

Background: Pharmacological inhibition of the NAD+-dependent deacetylase SIRT2 holds promise for cancer therapy by preventing deacetylation and inactivation of p53.
Results: We identified two novel SIRT2 inhibitors that induce apoptosis in a p53-dependent fashion and activate three p53 target genes.
Conclusion: Small-molecule inhibition of SIRT2 activates p53-dependent apoptosis in cancer cells.
Significance: The compounds reported here are promising lead candidates for use in cancer treatment.
Keywords: Cancer Biology, Histone Deacetylase Inhibitors, p53, SIRT, Sirt1, Sirtuins, Sirt2
Abstract
Sirtuin 2 (SIRT2) is an NAD+-dependent protein deacetylase whose targets include histone H4 lysine 16, p53, and α-tubulin. Because deacetylation of p53 regulates its effect on apoptosis, pharmacological inhibition of SIRT2-dependent p53 deacetylation is of great therapeutic interest for the treatment of cancer. Here, we have identified two structurally related compounds, AEM1 and AEM2, which are selective inhibitors of SIRT2 (IC50 values of 18.5 and 3.8 μm, respectively), but show only weak effects on other sirtuins such as SIRT1, SIRT3, and yeast Sir2. Interestingly, both compounds sensitized non-small cell lung cancer cell lines toward the induction of apoptosis by the DNA-damaging agent etoposide. Importantly, this sensitization was dependent on the presence of functional p53, thus establishing a link between SIRT2 inhibition by these compounds and p53 activation. Further, treatment with AEM1 and AEM2 led to elevated levels of p53 acetylation and to increased expression of CDKN1A, which encodes the cell cycle regulator p21WAF1, as well as the pro-apoptotic genes PUMA and NOXA, three transcriptional targets of p53. Altogether, our data suggest that inhibition of SIRT2 by these compounds causes increased activation of p53 by decreasing SIRT2-dependent p53 deacetylation. These compounds thus provide a good opportunity for lead optimization and drug development to target p53-proficient cancers.
Introduction
Sirtuin 2 (SIRT2)2 is one of seven members of the sirtuin family of proteins, whose members are homologous to the silencing protein Sir2 from Saccharomyces cerevisiae (1, 2) and possess NAD+-dependent histone and protein deacetylase activity (3–5). Sirtuin enzymes have received widespread attention over the last few years due to their diverse physiological roles in metabolism, aging, and age-related human disorders (6–8). SIRT2 is the closest homolog to Hst2 from S. cerevisiae, which is a cytoplasmic H4 lysine 16 (H4 K16) histone deacetylase that disrupts telomeric silencing and increases rDNA silencing upon overexpression (9, 10). SIRT2 deacetylates ϵ-N-acetyllysine residues on a variety of protein substrates (11), including histones H3 (12) and H4 (13), the transcription factors p53 (14), p65 (15), Foxo1 (16), and Foxo3a (17), as well as α-tubulin (18) (for review, see Ref. 19). Unlike other sirtuins, SIRT2 is mainly cytoplasmic, where it co-localizes with and deacetylates lysine 40 of α-tubulin (18). During G2/M phase, SIRT2 relocates to the nucleus and deacetylates histone H4 K16 (13, 20), thereby modulating chromatin condensation during metaphase (13, 21). SIRT2 levels increase in mitosis, and SIRT2 overexpression prolongs M phase and delays mitotic exit (21), thus demonstrating a role for SIRT2 in cell cycle regulation (22). In mice, the absence of SIRT2 leads to reduced activity of the anaphase-promoting complex/cyclosome through deacetylation of Cdh1 and Cdc20, which causes higher rates of aneuploidy. Consequently, SIRT2-deficient mice show an increased cancer incidence, suggesting that SIRT2 is a tumor suppressor.
However, as for other sirtuins (23), conflicting data exist regarding the role of SIRT2 in carcinogenesis. In contrast to its role as a potential tumor suppressor, SIRT2 deacetylates and thus inhibits the activity of p53 (14, 24), suggesting that SIRT2 inhibition may be useful for anticancer treatment. An efficient increase of in vivo acetylation of p53 in a breast carcinoma cell line requires inhibition of both SIRT2 and its homolog SIRT1 (14), which also deacetylates p53 (25). Consequently, simultaneous inhibition of both SIRT1 and SIRT2 induces apoptosis in some tumor cell lines and in Burkitt lymphoma xenografts (14, 26). In other cell lines, SIRT2 down-regulation alone is sufficient to cause apoptosis, and SIRT2 depletion leads to p53 accumulation by causing activation of the p38 MAP kinase, which leads to degradation of p300 and subsequent degradation of the negative p53 regulator MDM2 (27). Furthermore, another study reported increased SIRT2 expression in 6 of 11 human pancreatic adenocarcinomas (28), and SIRT2 was found to be up-regulated in human breast cancer and hepatocellular carcinoma (29). Altogether, the role of SIRT2 as an oncogene or a tumor suppressor may therefore vary depending on the cancer type and requires further investigation to develop SIRT2 inhibitors as therapeutic interventions for the treatment of selected cancer types.
Next to its role as an anticancer target, SIRT2 also holds promise as a target for the treatment of neurodegenerative disorders in that SIRT2 inhibition in primary neuronal and invertebrate models of Parkinson and Huntington diseases rescues neurotoxicity induced by α-synuclein and huntingtin proteins, respectively (30–32).
So far, only few inhibitors of SIRT2 have been identified, but they lack selectivity for SIRT2 versus other sirtuins or have suboptimal pharmacological properties (see “Discussion”). In this study, we report the identification of two novel, structurally related SIRT2 inhibitors, compounds AEM1 and AEM2. They show selective inhibition of SIRT2 with IC50 values of 18.5 and 3.8 μm, respectively, but no inhibition of the related sirtuins SIRT1, SIRT3, and yeast Sir2. Treatment of cancer cell lines with these compounds caused sensitization of the cells to etoposide-induced apoptosis. Furthermore, we show that the sensitization by compound AEM2 partially depends on the presence of functional p53. Furthermore, AEM1 and AEM2 caused increased acetylation of p53 and enhanced the induction of the canonical p53 target genes CDKN1A, PUMA, and NOXA. Thus, AEM2 constitutes a promising lead compound for the development of SIRT2 inhibitors as anticancer therapeutics.
EXPERIMENTAL PROCEDURES
Materials
SIRT1 (full-length) and SIRT1 (amino acids 235–664) were affinity-purified from Escherichia coli strains carrying polyhistidine-tagged full-length human SIRT1 (pET30z-SIRT1, a gift from T. Kouzarides) or SIRT1(235–664) (pAE1700) using standard methods. Sir2 from S. cerevisiae was affinity-purified from E. coli cells carrying polyhistidine-tagged Sir2 (pFX21, kindly provided by M. Grunstein). SIRT2 was purchased from Calbiochem. Compounds were purchased from ChemDiv (Moscow, Russia) or Asinex (Moscow, Russia). Compound AEM2 (ChemDiv 6423-0105) was subjected to analysis by liquid chromatography coupled to mass spectrometry (LC/MS) and 1H nuclear magnetic resonance (NMR) spectroscopy. It was found to have a purity of >98% and may consist of an enantiomer mixture (supplemental Figs. S1 and S2).
Fluorescence-based Deacetylation Assay with the Substrate MAL
Deacetylation assays using Boc(Ac)Lys-7-amino-4-methyl-coumarin (MAL; Bachem, Bubendorf, Switzerland) as a substrate were performed in a volume of 20 μl in 384-well low volume plates (Eppendorf) in a reaction buffer containing 25 mm Tris-HCl (pH 8.0), 137 mm NaCl, 1 mm MgCl2, 2.7 mm KCl, 1 mg/ml BSA, and 1 mm DTT. Enzymes were added at different concentrations to wells in a volume of 10 μl and were preincubated with inhibitors (volume 1 μl, diluted in dimethyl sulfoxide) or with dimethyl sulfoxide as a control for 10 min at room temperature. Subsequently, 10 μl 2× concentrated substrate solution containing 200 μm MAL and 2 mm NAD+ was added to initiate the reaction, which was incubated at 37 °C for 4 h. This allowed for ∼50% deacetylation of MAL. After incubation, 20 μl of trypsin solution (0.5 mg/ml) was added, and the trypsin cleavage reaction was allowed to proceed at 37 °C for 1 h. Fluorescence readings were obtained using a fluorescence reader (GENiosPro TECAN), with the excitation wavelength set to 360 nm and the emission set to 465 nm. IC50 values and curve fitting were performed using GraphPad Prism 5.04 with nonlinear regression analysis. The indicated values are the average of three replicates.
Potential autofluorescence of the compounds, which may confound the deacetylation assay, was controlled by measuring the fluorescence of reaction mixtures containing all components except the sirtuin enzyme with or without 250 μm compounds. None of the compounds presented here showed autofluorescence (data not shown). A potential effect of the compounds on the trypsin cleavage reaction was investigated by testing the ability of the compounds to inhibit trypsin cleavage of unacetylated MAL. No compound inhibition was observed (data not shown). The MAL deacetylation assay was used to screen a library of ∼18,000 compounds (ChemBioNet, Screening Unit of the Leibniz-Institute for Molecular Pharmacology, Berlin, Germany) for SIRT1 inhibitors.3
HPLC-based p53 Peptide Deacetylation Assay
A deacetylation assay using a p53 peptide substrate lacking a fluorophore was performed as follows. Assay conditions were as for the MAL deacetylation assay, but with a reaction volume of 50 μl. After preincubation of SIRT2 or SIRT1 with the inhibitors for 10 min, the reaction was started by adding the substrate, an acetylated p53 peptide (HLKSKKGQSTSRHKK(Ac)LMFK, synthesized by Biosyntan, Berlin, Germany), at 100 μm in reaction buffer containing 1 mm NAD+. The reaction was incubated for 30 min at 37 °C, which allows for 50% deacetylation, and the reaction was stopped by shock freezing in liquid nitrogen. 40 μl of the reaction mixture was injected into a reversed phase C18 column (Jupiter RP-C18; Phenomenex). The acetylated and deacetylated p53 peptides were separated using increasing concentrations (1–35%) of acetonitrile in 0.1% trifluoroacetic acid over 30 min. The peptides were detected and quantified using a UV detector at a wavelength of 214 nm. The percentage of deacetylation was calculated by dividing the area under the peak of the deacetylated product by the sum of the areas under the peak of both acetylated and deacetylated p53 peptides.
Cell Culture and Cell Cycle Analysis
Non-small cell lung cancer (NSCLC) cell lines were gifts from Dr. Ö. Türeci, Mainz. Cells were grown on tissue culture dishes (BD Falcon) in RPMI 1640 medium (Invitrogen) supplemented with 10% or 0.5% fetal bovine serum, glucose, l-glutamine, and penicillin/streptomycin in a humidified atmosphere at 5% CO2. Stable expression of a tamoxifen-inducible human p53 construct (pBabepuro-p53ERTM (33)) in H1299 cells was achieved by retroviral transduction as described previously (34). Measurements of cell cycle distribution were performed by flow cytometry (Calibur; BD Biosciences) as described previously (35). For cell viability measurements, cells were grown in flat-bottom 96-well plates in the presence or absence of compounds. After defined times, cells were treated with 10 μl of 12 mm 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) for 4 h at 37 °C. Subsequently, 100 μl of solubilization solution (0.01 m HCl, 10% SDS) was added and incubated overnight at 37 °C. The absorbance was determined using a spectrophotometer (550–600 nm). Levels of acetylated p53 were measured by Western blotting using whole cell protein extracts with an α-Ac-p53 antibody (p53-K382Ac; Millipore) and with α-tubulin (Abcam) as a loading control.
Quantification of mRNA Expression Levels
The expression level of endogenous CDKN1A, PUMA, NOXA, SIRT1, and SIRT2 was determined by reverse transcription of total RNA followed by quantitative PCR analysis. The NSCLC cell lines were grown in 6-cm dishes in the presence or absence of compounds for 6 h. Total RNA was extracted using TRIzol (Invitrogen) according to the manufacturer's protocol. 3 μg of total RNA was reverse-transcribed by extension of oligo(dT) primers using SuperScript III reverse transcriptase (Invitrogen). Real-time PCR of the cDNA using specific primers was performed using the Universal ProbeLibrary System (UPL; Roche Applied Science) for CDKN1A and SYBR Green (Qiagen) for PUMA, NOXA, SIRT1, and SIRT2.
RESULTS
Identification of Two SIRT2-specific Inhibitors
In the course of a screen for novel inhibitors of SIRT1, we identified one compound that showed mild inhibition of SIRT1, but which we subsequently found to be a potent inhibitor of SIRT2 and which was pursued in this study. For the inhibitor screen, an in vitro deacetylation assay with full-length SIRT1 and MAL as a substrate was used. MAL consists of an acetyllysine residue connected to the fluorophore 7-amino-4-methyl-coumarin and is a substrate for NAD+-dependent deacetylation by SIRT1 as well as other sirtuins. Candidate inhibitors of SIRT1 were subsequently tested for inhibition of other sirtuins. Interestingly, compound AEM1 (Fig. 1A) showed strong inhibition of SIRT2-dependent deacetylation of MAL, with an IC50 value of 18.5 μm (Fig. 1B). Conversely, compound AEM1 showed weak inhibition of SIRT1, with an IC50 value of 118.4 μm (Fig. 1C).
FIGURE 1.

Identification of a SIRT2-selective inhibitor, compound AEM1. A, chemical structure of the SIRT2 inhibitor AEM1 is shown. B, SIRT2-dependent MAL deacetylation was inhibited by compound AEM1. C, compound AEM1 showed weak inhibition of MAL deacetylation by SIRT1. D, compound AEM1 and AEM2 were potent inhibitors of p53 deacetylation by SIRT2, but not SIRT1. E, compound AEM1 showed weak inhibition of truncated SIRT1(Δ1–234) or the sirtuins SIRT3 and Sir2 from S. cerevisiae (ySir2). In all cases, deacetylation in a control reaction containing dimethyl sulfoxide was set to 100%. Results are given as the average ± S.D. (error bars) of three to six independent determinations.
Because previous studies have shown that the fluorophore on the deacetylation substrate influences sirtuin activity (36, 37), we sought to determine the inhibitory effect of compound AEM1 on a substrate lacking fluorophore 7-amino-4-methyl-coumarin. For this purpose, we used a peptide corresponding to amino acids 368–386 of p53 that carries an acetylated lysine (Lys-382) as a substrate and measured deacetylation by separating and quantifying the acetylated and unacetylated peptides by HPLC. Significantly, compound AEM1 inhibited SIRT2 activity to 15% at 50 μm and to 38% at 20 μm (Fig. 1D), which thus was in a range similar to that of inhibition of MAL deacetylation. Conversely, there was no detectable inhibition of SIRT1-mediated p53 deacetylation at 50 μm (Fig. 1D).
We next tested the inhibitory effect of compound AEM1 on other sirtuins by measuring its effect on MAL deacetylation by Sir2 from S. cerevisiae (ySir2) and SIRT3. Furthermore, the SIRT1 N terminus has previously been reported to affect its activity (38), and we therefore also tested inhibition of an N-terminally truncated SIRT1(Δ1–234). Inhibition of SIRT1(Δ1–234) as well as ySir2 and SIRT3 at 50 μm compound was relatively mild, with 65–80% remaining activity for MAL deacetylation (Fig. 1E), indicating that AEM1 had a high selectivity for inhibition of SIRT2.
We furthermore sought to perform an analysis of the relationship between the structure of compound AEM1 and its inhibitory activity on SIRT2. For this purpose, we obtained four derivatives (AEM2, compounds 1, 2, and 3) with different substituents on the carboxamide group compared with compound AEM1. Significantly, addition of a 3-methyl-pyridyl group (compound AEM2) led to an improved IC50 value of 3.8 μm in the MAL deacetylation assay (Fig. 2A). The presence of a methoxy-propyl group (compound 1) caused an inhibition similar to compound AEM1 itself (18.7 μm, Fig. 2B). Conversely, substitution of two other groups (compounds 2 and 3) caused a complete loss of SIRT2 inhibition (Fig. 2, C and D). Furthermore, compound AEM2 inhibited SIRT2-mediated deacetylation of the p53 peptide to 20% at 20 μm and to 10% at 50 μm (Fig. 1D) and thus was also more potent than compound AEM1 in this assay. Taken together, this showed that the presence of a heterocyclic aromatic ring at the carboxamide position led to an increased inhibitory effect on SIRT2 deacetylation.
FIGURE 2.

Relationship between chemical structure and SIRT2 inhibition of derivatives of compound AEM1. A, inhibition of SIRT2-dependent MAL deacetylation by compound AEM2. Representation is as in Fig. 1. B, inhibition of SIRT2 by compound 1. C and D, compounds 2 and 3 showing no SIRT2 inhibition.
Compounds AEM1 and AEM2 Sensitized Lung Cancer Cells to Etoposide-induced Apoptosis
Because sirtuin inhibitors have previously been shown to have anticancer potential due to sirtuin misregulation in cancer cells (for review, see Ref. 39), we sought to determine the effects in cancer cells of the novel SIRT2 inhibitors identified here. We therefore investigated their ability to induce apoptosis in two NSCLC cell lines, A549, which is p53-proficient, and H1299, a p53-negative (p53−/−) cancer cell line. Because both cell lines are resistant to the DNA-damaging agent etoposide (VP-16) (40), we were particularly interested to determine whether the compounds led to a sensitization of the cells to etoposide treatment and thus to increased etoposide-induced apoptosis. For this purpose, the cell lines were treated with different concentrations of AEM1 or AEM2 either in the absence or in the presence of a low concentration of etoposide (1 μm) for 48 h, and the extent of apoptosis was determined by measuring the fraction of cells with subdiploid DNA content (sub-G1) by FACS analysis. In the absence of etoposide, treatment with the compounds at 20 μm led to a mild increase of apoptosis in A549 cells (Fig. 3A). As reported earlier (40), treatment of A549 with etoposide alone caused a mild increase in apoptosis in A549. Significantly, however, the combined treatment with etoposide and the compounds caused a marked increase in apoptosis up to approximately 20%. This indicated that treatment of A549 cells with AEM1 or AEM2 sensitized the cells to the cytotoxic effect of etoposide.
FIGURE 3.

SIRT2 inhibition sensitized NSCLC cells to etoposide-mediated apoptosis in a p53-dependent fashion. A and B, the cell lines A549 (p53+/+) (A) and H1299 (p53−/−) (B) were treated with increasing concentrations of compound AEM1 (left) or AEM2 (right) in the absence (dark bars) or presence of 1 μm etoposide (VP-16, light bars). After 48 h, the percentage of cells with subdiploid DNA content (sub-G1) was measured by flow cytometry as an indicator of apoptosis. Values of three independent determinations ± S.D. (error bars) are shown. C, p53−/− H1299 cells stably transduced with a tamoxifen-inducible p53-ERTM fusion (H1299 p53-ERTM) were treated as in B with compound AEM1 or AEM2 in the absence or presence of etoposide (VP-16), and apoptosis was measured as in B. D, p53 function in H1299 p53-ERTM cells was induced with 100 nm tamoxifen in parallel, and the effect of treatment with compounds AEM1 and AEM2 in combination with etoposide was measured as in A.
We next asked whether compounds AEM1 and AEM2 displayed a similar effect in the p53−/− cell line H1299. Strikingly, whereas treatment with AEM1 or AEM2 alone led to a similar increase of apoptosis as in the p53-proficient A549 cell line, it did not lead to a sensitization of the cells to etoposide. Specifically, the fraction of apoptotic H1299 cells upon etoposide treatment was approximately 10%, regardless of whether they were also exposed to the compounds or not (Fig. 3B). As H1299 cells lack the p53 tumor suppressor, these results suggested that the ability of compounds AEM1 and AEM2 to enhance etoposide-mediated apoptosis depended on p53. Because p53 is a target for SIRT2 deacetylation (14), this suggested that compounds AEM1 and AEM2 inhibited SIRT2 in vivo and that this led to increased p53 acetylation and thus to higher p53 activity and concurrent etoposide-mediated cell death. On their own, both compounds reduced cell viability of the cells to ∼50% at 20 μm and reduced cell proliferation (data not shown).
The Cytotoxic Effect of the SIRT2 Inhibitors Was Partially Dependent on Functional p53
There are arguably more genetic differences between the A549 and the H1299 cell lines than their p53 status. Therefore, to investigate the connection between the effect of compounds AEM1 and AEM2 and p53, we constructed H1299-derived cell lines with an inducible p53 allele (H1299 p53-ERTM cell line), thus eliminating any other possible differences in the background of the cell line. The H1299 p53-ERTM cell line carries a transgene encoding a fusion of human p53 to a modified murine estrogen receptor ligand-binding domain (ERTM, estrogen receptortamoxifen mutant). The p53-ERTM fusion protein is expressed in these cells but only becomes activated upon addition of 4-hydroxytamoxifen (41). In the absence of tamoxifen, the H1299 p53-ERTM cells showed a response similar to that of the original H1299 cell line to treatment with etoposide and compound AEM1 or AEM2 (Fig. 3C). Significantly, however, upon activation of p53-ERTM function with 4-hydroxytamoxifen, compound AEM2 significantly stimulated etoposide-induced cellular apoptosis (Fig. 3D). This clearly indicated that the induction of apoptosis by compound AEM2 required the presence of functional p53. In contrast to compound AEM2, compound AEM1 did not increase etoposide-mediated apoptosis at concentrations up to 20 μm (Fig. 3D), perhaps because of its weaker inhibitory effect on SIRT2. In summary, these results established a functional link between pharmacological SIRT2 inhibition and p53-mediated apoptosis.
Treatment with SIRT2 Inhibitors Caused Increased p53 Acetylation and Induced Expression of the Canonical p53 Target Genes CDKN1A, PUMA, and NOXA
The effects of AEM1 and AEM2 on p53-mediated apoptosis above raised the question whether these compounds, which we identified as in vitro SIRT2 inhibitors, also caused a measurable increase in p53 acetylation in vivo. To test this, the p53-proficient cell line A549 was treated with AEM1 or AEM2 in the presence or absence of etoposide, and p53 acetylation levels were measured by Western blotting with an α-p53-K382Ac antibody. As expected, treatment with etoposide alone led to p53 activation and thus to an induction of p53 acetylation (Fig. 4A). Significantly, concomitant treatment with either AEM1 or AEM2 enhanced the etoposide-induced p53 acetylation, whereas the SIRT2 inhibitors alone showed no effect. Of note, neither compound led to a reduction of SIRT2 or SIRT1 expression (Fig. 4B), which might also have explained higher p53 acetylation levels. Therefore, because both compounds are in vitro SIRT2 inhibitors, this indicated that they directly inhibit SIRT2 in vivo, which leads to increased p53 acetylation levels.
FIGURE 4.
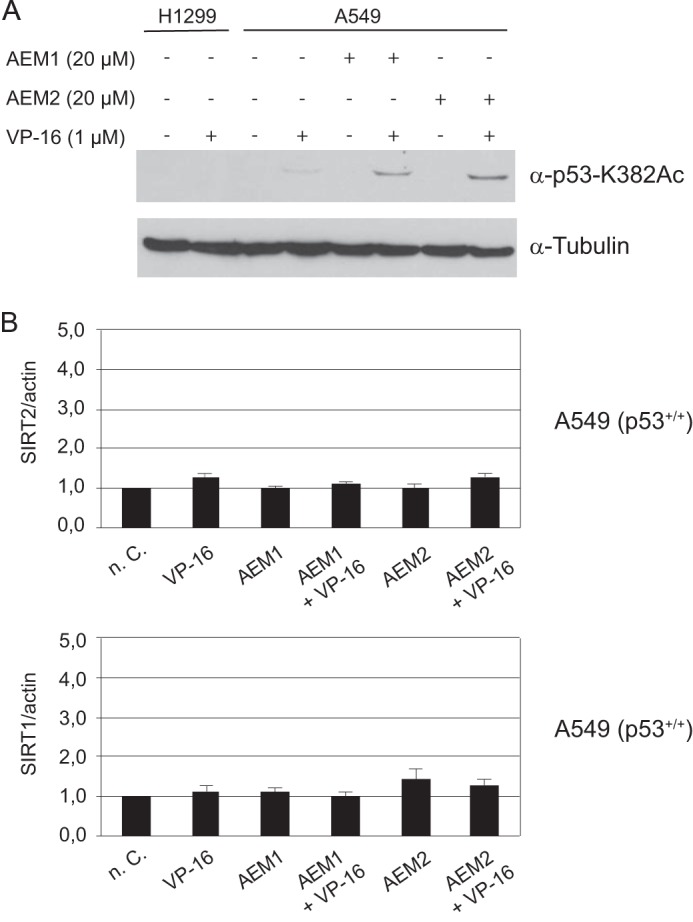
Enhancement of p53 acetylation signal by pharmacological SIRT2 inhibition. A, the cell line A549 (p53+/+) was treated with 20 μm compound AEM1 or AEM2 in the absence or presence of 1 μm etoposide (VP-16). The acetylation level of p53 protein was determined by Western blotting using an α-p53-K382Ac antibody. Tubulin served as a loading control. The cell line H1299 (p53−/−) was used as a negative control. B, expression levels of SIRT2 and SIRT1 were unaffected by compounds AEM1 and AEM2. mRNA expression levels were measured by real-time PCR analysis. Expression levels of untreated cells (n. C.) was set to 1 and normalized to actin mRNA in individual experiments. Values of three independent determinations ± S.D. (error bars) are shown.
To further evaluate the link between SIRT2 inhibition by AEM1 and AEM2 and the induction of p53 activity, we tested whether SIRT2 inhibition by these compounds caused induction of three transcriptional targets of p53, CDKN1A (42), NOXA (43), and PUMA (44). For this purpose, p53-proficient A549 cells were treated with the compounds alone or in combination with etoposide, and the level of CDKN1A, PUMA, and NOXA RNA expression was measured. Etoposide treatment alone causes p53 induction, which led to increased expression of CDKN1A and PUMA, but not NOXA (Fig. 5). Importantly, induction of all three p53 target genes was potentiated upon simultaneous treatment with AEM2 (and to a lesser degree with AEM1). These results further corroborated the notion that compound AEM2 caused inhibition of SIRT2 within the cell, which in turn elicited activation of p53 and thus stronger transcriptional activation of p53 target genes, which in turn elicits p53-dependent apoptosis via pro-apoptotic BH3 molecules encoded by PUMA and NOXA.
FIGURE 5.
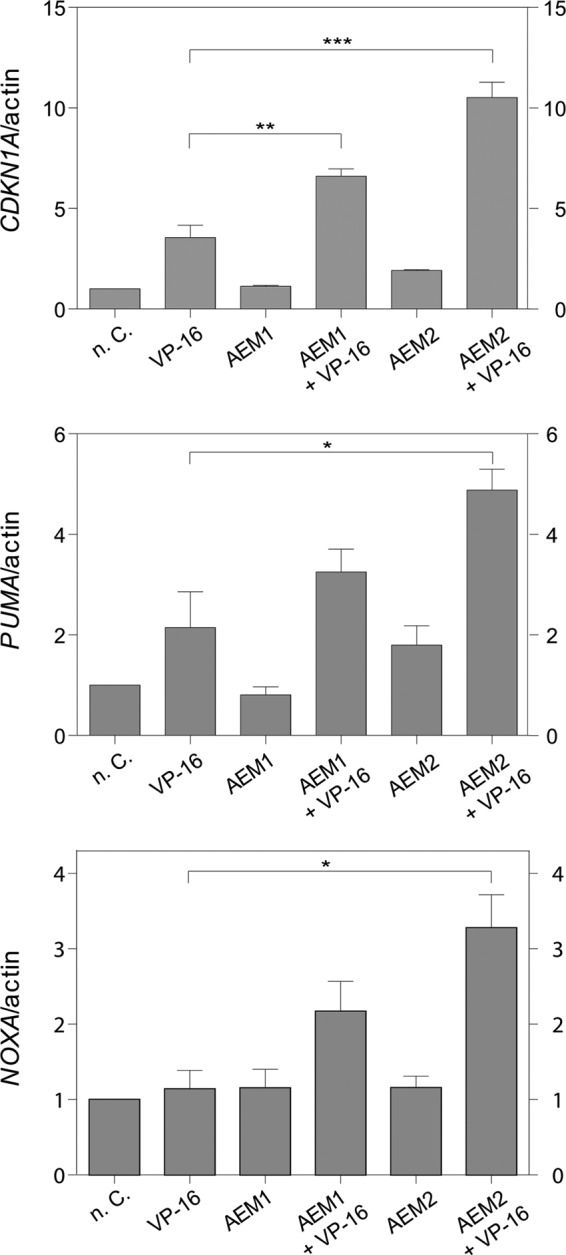
SIRT2 inhibition induced CDKN1A, PUMA, and NOXA expression in response to etoposide in the NSCLC cell line A549. The cell line A549 was treated with 20 μm AEM1 or AEM2 in the absence or presence of 1 μm etoposide (VP-16). After 6 h, the expression level of each gene was measured by real-time PCR analysis. RNA expression of untreated cells (n. C.) was set to 1 and normalized to actin mRNA in individual experiments. Values of at least three independent determinations ± S.D. (error bars) are shown. p values were determined by an unpaired, two-tailed Student's t test (*, p < 0.1; **, p < 0.01; ***, p < 0.001).
DISCUSSION
In recent years, SIRT2 inhibition has emerged as a strategy for the treatment of age-related disorders, including neurological diseases and cancer (19). In this study, we have identified two novel SIRT2 inhibitors, AEM1 and AEM2, that show improved selectivity toward SIRT2 compared with other sirtuin inhibitors, induce apoptosis in two cancer cell lines, and enhance etoposide-mediated apoptosis. Significantly, we demonstrate that AEM2 exerts its effect on etoposide-mediated cell death in a manner that is dependent on p53, and it causes increased acetylation of p53 in vivo. Furthermore, AEM2 leads to enhanced activation of the p53 target genes CDKN1A, PUMA, and NOXA, thus providing a functional link between pharmacological inhibition of p53 acetylation and selective cytotoxicity of a SIRT2 inhibitor in cancer cells.
We note that only few sirtuin inhibitors with selectivity for SIRT2 have been described so far (Table 1). Compound AEM2 shows an ∼30-fold preference for SIRT2 inhibition over SIRT1. EX527 also inhibits SIRT2, but is a more potent inhibitor of SIRT1. Nonetheless, EX527 lacks an in vivo effect on apoptosis (14). Furthermore, compound AEM2 shows similar inhibition of SIRT2 as AC-93253, but is more selective because it shows less inhibition of SIRT1. However, AC-93253 showed high cytotoxicity in several cancer cell lines at nanomolar concentrations (45), suggesting that its toxicity may result from the inactivation of proteins other than SIRT2 targets. Notably, compound AEM2 shows somewhat higher inhibition of SIRT2 than the SIRT2 inhibitor AK-7, and AK-7 is similarly selective. Therefore, the two compounds identified here constitute improved lead candidates for the development of SIRT2-specific inhibitors for therapeutic use.
TABLE 1.
Comparison of IC50 and selectivity of SIRT2 inhibitors
| Compounda | IC50 for SIRT1 | IC50 for SIRT2 | Reference |
|---|---|---|---|
| μm | μm | ||
| AC-93253 | 45.3 | 6 | (45) |
| AGK2 | 40 | 3.5 | (31) |
| AK-1 | >50 | 12.5 | (31) |
| AK-7 | >50 | 15.5 | (48) |
| Cambinol | 56 | 59 | (26) |
| Compound AEM1 | >100 | 18.5 | This study |
| Compound AEM2 | >100 | 3.8 | This study |
| EX527 | 0.38 | 32.6 | (14) |
| Salermide | 76.2 | 45 | (14) |
| Sirtinol | 37.6 | 103.4 | (14) |
a Compounds are sorted alphabetically.
Acetylation of p53 by p300/CBP that is induced by cellular stress stimulates the DNA-binding capacity of p53 and enhances its biological function in vivo (46). Deacetylation of p53 by SIRT1 and SIRT2 therefore counteracts p53-dependent cell cycle arrest (25), and SIRT1/SIRT2 inhibition is therefore expected to enhance p53-mediated apoptosis. Our results with compound AEM2 are in good agreement with this. Etoposide treatment leads to cellular stress and to the stabilization of p53, which may result in cell cycle arrest and apoptosis. We observed that apoptosis was enhanced by the presence of compound AEM2, but only in p53-proficient cells, and this enhancement was accompanied by increased levels of acetylated p53. Our observations are consistent with the interpretation that compound AEM2 directly inhibits p53 deacetylation by SIRT2 and that p53 therefore is more active on promoters of its downstream effectors. The latter is supported by the potentiation of the induction of the canonical p53 target gene CDKN1A and the pro-apoptotic genes PUMA and NOXA.
It should, however, be noted that both compound AEM1 and AEM2 had etoposide-independent effects on cell viability, which is consistent with the fact that SIRT2 has other cellular targets apart from p53. Whether these effects are mediated by α-tubulin or histone deacetylation, or whether other unknown targets are responsible, remains to be determined. It is also possible that the compounds exert off-target effects that contribute to their effect on cellular viability.
Several sirtuin inhibitors have been tested for their anticancer potential. Heltweg et al. (26) developed the β-naphthol derivative cambinol, which inhibits both SIRT1 and SIRT2. Treatment with cambinol sensitized cancer cells to DNA-damaging agents, but this effect was independent of the presence of the SIRT1 target proteins p53, Ku70, and Foxo3a, suggesting that in this context, reduced deacetylation of other SIRT1 and SIRT2 targets was important for cambinol-induced cell death. In a second study, Peck et al. (14) investigated the effect of the three sirtuin inhibitors, sirtinol, salermide, and EX527, in a breast cancer cell line. Based on the different inhibitory activities of the three compounds and on experiments down-regulating both sirtuins, they concluded that the simultaneous inhibition of both SIRT1 and SIRT2 was required to induce cell death and to increase acetylation of p53 and α-tubulin. However, in contrast to the results with cambinol (26), in this case the induction of apoptosis by sirtinol and salermide required the presence of functional p53 (14). Third, the SIRT1/SIRT2 inhibitor AC-93253 caused varying degrees of cytotoxicity in a panel of cancer cell lines, although, as noted above, this may be due to a generally high toxicity of the compound (45). Altogether, the effect of SIRT1/2 inhibitors appears to be complex and strongly dependent on the cell type and the genetic context. Our results support this view and show that SIRT2 inhibition alone can induce cell death in NSCLC cell lines and that cytotoxicity may be enhanced in the presence of functional p53. Thus, therapeutic SIRT2 inhibition may be beneficial for the treatment of selected cancer types. One possibility is to combine SIRT2 inhibitors with other (histone) deacetylase inhibitors such as trichostatin A or vorinostat to inhibit parallel functional pathways. In other scenarios, a more selective inhibition of SIRT2 may be preferable over the pleiotropic effects of inhibition of the “classical” histone deacetylase classes I, II, and IV (47).
Next to their potential in cancer treatment, SIRT2 inhibitors are being developed to counteract neurodegenerative disorders such as Huntington, Parkinson, and Alzheimer disease (19). Initial reports identified the compound AGK2 as a selective SIRT2 inhibitor that rescued α-synuclein-mediated toxicity in an in vitro model of Parkinson (31). Furthermore, AGK2 provided neuroprotection in an in vitro model of Huntington disease, and genetic inhibition of a sirtuin in a Drosophila model of Huntington disease elicited neuroprotection (30). Unfortunately, AGK2 lacks brain-permeable properties, thus precluding its use for the treatment of human neurological disorders. A later study identified the compound AK-7 as a potent and selective in vitro inhibitor of SIRT2 (48). AK-7 is brain-permeable, and its application in mouse models of Huntington disease resulted in a significant improvement of neurological phenotypes (49). As noted above, the inhibitors AEM1 and AEM2 show a similar degree of SIRT2 inhibition and similar selectivity as AK-7. Thus, in addition their possible use for anticancer therapy, they also hold potential for the treatment of neurological disorders.
Supplementary Material
Acknowledgments
We thank T. Kouzarides and M. Grunstein for providing plasmids; J. Oeljeklaus and M. Kaiser for LC/MS and NMR analysis of AEM2 and advice; K. Jänen, J. Haremza, J. Markowetz, and M. Rübeling for technical support; and all laboratory members for valuable discussions.
This work was supported by Deutsche Forschungsgemeinschaft Graduate Research Training Program Grants GRK1431 (to A. E.-M.) and SCHU1541/5-1 (to M. S.) and by the University of Duisburg-Essen.

This article contains supplemental Figs. S1 and S2.
G. Hoffmann, F. Breitenbücher, M. Schuler, and A. E. Ehrenhofer-Murray, unpublished data.
- SIRT2
- sirtuin 2
- MAL
- Boc(Ac)Lys-7-amino-4-methyl-coumarin
- NSCLC
- non-small cell lung cancer
- TM
- tamoxifen mutant.
REFERENCES
- 1. Frye R. A. (1999) Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 260, 273–279 [DOI] [PubMed] [Google Scholar]
- 2. Frye R. A. (2000) Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 273, 793–798 [DOI] [PubMed] [Google Scholar]
- 3. Imai S., Armstrong C. M., Kaeberlein M., Guarente L. (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 [DOI] [PubMed] [Google Scholar]
- 4. Landry J., Sutton A., Tafrov S. T., Heller R. C., Stebbins J., Pillus L., Sternglanz R. (2000) The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. U.S.A. 97, 5807–5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith J. S., Brachmann C. B., Celic I., Kenna M. A., Muhammad S., Starai V. J., Avalos J. L., Escalante-Semerena J. C., Grubmeyer C., Wolberger C., Boeke J. D. (2000) A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. U.S.A. 97, 6658–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Milne J. C., Denu J. M. (2008) The Sirtuin family: therapeutic targets to treat diseases of aging. Curr. Opin. Chem. Biol. 12, 11–17 [DOI] [PubMed] [Google Scholar]
- 7. Haigis M. C., Sinclair D. A. (2010) Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5, 253–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Longo V. D., Kennedy B. K. (2006) Sirtuins in aging and age-related disease. Cell 126, 257–268 [DOI] [PubMed] [Google Scholar]
- 9. Perrod S., Cockell M. M., Laroche T., Renauld H., Ducrest A. L., Bonnard C., Gasser S. M. (2001) A cytosolic NAD-dependent deacetylase, Hst2p, can modulate nucleolar and telomeric silencing in yeast. EMBO J. 20, 197–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao K., Chai X., Marmorstein R. (2003) Structure of the yeast Hst2 protein deacetylase in ternary complex with 2′-O-acetyl ADP ribose and histone peptide. Structure 11, 1403–1411 [DOI] [PubMed] [Google Scholar]
- 11. Sauve A. A., Wolberger C., Schramm V. L., Boeke J. D. (2006) The biochemistry of sirtuins. Annu. Rev. Biochem. 75, 435–465 [DOI] [PubMed] [Google Scholar]
- 12. Das C., Lucia M. S., Hansen K. C., Tyler J. K. (2009) CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 459, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vaquero A., Scher M. B., Lee D. H., Sutton A., Cheng H. L., Alt F. W., Serrano L., Sternglanz R., Reinberg D. (2006) SirT2 is a histone deacetylase with preference for histone H4 Lys-16 during mitosis. Genes Dev. 20, 1256–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Peck B., Chen C. Y., Ho K. K., Di Fruscia P., Myatt S. S., Coombes R. C., Fuchter M. J., Hsiao C. D., Lam E. W. (2010) SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol. Cancer Ther. 9, 844–855 [DOI] [PubMed] [Google Scholar]
- 15. Rothgiesser K. M., Erener S., Waibel S., Lüscher B., Hottiger M. O. (2010) SIRT2 regulates NF-κB-dependent gene expression through deacetylation of p65 Lys-310. J. Cell Sci. 123, 4251–4258 [DOI] [PubMed] [Google Scholar]
- 16. Jing E., Gesta S., Kahn C. R. (2007) SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 6, 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang F., Nguyen M., Qin F. X., Tong Q. (2007) SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6, 505–514 [DOI] [PubMed] [Google Scholar]
- 18. North B. J., Marshall B. L., Borra M. T., Denu J. M., Verdin E. (2003) The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 11, 437–444 [DOI] [PubMed] [Google Scholar]
- 19. de Oliveira R. M., Sarkander J., Kazantsev A. G., Outeiro T. F. (2012) SIRT2 as a therapeutic target for age-related disorders. Front. Pharmacol. 3, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. North B. J., Verdin E. (2007) Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PloS One 2, e784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dryden S. C., Nahhas F. A., Nowak J. E., Goustin A. S., Tainsky M. A. (2003) Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol. Cell. Biol. 23, 3173–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Inoue T., Hiratsuka M., Osaki M., Oshimura M. (2007) The molecular biology of mammalian SIRT proteins: SIRT2 in cell cycle regulation. Cell Cycle 6, 1011–1018 [DOI] [PubMed] [Google Scholar]
- 23. Bosch-Presegué L., Vaquero A. (2011) The dual role of sirtuins in cancer. Genes Cancer 2, 648–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jin Y. H., Kim Y. J., Kim D. W., Baek K. H., Kang B. Y., Yeo C. Y., Lee K. Y. (2008) Sirt2 interacts with 14-3-3 β/γ and down-regulates the activity of p53. Biochem. Biophys. Res. Commun. 368, 690–695 [DOI] [PubMed] [Google Scholar]
- 25. Luo J., Nikolaev A. Y., Imai S., Chen D., Su F., Shiloh A., Guarente L., Gu W. (2001) Negative control of p53 by Sir2α promotes cell survival under stress. Cell 107, 137–148 [DOI] [PubMed] [Google Scholar]
- 26. Heltweg B., Gatbonton T., Schuler A. D., Posakony J., Li H., Goehle S., Kollipara R., Depinho R. A., Gu Y., Simon J. A., Bedalov A. (2006) Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 66, 4368–4377 [DOI] [PubMed] [Google Scholar]
- 27. Li Y., Matsumori H., Nakayama Y., Osaki M., Kojima H., Kurimasa A., Ito H., Mori S., Katoh M., Oshimura M., Inoue T. (2011) SIRT2 down-regulation in HeLa can induce p53 accumulation via p38 MAPK activation-dependent p300 decrease, eventually leading to apoptosis. Genes Cells 16, 34–45 [DOI] [PubMed] [Google Scholar]
- 28. Ouaïssi M., Sielezneff I., Silvestre R., Sastre B., Bernard J. P., Lafontaine J. S., Payan M. J., Dahan L., Pirrò N., Seitz J. F., Mas E., Lombardo D., Ouaissi A. (2008) High histone deacetylase 7 (HDAC7) expression is significantly associated with adenocarcinomas of the pancreas. Ann. Surg. Oncol. 15, 2318–2328 [DOI] [PubMed] [Google Scholar]
- 29. Kim H. S., Vassilopoulos A., Wang R. H., Lahusen T., Xiao Z., Xu X., Li C., Veenstra T. D., Li B., Yu H., Ji J., Wang X. W., Park S. H., Cha Y. I., Gius D., Deng C. X. (2011) SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 20, 487–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luthi-Carter R., Taylor D. M., Pallos J., Lambert E., Amore A., Parker A., Moffitt H., Smith D. L., Runne H., Gokce O., Kuhn A., Xiang Z., Maxwell M. M., Reeves S. A., Bates G. P., Neri C., Thompson L. M., Marsh J. L., Kazantsev A. G. (2010) SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 107, 7927–7932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Outeiro T. F., Kontopoulos E., Altmann S. M., Kufareva I., Strathearn K. E., Amore A. M., Volk C. B., Maxwell M. M., Rochet J. C., McLean P. J., Young A. B., Abagyan R., Feany M. B., Hyman B. T., Kazantsev A. G. (2007) Sirtuin 2 inhibitors rescue α-synuclein-mediated toxicity in models of Parkinson's disease. Science 317, 516–519 [DOI] [PubMed] [Google Scholar]
- 32. Pallos J., Bodai L., Lukacsovich T., Purcell J. M., Steffan J. S., Thompson L. M., Marsh J. L. (2008) Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington's disease. Hum. Mol. Genet. 17, 3767–3775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Littlewood T. D., Hancock D. C., Danielian P. S., Parker M. G., Evan G. I. (1995) A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 23, 1686–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schuler M., Maurer U., Goldstein J. C., Breitenbücher F., Hoffarth S., Waterhouse N. J., Green D. R. (2003) p53 triggers apoptosis in oncogene-expressing fibroblasts by the induction of Noxa and mitochondrial Bax translocation. Cell Death Differ. 10, 451–460 [DOI] [PubMed] [Google Scholar]
- 35. Nicoletti I., Migliorati G., Pagliacci M. C., Grignani F., Riccardi C. (1991) A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 139, 271–279 [DOI] [PubMed] [Google Scholar]
- 36. Beher D., Wu J., Cumine S., Kim K. W., Lu S. C., Atangan L., Wang M. (2009) Resveratrol is not a direct activator of SIRT1 enzyme activity. Chem. Biol. Drug Des. 74, 619–624 [DOI] [PubMed] [Google Scholar]
- 37. Kaeberlein M., McDonagh T., Heltweg B., Hixon J., Westman E. A., Caldwell S. D., Napper A., Curtis R., DiStefano P. S., Fields S., Bedalov A., Kennedy B. K. (2005) Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 280, 17038–17045 [DOI] [PubMed] [Google Scholar]
- 38. Pan M., Yuan H., Brent M., Ding E. C., Marmorstein R. (2012) SIRT1 contains N- and C-terminal regions that potentiate deacetylase activity. J. Biol. Chem. 287, 2468–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fraga M. F., Esteller M. (2007) Epigenetics and aging: the targets and the marks. Trends Genet. 23, 413–418 [DOI] [PubMed] [Google Scholar]
- 40. Meiler J., Guyot M., Hoffarth S., Wesarg E., Höhn Y., Breitenbuecher F., Schuler M. (2012) Individual dose and scheduling determine the efficacy of combining cytotoxic anticancer agents with a kinase inhibitor in non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 138, 1385–1394 [DOI] [PubMed] [Google Scholar]
- 41. Vater C. A., Bartle L. M., Dionne C. A., Littlewood T. D., Goldmacher V. S. (1996) Induction of apoptosis by tamoxifen-activation of a p53-estrogen receptor fusion protein expressed in E1A and T24 H-ras transformed p53−/− mouse embryo fibroblasts. Oncogene 13, 739–748 [PubMed] [Google Scholar]
- 42. el-Deiry W. S., Tokino T., Velculescu V. E., Levy D. B., Parsons R., Trent J. M., Lin D., Mercer W. E., Kinzler K. W., Vogelstein B. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 [DOI] [PubMed] [Google Scholar]
- 43. Oda E., Ohki R., Murasawa H., Nemoto J., Shibue T., Yamashita T., Tokino T., Taniguchi T., Tanaka N. (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288, 1053–1058 [DOI] [PubMed] [Google Scholar]
- 44. Yu J., Zhang L., Hwang P. M., Kinzler K. W., Vogelstein B. (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 7, 673–682 [DOI] [PubMed] [Google Scholar]
- 45. Zhang Y., Au Q., Zhang M., Barber J. R., Ng S. C., Zhang B. (2009) Identification of a small molecule SIRT2 inhibitor with selective tumor cytotoxicity. Biochem. Biophys. Res. Commun. 386, 729–733 [DOI] [PubMed] [Google Scholar]
- 46. Gu W., Roeder R. G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606 [DOI] [PubMed] [Google Scholar]
- 47. New M., Olzscha H., La Thangue N. B. (2012) HDAC inhibitor-based therapies: can we interpret the code? Mol. Oncol. 6, 637–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Taylor D. M., Balabadra U., Xiang Z., Woodman B., Meade S., Amore A., Maxwell M. M., Reeves S., Bates G. P., Luthi-Carter R., Lowden P. A., Kazantsev A. G. (2011) A brain-permeable small molecule reduces neuronal cholesterol by inhibiting activity of sirtuin 2 deacetylase. ACS Chem. Biol. 6, 540–546 [DOI] [PubMed] [Google Scholar]
- 49. Chopra V., Quinti L., Kim J., Vollor L., Narayanan K. L., Edgerly C., Cipicchio P. M., Lauver M. A., Choi S. H., Silverman R. B., Ferrante R. J., Hersch S., Kazantsev A. G. (2012) The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington's disease mouse models. Cell Rep. 2, 1492–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.