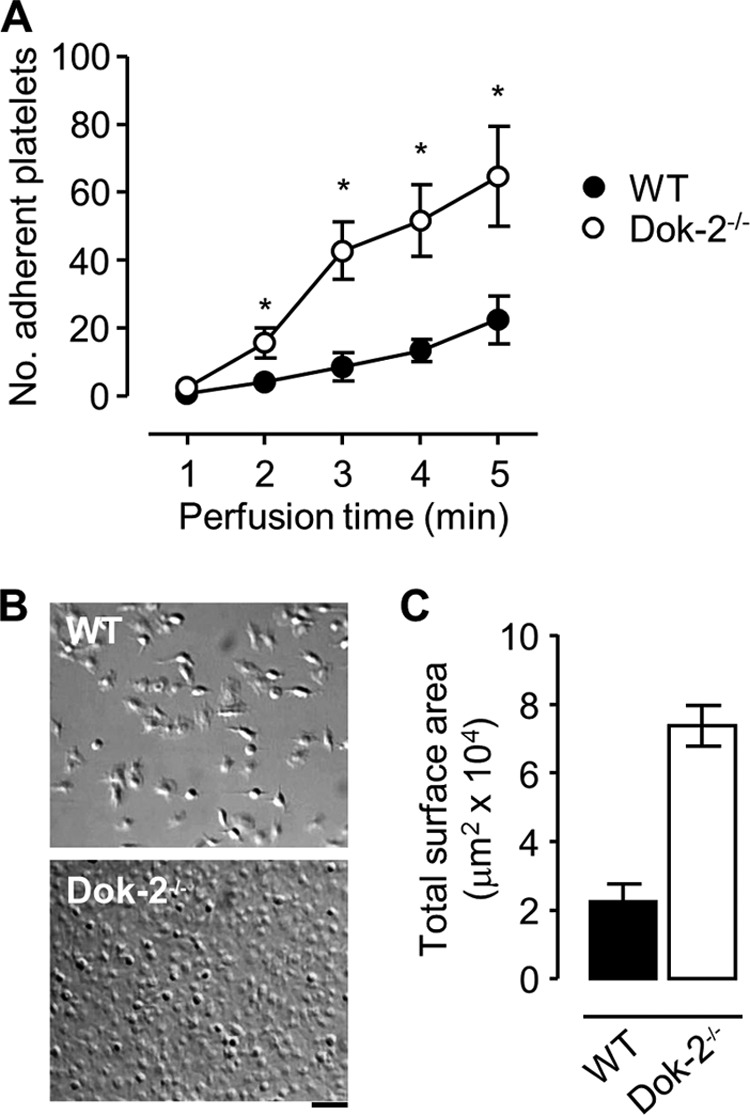
Dok-2 negatively regulates integrin αIIbβ3-dependent platelet adhesion under shear conditions. Anticoagulated whole blood from WT or Dok-2−/− mice was perfused through fibrinogen (20 or 100 μg/ml)-coated microslides at 600 s−1. A, the number of WT (●) or Dok-2−/− (○) platelets adherent (≥2 s) to fibrinogen (20 μg/ml) during whole blood perfusion (mean ± S.E.; n = 4; *, p < 0.05). Quantification of platelet adhesion was performed as described under “Experimental Procedures.” B, representative cropped images taken from one paired flow on a fibrinogen matrix (100 μg/ml) following modified Tyrode buffer washout. Scale bar = 10 μm. C, the total surface area coverage of adherent platelets. These data are taken from three random fields per experiment and are representative of five independent experiments (mean ± S.E.).
Dok-2 Deficiency Enhances the Shear-dependent Adhesion of Discoid Platelets
To gain insight into the mechanism by which Dok-2 regulates platelet adhesive function under flow, we performed high-magnification, real-time imaging of mouse platelets interacting with immobilized fibrinogen. At 600 s, the majority of WT mouse platelets adhered to immobilized fibrinogen in a reversible manner, with ∼60% of platelets translocating or rolling on the fibrinogen surface in a sliding or flip-flop rotational manner (Fig. 4) (34). In control studies, we confirmed that mouse translocation was not due to contaminating von Willebrand factor in the fibrinogen preparation because it was not significantly altered by blocking the ligand-binding function of platelet GPIb (supplemental Fig. S5).3 The translocation behavior of Dok-2−/− platelets was distinct from WT controls in that twice as many Dok-2−/− platelets formed sustained adhesion contacts with the fibrinogen substrate (Fig. 4B), resulting in a 3-fold lower translocation velocity (Fig. 4C). In addition to altered translocation dynamics, Dok-2−/− platelets exhibited greater stability on the fibrinogen substrate, with 41.79 ± 3.14% of Dok-2−/− platelets detaching from the fibrinogen substrate versus 80.86 ± 3.651% for WT controls (p < 0.0001) (Fig. 4D).
FIGURE 4.
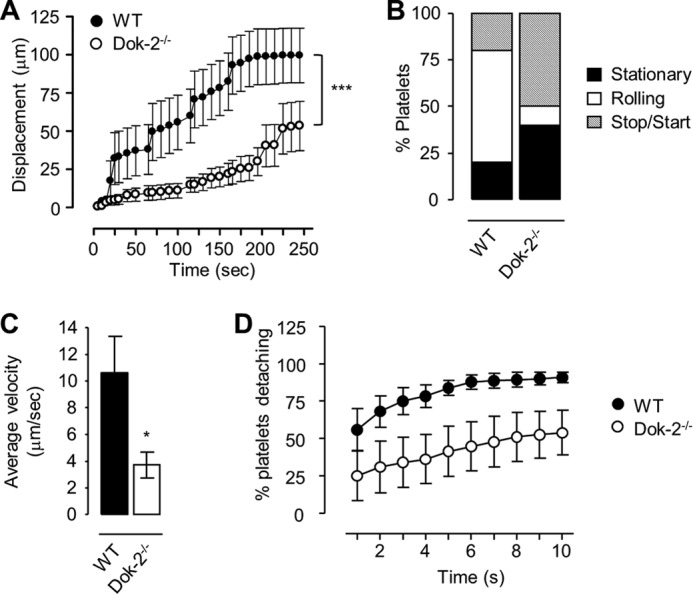
Mouse platelets translocate over a fibrinogen matrix, with Dok-2−/− platelets demonstrating increased stability and stationary adhesion. Anticoagulated whole blood from WT (●) or Dok-2−/− (○) mice was perfused over fibrinogen (20 μg/ml)-coated microslides at 600 s−1. A, the displacement (mean ± S.E.) of WT and Dok-2−/− platelets, every 5 frames, observed over a 10-s period (250 frames), after 4 min of flow. ***, p < 0.001. B, the percentage of platelets exhibiting rolling, stop/start, or stationary translocation dynamics. C, the average velocity of WT and Dok-2−/− platelets. *, p < 0.05. D, the percentage of WT (●) or Dok-2−/− (○) platelets detaching from the fibrinogen matrix (20 μg/ml) over a 10-s time interval. Data represent mean ± S.E. (n = 3).
Although high-magnification imaging revealed no significant differences in the morphology of Dok-2−/− or WT platelets during the initial stages of surface translocation on immobilized fibrinogen, SEM confirmed the presence of membrane tethers in a high proportion of discoid platelets adhering to the fibrinogen substrate (Fig. 5A). Although there was no measurable difference in tether width between WT and Dok-2−/− platelets (WT, 0.1659 ± 0.01155 μm, n = 28; Dok-2−/−, 0.1784 ± 0.01843 μm, n = 21), there was a significant difference in tether length, with Dok-2−/− tethers ∼25% longer than their WT counterparts (WT, 1.678 ± 0.1245 μm, n = 28; Dok-2−/−, 2.213 ± 0.1692 μm, n = 21, p < 0.05).
FIGURE 5.
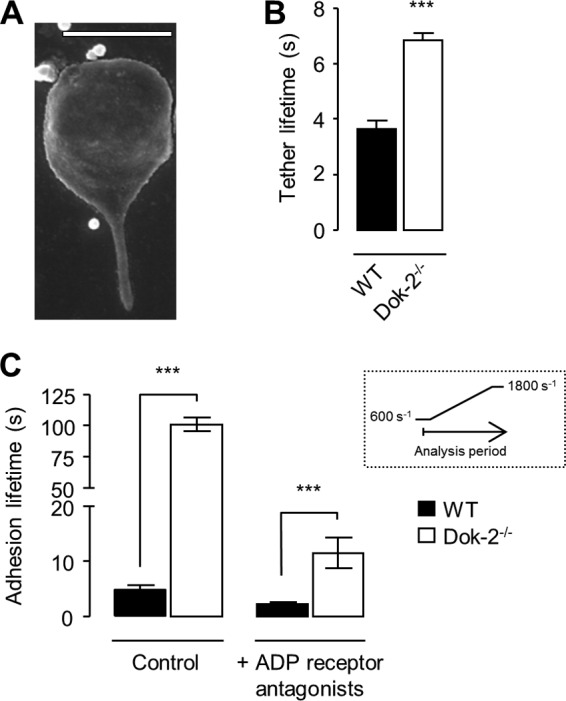
Dok-2 deficiency leads to an increased adhesion lifetime of platelet membrane tethers. Anticoagulated whole blood from WT or Dok-2−/− mice was perfused over fibrinogen (20 μg/ml)-coated microslides at 600 s−1. A, a cropped SEM image of a representative murine platelet tether (WT). The image was taken from one experiment representative of three. Scale bar = 2 μm. B, tether adhesion lifetime was determined by counting the number of frames for which a platelet was tethered to the matrix, as described under “Experimental Procedures.” ***, p < 0.001. C, whole blood was perfused over fibrinogen in the presence or absence of ADP receptor antagonists (MRS2179 (400 μm) and 2-methylthioadenosine 5′-monophosphate triethylammonium salt (2MeSAMP) (40 μm)) and apyrase (0.02 units/ml) at 600 s−1 for 2 min. The shear rate was increased to 1800 s−1 for 3 min. The histogram shows the time (s) platelets remained attached (adhesion time) during the shear rate increase as a measure of bond strength. The inset shows a schematic of the analysis period. Data represent the mean ± S.E., n = 3. ***, p < 0.001.
Analysis of the adhesion lifetime of membrane tethers to the fibrinogen matrix revealed an ∼80% increase in tether lifetimes in Dok-2−/− platelets relative to WT controls (Fig. 5B), consistent with more stable platelet adhesion. To test this hypothesis further, the effects of exposing WT and Dok-2−/− adherent platelets to sudden shear increases (from 600–1800 s) was investigated (Fig. 5C). Analysis of the number of platelets that resisted detachment from the matrix revealed a marked difference between WT and Dok-2−/− platelets, with Dok-2−/− platelets exhibiting an ∼20-fold increase in the duration of stationary adhesion relative to WT controls (Fig. 5C). Adhesion stability of platelets on a fibrinogen matrix is partly regulated by the release of dense granule ADP (31), and, although adhesion stability was reduced by pretreating platelets with ADP receptor antagonists (Fig. 5C), the relative difference in adhesion between WT and Dok-2−/− platelets remained. These findings suggest that Dok-2 plays an important role in regulating the stability of integrin αIIbβ3-fibrinogen interactions under flow.
Dysregulated Calcium Flux and PtdIns(3,4)P2 Accumulation in Dok-2−/− Platelets
We have demonstrated previously that integrin αIIbβ3 adhesion contacts are negatively regulated by the Src family member Lyn kinase and the Src homology 2 domain-containing inositol 5-phosphatase (SHIP1) (30), both of which have been linked previously to Dok-2 function in lymphocytes (35–38). To investigate whether Dok-2 phosphorylation in platelets is regulated by Lyn kinase, anti-phosphotyrosine immunoprecipitation studies were performed on Lyn-deficient platelets. Phosphotyrosine-containing proteins were immunoprecipitated from Lyn+/+ and Lyn−/− platelet lysates, and the levels of Dok-2 in these immunoprecipitates were determined by immunoblot analysis. These studies revealed a major reduction in the level of tyrosine-phosphorylated Dok-2 in Lyn−/− platelets (Fig. 6A).
FIGURE 6.

Dok-2 regulates platelet calcium flux and PI(3,4,5)P3 metabolism. A, washed Lyn+/+ and Lyn−/− mouse platelets (5.0 × 108/ml) were left untreated (Rest) or stimulated with thrombin (Thr, 1 unit/ml). Tyrosine-phosphorylated proteins were immunoprecipitated (4G10) from whole cell lysates and blotted for Dok-2 and Src (using 4G10) as described under “Experimental Procedures.” The immunoblot was taken from one experiment representative of three independent experiments, with densitometric quantification presented in the histogram (mean ± S.E., n = 3). B, washed WT and Dok-2−/− platelets were loaded with 32P, stimulated with thrombin (1 unit/ml, 2 min), lysed, and then 32P-labeled phospholipids were extracted and analyzed by strong anion exchange-HPLC as described under “Experimental Procedures.” The histogram depicts the ratio of PtdIns(3,4)P2 to PtdIns(3,4,5)P3 (mean ± S.E., n = 3). *, p < 0.05. C–E, washed WT and Dok-2−/− mouse platelets (2.5 × 108/ml) were loaded with calcium dyes (Oregon green 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid and FuraRed), and their calcium concentrations (nanomolar) determined under basal and agonist-stimulated (C) or shear (D and E) conditions, as described under “Experimental Procedures.” C, platelets were allowed to rest in the presence or absence of calcium-chelating agents (EGTA (1 mm) and EDTA (1 mm)) and/or the inositol 1,4,5-trisphosphate receptor inhibitor 2-APB (10 μm). In some experiments, platelets were stimulated with thrombin (0.1 units/ml) prior to the determination of cytosolic calcium levels (mean ± S.E., n = 3). **, p < 0.01. D and E, washed platelets were reconstituted with red blood cells, perfused over fibrinogen (100 μg/ml) at 600 s−1, and then the calcium concentration of adherent platelets was determined as described under “Experimental Procedures.” Representative tracings of calcium flux are shown for individual platelets (D). Tracings are from one experiment representative of five. E, the number of calcium peaks ≥800 nm. Data are from 10 platelets/genotype (mean ± S.E., n = 5). *, p < 0.05.
Tyrosine phosphorylation of Dok-2 regulates its interaction with SHIP1 (37), the major 5-phosphatase in platelets regulating the conversion of PtdIns(3,4,5)P3 to PtdIns(3,4)P2. To investigate whether Dok-2 deficiency impacted the metabolic conversion of PtdIns(3,4,5)P3 to PtdIns(3,4)P2, 32P-labeled phospholipids were extracted from resting and thrombin-stimulated platelets, and the levels of 3-phosphorylated phosphoinositides quantified by strong anion exchange-HPLC (30, 39). These studies revealed that the conversion of PtdIns(3,4,5)P3 to PtdIns(3,4)P2 was less efficient in Dok-2−/− platelets (Fig. 6B), similar in magnitude to that reported in lymphocytes (38).
Changes in phosphatidylinositol 3-kinase lipid products lead to dysregulated integrin αIIbβ3 adhesive function and calcium signaling under flow (31). To examine potential alterations in cytosolic calcium levels in Dok-2−/− platelets, washed platelets were loaded with the calcium indicator dyes Oregon green 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid and FuraRed, and fluorescence changes were monitored by confocal microscopy (31, 40). As demonstrated in Fig. 6C, Dok-2−/− platelets displayed a 2-fold increase in basal calcium levels relative to WT controls. This increase was partially prevented by chelating extracellular calcium or by blocking the platelet inositol 1,4,5-trisphosphate receptor with 2-aminoethoxydiphenyl borate (2-APB) (Fig. 6C). However, complete reversal of the elevated calcium required the concurrent treatment of platelets with both EGTA and 2-APB (Fig. 6C). Analysis of cytosolic calcium flux during shear-dependent adhesion to fibrinogen revealed that Dok-2−/− platelets exhibited a greater frequency of cytosolic calcium flux (calcium ≥800 nm) relative to WT controls (Fig. 6, D and E). This latter difference in calcium dynamics was specific to shear-activated platelets because the extent of cytosolic calcium flux in platelets stimulated with soluble agonists, including thrombin, collagen-related peptide, or ADP, was identical between WT and Dok-2−/− platelets (supplemental Fig. S6). These findings suggest an important role for Dok-2 in regulating shear-dependent calcium flux in platelets.
Dok-2 Deficiency Stabilizes Discoid Platelet Aggregates and Accelerates Thrombus Growth in Vivo
To investigate the potential pathophysiological significance of our in vitro findings, we examined platelet thrombus formation in Dok-2−/− mice using several distinct in vivo thrombosis models (27, 28, 41, 42). Analysis of thrombus development in a mouse carotid artery electrolytic thrombosis model (28) revealed that Dok-2−/− mice developed thrombi more rapidly than their WT counterparts, as indicated by a more rapid reduction in blood flow following electrolytic injury (Fig. 7A, time 0). Full vascular occlusion was also more rapid in Dok-2−/− mice with cessation of blood flow after ∼10 min of injury compared with ∼17 min in WT controls.
FIGURE 7.

Dok-2−/− mice show increased thrombus formation in vivo. A, mean carotid artery blood flow (milliliters/minute/100 grams) in WT or Dok-2−/− mice was monitored following electrolytic injury (Injury & stasis), with data depicting the mean ± S.E. (WT, n = 7; Dok-2−/−, n = 8). B, discoid platelet aggregate formation in murine mesenteric venules (imaged using intravital microscopy) was induced by rose bengal-mediated photoactivation (see “Experimental Procedures”). The dot plot shows the time taken (in increments of 10 s) for complete disaggregation of discoid platelets to occur (i.e. the first time, when no platelets remained adherent to the site of injury). We assigned an arbitrary maximum value of 300 s. Data are mean ± S.E. *, p < 0.05, n = 6–8 mice/genotype (as indicated). C and D, platelet aggregate formation was induced by a mechanical needle puncture, and subsequent platelet accrual was monitored by intravital microscopy (see “Experimental Procedures”). C, representative images taken from WT and Dok-2−/− vessels at 0, 125, and 250 s after injury. For clarity, platelet aggregates are demarcated. D, the cumulative size of forming aggregates (square micrometers) measured over 10-s intervals for a maximum of 250 s (1 frame = 1 s). Data depict the mean ± S.E. WT = 25 injuries (n = 6), Dok-2−/− = 23 injuries (n = 5). ns, p > 0.05; **, p < 0.01; ***, p < 0.001.
To investigate whether enhanced thrombus growth in Dok-2−/− mice is related to enhanced stability of discoid platelet aggregates, we employed high-magnification intravital microscopy to monitor platelet aggregation dynamics in the mouse mesenteric microcirculation. We chose photoactivation of systemically administered rose bengal to induce platelet aggregation because we have demonstrated previously that this thrombosis model induces prominent aggregation of discoid platelets during the earliest phases of thrombus development (42). In postcapillary venules, the adhesion and aggregation of discoid platelets at sites of photochemical injury was rapid, with significant aggregates forming within 30 s of photoactivation. In WT mice, these aggregates were very transient, with an approximate 45% reduction in aggregate size within the first 10 s of the cessation of photoillumination (124.4 ± 31.88 platelets at time 0 and 81.13 ± 31.55 platelets at time 10 s) compared with a 12% reduction in Dok-2−/− mice (195.0 ± 29.72 platelets at time 0 and 172.0 ± 25.18 platelets at time 10 s, p < 0.05). Furthermore, although discoid platelet aggregates in both WT and Dok-2−/− mice remained unstable, the mean time to complete disaggregation was significantly higher in Dok-2−/− mice relative to WT controls so that >80% of residual Dok-2−/− platelet aggregates persisted for at least 300 s following cessation of photoillumination compared with <20% for WT mice (Fig. 7B). In control studies, we established that the rose bengal-induced aggregates in WT and Dok-2−/− mice were primarily composed of P-selectin-negative platelets,3 consistent with previous findings of a low level of platelet stimulation during the early stages of thrombus development (41).
To further investigate the role of Dok-2 in regulating discoid platelet aggregation at sites of vascular injury, we employed a microinjector needle injury model in the mesenteric circulation of mice that leads to the initial rapid formation of discoid platelet aggregates at sites of vessel puncture. As demonstrated in Fig. 7, C and D, Dok-2 deficiency resulted in a marked increase in the rate and extent of platelet aggregation. Moreover, similar to our findings in the rose bengal model, these discoid platelet aggregates were more stable than WT controls, leading to the formation of persistent, large platelet aggregates (Fig. 7, C and D) (supplemental Movie 2). These findings, in combination with our in vitro studies, support an important role for Dok-2 in regulating discoid platelet aggregation and subsequent thrombus growth.
DISCUSSION
A growing body of experimental evidence suggests that platelet aggregation can be induced by two distinct, complementary mechanisms: a biomechanical platelet aggregation mechanism induced by hemodynamic shear stress and a soluble agonist-dependent mechanism (43). The former is particularly sensitive to microscale shear gradients and involves the aggregation of discoid, non-degranulated platelets. This aggregation mechanism is dynamic and reversible and is critically dependent on the biomechanical adhesive function of both GPIb and integrin αIIbβ3. In contrast, soluble agonist-induced platelet aggregation occurs between shape-changed platelets, is primarily mediated by high-affinity integrin αIIbβ3 bonds, and typically leads to platelet degranulation and the development of more stable platelet aggregates. This study define a new prothrombotic platelet phenotype in Dok-2-deficient mice that involves enhanced integrin αIIbβ3-dependent, biomechanical platelet activation, leading to more stable discoid platelet aggregation and an accelerated rate of thrombus growth in vivo. Notably, this prothrombotic phenotype occurred independently of changes in soluble agonist-dependent platelet aggregation and was, therefore, not detected by conventional platelet functional assays.
By employing flow-based platelet functional assays, we demonstrated an important role for Dok-2 in negatively regulating the stability of integrin αIIbβ3 adhesion contacts, leading to increased membrane tether lifetimes and more efficient recruitment of discoid platelets onto the surface of forming thrombi. Mechanistically, our studies suggest that the increased reactivity of Dok-2−/− platelets is due to enhanced integrin αIIbβ3 bond stability and more stable discoid platelet aggregation that is coincident with an exaggerated cytosolic calcium response. Several lines of evidence suggest that the principal alteration in platelet adhesive function in Dok-2−/− mice is related to integrin αIIbβ3. First, we demonstrated a shear-specific increase in platelet adhesion to the integrin αIIbβ3-selective ligand, fibrinogen. Second, we observed no difference in shear-dependent platelet tethering through the von Willebrand factor-GPIb interaction or subsequent GPVI- and integrin α2β1-dependent firm adhesion to immobilized collagen in Dok-2−/− mice. Third, Dok-2−/− platelets exhibit a shear-specific increase in stable adhesion to platelet monolayers and to the surface of formed thrombi in vitro through an adhesive process dependent on integrin αIIbβ3. Notably, there was no difference in the tethering of discoid Dok-2−/− platelets to spread platelet monolayers or thrombi, consistent with normal von Willebrand factor-GPIb adhesive function in these mice. Furthermore, all shear-specific increases in adhesive function of Dok-2−/− platelets were observed with discoid platelets. Thus, given that biomechanical platelet activation processes relevant to shear-dependent aggregation of discoid platelets occurs primarily through GPIb and integrin αIIbβ3 (41, 43), the most likely explanation for our experimental findings is that Dok-2 is primarily regulating the biomechanical adhesive function of integrin αIIbβ3.
Recent experimental evidence supports an important role for discoid platelet aggregates in promoting thrombus growth in vivo (26, 41). These aggregates appear to be dependent on the formation of membrane tethers (smooth cylinders of lipid bilayer pulled from the rim of discoid platelets under the influence of hemodynamic forces (26, 41)). Membrane tethers appear to play a major role in regulating the adhesive function of discoid platelets by acting as sites of localized cell attachment and activation (41). This study suggest that Dok-2 plays an important role in regulating the lifetime of membrane tether bonds, thereby promoting more efficient discoid platelet aggregation under flow. Our current working hypothesis is that integrin αIIbβ3 adhesion bonds formed on the surface of Dok-2−/− discoid platelets have greater tensile strength, presumably because of localized changes in the number or stability of integrin bonds, resulting in increased tether bond lifetimes and more elongated membrane tethers. This may, in turn, contribute to an increase in mechanical resistance inferred on the Dok-2−/− tethers, which prolongs their lifetime and the stability of the platelet-fibrinogen interaction. Ongoing studies in our laboratory are attempting to address this issue.
Several lines of evidence indicate that the enhanced aggregation and thrombotic response of Dok-2−/− platelets primarily reflect a shear-specific enhancement in integrin αIIbβ3 adhesive function rather than a global up-regulation in integrin αIIbβ3 affinity. For example, integrin αIIbβ3-dependent functional responses under static or low-shear conditions, such as aggregation in an aggregometer (supplemental Fig. S2), spreading (supplemental Fig. S3), or clot retraction,3 were not significantly altered in Dok-2−/− platelets. In contrast, integrin αIIbβ3-dependent platelet adhesion to immobilized fibrinogen, onto the surface of spread platelets or to preformed thrombi, was always enhanced under shear conditions. Notably, the alterations in integrin αIIbβ3 adhesive function in Dok-2−/− platelets appeared to be related to discoid platelets and most likely reflect alterations in post-ligand binding events (outside-in signaling) rather than changes in integrin αIIbβ3 affinity (inside-out signaling). Consistent with this, the initial formation of platelet-fibrinogen contacts under flow (platelet tethering) was similar in WT and Dok-2−/− platelets (data not shown). Similarly, agonist-induced affinity regulation of integrin αIIbβ3 and the rate and extent of platelet aggregation in aggregometry assays was normal in Dok2−/− platelets, indicating a minimal contribution of Dok2 to integrin αIIbβ3 affinity regulation. This conclusion was further supported by competition binding assays wherein excess unlabeled fibrinogen was equally effective at displacing JON/A binding to activated integrin αIIbβ3 on the surface of WT and Dok-2−/− platelets.3 Overall, our findings are more consistent with a role for Dok-2 in regulating biomechanical signaling processes linked to integrin αIIbβ3 adhesive function that serves to regulate the stability of localized integrin αIIbβ3 adhesion contacts under flow.
The demonstration that Dok-2−/− platelets have increased basal cytosolic calcium levels and an increased frequency of calcium transients following shear-dependent adhesion suggests a role for Dok-2 in regulating cytosolic calcium flux. Such findings support a growing body of evidence that Dok family members modulate cytosolic calcium levels in multiple cell types through the formation of multimolecular signaling complexes (37). For example, Dok-3 has been demonstrated to modulate cytosolic calcium flux in B cells and DT40 cells (44, 45) through an association with the adaptor protein Grb2, leading to the localized negative regulation of Btk and the impaired activation of PLCγ2 (45). Although Dok-1 and Dok-3 have been demonstrated to associate with Grb2 in platelets (17), whether a Dok-2/Grb2 complex regulates similar signaling processes downstream of integrin αIIbβ3 in platelets is unclear. Dok-2 has also been reported to associate with the tyrosine kinase Lyn (46) and the SH2 domain-containing inositol 5-phosphatase SHIP1 (17, 37, 38, 46, 47), with such an association demonstrated to negatively regulate CD4-mediated signaling in T cells, leading to modulation of calcium flux and PtdIns(3,4,5)P3 levels (38). The findings presented here provide evidence that such a signaling complex may be responsible for the negative modulation of integrin αIIbβ3 in platelets. For example, the phenotype displayed by Dok-2−/− platelets, with increased calcium flux and enhanced integrin αIIbβ3 function, is consistent with the findings from SHIP1- or Lyn-deficient mice (30, 48), suggestive of a functionally relevant signaling complex between these molecules. Furthermore, tyrosine phosphorylation of SHIP1 is reduced by >70% in Lyn−/− platelets (30), concomitant with reduced SHIP1 activity and PtdIns(3,4,5)P3 metabolism. We also demonstrate that Dok-2 association with other tyrosine phosphorylated proteins is reduced in Lyn−/− platelets along with a concomitant transient reduction in PtdIns(3,4,5)P3 metabolism. When taken together with the well defined role of PtdIns(3,4,5)P3 in regulating calcium flux (49, 50) and the similarities in platelet phenotype between SHIP1-, Lyn-, and Dok-2-deficient mice (30, 37), these findings all point toward an important cooperative role for Dok-2, SHIP1, and Lyn in modulating calcium flux and integrin function.
Overall, our studies have demonstrated an important functional role for Dok-2 in regulating the biomechanical adhesive function of discoid platelets, serving as an endogenous negative regulator of integrin αIIbβ3. Notably, all other reported negative regulators of integrin αIIbβ3 (30, 51, 52) influence integrin αIIbβ3 adhesive function under both static and shear conditions, suggesting that Dok-2 represents a bona fide mechanosensory platelet signaling molecule. A similar concept has been developed for the signaling modulator adhesion and degranulation promoting adaptor protein (ADAP), which primarily regulates integrin αIIbβ3 outside-in biomechanical signals, although in contrast to Dok-2, the adhesion and degranulation promoting adaptor protein serves as a positive regulator of integrin αIIbβ3 signaling (53). Our findings of an exaggerated biomechanical activation response in Dok-2-deficient platelets may have potentially important implications for the identification of prothrombotic platelet phenotypes. A link between increased platelet reactivity and adverse cardiovascular events is well established (reviewed in Ref. 54). More specifically, enhanced integrin αIIbβ3 activation, platelet aggregation, P-selectin expression, and thromboxane A2 generation following agonist stimulation are associated with an increased risk of arterial thrombosis and cardiovascular disease (54). Typically, these changes in platelet reactivity are detected using routine platelet functional assays, including platelet aggregometry and flow cytometry. Our findings raise the interesting possibility that shear-dependent platelet functional assays may be required to identify dysregulated biomechanical platelet activation mechanisms linked to a prothrombotic phenotype. Such assays may uncover a broader range of platelet hyperactivity disorders than currently appreciated.
Supplementary Material
Acknowledgments
We thank Alfred Medical Research and Education Precinct Animal Services staff for breeding, caring for, and maintaining the Dok-2−/− breeding colony; Dr. Margaret Hibbs for Lyn−/− mice; Joan Clarke of Monash Micro Imaging (Monash University, Clayton, Victoria, Australia) for assistance with scanning electron microscopy images; and Dr. David Bark, Dr. Saheb Al-Daher, Amrita Sran, and Dr. Fu Jia for technical assistance.
This work was supported by a National Health and Medical Research Council Australia project grant (to S. C. H. and W. S. N.), by a C. J. Martin fellowship (to S. C. H.), by a career development award (biomedical) (to S. C. H.), and by an Australia fellowship (to S. P. J.).

This article contains supplemental Figs. S1–S6, Movies 1 and 2, Experimental Procedures, and References.
S. C. Hughan, S. Sturgeon, S. M. Schoenwaelder, and S. P. Jackson, unpublished observations.
- SH2
- Src homology domain 2
- 2-APB
- 2-aminoethoxydiphenyl borate
- PtdIns
- phosphatidylinositol.
REFERENCES
- 1. Ruggeri Z. M. (2002) Platelets in atherothrombosis. Nat. Med. 8, 1227–1234 [DOI] [PubMed] [Google Scholar]
- 2. Bhatt D. L., Topol E. J. (2003) Scientific and therapeutic advances in antiplatelet therapy. Nat. Rev. Drug Discov. 2, 15–28 [DOI] [PubMed] [Google Scholar]
- 3. Jackson S. P., Schoenwaelder S. M. (2003) Antiplatelet therapy. In search of the “magic bullet”. Nat. Rev. Drug Discov. 2, 775–789 [DOI] [PubMed] [Google Scholar]
- 4. De Meyer S. F., Vanhoorelbeke K., Broos K., Salles I. I., Deckmyn H. (2008) Antiplatelet drugs. Br. J. Haematol. 142, 515–528 [DOI] [PubMed] [Google Scholar]
- 5. Coller B. S., Shattil S. J. (2008) The GPIIb/IIIa (integrin αIIbβ3) odyssey. A technology-driven saga of a receptor with twists, turns, and even a bend. Blood 112, 3011–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kasirer-Friede A., Kahn M. L., Shattil S. J. (2007) Platelet integrins and immunoreceptors. Immunol. Rev. 218, 247–264 [DOI] [PubMed] [Google Scholar]
- 7. Shattil S. J., Newman P. J. (2004) Integrins. Dynamic scaffolds for adhesion and signaling in platelets. Blood 104, 1606–1615 [DOI] [PubMed] [Google Scholar]
- 8. Di Cristofano A., Carpino N., Dunant N., Friedland G., Kobayashi R., Strife A., Wisniewski D., Clarkson B., Pandolfi P. P., Resh M. D. (1998) Molecular cloning and characterization of p56dok-2 defines a new family of RasGAP-binding proteins. J. Biol. Chem. 273, 4827–4830 [DOI] [PubMed] [Google Scholar]
- 9. Lemay S., Davidson D., Latour S., Veillette A. (2000) Dok-3, a novel adapter molecule involved in the negative regulation of immunoreceptor signaling. Mol. Cell. Biol. 20, 2743–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cai D., Dhe-Paganon S., Melendez P. A., Lee J., Shoelson S. E. (2003) Two new substrates in insulin signaling, IRS5/DOK4 and IRS6/DOK5. J. Biol. Chem. 278, 25323–25330 [DOI] [PubMed] [Google Scholar]
- 11. Carpino N., Wisniewski D., Strife A., Marshak D., Kobayashi R., Stillman B., Clarkson B. (1997) p62(dok). A constitutively tyrosine-phosphorylated, GAP-associated protein in chronic myelogenous leukemia progenitor cells. Cell 88, 197–204 [DOI] [PubMed] [Google Scholar]
- 12. Grimm J., Sachs M., Britsch S., Di Cesare S., Schwarz-Romond T., Alitalo K., Birchmeier W. (2001) Novel p62dok family members, dok-4 and dok-5, are substrates of the c-Ret receptor tyrosine kinase and mediate neuronal differentiation. J. Cell Biol. 154, 345–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Okada K., Inoue A., Okada M., Murata Y., Kakuta S., Jigami T., Kubo S., Shiraishi H., Eguchi K., Motomura M., Akiyama T., Iwakura Y., Higuchi O., Yamanashi Y. (2006) The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science 312, 1802–1805 [DOI] [PubMed] [Google Scholar]
- 14. Calderwood D. A., Fujioka Y., de Pereda J. M., García-Alvarez B., Nakamoto T., Margolis B., McGlade C. J., Liddington R. C., Ginsberg M. H. (2003) Integrin β cytoplasmic domain interactions with phosphotyrosine-binding domains. A structural prototype for diversity in integrin signaling. Proc. Natl. Acad. Sci. U.S.A. 100, 2272–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. García A., Prabhakar S., Hughan S., Anderson T. W., Brock C. J., Pearce A. C., Dwek R. A., Watson S. P., Hebestreit H. F., Zitzmann N. (2004) Differential proteome analysis of TRAP-activated platelets. Involvement of DOK-2 and phosphorylation of RGS proteins. Blood 103, 2088–2095 [DOI] [PubMed] [Google Scholar]
- 16. Hughan S. C., Watson S. P. (2007) Differential regulation of adapter proteins Dok2 and Dok1 in platelets, leading to an association of Dok2 with integrin αIIbβ3. J. Thromb. Haemost. 5, 387–394 [DOI] [PubMed] [Google Scholar]
- 17. Senis Y. A., Antrobus R., Severin S., Parguiña A. F., Rosa I., Zitzmann N., Watson S. P., García A. (2009) Proteomic analysis of integrin αIIbβ3 outside-in signaling reveals Src-kinase-independent phosphorylation of Dok-1 and Dok-3 leading to SHIP-1 interactions. J. Thromb. Haemost. 7, 1718–1726 [DOI] [PubMed] [Google Scholar]
- 18. Bedirian A., Baldwin C., Abe J., Takano T., Lemay S. (2004) Pleckstrin homology and phosphotyrosine-binding domain-dependent membrane association and tyrosine phosphorylation of Dok-4, an inhibitory adapter molecule expressed in epithelial cells. J. Biol. Chem. 279, 19335–19349 [DOI] [PubMed] [Google Scholar]
- 19. Gérard A., Favre C., Garçon F., Némorin J. G., Duplay P., Pastor S., Collette Y., Olive D., Nunès J. A. (2004) Functional interaction of RasGAP-binding proteins Dok-1 and Dok-2 with the Tec protein tyrosine kinase. Oncogene 23, 1594–1598 [DOI] [PubMed] [Google Scholar]
- 20. Niki M., Di Cristofano A., Zhao M., Honda H., Hirai H., Van Aelst L., Cordon-Cardo C., Pandolfi P. P. (2004) Role of Dok-1 and Dok-2 in leukemia suppression. J. Exp. Med. 200, 1689–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shinohara H., Inoue A., Toyama-Sorimachi N., Nagai Y., Yasuda T., Suzuki H., Horai R., Iwakura Y., Yamamoto T., Karasuyama H., Miyake K., Yamanashi Y. (2005) Dok-1 and Dok-2 are negative regulators of lipopolysaccharide-induced signaling. J. Exp. Med. 201, 333–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yamanashi Y., Tamura T., Kanamori T., Yamane H., Nariuchi H., Yamamoto T., Baltimore D. (2000) Role of the rasGAP-associated docking protein p62(dok) in negative regulation of B cell receptor-mediated signaling. Genes Dev. 14, 11–16 [PMC free article] [PubMed] [Google Scholar]
- 23. Yasuda T., Bundo K., Hino A., Honda K., Inoue A., Shirakata M., Osawa M., Tamura T., Nariuchi H., Oda H., Yamamoto T., Yamanashi Y. (2007) Dok-1 and Dok-2 are negative regulators of T cell receptor signaling. Int. Immunol. 19, 487–495 [DOI] [PubMed] [Google Scholar]
- 24. Yasuda T., Shirakata M., Iwama A., Ishii A., Ebihara Y., Osawa M., Honda K., Shinohara H., Sudo K., Tsuji K., Nakauchi H., Iwakura Y., Hirai H., Oda H., Yamamoto T., Yamanashi Y. (2004) Role of Dok-1 and Dok-2 in myeloid homeostasis and suppression of leukemia. J. Exp. Med. 200, 1681–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao M., Janas J. A., Niki M., Pandolfi P. P., Van Aelst L. (2006) Dok-1 independently attenuates Ras/mitogen-activated protein kinase and Src/c-myc pathways to inhibit platelet-derived growth factor-induced mitogenesis. Mol. Cell. Biol. 26, 2479–2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maxwell M. J., Westein E., Nesbitt W. S., Giuliano S., Dopheide S. M., Jackson S. P. (2007) Identification of a 2-stage platelet aggregation process mediating shear-dependent thrombus formation. Blood 109, 566–576 [DOI] [PubMed] [Google Scholar]
- 27. Schoenwaelder S. M., Ono A., Sturgeon S., Chan S. M., Mangin P., Maxwell M. J., Turnbull S., Mulchandani M., Anderson K., Kauffenstein G., Rewcastle G. W., Kendall J., Gachet C., Salem H. H., Jackson S. P. (2007) Identification of a unique co-operative phosphoinositide 3-kinase signaling mechanism regulating integrin α IIb β 3 adhesive function in platelets. J. Biol. Chem. 282, 28648–28658 [DOI] [PubMed] [Google Scholar]
- 28. Sturgeon S. A., Jones C., Angus J. A., Wright C. E. (2006) Adaptation of the Folts and electrolytic methods of arterial thrombosis for the study of anti-thrombotic molecules in small animals. J. Pharmacol. Toxicol. Methods 53, 20–29 [DOI] [PubMed] [Google Scholar]
- 29. Hibbs M. L., Tarlinton D. M., Armes J., Grail D., Hodgson G., Maglitto R., Stacker S. A., Dunn A. R. (1995) Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell 83, 301–311 [DOI] [PubMed] [Google Scholar]
- 30. Maxwell M. J., Yuan Y., Anderson K. E., Hibbs M. L., Salem H. H., Jackson S. P. (2004) SHIP1 and Lyn kinase negatively regulate integrin α IIb β 3 signaling in platelets. J. Biol. Chem. 279, 32196–32204 [DOI] [PubMed] [Google Scholar]
- 31. Goncalves I., Hughan S. C., Schoenwaelder S. M., Yap C. L., Yuan Y., Jackson S. P. (2003) Integrin αIIb β 3-dependent calcium signals regulate platelet-fibrinogen interactions under flow. Involvement of phospholipase C γ 2. J. Biol. Chem. 278, 34812–34822 [DOI] [PubMed] [Google Scholar]
- 32. Dopheide S. M., Maxwell M. J., Jackson S. P. (2002) Shear-dependent tether formation during platelet translocation on von Willebrand factor. Blood 99, 159–167 [DOI] [PubMed] [Google Scholar]
- 33. Phillips D. R., Nannizzi-Alaimo L., Prasad K. S. (2001) β3 tyrosine phosphorylation in αIIbβ3 (platelet membrane GP IIb-IIIa) outside-in integrin signaling. Thromb. Haemost. 86, 246–258 [PubMed] [Google Scholar]
- 34. Savage B., Saldívar E., Ruggeri Z. M. (1996) Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 84, 289–297 [DOI] [PubMed] [Google Scholar]
- 35. Dong S., Corre B., Foulon E., Dufour E., Veillette A., Acuto O., Michel F. (2006) T cell receptor for antigen induces linker for activation of T cell-dependent activation of a negative signaling complex involving Dok-2, SHIP-1, and Grb-2. J. Exp. Med. 203, 2509–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lock P., Casagranda F., Dunn A. R. (1999) Independent SH2-binding sites mediate interaction of Dok-related protein with RasGTPase-activating protein and Nck. J. Biol. Chem. 274, 22775–22784 [DOI] [PubMed] [Google Scholar]
- 37. Mashima R., Hishida Y., Tezuka T., Yamanashi Y. (2009) The roles of Dok family adapters in immunoreceptor signaling. Immunol. Rev. 232, 273–285 [DOI] [PubMed] [Google Scholar]
- 38. Waterman P. M., Marschner S., Brandl E., Cambier J. C. (2012) The inositol 5-phosphatase SHIP-1 and adaptors Dok-1 and 2 play central roles in CD4-mediated inhibitory signaling. Immunol. Lett. 143, 122–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jackson S. P., Schoenwaelder S. M., Goncalves I., Nesbitt W. S., Yap C. L., Wright C. E., Kenche V., Anderson K. E., Dopheide S. M., Yuan Y., Sturgeon S. A., Prabaharan H., Thompson P. E., Smith G. D., Shepherd P. R., Daniele N., Kulkarni S., Abbott B., Saylik D., Jones C., Lu L., Giuliano S., Hughan S. C., Angus J. A., Robertson A. D., Salem H. H. (2005) PI 3-kinase p110β. A new target for antithrombotic therapy. Nat. Med. 11, 507–514 [DOI] [PubMed] [Google Scholar]
- 40. Yuan Y., Kulkarni S., Ulsemer P., Cranmer S. L., Yap C. L., Nesbitt W. S., Harper I., Mistry N., Dopheide S. M., Hughan S. C., Williamson D., de la Salle C., Salem H. H., Lanza F., Jackson S. P. (1999) The von Willebrand factor-glycoprotein Ib/V/IX interaction induces actin polymerization and cytoskeletal reorganization in rolling platelets and glycoprotein Ib/V/IX-transfected cells. J. Biol. Chem. 274, 36241–36251 [DOI] [PubMed] [Google Scholar]
- 41. Nesbitt W. S., Westein E., Tovar-Lopez F. J., Tolouei E., Mitchell A., Fu J., Carberry J., Fouras A., Jackson S. P. (2009) A shear gradient-dependent platelet aggregation mechanism drives thrombus formation. Nat. Med. 15, 665–673 [DOI] [PubMed] [Google Scholar]
- 42. Kulkarni S., Dopheide S. M., Yap C. L., Ravanat C., Freund M., Mangin P., Heel K. A., Street A., Harper I. S., Lanza F., Jackson S. P. (2000) A revised model of platelet aggregation. J. Clin. Invest. 105, 783–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jackson S. P., Nesbitt W. S., Westein E. (2009) Dynamics of platelet thrombus formation. J. Thromb. Haemost. 7, 17–20 [DOI] [PubMed] [Google Scholar]
- 44. Ng C. H., Xu S., Lam K. P. (2007) Dok-3 plays a nonredundant role in negative regulation of B-cell activation. Blood 110, 259–266 [DOI] [PubMed] [Google Scholar]
- 45. Stork B., Neumann K., Goldbeck I., Alers S., Kähne T., Naumann M., Engelke M., Wienands J. (2007) Subcellular localization of Grb2 by the adaptor protein Dok-3 restricts the intensity of Ca2+ signaling in B cells. EMBO J. 26, 1140–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Abramson J., Rozenblum G., Pecht I. (2003) Dok protein family members are involved in signaling mediated by the type 1 Fcϵ receptor. Eur. J. Immunol. 33, 85–91 [DOI] [PubMed] [Google Scholar]
- 47. Latour S., Veillette A. (2001) Proximal protein tyrosine kinases in immunoreceptor signaling. Curr. Opin. Immunol. 13, 299–306 [DOI] [PubMed] [Google Scholar]
- 48. Quek L. S., Pasquet J. M., Hers I., Cornall R., Knight G., Barnes M., Hibbs M. L., Dunn A. R., Lowell C. A., Watson S. P. (2000) Fyn and Lyn phosphorylate the Fc receptor γ chain downstream of glycoprotein VI in murine platelets, and Lyn regulates a novel feedback pathway. Blood 96, 4246–4253 [PubMed] [Google Scholar]
- 49. Clayton E., Bardi G., Bell S. E., Chantry D., Downes C. P., Gray A., Humphries L. A., Rawlings D., Reynolds H., Vigorito E., Turner M. (2002) A crucial role for the p110δ subunit of phosphatidylinositol 3-kinase in B cell development and activation. J. Exp. Med. 196, 753–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mazzucato M., Pradella P., Cozzi M. R., De Marco L., Ruggeri Z. M. (2002) Sequential cytoplasmic calcium signals in a 2-stage platelet activation process induced by the glycoprotein Iα mechanoreceptor. Blood 100, 2793–2800 [DOI] [PubMed] [Google Scholar]
- 51. Huang C. L., Cheng J. C., Liao C. H., Stern A., Hsieh J. T., Wang C. H., Hsu H. L., Tseng C. P. (2004) Disabled-2 is a negative regulator of integrin α(IIb)β(3)-mediated fibrinogen adhesion and cell signaling. J. Biol. Chem. 279, 42279–42289 [DOI] [PubMed] [Google Scholar]
- 52. Rathore V., Stapleton M. A., Hillery C. A., Montgomery R. R., Nichols T. C., Merricks E. P., Newman D. K., Newman P. J. (2003) PECAM-1 negatively regulates GPIb/V/IX signaling in murine platelets. Blood 102, 3658–3664 [DOI] [PubMed] [Google Scholar]
- 53. Kasirer-Friede A., Ruggeri Z. M., Shattil S. J. (2010) Role for ADAP in shear flow-induced platelet mechanotransduction. Blood 115, 2274–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tsiara S., Elisaf M., Jagroop I. A., Mikhailidis D. P. (2003) Platelets as predictors of vascular risk. Is there a practical index of platelet activity? Clin. Appl. Thromb. Hemost. 9, 177–190 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.