

Background: Regulation of vascular permeability by microtubule (MT)-associated proteins is not well understood.
Results: ANP promoted MT peripheral growth and protected endothelial barrier via Rac-PAK1-dependent inactivation of GEF-H1 and consequent suppression of Rho signaling.
Conclusion: A PAK1-GEF-H1 dependent mechanism mediates endothelial barrier protection by ANP.
Significance: Modulation of GEF-H1 activity represents a novel approach in prevention of pathologic vascular leak.
Keywords: Endothelial Dysfunction, Lung Injury, Microtubules, Permeability, Rho GTPases, Signal Transduction, Vascular Biology
Abstract
Microtubule (MT) dynamics is involved in a variety of cell functions, including control of the endothelial cell (EC) barrier. Release of Rho-specific nucleotide exchange factor GEF-H1 from microtubules activates the Rho pathway of EC permeability. In turn, pathologic vascular leak can be prevented by treatment with atrial natriuretic peptide (ANP). This study investigated a novel mechanism of vascular barrier protection by ANP via modulation of GEF-H1 function. In pulmonary ECs, ANP suppressed thrombin-induced disassembly of peripheral MT and attenuated Rho signaling and cell retraction. ANP effects were mediated by the Rac1 GTPase effector PAK1. Activation of Rac1-PAK1 promoted PAK1 interaction with the Rho activator GEF-H1, inducing phosphorylation of total and MT-bound GEF-H1 and leading to attenuation of Rho-dependent actin remodeling. In vivo, ANP attenuated lung injury caused by excessive mechanical ventilation and TRAP peptide (TRAP/HTV), which was further exacerbated in ANP−/− mice. The protective effects of ANP against TRAP/HTV-induced lung injury were linked to the increased pool of stabilized MT and inactivation of Rho signaling via ANP-induced, PAK1-dependent inhibitory phosphorylation of GEF-H1. This study demonstrates a novel protective mechanism of ANP against pathologic hyperpermeability and suggests a novel pharmacological intervention for the prevention of increased vascular leak via PAK1-dependent modulation of GEF-H1 activity.
Introduction
Increased capillary endothelial permeability as well as reduced alveolar liquid clearance capacity are major pathologic mechanisms of pulmonary edema, acute lung injury (ALI),2 and its life-threatening complication, the acute respiratory distress syndrome. Mechanisms of endothelial cell (EC) permeability involve dynamic cytoskeletal changes, assembly and disassembly of cell-cell junctions, and signaling cross-talk between various cytoskeletal compartments, such as actin networks and microtubules (MTs) (1–3).
Previous reports have demonstrated partial microtubule disassembly induced by barrier disruptive agonists, such as thrombin, TNFα, and TGFβ (3–6). In ECs, microtubule depolymerization induces activation of small GTPase, RhoA, and its effector, Rho-kinase; this leads to increased myosin light chain (MLC) phosphorylation, actin stress fiber formation, and increased permeability (3).
Microtubule-associated guanine nucleotide exchange factor H1 (GEF-H1) has been implicated in regulation of Rho activity by microtubules. In the microtubule-bound state, the guanine exchange activity of GEF-H1 is suppressed, whereas GEF-H1 release caused by microtubule disruption stimulates Rho-specific GEF activity (7). GEF-H1 activity may be additionally regulated via phosphorylation by PAK family kinases (8, 9) and by other pathways (10, 11).
Natriuretic peptides (atrial, brain, and C-type) belong to a family of mediators that regulate a variety of physiological functions, including vascular tone, plasma volume, and renal function (12). In addition, atrial natriuretic peptide (ANP) exhibits other activities, such as protection of the vascular barrier in the in vivo models of lung injury induced by Gram-negative and Gram-positive pathogens (13–16). ANP also protects endothelial barrier in the models of endothelial hyperpermeability induced by lysophosphatidylcholine (17), hypoxia, TNFα (14, 18), and thrombin (19, 20). Signaling pathways of ANP-mediated endothelial barrier involve the activation of Epac-Rap1 GTPase cascade, leading to stimulation of Rac GTPase and its downstream effector, PAK1 (19).
This study tested a hypothesis that the protective effect of ANP in the pathologic settings of vascular endothelial barrier dysfunction is directed by Rac-PAK1-dependent GEF-H1 phosphorylation and resultant inactivation and down-regulation of barrier-disruptive Rho signaling. We used functional, imaging, biochemical, and molecular approaches as well as a two-hit model of acute lung injury to investigate ANP effects on MT dynamics, GEF-H1 microtubule association, PAK1-GEF-H1 functional interactions, Rho signaling, and vascular permeability in vitro and in vivo.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
Human pulmonary artery endothelial cells (HPAECs) were obtained from Lonza (Allendale, NJ). Cells were maintained in a complete culture medium according to the manufacturer's recommendations and used for experiments at passages 5–8. HEK293T cells (ATTC, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Invitrogen). ANP and TRAP6 were purchased from Ana Spec (San Jose, CA). Antibodies to PAK1, HA, and cMyc tags were purchased from Santa Cruz Biotechnology, Inc.; antibody to end-binding protein-1 (EB1) was from BD Transduction Laboratories; anti-phospho-GEF-H1 was from Abcam (Cambridge, MA); and GEF-H1, diphospho-MLC, and phospho-PAK1 antibodies were from Cell Signaling Inc. (Beverly, MA). Reagents for immunofluorescence were purchased from Molecular Probes, Inc. (Eugene, OR). Unless specified, biochemical reagents were obtained from Sigma.
siRNA and DNA Transfections
For PAK1 knockdown, predesigned human siRNA was ordered from Ambion (Austin, TX) in purified, desalted, deprotected, and annealed double strand form. Transfection of EC with siRNA was performed as described previously (21). After 72 h of transfection, cells were used for experiments or harvested for Western blot verification of specific protein depletion. Nonspecific RNA (Dharmacon, Lafayette, CO) was used as a control treatment. Plasmids encoding human wild type GEF-H1, wild type PAK1, and dominant negative PAK1 (H83L/H86L/K299R) constructs (22) were kindly provided by G. Bokoch; GEF-H1-S885A was kindly provided by K. Szaszi (St. Michael's Hospital, Toronto, Canada); and plasmids encoding Rac1 constitutively active (G12V) and dominant negative (T17N) mutants were purchased from the Missouri University of Science and Technology (Rolla, MO) and used for transient transfections according to protocols described previously (3, 21). Control transfections were performed with empty vectors. After 24–48 h of transfection, cells were treated with agonist of interest and used for permeability measurements or subjected to immunofluorescence analysis.
Measurement of Endothelial Permeability
The cellular barrier properties were analyzed by measurements of transendothelial electrical resistance across confluent human pulmonary artery endothelial monolayers using an electrical cell-substrate impedance-sensing system (Applied Biophysics, Troy, NY) as described previously (3, 23). An express micromolecule permeability testing (XPerT) assay was recently developed in our group (24) and is now available from Millipore (Vascular Permeability Imaging Assay, catalog no. 17-10398). This assay is based on high affinity binding of avidin-conjugated FITC-labeled tracer to the biotinylated extracellular matrix proteins immobilized on the bottom of culture dishes after the EC barrier is compromised by treatment with a barrier-disruptive agonist. Permeability assays were performed in 96-well plates. Briefly, HPAECs were seeded on biotinylated glass coverslips or gelatin-coated plates and grown for 48–72 h prior to testing. FITC-avidin solution was added directly to the culture medium for 3 min before termination of the experiment. Unbound FITC-avidin was washed out with PBS (pH 7.4, 37 °C), and the fluorescence of matrix-bound FITC-avidin was measured on a Victor X5 multilabel plate reader (PerkinElmer Life Sciences). In permeability visualization experiments, cells were fixed with 3.7% formaldehyde in PBS (10 min, room temperature), and immunofluorescence staining of VE-cadherin was performed as described elsewhere (21). Images were acquired using the Nikon video imaging system Eclipse TE 300 (Nikon, Tokyo, Japan) equipped with a digital camera (DKC 5000, Sony, Tokyo, Japan); 10× objective lenses were used. Images were processed with Adobe Photoshop version 7.0 software (Adobe Systems, San Jose, CA).
Immunofluorescence and Live Cell Imaging
Endothelial monolayers plated on glass coverslips were subjected to immunofluorescence staining with Texas Red phalloidin to visualize F-actin (21, 23). For microtubule quantification, cells were fixed with −20 °C methanol, and immunostaining was carried out with β-tubulin or EB1 antibodies as described previously (25, 26). Briefly, the cell boundaries were outlined by VE-cadherin staining, and concentric outline shapes reduced to 70% were applied to the images to mark peripheral (outer 30% of diameter) and central (inner 70% of diameter) regions. The integrated fluorescence density in the peripheral area was measured using MetaMorph software and was calculated as a percentage of the integrated fluorescence density in the total cell area. The results were normalized in each experiment. Similar methods were applied to EB1 quantification in fixed cells except that EB1 immunoactivity was manually counted, and results were not normalized. For time lapse microtubule plus-end tracking, cells were seeded on MatTek dishes (MatTek, Ashland, MA) and transfected with GFP-EB1. Images were acquired with a ×100, numerical aperture 1.45 oil objective in a 3I Marianas Yokogawa-type spinning disk confocal system equipped with a CO2 chamber and a heated stage. Time lapse images were taken with 2-s intervals for 40–60 s. 20 consecutive images in each condition were used for projection analysis using ImageJ software. For tracking analysis, EB1 in the cell margin area (2–10 μm from the cell border) was tracked with the Manual Tracking plug-in in ImageJ software. The median track length was calculated using Excel software.
Immunoblotting
After stimulation, cells were lysed, and protein extracts were separated by SDS-PAGE, transferred to nitrocellulose membrane, and probed with specific antibodies, as described previously (5). Intensities of immunoreactive protein bands were quantified using ImageQuant software.
Co-immunoprecipitation studies and Western blot analysis were performed using confluent HPAEC or HEK293 cell line treated with vehicle or stimulated with the agonist of interest as described previously (27). Protein extracts were separated by SDS-PAGE, transferred to PVDF membrane, and probed with specific antibodies as described previously (28).
Animal Studies
All animal care and treatment procedures were approved by the University of Chicago Institutional Animal Care and Use Committee. Animals were handled according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. C57BL/6J mice and mice with a targeted disruption of the ANP gene (strain B6.129P2-Nppatm1Unc/J) were purchased from Jackson Laboratories (Bar Harbor, ME). The two-hit model of ventilator-induced lung injury was performed as described previously (21). In brief, mice were given a single dose of thrombin-related activating peptide, TRAP6 (1.5 × 10−5 mol/kg, intratracheal instillation), followed by 4 h of mechanical ventilation at high tidal volume (HTV; 30 ml/kg) ventilation. In experiments with ANP, animals were injected with ANP (2 μg/kg, intravenously) or sterile saline twice: immediately before and 2 h after the onset of mechanical ventilation. Parameters of lung injury were evaluated by measurements of bronchoalveolar lavage (BAL) cell count and protein concentration, determination of myeloperoxidase activity in the lung homogenates, staining of lung sections for hematoxylin and eosin, and evaluation of Evans blue accumulation in the lung tissue, as described previously (15, 21).
Statistical Analysis
Results are expressed as mean ± S.D. of 3–6 independent experiments. Experimental samples were compared with controls by unpaired Student's t test. For multiple-group comparisons, a one-way analysis of variance and post hoc multiple-comparison tests were used. p < 0.05 was considered statistically significant.
RESULTS
ANP Effect on Thrombin-induced Endothelial Permeability in Vitro
EC monolayer macromolecular permeability was analyzed using a permeability assay recently developed by our group (24). ANP did not affect the basal endothelial permeability but significantly attenuated thrombin-induced EC permeability (Fig. 1, A and B). ANP also preserved the pool of acetylated tubulin, which was decreased in EC treated with thrombin alone (Fig. 1C). These results suggest the role of ANP in the modulation of MT dynamics. This potential mechanism of ANP-dependent endothelial barrier protection in ALI was investigated in greater detail.
FIGURE 1.

Effects of ANP on thrombin-induced increase in EC macromolecular permeability and pool of stabilized MTs. A and B, HPAECs grown in 96-well plates (A) or on glass coverslips (B) with immobilized biotinylated gelatin (0.25 mg/ml) were pretreated with ANP (100 nm, 20 min) followed by challenge with thrombin (0.5 units/ml, 15 min) and the addition of FITC-avidin (25 μg/ml, 3 min). Unbound FITC-avidin was removed, and FITC fluorescence was measured. *, p < 0.05 versus thrombin alone. Bar, 50 μm. C, Western blot analysis of acetylated tubulin in the lysates from thrombin-challenged HPAECs with or without ANP pretreatment. Equal protein loading was confirmed by membrane reprobing for total β-tubulin. Bar graphs depict the quantitative densitometry analysis of Western blot densitometry data. *, p < 0.05 versus thrombin alone. AU, absorbance units. Error bars, S.D.
ANP Attenuates Microtubule Disassembly, Actin Remodeling, and Reduction in MT Growth Induced by Thrombin
The role of ANP in prevention of thrombin-induced actin stress fiber formation and reduction in peripheral microtubules was examined by immunofluorescence staining of EC monolayers. Immunofluorescent analysis of MT structure showed preservation of the peripheral microtubule network by ANP in the thrombin-treated EC (Fig. 2A). In addition, ANP abolished thrombin-induced stress fiber formation driven by Rho-dependent mechanism (29). Quantitative analysis confirmed the protective effects of ANP against thrombin-induced reduction of the peripheral microtubule pool (Fig. 2B).
FIGURE 2.

ANP prevents thrombin-induced disassembly of peripheral MTs. A, cells grown on coverslips were stimulated with thrombin (0.5 units/ml, 15 min) with or without ANP pretreatment (100 nm, 20 min) followed by immunofluorescent staining with an antibody against β-tubulin (top) or Texas Red-phalloidin to visualize F-actin (middle). Merged images of β-tubulin and F-actin staining are shown in the bottom panel. Bar, 5 μm. B, quantification of peripheral microtubules. *, p < 0.05, thrombin + ANP versus thrombin. Error bars, S.D.
Alternative analysis of thrombin and ANP effects on microtubule growth was performed by quantitative evaluation of the EB1, a marker of microtubule plus-ends, at the peripheral areas of methanol-fixed EC. The results show a decreased fraction of peripheral EB1 immunoreactivity in thrombin-treated EC (Fig. 3A). Quantitative analysis of the peripheral EB1 immunoreactivity is shown in Fig. 3B. This reduction of peripheral EB1 presence was abolished by ANP.
FIGURE 3.

ANP prevents peripheral MT network disassembly caused by thrombin. A, cells were pretreated with ANP (100 nm, 20 min) followed by thrombin (0.5 units/ml) stimulation for 15 min. Endogenous EB1 was visualized by immunostaining with anti-EB1 antibody. Bar, 5 μm. B, quantification of peripheral EB1 in methanol-fixed HPAEC monolayers; *, p < 0.05, thrombin + ANP versus thrombin. Error bars, S.D.
Effects of ANP treatment on microtubule dynamics in control and thrombin-stimulated cells were further examined using a live imaging approach. EC were transfected with GFP-tagged EB1, which tracks the growing plus-end of microtubules. EB1 tracking in live cells was performed as described previously (25, 26). Projection images were generated as described under “Experimental Procedures.” Thrombin stimulation caused reduction of the length of EB1 tracks, which represents episodes of uninterrupted microtubule growth (Fig. 4A). These thrombin effects were completely abolished by ANP pretreatment (Fig. 4B). These results demonstrate that ANP blocks thrombin-induced reduction in both the length of microtubule continuous growth and the number of growing ends at the cell margin.
FIGURE 4.

ANP blocks thrombin-induced inhibition of microtubule growth. Live cell imaging of HPAECs labeled with GFP-EB1 and treated with thrombin (0.5 units/ml) (A) or ANP (100 nm, 20 min) (B), followed by thrombin stimulation for 15 min. Projection analysis of 20 consecutive images before and after thrombin treatment is shown. Bar, 2 μm. Graphs depict quantification of GFP-EB1 track length. Each pair of dots represents the median track length in a cell before and after thrombin treatment. *, p < 0.05 versus control.
ANP Preserves the Dynamics of Microtubule Growth to Cell Periphery in Thrombin-treated Cells via a PAK1-dependent Mechanism
Involvement of PAK1 in the mechanism of ANP-mediated preservation of MT growth in thrombin-challenged EC was tested by an siRNA-based knockdown approach. In contrast to control EC transfected with nonspecific RNA duplex and treated with ANP and thrombin (Fig. 5A), transfection with PAK1-specific siRNA abrogated ANP-induced protection of microtubule continuous growth (Fig. 5B). Furthermore, PAK1 depletion had no effect on MLC phosphorylation induced by thrombin, but it suppressed inhibitory effects of ANP on MLC phosphorylation (Fig. 5C). The role of PAK1 activity in the ANP effects on thrombin-induced changes in microtubules, actin cytoskeleton, and endothelial permeability was tested in the following experiments.
FIGURE 5.

Effects of PAK1 depletion on the ANP-dependent attenuation of thrombin induced MLC phosphorylation and microtubule dynamics in pulmonary ECs. ECs were transfected with nonspecific RNA or PAK1-specific siRNA prior to agonist treatments. A, live cell imaging of control HPAECs expressing GFP-EB1; B, PAK1-depleted ECs expressing GFP-EB1 preincubated with ANP (20 min) prior to thrombin treatment. Projection analysis of 20 consecutive images before and after thrombin treatment (0.5 units/ml, 15 min) is shown. Bar, 2 μm. Graphs depict quantification of GFP-EB1 track length. Each pair of dots represents the median track length in a cell before and after thrombin treatment. *, p < 0.05 versus control. C, cells were treated with ANP (100 nm, 20 min) prior to thrombin (0.5 units/ml, 15 min) challenge. MLC phosphorylation in cell lysates was analyzed by Western blot with phosphospecific antibodies. Efficiency of siRNA-induced PAK1 depletion and equal protein loading were confirmed by membrane reprobing with PAK1 and total β-tubulin antibodies, respectively. Bar graphs depict the quantitative densitometry analysis of Western blot data. *, p < 0.05 versus thrombin alone. Error bars, S.D.
Dominant Negative PAK1 Attenuates Protective Effects of ANP on Thrombin-induced Cytoskeletal Remodeling and EC Permeability
Pulmonary ECs expressing dominant negative PAK1 mutant (PAK1-DN) were treated with ANP, thrombin, or both. PAK1-DN exogenous expression abrogated microtubule preservation in thrombin-challenged cells pretreated with ANP (Fig. 6A). Insets 1 and 2 depict higher magnification images of peripheral regions of non-transfected and PAK1-DN-transfected EC, respectively, showing a dramatic reduction of the peripheral MT network in PAK1-DN-expressing cells. Exogenous expression of PAK1-DN also suppressed the inhibitory effects of ANP pretreatment on thrombin-induced stress fiber formation (Fig. 6B). Insets 3 and 4 depict higher magnification images of peripheral regions of non-transfected and PAK1-DN-transfected ECs, respectively, showing details of F-actin arrangement.
FIGURE 6.

Molecular inhibition of PAK1 abrogates protective effects of ANP on thrombin-induced peripheral MT disassembly, actin stress fiber formation, and endothelial permeability. Analysis of MT remodeling (A) and F-actin remodeling (B) in ECs expressing dominant negative PAK1 (myc-PAK1-DN) was performed by double immunofluorescence staining with either β-tubulin or Texas Red phalloidin and Myc antibody to detect PAK1-DN-expressing cells. Bar, 10 μm. Magnified images (insets) show details of MT and actin structure in non-transfected and PAK1-DN-expressing cells. Bar graphs depict the quantitative analysis of F-actin content. *, p < 0.05 versus transfected cells. C, transendothelial resistance reflecting EC monolayer barrier properties was monitored over 3 h in control and PAK1-DN-transfected ECs stimulated with thrombin or ANP + thrombin. Data are expressed as mean ± S.D. (error bars).
We further tested effects of dominant negative PAK1 expression on changes in HPAEC permeability caused by thrombin challenge with and without ANP pretreatment. After 24 h of transfection, HPAECs were treated with thrombin with or without ANP pretreatment, and transendothelial electrical resistance was monitored over time. Transfection with PAK1-DN abrogated protective effects of ANP against thrombin-induced transendothelial electrical resistance decline, suggesting PAK1 involvement in the ANP-mediated EC barrier-protective effects (Fig. 6C).
Effects of Thrombin and ANP on the GEF-H1 Interactions with Microtubules and PAK1 in Endothelial Cells
GEF-H1 localizes to microtubules and serves as an important regulator of Rho activity (7). Existing data indicate that GEF-H1 activity may be regulated in different systems by phosphorylation mediated by PAK1, PAK2, or PAK4 (9, 10, 30). We investigated the effects of thrombin and ANP treatment on the PAK1-GEF-H1 association and PAK1 and GEF-H1 site-specific phosphorylation.
Analysis of site-specific phosphorylation of GEF-H1 immunoprecipitated from control, thrombin-stimulated, and ANP-stimulated HPAECs shows GEF-H1 phosphorylation at the PAK-specific Ser885 site in ECs stimulated with ANP but not with thrombin (Fig. 7A). We next examined whether ANP induces phosphorylation of GEF-H1 associated with microtubules. The results show low levels of GEF-H1 phosphorylation in the MT fractions isolated from control and thrombin-treated ECs but significant elevation of Ser885 phosphorylation induced by ANP (Fig. 7B). We next examined potential functional association between GEF-H1 and PAK1 induced by ANP. The results show that ANP increased the presence of GEF-H1 in the PAK1 immunoprecipitated complexes (Fig. 7C). These data suggest a PAK1-dependent mechanism of GEF-H1 phosphorylation induced by ANP. In the next experiments, we used co-expression of GEF-H1 and PAK1 mutants in the HEK cell expression system to more precisely define the PAK1-GEF-H1 functional interactions.
FIGURE 7.
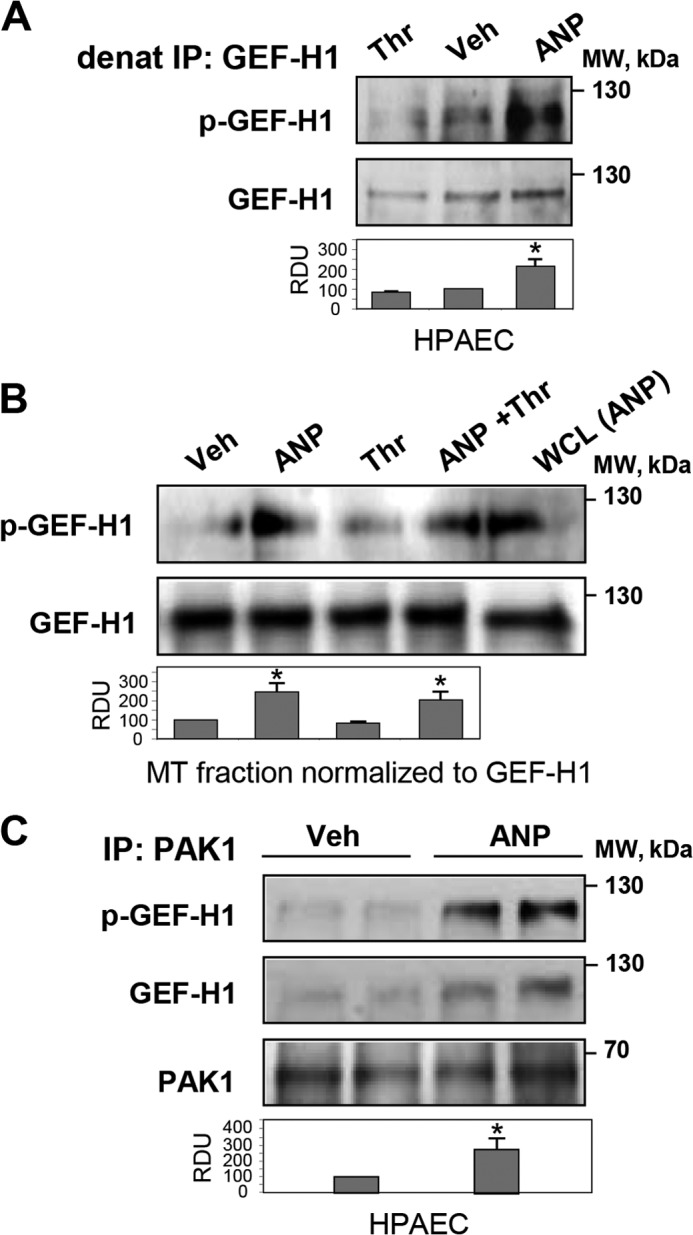
Effects of ANP and thrombin on GEF-H1-MT and GEF-H1-PAK1 interactions in pulmonary ECs. HPAECs were stimulated with ANP (100 nm) or thrombin (0.5 units/ml) alone or were pretreated with ANP followed by thrombin challenge. A, analysis of GEF-H1 phosphorylation in control, thrombin-treated, and ANP-treated cells was performed after GEF-H1 immunoprecipitation (IP) under denaturing conditions using GEF-H1 phospho-Ser885 antibodies. Staining of total GEF-H1 was used as a control. *, p < 0.05 versus vehicle. B, detection of phospho-GEF-H1 in MT fractions isolated from control and agonist-stimulated ECs. Protein loadings were equalized by GEF-H1 content in the MT fractions; *, p < 0.05 versus thrombin alone. C, immunoprecipitation from vehicle- and ANP-stimulated HPAECs using PAK1 antibody followed by Western blot detection of total and phosphorylated GEF-H1. Membrane reprobing with PAK1 antibody was used as a control; *, p < 0.05 versus vehicle. Error bars, S.D. WCL, whole cell lysates.
Role of PAK1 in GEF-H1 Phosphorylation
PAK1 is a downstream effector of Rac1 GTPase, which may become activated in response to chemical and mechanical stimuli. In HEK cells, Rac activation can be achieved by treatment with EGF (31). The EGF stimulation was used in the following experiments to activate Rac1-PAK1 signaling. EGF treatment induced association between exogenously expressed wild type GEF-H1 and wild type PAK1 monitored in coimmunoprecipitation assays. In contrast, EGF failed to increase GEF-H1 association with dominant negative PAK1 mutant in EGF-stimulated cells (Fig. 8A). EGF stimulation of cells co-expressing GEF-H1 and wild type PAK1 induced GEF-H1 phosphorylation at Ser885 (Fig. 8B). In contrast, EGF-induced GEF-H1 phosphorylation was not observed in cells expressing dominant negative PAK1 mutant. Involvement of the Rac1-PAK1 mechanism in the GEF-H1 phosphorylation was further verified in experiments with co-expression of GEF-H1 and constitutively activated (Rac1-CA) or dominant negative (Rac1-DN) Rac1 mutants. Increased GEF-H1 phosphorylation was observed in cells expressing Rac1-CA but not in cells expressing Rac1-DN (Fig. 8C).
FIGURE 8.
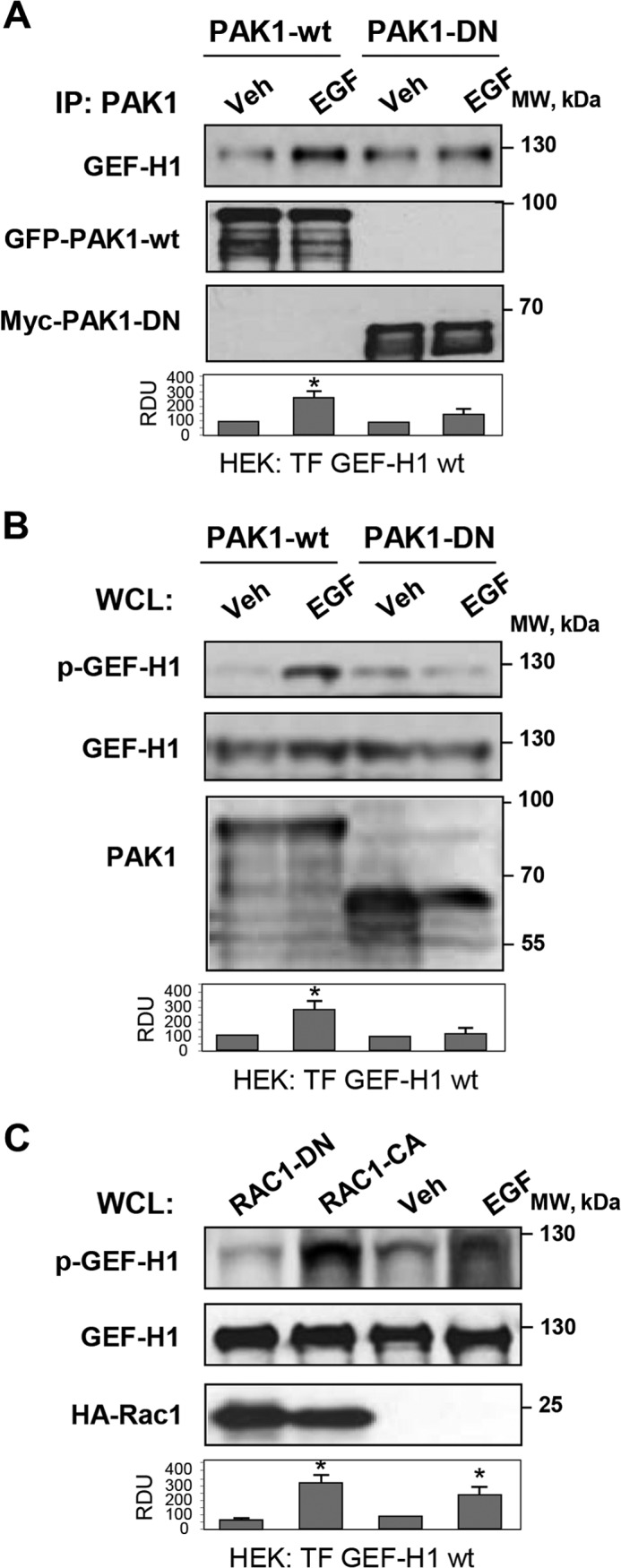
Analysis of Rac1/Pak1-GEF-H1 functional interactions. HEK293T cell line was used for coexpression of GEF-H1, PAK1, and Rac1 mutants. A, GEF-H1 was coexpressed with wild type or dominant negative PAK1, and cells were stimulated with EGF (500 ng/ml, 15 min) or left untreated. Immunoprecipitation (IP) was performed using PAK1 antibody, followed by Western blot detection of GEF-H1. Expression of wild type PAK1 (PAK1-wt) was detected by GFP tag antibody, and expression of PAK1-DN was verified by Myc tag antibody. Bar graphs depict the quantitative densitometry analysis of Western blot data. *, p < 0.05 versus transfected cells. B, HEK cells were cotransfected with wild type PAK1 or PAK1-DN mutant and GEF-H1. After EGF stimulation (500 ng/ml, 15 min), GEF-H1 phosphorylation in total cell lysates (WCL) was detected by Western blot with phospho-Ser885 GEF-H1 antibody. Control membrane reprobing was performed to verify expression of GEF-H1 and PAK1 constructs. *, p < 0.05 versus transfected cells. C, HEK cells were cotransfected with activated (Rac1-CA) or dominant negative (Rac1-DN) Rac1 and GEF-H1, followed by Western blot analysis of phospho-Ser885 GEF-H1 levels in total cell lysates. EGF stimulation of GEF-H1-expressing cells was used as a control. Expression of Rac1 constructs was detected by membrane reprobing with HA tag antibody. *, p < 0.05 versus vehicle. Error bars, S.D.
PAK1 and Rac1 Mediate GEF-H1 Phosphorylation in Pulmonary ECs Induced by ANP and Thrombin Stimulation
The findings made in the HEK cell model were further verified in the agonist-stimulated primary human pulmonary artery endothelial cells. ANP stimulation caused GEF-H1 phosphorylation in ECs with ectopic expression of wild type PAK1, whereas ectopic expression of PAK1-DN mutant blocked ANP effect (Fig. 9A). In turn, expression of constitutively activated Rac1 and wild type PAK1 induced significant GEF-H1 phosphorylation even in the absence of agonist stimulation (Fig. 9B). In cells expressing wild type PAK1, stimulation with ANP was required to induce GEF-H1 phosphorylation. ANP also stimulated GEF-H1 phosphorylation in thrombin-treated ECs expressing wild type PAK1 (Fig. 9B).
FIGURE 9.

Role of Rac1-Pak1 in the ANP-induced GEF-H1 phosphorylation in pulmonary ECs. A, HPAECs transfected with wild type or dominant negative PAK1 were stimulated with ANP or left untreated. Analysis of phospho-Ser885 GEF-H1 levels in total cell lysates was performed by Western blot. Staining for GEF-H1 and β-tubulin was used as normalization control. Bar graphs depict the quantitative densitometry analysis of Western blot data. *, p < 0.05 versus transfected cells. B, HPAECs transfected with activated Rac (Rac1-CA) and/or wild type PAK1 were treated with ANP, thrombin, or their combination, and Western blot analysis of phospho-Ser885 GEF-H1 in total cell lysates was performed. Expression of GEF-H1, PAK1, and Rac-CA was verified by Western blot. Equal protein loading was confirmed by membrane reprobing for total β-tubulin. *, p < 0.05 versus vehicle. Error bars, S.D.
Role of GEF-H1 Phosphorylation on ANP Protective Effects
To study the functional significance of GEF-H1 phosphorylation for ANP effects, endothelial cells were transfected with the Ser885 phosphorylation-deficient mutant, GEF-H1-S885A, or the wild type GEF-H1. After 24 h of transfection, HPAECs were treated with thrombin with or without ANP pretreatment, and permeability changes were monitored over time. Transfection with GEF-H1-S885A abolished protective effects of ANP against thrombin-induced hyperpermeability (Fig. 10A). Furthermore, ectopic expression of phospho-deficient GEF-H1 mutant suppressed inhibitory effects of ANP on thrombin-induced MLC phosphorylation, suggesting the role of GEF-H1 inhibitory phosphorylation in the ANP-mediated EC barrier-protective effects (Fig. 10B).
FIGURE 10.

Expression of GEF-H1 phospho-deficient mutant abolished protective effects of ANP in the thrombin model of EC barrier dysfunction. HPAECs transfected with the wild type GEF-H1 or the GEF-H1-S885A mutant were stimulated with ANP (100 nm, 20 min) prior to thrombin (0.5 units/ml) challenge. A, transendothelial resistance reflecting EC monolayer barrier properties was monitored over 2 h in transfected ECs stimulated with thrombin or ANP + thrombin. Data are expressed as mean ± S.D. Inset, expression of GFP-GEF-H1 wild type or GFP-GEF-H1-S885A in HPAECs. B, MLC phosphorylation in cell lysates was analyzed by Western blot with phosphospecific antibodies. Efficiency of siRNA-induced PAK1 depletion and equal protein loading were confirmed by membrane reprobing with PAK1 and total β-tubulin antibodies, respectively. *, p < 0.05 versus thrombin alone. Error bars, S.D.
ANP Attenuates Lung Vascular Leak in the Two-hit Model of Acute Injury
In the next experiments, we used animal model of lung injury to characterize MT-associated signaling involved in ANP effects. Mice exposed to HTV mechanical ventilation were injected with TRAP to recapitulate the two-hit model of ALI (32). Evaluation of the BAL samples from wild type mice after 4 h of TRAP/HTV revealed a significant increase in BAL protein content as compared with the non-ventilated control mice or mice treated with ANP alone (Fig. 11A). This increase was significantly attenuated by ANP pretreatment.
FIGURE 11.

ANP prevents lung vascular leak in the two-hit model of acute lung injury and stimulates PAK1 signaling and microtubule stability. Mice were subjected to mechanical ventilation at HTV (30 ml/kg, 4 h) with or without TRAP6 injection or left spontaneously ventilated. Animals were injected with ANP or sterile saline, immediately followed by mechanical ventilation. A, lung barrier dysfunction was evaluated by measurements of protein concentration in BAL. *, p < 0.05 versus TRAP/HTV. B, lung vascular leak was evaluated visually by Evans blue accumulation in the lung tissue samples. C, quantitative spectrophotometric analysis of Evans blue extracted from the lung tissue samples. *, p < 0.05 versus TRAP/HTV. Error bars, S.D.
TRAP/HTV-induced lung vascular leak was also detected by Evans blue dye accumulation in the lung parenchyma, which was evident in mice after 4 h of TRAP/HTV. Images of original lung preparations showing Evans blue extravasation are shown in Fig. 11B. In turn, pretreatment with ANP substantially decreased the Evans blue accumulation shown in the images of lungs collected from control and TRAP/HTV-exposed animals. Quantitative analysis of Evans blue-labeled albumin extravasation further confirmed these results (Fig. 11C).
ANP Attenuates Lung Vascular Leak and Inflammation in the Two-hit Model of Lung Injury
Exposure to 4 h of TRAP/HTV caused a significant increase in total cell count as well as elevated numbers of neutrophils compared with the non-ventilated controls (Fig. 12A). Measurement of myeloperoxidase activity in the lung tissue and morphological analysis of lung injury by H&E staining were used as additional parameters of neutrophilic infiltration and inflammation. ANP caused a marked attenuation of TRAP/HTV-induced lung inflammation (Fig. 12, B and C). These protective effects were accompanied by attenuation of Rho signaling detected by decreased levels of MLC phosphorylation in the lung tissue. Importantly, ANP treatment increased the pool of acetylated β-tubulin representing stable MTs and stimulated site-specific phosphorylation of PAK1 and GEF-H1 known to affect their functional activity (Fig. 12, D and E).
FIGURE 12.

ANP prevents lung dysfunction in the two-hit model of acute lung injury and stimulates PAK1 signaling and microtubule stability. Mice were injected with ANP or saline, followed by mechanical ventilation at HTV (30 ml/kg, 4 h) with or without TRAP6 injection or left spontaneously ventilated. A, total cell count and neutrophil counts were determined in BAL collected after TRAP/HTV exposure. *, p < 0.05 versus TRAP/HTV. B, myeloperoxidase (MPO) activity was measured in lung tissue homogenates; *, p < 0.05 versus TRAP/HTV. C, lung sections from control and TRAP6/HTV-treated mice with or without ANP. Micrographs show hematoxylin and eosin staining; magnification, ×40. D, PAK1 and MLC phosphorylation and the amount of acetylated tubulin after 4-h exposure of mice to TRAP/HTV with or without ANP were determined by Western blot analysis of lung tissue homogenates. Bar graphs depict the quantitative densitometry analysis of Western blot data. *, p < 0.05 versus TRAP/HTV. E, site-specific GEF-H1 phosphorylation in the lungs of mice treated with ANP alone was determined by Western blot analysis. Equal protein loading was confirmed by membrane reprobing for total β-tubulin; *, p < 0.05 versus saline. Error bars, S.D.
Next, we evaluated parameters of ALI induced by TRAP/HTV in ANP knock-out mice (strain B6.129P2-Nppatm1Unc/J). After 4 h of mechanical ventilation, parameters of lung injury in ANP−/− mice and matched controls were analyzed by measurements of BAL protein content and cell count. In comparison with wild type controls, ANP−/− mice developed more severe lung injury manifested by a 1.5-fold increase in BAL protein concentration and a nearly 2-fold increase in total cell count and neutrophil count (Fig. 13, A–C).
FIGURE 13.

Analysis of TRAP/HTV-induced lung injury in ANP knock-out mice. ANP knock-out mice (Nppa−/−) or matched controls were exposed to TRAP6 and HTV (30 ml/kg) for 4 h. Protein concentration (A), total cell count (B), and neutrophil content (C) were measured in BAL fluid. D, TRAP/HTV-induced vascular leak was assessed visually and further quantified by spectrometric determination of Evans blue-labeled albumin extravasation into the lung tissue. E, TRAP/HTV-induced neutrophilic infiltration was evaluated by histological analysis of lung sections from wild type and Nppa−/− mice. F, PAK1 and MLC phosphorylation and the amount of acetylated tubulin in lung samples from TRAP/HTV exposed wild type and Nppa−/− mice were analyzed by Western blot with phosphospecific antibodies. Bar graphs depict the quantitative analysis of Western blot densitometry data. *, p < 0.05 versus wild type. G, MLC phosphorylation in Nppa−/− mice exposed to TRAP/HTV with or without ANP treatment. Equal protein loading was confirmed by probing of membranes with total β-tubulin antibodies. *, p < 0.05 versus TRAP/HTV. Error bars, S.D.
Effects of ANP on the lung vascular leak induced by HTV/TRAP were evaluated by measurements of Evans blue extravasation into the lung tissue. Evans blue accumulation in the parenchyma was significantly increased in the ANP−/− mice (Fig. 13D). Histological analysis of lung sections stained with hematoxylin and eosin revealed that TRAP6/HTV caused more severe inflammatory cell infiltration in the lungs of ANP−/− mice (Fig. 13E). Biochemical analysis of tissue samples showed higher levels of MLC phosphorylation and decreased content of phospho-PAK1 and acetylated tubulin in ANP−/− mice (Fig. 13F). Rescue experiments with ANP administration demonstrated inhibitory effects of ANP on Rho pathway activation in the TRAP/HTV model (Fig. 13G). These results demonstrate that ANP−/− mice develop more severe injury in response to TRAP/HTV. Taken together, in vivo data reveal strong protective effect of ANP against lung vascular dysfunction and suggest the involvement of microtubule-dependent signaling in the ANP protective response.
DISCUSSION
This study demonstrates a novel mechanism of ANP barrier-protective effects in endothelial cells and a two-hit model of TRAP/HTV induced lung injury. In this mechanism, ANP exerts barrier-protective effects through stabilization of MTs, Rac1-PAK1 dependent phosphorylation of GEF-H1, and resultant down-regulation of Rho signaling. This mechanism was supported by experiments with expression of activated or dominant negative Rac, which correspondingly promoted or abolished PAK1-mediated GEF-H1 phosphorylation.
Direct stabilization of microtubules by plant alkaloid taxol attenuated lung injury induced by LPS (33) and inhibited the Rho pathway of endothelial permeability caused by thrombin and TGF-β1 (3, 5). GEF-H1 association with MTs is known to block GEF-H1 Rho-specific nucleotide exchange activity (7). These published data are consistent with our observation of a thrombin-induced decrease in GEF-H1 association with the MT fraction, which was abolished by ANP pretreatment (data not shown). We also observed preservation of peripheral MT growth by ANP in thrombin-stimulated ECs. Tracking MTs in ECs stimulated with ANP and thrombin or in cells expressing dominant negative PAK1 showed that ANP not only stabilizes existing MTs but also restores in a PAK1-dependent manner the MT growth to cell periphery affected by thrombin. PAK1 inhibition also abrogated ANP-induced MT preservation and inhibited ANP protection against thrombin-induced actin stress fiber formation and Rho-dependent MLC phosphorylation. Furthermore, ANP stimulated PAK1-GEF-H1 association, leading to GEF-H1 phosphorylation at Ser885. Importantly, expression of Ser885 phosphorylation-deficient GEF-H1 mutant suppressed protective effects of ANP against thrombin-induced EC barrier disruption.
Phosphorylation of Ser885 was shown by a number of kinases, including PAK1, PAK2, PAK4, Aurora A/B, Par1b, PKA, and MAPK (8–11, 30, 34). GEF-H1 phosphorylation at Ser885 was also shown to regulate GEF-H1 binding to microtubules. However, conflicting reports in the literature regarding the functional significance of site-specific GEF-H1 phosphorylation suggest more complicated mechanism of GEF-H1 functional control. For example, phosphorylation of GEF-H1 at Ser885 by PAK1 or PKA induces 14-3-3 binding to the exchange factor and relocation of 14-3-3 to microtubules and inhibits GEF-H1-associated nucleotide exchange activity in cells and suppresses GEF-H1-dependent, Rho-mediated stress fiber formation in fibroblasts (9, 35). GEF-H1 phosphorylation at Ser885/Ser959 also inhibits RhoA-specific GEF activity of GEF-H1. As a consequence, GEF-H1 phosphorylated on both of the serine residues loses the ability to stimulate RhoA and fails to induce RhoA-dependent stress fiber formation (11). In contrast, another study shows that ERK-dependent GEF-H1 phosphorylation at Thr678 leads to activation of Rho, whereas both Rac and Rho activation require GEF-H1 phosphorylation at Ser885 (34).
In silico analysis (by PhosphoSite) suggests that GEF-H1 can undergo phosphorylation at 36 residues, indicating sophisticated phosphorylation-dependent regulation of GEF-H1 activity (36). In addition to phosphorylation at Ser885 in the N-terminal domain, PAR1b also phosphorylates GEF-H1 at C terminus on Ser143, Ser172, and Ser186. This phosphorylation causes dissociation of GEF-H1 from microtubules, which could activate GEF-H1 (37). Collectively, these data suggest that phosphorylation of GEF-H1 at multiple sites may have both positive and negative effects on GEF-H1 activity and association with MTs depending on the biological context. Thus, unique sets of signaling kinases activated by different agonists or changes in mechanical microenvironment may explain the different modes of regulation of GEF-H1 functional activity by phosphorylation in physiologic and pathologic settings.
Our data show that PAK1 is critical for ANP-induced GEF-H1 phosphorylation and association with MTs, MT stability, growth dynamics, and protective effects of ANP against thrombin-induced EC permeability. Both the ectopic expression of dominant negative PAK1 and siRNA-induced PAK1 knockdown prevented GEF-H1 phosphorylation. Because ANP also stimulates PKA in pulmonary ECs (19), we also cannot exclude additional PKA-dependent phosphorylation of Ser885 or other putative sites within GEF-H1. Whether PKA phosphorylates other sites in the GEF-H1 remains to be determined. ANP-induced GEF-H1 phosphorylation was abolished by overexpression of dominant negative PAK1. These data further support the role of PAK1-induced GEF-H1 phosphorylation in the ANP-mediated mechanism of MT stabilization, attenuation of Rho-dependent stress fiber formation, and increased endothelial permeability. Although we cannot exclude PAK2 and PAK4 roles in the ANP effect, our experiments with ectopic expression of PAK1 suggest potential redundancy in GEF-H1 regulation by PAK variants.
Of note, thrombin reduced the pool of stable MTs (Fig. 1C) and inhibited the peripheral growth of EB-1-positive MTs, which was restored by ANP co-treatment. These findings are consistent with increased GEF-H1 targeting to the MTs caused by PAK1 phosphorylation. Additional support for our observation came from a recent report by Nagae et al. (38), who described the existence of two populations: centrosomal and non-centrosomal MTs. These MT populations were shown to have different biochemical characteristics, mechanisms of stabilization, and preference for GEF-H1 binding. Assuming that GEF-H1 is preferentially localized on peripheral MTs that are prone to thrombin-induced disassembly, most of the GEF-H1 becomes released upon agonist stimulation. Thus, normalized MT fractions of control, ANP-stimulated, and thrombin-stimulated cells may represent different pools of MTs containing different levels of bound GEF-H1.
The proposed PAK1-GEF-H1-MT mechanism of ANP barrier-protective effects was also supported by an animal model of acute lung injury. ANP suppressed vascular leak and lung injury caused by TRAP/HTV. These effects were accompanied by increased PAK1 phosphorylation and tubulin acetylation in the lung homogenates. ANP barrier-protective effects in pulmonary endothelial culture were associated with increased PAK1 and GEF-H1 phosphorylation, an increased pool of stable MTs, and decreased levels of phospho-MLC. These results support the PAK-GEF-H1 pathway as an additional mechanism contributing to the ANP-mediated protection of the lung against inflammatory agents and injurious mechanical stimuli. Thus, in addition to the previously reported attenuation of ventilator-induced lung injury in mice by siRNA-induced GEF-H1 depletion (21), this study describes the ANP-induced protection in the TRAP-VILI model via more delicate regulation of GEF-H1 function by PAK-dependent phosphorylation.
The pathologic role of Rho activation in animal models of acute lung injury and EC cultures stimulated with ALI-relevant agonists or high magnitude cyclic stretch is well recognized (29, 39, 40), and pharmacologic inhibition of the Rho pathway by prostanoids, simvastatin, S1P receptor agonist FTY720, or Rho kinase inhibitor Y-27632 has been proposed as a potential treatment of ALI (41–45). However, endogenous regulators of excessive Rho activation in ALI are not well characterized. We show ANP as a potential agent that protects lung dysfunction caused by TRAP/HTV. Thus, increased ANP levels observed in ALI patients may represent an important compensatory mechanism aimed at attenuation of injury and lung barrier dysfunction. This notion is supported by data that show that ANP knock-out increases the severity of lung inflammation and barrier dysfunction caused by bacterial pathogens (15, 16) or thrombin and excessive mechanical ventilation (this study).
In conclusion, we propose a mechanism of ANP-mediated EC barrier protection in the model of acute lung injury (Fig. 14). A pathologic regional mechanical strain associated with mechanical ventilation and circulating inflammatory mediators, such as thrombin, activate the Rho pathway of vascular endothelial barrier dysfunction. The Rho-Rho kinase cascade triggers actomyosin contraction and EC retraction, leading to the formation of paracellular gaps and vascular leak. In concert with this, Rho-Rho kinase induces phosphorylation and inactivation of MT-stabilizing proteins, such as Tau protein, leading to depolymerization of peripheral MTs (46–48), which causes release and activation of GEF-H1 and further propagation of barrier-disruptive Rho signaling. In turn, ANP inactivates GEF-H1 via Rac-PAK1-mediated phosphorylation at Ser885 and possibly other sites, thus leading to Rho down-regulation, MT stabilization, and protection of EC barrier function. These events may lead to attenuation of lung injury. However, the reported mechanism of PAK1-GEF-H1-MT-mediated protective effects of ANP may be relevant to many other pathologic conditions associated with increased endothelial permeability, including sepsis, inflammation, prolonged mechanical ventilation, burn, and trauma.
FIGURE 14.
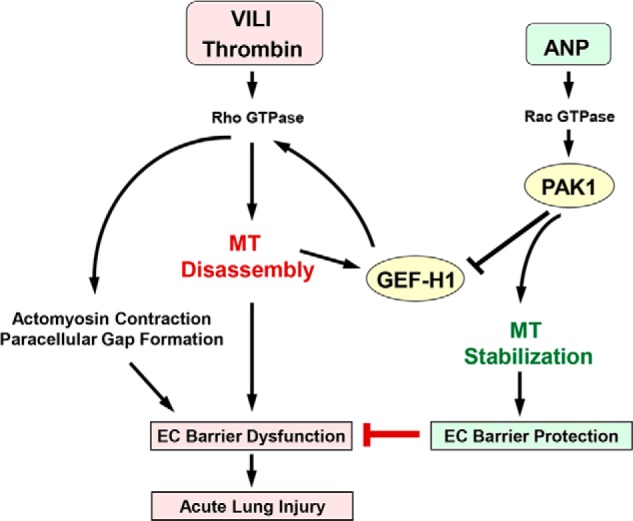
PAK1 and GEF-H1 in ANP-induced microtubule stabilization and attenuation of thrombin- and ventilator-induced endothelial hyperpermeability and acute lung injury. Thrombin or HTV-related pathologic mechanical forces induce peripheral microtubule disassembly, activation of microtubule-released Rho-specific GEF-H1, and stimulation of the Rho-dependent pathway of endothelial permeability and lung barrier dysfunction. ANP stimulates Rac GTPase and its effector PAK1, which down-regulates GEF-H1 activity via phosphorylation at Ser885. In addition, PAK1 stabilizes microtubules and promotes GEF-H1 recruitment to MT and further GEF-H1 inactivation. As a result, ANP-induced GEF-H1 inactivation and MT stabilization reduce RhoA signaling, abrogate barrier-disruptive effects of pathologic mechanical and chemical stimulation, and prevent increased vascular leak associated with ALI/ARDS.
Acknowledgments
We thank Oleksii Dubrovskyi for technical assistance with measurements of endothelial permeability using the XPerT assay and K. Szaszi (St. Michael's Hospital, Toronto, Canada) for sharing the GEF-H1-S885A construct.
This work was supported, in whole or in part, by National Institutes of Health, NHLBI, Public Health Service Grants HL89257 and HL107920.
- ALI
- acute lung injury
- EC
- endothelial cell
- MT
- microtubule
- MLC
- myosin light chain
- GEF
- guanine nucleotide exchange factor
- ANP
- atrial natriuretic peptide
- HPAEC
- human pulmonary artery endothelial cell
- XPerT
- express micromolecule permeability testing
- HTV
- high tidal volume
- EB1
- end-binding protein-1
- CA
- constitutively activated
- DN
- dominant negative
- RDU
- relative density units.
REFERENCES
- 1. Mehta D., Malik A. B. (2006) Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 86, 279–367 [DOI] [PubMed] [Google Scholar]
- 2. Lucas R., Verin A. D., Black S. M., Catravas J. D. (2009) Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem. Pharmacol. 77, 1763–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Birukova A. A., Birukov K. G., Smurova K., Adyshev D., Kaibuchi K., Alieva I., Garcia J. G., Verin A. D. (2004) Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J. 18, 1879–1890 [DOI] [PubMed] [Google Scholar]
- 4. Kakiashvili E., Speight P., Waheed F., Seth R., Lodyga M., Tanimura S., Kohno M., Rotstein O. D., Kapus A., Szászi K. (2009) GEF-H1 mediates tumor necrosis factor-α-induced Rho activation and myosin phosphorylation. Role in the regulation of tubular paracellular permeability. J. Biol. Chem. 284, 11454–11466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Birukova A. A., Birukov K. G., Adyshev D., Usatyuk P., Natarajan V., Garcia J. G., Verin A. D. (2005) Involvement of microtubules and Rho pathway in TGF-β1-induced lung vascular barrier dysfunction. J. Cell Physiol. 204, 934–947 [DOI] [PubMed] [Google Scholar]
- 6. Shivanna M., Srinivas S. P. (2009) Microtubule stabilization opposes the (TNF-α)-induced loss in the barrier integrity of corneal endothelium. Exp. Eye Res. 89, 950–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krendel M., Zenke F. T., Bokoch G. M. (2002) Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 4, 294–301 [DOI] [PubMed] [Google Scholar]
- 8. Callow M. G., Zozulya S., Gishizky M. L., Jallal B., Smeal T. (2005) PAK4 mediates morphological changes through the regulation of GEF-H1. J. Cell Sci. 118, 1861–1872 [DOI] [PubMed] [Google Scholar]
- 9. Zenke F. T., Krendel M., DerMardirossian C., King C. C., Bohl B. P., Bokoch G. M. (2004) p21-activated kinase 1 phosphorylates and regulates 14-3-3 binding to GEF-H1, a microtubule-localized Rho exchange factor. J. Biol. Chem. 279, 18392–18400 [DOI] [PubMed] [Google Scholar]
- 10. Birkenfeld J., Nalbant P., Bohl B. P., Pertz O., Hahn K. M., Bokoch G. M. (2007) GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev. Cell 12, 699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamahashi Y., Saito Y., Murata-Kamiya N., Hatakeyama M. (2011) Polarity-regulating kinase partitioning-defective 1b (PAR1b) phosphorylates guanine nucleotide exchange factor H1 (GEF-H1) to regulate RhoA-dependent actin cytoskeletal reorganization. J. Biol. Chem. 286, 44576–44584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baxter G. F. (2004) The natriuretic peptides. Basic Res. Cardiol. 99, 71–75 [DOI] [PubMed] [Google Scholar]
- 13. Fürst R., Brueckl C., Kuebler W. M., Zahler S., Krötz F., Görlach A., Vollmar A. M., Kiemer A. K. (2005) Atrial natriuretic peptide induces mitogen-activated protein kinase phosphatase-1 in human endothelial cells via Rac1 and NAD(P)H oxidase/Nox2-activation. Circ. Res. 96, 43–53 [DOI] [PubMed] [Google Scholar]
- 14. Irwin D. C., Tissot van Patot M. C., Tucker A., Bowen R. (2005) Direct ANP inhibition of hypoxia-induced inflammatory pathways in pulmonary microvascular and macrovascular endothelial monolayers. Am. J. Physiol. Lung Cell Mol. Physiol. 288, L849–L859 [DOI] [PubMed] [Google Scholar]
- 15. Birukova A. A., Xing J., Fu P., Yakubov B., Dubrovskyi O., Fortune J. A., Klibanov A. M., Birukov K. G. (2010) Atrial natriuretic peptide attenuates LPS-induced lung vascular leak. Role of PAK1. Am. J. Physiol. Lung Cell Mol. Physiol. 299, L652–L663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xing J., Moldobaeva N., Birukova A. A. (2011) Atrial natriuretic peptide protects against Staphylococcus aureus-induced lung injury and endothelial barrier dysfunction. J. Appl. Physiol. 110, 213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Murohara T., Kugiyama K., Ota Y., Doi H., Ogata N., Ohgushi M., Yasue H. (1999) Effects of atrial and brain natriuretic peptides on lysophosphatidylcholine-mediated endothelial dysfunction. J. Cardiovasc. Pharmacol. 34, 870–878 [DOI] [PubMed] [Google Scholar]
- 18. Kiemer A. K., Weber N. C., Fürst R., Bildner N., Kulhanek-Heinze S., Vollmar A. M. (2002) Inhibition of p38 MAPK activation via induction of MKP-1. Atrial natriuretic peptide reduces TNF-α-induced actin polymerization and endothelial permeability. Circ. Res. 90, 874–881 [DOI] [PubMed] [Google Scholar]
- 19. Birukova A. A., Zagranichnaya T., Alekseeva E., Bokoch G. M., Birukov K. G. (2008) Epac/Rap and PKA are novel mechanisms of ANP-induced Rac-mediated pulmonary endothelial barrier protection. J. Cell Physiol. 215, 715–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klinger J. R., Warburton R., Carino G. P., Murray J., Murphy C., Napier M., Harrington E. O. (2006) Natriuretic peptides differentially attenuate thrombin-induced barrier dysfunction in pulmonary microvascular endothelial cells. Exp. Cell Res. 312, 401–410 [DOI] [PubMed] [Google Scholar]
- 21. Birukova A. A., Fu P., Xing J., Yakubov B., Cokic I., Birukov K. G. (2010) Mechanotransduction by GEF-H1 as a novel mechanism of ventilator-induced vascular endothelial permeability. Am. J. Physiol. Lung Cell Mol. Physiol. 298, L837–L848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sells M. A., Knaus U. G., Bagrodia S., Ambrose D. M., Bokoch G. M., Chernoff J. (1997) Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 7, 202–210 [DOI] [PubMed] [Google Scholar]
- 23. Birukova A. A., Adyshev D., Gorshkov B., Bokoch G. M., Birukov K. G., Verin A. D. (2006) GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 290, L540–L548 [DOI] [PubMed] [Google Scholar]
- 24. Dubrovskyi O., Birukova A. A., Birukov K. G. (2013) Measurement of local permeability at subcellular level in cell models of agonist- and ventilator-induced lung injury. Lab. Invest. 93, 254–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Komarova Y., De Groot C. O., Grigoriev I., Gouveia S. M., Munteanu E. L., Schober J. M., Honnappa S., Buey R. M., Hoogenraad C. C., Dogterom M., Borisy G. G., Steinmetz M. O., Akhmanova A. (2009) Mammalian end binding proteins control persistent microtubule growth. J. Cell Biol. 184, 691–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tian X., Tian Y., Sarich N., Wu T., Birukova A. A. (2012) Novel role of stathmin in microtubule-dependent control of endothelial permeability. FASEB J. 26, 3862–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Birukova A. A., Malyukova I., Poroyko V., Birukov K. G. (2007) Paxillin-β-catenin interactions are involved in Rac/Cdc42-mediated endothelial barrier-protective response to oxidized phospholipids. Am. J. Physiol. Lung Cell Mol. Physiol. 293, L199–L211 [DOI] [PubMed] [Google Scholar]
- 28. Xing J., Birukova A. A. (2010) ANP attenuates inflammatory signaling and Rho pathway of lung endothelial permeability induced by LPS and TNFα. Microvasc. Res. 79, 26–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Nieuw Amerongen G. P., van Delft S., Vermeer M. A., Collard J. G., van Hinsbergh V. W. (2000) Activation of RhoA by thrombin in endothelial hyperpermeability. Role of Rho kinase and protein tyrosine kinases. Circ. Res. 87, 335–340 [DOI] [PubMed] [Google Scholar]
- 30. Kosoff R., Chow H. Y., Radu M., Chernoff J. (2013) Pak2 kinase restrains mast cell FcϵRI receptor signaling through modulation of Rho protein guanine nucleotide exchange factor (GEF) activity. J. Biol. Chem. 288, 974–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dubrovskyi O., Tian X., Poroyko V., Yakubov B., Birukova A. A., Birukov K. G. (2012) Identification of paxilin domains interacting with β-catenin. FEBS Lett. 586, 2294–2299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Birukova A. A., Fu P., Xing J., Cokic I., Birukov K. G. (2010) Lung endothelial barrier protection by iloprost in the 2-hit models of ventilator-induced lung injury (VILI) involves inhibition of Rho signaling. Transl. Res. 155, 44–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mirzapoiazova T., Kolosova I. A., Moreno L., Sammani S., Garcia J. G., Verin A. D. (2007) Suppression of endotoxin-induced inflammation by taxol. Eur. Respir. J. 30, 429–435 [DOI] [PubMed] [Google Scholar]
- 34. Waheed F., Dan Q., Amoozadeh Y., Zhang Y., Tanimura S., Speight P., Kapus A., Szászi K. (2013) Central role of the exchange factor GEF-H1 in TNF-α-induced sequential activation of Rac, ADAM17/TACE, and RhoA in tubular epithelial cells. Mol. Biol. Cell 24, 1068–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meiri D., Greeve M. A., Brunet A., Finan D., Wells C. D., LaRose J., Rottapel R. (2009) Modulation of Rho guanine exchange factor Lfc activity by protein kinase A-mediated phosphorylation. Mol. Cell Biol. 29, 5963–5973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Birkenfeld J., Nalbant P., Yoon S. H., Bokoch G. M. (2008) Cellular functions of GEF-H1, a microtubule-regulated Rho-GEF. Is altered GEF-H1 activity a crucial determinant of disease pathogenesis? Trends Cell Biol. 18, 210–219 [DOI] [PubMed] [Google Scholar]
- 37. Yoshimura Y., Miki H. (2011) Dynamic regulation of GEF-H1 localization at microtubules by Par1b/MARK2. Biochem. Biophys. Res. Commun. 408, 322–328 [DOI] [PubMed] [Google Scholar]
- 38. Nagae S., Meng W., Takeichi M. (2013) Non-centrosomal microtubules regulate F-actin organization through the suppression of GEF-H1 activity. Genes Cells 18, 387–396 [DOI] [PubMed] [Google Scholar]
- 39. Essler M., Staddon J. M., Weber P. C., Aepfelbacher M. (2000) Cyclic AMP blocks bacterial lipopolysaccharide-induced myosin light chain phosphorylation in endothelial cells through inhibition of Rho/Rho kinase signaling. J. Immunol. 164, 6543–6549 [DOI] [PubMed] [Google Scholar]
- 40. Birukova A. A., Tian Y., Meliton A., Leff A., Wu T., Birukov K. G. (2012) Stimulation of Rho signaling by pathologic mechanical stretch is a “second hit” to Rho-independent lung injury induced by IL-6. Am. J. Physiol. Lung Cell Mol. Physiol. 302, L965–L975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Groeneveld A. B. (2002) Vascular pharmacology of acute lung injury and acute respiratory distress syndrome. Vascul. Pharmacol. 39, 247–256 [DOI] [PubMed] [Google Scholar]
- 42. Dudek S. M., Camp S. M., Chiang E. T., Singleton P. A., Usatyuk P. V., Zhao Y., Natarajan V., Garcia J. G. (2007) Pulmonary endothelial cell barrier enhancement by FTY720 does not require the S1P1 receptor. Cell. Signal. 19, 1754–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tasaka S., Koh H., Yamada W., Shimizu M., Ogawa Y., Hasegawa N., Yamaguchi K., Ishii Y., Richer S. E., Doerschuk C. M., Ishizaka A. (2005) Attenuation of endotoxin-induced acute lung injury by the Rho-associated kinase inhibitor, Y-27632. Am. J. Respir. Cell Mol. Biol. 32, 504–510 [DOI] [PubMed] [Google Scholar]
- 44. Chen W., Pendyala S., Natarajan V., Garcia J. G., Jacobson J. R. (2008) Endothelial cell barrier protection by simvastatin. GTPase regulation and NADPH oxidase inhibition. Am. J. Physiol. Lung Cell Mol. Physiol. 295, L575–L583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jacobson J. R., Garcia J. G. (2007) Novel therapies for microvascular permeability in sepsis. Curr. Drug Targets 8, 509–514 [DOI] [PubMed] [Google Scholar]
- 46. Amano M., Kaneko T., Maeda A., Nakayama M., Ito M., Yamauchi T., Goto H., Fukata Y., Oshiro N., Shinohara A., Iwamatsu A., Kaibuchi K. (2003) Identification of Tau and MAP2 as novel substrates of Rho-kinase and myosin phosphatase. J. Neurochem. 87, 780–790 [DOI] [PubMed] [Google Scholar]
- 47. Drechsel D. N., Hyman A. A., Cobb M. H., Kirschner M. W. (1992) Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 3, 1141–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gupta R. P., Abou-Donia M. B. (1999) Tau phosphorylation by diisopropyl phosphorofluoridate (DFP)-treated hen brain supernatant inhibits its binding with microtubules. Role of Ca2+/calmodulin-dependent protein kinase II in tau phosphorylation. Arch. Biochem. Biophys. 365, 268–278 [DOI] [PubMed] [Google Scholar]