

Background: Mycothiol, the major low-molecular weight thiol of Mycobacterium tuberculosis, is important for peroxide detoxification and virulence.
Results: Mycothiol, together with mycoredoxin-1, a glutaredoxin-like protein, reduces the one-cysteine peroxiredoxin AhpE from the bacterium.
Conclusion: Mycobacterium tuberculosis AhpE is a mycothiol/mycoredoxin-1-dependent peroxidase.
Significance: Our results provide the first molecular link between a thiol-dependent peroxidase and the mycothiol/mycoredoxin-1 pathway in Mycobacteria.
Keywords: Hydrogen Peroxide, Mycobacterium tuberculosis, Peroxiredoxin, Redox Signaling, Thiol, Mycoredoxin, Mycothiol
Abstract
Mycobacterium tuberculosis (M. tuberculosis), the pathogen responsible for tuberculosis, detoxifies cytotoxic peroxides produced by activated macrophages. M. tuberculosis expresses alkyl hydroxyperoxide reductase E (AhpE), among other peroxiredoxins. So far the system that reduces AhpE was not known. We identified M. tuberculosis mycoredoxin-1 (MtMrx1) acting in combination with mycothiol and mycothiol disulfide reductase (MR), as a biologically relevant reducing system for MtAhpE. MtMrx1, a glutaredoxin-like, mycothiol-dependent oxidoreductase, directly reduces the oxidized form of MtAhpE, through a protein mixed disulfide with the N-terminal cysteine of MtMrx1 and the sulfenic acid derivative of the peroxidatic cysteine of MtAhpE. This disulfide is then reduced by the C-terminal cysteine in MtMrx1. Accordingly, MtAhpE catalyzes the oxidation of wt MtMrx1 by hydrogen peroxide but not of MtMrx1 lacking the C-terminal cysteine, confirming a dithiolic mechanism. Alternatively, oxidized MtAhpE forms a mixed disulfide with mycothiol, which in turn is reduced by MtMrx1 using a monothiolic mechanism. We demonstrated the H2O2-dependent NADPH oxidation catalyzed by MtAhpE in the presence of MR, Mrx1, and mycothiol. Disulfide formation involving mycothiol probably competes with the direct reduction by MtMrx1 in aqueous intracellular media, where mycothiol is present at millimolar concentrations. However, MtAhpE was found to be associated with the membrane fraction, and since mycothiol is hydrophilic, direct reduction by MtMrx1 might be favored. The results reported herein allow the rationalization of peroxide detoxification actions inferred for mycothiol, and more recently, for Mrx1 in cellular systems. We report the first molecular link between a thiol-dependent peroxidase and the mycothiol/Mrx1 pathway in Mycobacteria.
Introduction
Mycobacterium tuberculosis is the causative agent of tuberculosis disease (TB).6 Despite the efforts made during the last two decades to reduce new TB cases and deaths, the global burden of the disease remains enormous (1). Moreover, the emergency of multi- and extensively drug resistant strains underlines the need for the urgent development of new therapeutic approaches (1, 2). However, since many aspects of the pathogenic mechanisms and virulence of M. tuberculosis are still unknown, the identification of novel drug targets has been very challenging (2).
M. tuberculosis survives inside the hostile environment of host cells with diverse defense strategies. These antioxidant defenses allow the pathogen to detoxify reactive oxygen and nitrogen species formed by activated macrophages (3–5). One particular feature of the antioxidant systems of M. tuberculosis as well as other Actinomycetes is the absence of glutathione, and the presence of millimolar concentrations of 1-d-myo-inosityl 2-(N-acetyl-l-cysteinyl)amido-2-deoxy-α-d-glucopyranoside (mycothiol or MSH) (6, 7) as the main low molecular weight thiol. Mycothiol is maintained in its reduced state by a mycothiol disulfide reductase that uses NADPH as electron donor (8) and plays a role in peroxide detoxification in vivo, as evidenced by the increased susceptibility to hydrogen peroxide (H2O2), menadione and tert-butyl hydroperoxide in M. smegmatis and M. tuberculosis mutant strains disrupted in the genes required for mycothiol biosynthesis (9–11). Compensatory overexpression of an organic hydroperoxide resistance protein (Ohr) in M. smegmatis lacking MSH suggests the existence of a MSH-dependent organic hydroperoxide peroxidase (12). Although these data are only indirect evidence for the presence of a MSH-dependent peroxidase in these bacteria, the molecular link between MSH and peroxidase activity has not been clearly established yet, and purified peroxidases studied so far failed in using MSH as reducing substrate (13). Despite the absence of glutathione, the M. tuberculosis genome encodes different glutaredoxin-like proteins (14). Among them, mycoredoxin 1 (MtMrx1 EC 1.20.4.3) has recently been described as a 10-kDa protein with a CGYC catalytic motif that accepts electrons from MSH and reduces MSH-containing mixed disulfides by a monothiolic mechanism (7, 15). Strains of M. smegmatis lacking Mrx1 are more susceptible to different forms of oxidative stress (15). Moreover, in Corynebacterium glutamicum, this protein participates in the arsenate resistance system by reducing the mycothiol arsenate adduct (16). More recently, twenty-five proteins of C. glutamicum including thiol peroxidase (TPx) were found mycothiolated under hypochloric stress conditions (17). The S-mycothiolation of Tpx inhibits its peroxidase activity, but could be restored after treatment with CgMrx1. So far no MtMrx1-dependent protein reduction has been described in Mycobacteria. M. tuberculosis expresses a heme-dependent peroxidase (catalase peroxidase, KatG, EC 1.11.1.6) and several thiol-dependent peroxidases of the peroxiredoxin (Prx, EC 1.11.1.15) type, including alkyl hydroperoxide reductase C, TPx, two bacterioferritin comigratory proteins (Bcp andBcpB) and alkyl hydroperoxide reductase E (MtAhpE) (13, 18).7 As a one-cysteine peroxidase, MtAhpE lacks a resolving cysteine. However, the structure and sequence of MtAhpE show greater similarity with two-cysteine Prxs than with other one-cysteine Prxs, and is considered as the prototype of a novel Prx subfamily (19–21). We have previously investigated the peroxidase activity of MtAhpE, the only Prx of the AhpE subfamily to be functionally characterized. It is a broad-spectrum peroxidase with higher catalytic efficiency for fatty acid hydroperoxides and peroxynitrite than for H2O2. Upon reaction with the peroxide substrate (ROOH), the thiolate form of the peroxidatic cysteine (CP) is oxidized to a sulfenic acid (22, 23) as in Equation 1.
MtAhpE catalytically reduces hydrogen peroxide (H2O2) in the presence of dithiothreitol (DTT) or thionitrobenzoate (TNB) as reducing agents (22). However, the physiological reducing substrate(s) for this enzyme (as well as for AhpE-like Prxs from several other bacteria) is/are still unknown. Neither N-acetylcysteine nor glutathione reduced oxidized MtAhpE, but led to the formation of mixed disulfides (22) in Equation 2.
Similar disulfide formation involving the main low molecular weight thiol of the bacteria, mycothiol, had not been addressed so far.
Herein, we report that MtMrx1 and MtMrx1 in combination with mycothiol acts as a reducing substrate for MtAhpE. Our data not only constitute the first report for biologically relevant routes of reduction for an AhpE-like Prx, but also describe the first acceptor for the electrons provided by a mycoredoxin in Mycobacteria. We also propose a functional link between the mycothiol/mycoredoxin-1 pathway and the bacterial peroxide detoxification systems, which helps to rationalize the increased peroxide-dependent cytotoxicity in Mycobacteria with a reduced MSH content (9–11).
EXPERIMENTAL PROCEDURES
Chemicals
H2O2 was from Mallinckrodt Chemicals. Dithiotreitol (DTT), N-ethylmaleimide (NEM), β-mercaptoethanol (β-ME), diethylenetriaminepentaacetic acid (DTPA), methoxypolyethylene glycol maleimide (PEG-maleimide), sodium dodecyl sulfate (SDS), 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid, N-(2-hydroxyethyl) piperazine-N′-(2-ethanesulfonic acid), (HEPES), and horseradish peroxidase (HRP) were from Sigma. β-Nicotinamide adenine dinucleotide phosphate reduced tetrasodium salt (NADPH) was from Applichem. PageRuler™ pre-stained protein ladder was from Fermentas. Amplex® red was from Invitrogen. All other reagents were obtained from standard commercial sources and used as received. All experiments were performed in 100 mm phosphate buffer, 0.1 mm DTPA, pH 7.4, and 25 °C, except if otherwise indicated.
Mycothiol Purification
MSH was purified from M. smegmatis as described (7).
Protein Expression and Purification
Mycothiol disulfide reductase from either M. tuberculosis (MtMR) or C. glutamicum (CgMR) were expressed and purified as previously described (15, 16). The CgMR preparation was more robust and active compared with MtMR, and therefore it was preferentially utilized. MtAhpE was expressed in Escherichia coli BL21 (DE3) (expression vector pDEST17) as a recombinant His-tagged protein and purified as previously described (22). The enzyme was stored at −80 °C under argon atmosphere. Wild type MtMrx1 (MtMrx1wt) and a mutant form at the second cysteine residue (MtMrx1CXXA) were expressed in Escherichia coli BL21 Star and purified as previously described (15). Following the immobilized metal ion affinity chromatography step, MtMrx1CXXA was further purified by size exclusion chromatography using a Superdex 75 10/300 column in 100 mm phosphate buffer, 0.1 mm DTPA, pH 7.4. Both MtMrx1wt and MtMrx1CXXA were stored at −20 °C in the presence of 5 mm DTT. Excess reductant was removed by gel filtration using a HiTrap desalting column (Amersham Bioscience) with UV-vis detection at 280 nm immediately before use.
Protein Thiol Reduction
MtAhpE was reduced before use by incubation with 1 mm DTT for 30 min at 4 °C. Excess reductant was removed from all proteins immediately before use by gel filtration as above indicated.
Peroxide, Protein, and Thiol Quantitation
The concentration of H2O2 stock solutions was measured at 240 nm (λ240 = 43.6 m−1cm−1) (24). MtAhpE, MtMrx1wt, and MtMrx1CXXA concentrations were determined spectrophotometrically at 280 nm, using molar absorption coefficients of 23,950 (22), 9,974 and 9,942 m−1 cm−1, respectively, calculated from amino acidic composition. Protein concentrations calculated refer to those of monomers. The concentration of HRP was determined by its absorption at the Soret band (λ 403 = 1.02 × 105 m−1cm−1 (25)). Protein thiol content was measured by Ellman's assay (λ412 = 14,150 m−1cm−1) (26, 27). The concentration of MR was estimated by the absorption of the FAD cofactor at 463 nm (ϵ = 11,300 m−1s−1) as previously (8).
Protein Thiol Alkylation by PEG-maleimide and Electrophoretic Analysis
Reaction mixtures (100 μl) containing reduced and/or oxidized MtAhpE in the presence or absence of MSH, MtMrx1wt, or MtMrx1CXXA at indicated concentrations were precipitated by addition of 10 μl of trichloroacetic acid (TCA) (10% (w/v)) and kept on ice for 30 min. Protein precipitates were pelleted, washed with 100 μl of ice-cold acetone, dried at 37 °C, resuspended in 15 μl of 3 mm PEG-maleimide (in 50 mm Tris, 10 mm EDTA, 0.1% SDS, pH 7.5) and incubated at 45 °C for 45 min. Samples were immediately loaded on a 15% SDS-PAGE in the absence of β-ME, and proteins were stained with Coomassie Brilliant Blue.
Mass Spectrometrical Identification of Reaction Products
Protein samples were diluted to ∼5 μm in 50% acetonitrile 0.1% acetic acid and intact protein mass measurements were performed by direct infusion in a microelectrospray ionization ion trap mass spectrometer (LTQ XL, ThermoFisher Scientific, San José, CA). The mass spectrometer was operated manually in positive ion mode with a source voltage set at 3.8 kV and the ion transfer tube at 220 °C The parent ions were submitted to in-source dissociation (SID) using the minimal energy to promote efficient declustering of water molecules and salts adducts. The mass spectra were deconvoluted using the software ProMass Deconvolution from ThermoFisher Scientific.
For the identification of MtAhpE-MtMrx1CXXA mixed disulfide, the expected corresponding band was in-gel digested with sequencing grade trypsin (0.5 μg) overnight at 30 °C. The reaction was stopped by adding 0.1% trifluoroacetic acid. The peptides were analyzed by LC-MS/MS as described (28). The mass spectrometer was operated in the data-dependent-mode and switched automatically between MS, Zoom Scan for charge state determination and MS/MS for the most abundant ion. Each MS scan was followed by a maximum of five MS/MS using collision energy of 35%. Dynamic exclusion was enabled to allow analysis of co-eluting peptides. For peptide identification peak lists were generated using the application spectrum selector in the Proteome Discoverer 1.3 package. The resulting peak lists were searched using Sequest against a homemade protein database containing the MtMrx1 and MtAhpE sequences (Uniprot Q8VJ51 and Q73YJ5). The following parameters were used: trypsin was selected with cleavage only after lysine and arginine; the number of internal cleavage sites was set to 1; the mass tolerance for precursor and fragment ions was 1.1 Da and 1.0 Da, respectively; and the considered dynamic modifications were +15.99 Da for oxidized methionine and +71.0 Da for acrylamide addition to cysteine. Peptide matches were filtered using charge-state versus cross-correlation scores (Xcorr). The mixed disulfide peptide between MtMrx1 and MtAhpE was identified by the use of DBond software (29) and manually validated.
Rate Constant Determination of AhpE-mycothiol (AhpE-SS-M) Disulfide Formation
The kinetics of oxidized MtAhpE reaction with mycothiol to form the mixed disulfide described above was determined by a competition approach, following MtAhpE overoxidation to sulfinic acid by excess H2O2 in the absence and presence of mycothiol, which does not compete for initial oxidation of the peroxidatic cysteine due to the high rate constant of the latter enzyme. Overoxidation was measured following the accompanying intrinsic fluorescence increase, using an Aminco Bowman Series 2 luminescence spectrophotometer as previously (22), as in Equation 3.
Reduced MtAhpE (2 μm) was mixed with H2O2 (150 μm) in the absence or presence of MSH (11–33 μm) producing a rapid decrease in the enzyme intrinsic fluorescence intensity, corresponding to its oxidation to sulfenic acid (k = 8.2 × 104 m−1 s−1, reaction half-life under this conditions is ∼0.06 s), followed by an increase in fluorescence corresponding to the enzyme overoxidation (k1 = 40 m−1 s−1, reaction half-life under this conditions is 115 s), as reported previously (22). Observed rate constants of fluorescence increase (kobs) were determined by fitting experimental data to single exponentials. In the presence of mycothiol, in Equation 4,
where k2 is the second order rate constant for the reaction between oxidized MtAhpE and reduced mycothiol. k2 at pH 7.4 and 25 °C was obtained from the slope of kobs versus MSH concentration, and the offset corresponds to k1 × [H2O2].
Reduction of MtAhpE-SS-M by MtMrx1wt
Reaction mixtures containing reduced MtAhpE (20 μm) were treated with mycothiol disulfide (MSSM, 20 μm) or oxidized with an equimolar concentration of H2O2. Immediately after oxidation, reduced MSH was added to reach a final concentration of 20 μm and incubated for 30 min to form MtAhpE-SS-M. Time of incubation was selected to allow ≥90% mixed disulfide formation as calculated using Gepasy 3 software (30) and the rate constant of the reaction reported below. Then, MtMrx1 wt was added to one of the MtAhpE-SS-M containing samples (20 μm Mrx1 and 10 μm MtAhpE-SS-M final concentrations). After 15 min, samples were precipitated with TCA and analyzed by alkylation with PEG-maleimide.
Reduction of MtAhpE-SOH by MtMrx1wt
Oxidized MtAhpE was prepared by incubation of reduced MtAhpE (17 μm) with an equimolar concentration of H2O2 for 1 min immediately after the oxidation step, reduced MtMrx1 was added to reach a final concentration of 16 μm MtMrx1 and 10 μm MtAhpE. Aliquots (90 μl) were taken at different times of incubation (0.2, 0.5, and 4 min) and were directly added into tubes containing 10 μl of 100% TCA to stop the reaction and to precipitate the proteins. Samples before H2O2 addition (reduced MtAhpE) and after oxidation and before MtMrx1 addition (oxidized MtAhpE) were used as positive and negative controls, respectively. Samples were analyzed by alkylation with PEG-maleimide.
Reaction of Oxidized MtAhpE with the Nucleophilic Thiol in MtMrx1
Reaction mixtures containing reduced MtAhpE (10 μm) in the absence or presence of MtMrx1wt or MtMrx1CXXA (30 μm) were treated with H2O2 (10 μm). After 15 min, TCA was added, and samples were analyzed by SDS-PAGE.
Reaction of MtAhpE-SS-M with MtMrx1wt or MtMrx1CXXA
Reaction mixtures containing reduced MtAhpE (16 μm) in the absence or presence of MSH (30 μm) were treated with H2O2 (16 μm), to form MtAhpE-SOH and MtAhpE-SS-M, respectively. After 30 min, MtMrx1 wt or MtMrx1CXXA was added (20 μm MtMrx1wt or CXXA and 10 μm MtAhpE-SOH or MtAhpE-SS-M final concentrations). After 30 min, 10 mm NEM was added and incubated for 15 min, and samples were analyzed by SDS-electrophoresis in the absence of reductant.
Kinetics of Reaction of Oxidized MtAhpE with the Nucleophilic Thiol in MtMrx1CXXA
For the determination of the second-order rate constant of reaction between MtAhpE-SOH and MtMrx1 CXXA, reaction mixtures containing 1 μm MtAhpE and increasing concentrations of MtMrx1CXXA (10, 25 and 40 μm) were treated with 1 μm H2O2. Because of the high reactivity of the peroxidatic thiol in MtAhpE (8 × 104 m−1 s−1 (22)) compared with the thiol groups of MtMrx1 (6.6 m−1 s−1, see below), H2O2 is reduced by the former. 90-μl aliquots were taken at different time points and pipetted into Eppendorf tubes containing 10 μl of 100% TCA to stop the reaction and to precipitate the proteins. Samples were analyzed by SDS-PAGE, and Coomassie-stained gels were scanned in an Oddysey® LI-COR at 700 nm. Band intensity corresponding to the MtAhpE-MtMrx1CXXA mixed disulfide (as identified by in gel digestion and mass spectrometry analysis, see below) was normalized against MtMrx1CXXA intensity, which was in ≥10-fold excess over MtAhpE and therefore, should not be appreciably consumed during the assays. Relative disulfide intensity was plotted as a function of time, and plots were fitted to exponential growth curves. The second-order rate constant of the reaction of MtAhpE-SOH with MtMrx1CXXA was obtained from the slope of the plot of the observed rate constants of mixed disulfide formation versus MtMrx1CXXA concentration.
Determination of the pKa of the Thiols in Nucleophylic Cysteines of MtMrx1wt and CXXA
The pKa of the nucleophilic cysteines of MtMrx1wt and the MtMrx1CXXA mutant were determined spectrophotometrically as described (31). Note that we used alkylated protein instead of oxidized (S-S) protein to correct for the background. The proteins were alkylated with 10 mm iodoacetamide for 30 min at room temperature. Excess of iodoacetamide was removed using size exclusion chromatography on Superdex75 10/300.
Kinetics of MtMrx1 Oxidation by H2O2
The intrinsic fluorescence intensity of MtMrx1 (λexc = 295 nm, λem = 335 nm) decreased upon oxidation by H2O2, in a way that was fully reversible by DTT-treatment. We took advantage of this spectral change to measure the kinetics of MtMrx1 reaction with H2O2, as previously reported for Trypanosoma brucei tryparedoxin 1 oxidation (32). Reduced MtMrx1 (10 μm) was mixed with an excess of H2O2 (0.25–1.0 mm) in an Aminco Bowman Series 2 luminescence spectrophotometer, and time courses of intrinsic fluorescence change were registered. Observed rate constants of fluorescence decrease (kobs) were determined by fitting experimental data to single exponentials. The second order rate constant for the reaction between reduced MtMrx1 and H2O2 at pH 7.4 and 25 °C was obtained from the slope of the plot of kobs versus H2O2 concentration.
Catalytic Consumption of H2O2 by MtAhpE in the Presence of MtMrx1
H2O2 was slowly delivered into solutions containing 2 μm MtAhpE and/or reduced wild type or CXXA MtMrx1 (50 μm) using a motor-driven syringe system (KD Scientific) under continuous stirring (flux = 1 μm/min during 15 min). Aliquots of 20 μl were taken at different times and directly pipetted into plate wells (Fisherbrand® flat bottom well plate, clear) containing 180 μl of a solution of 1 μm HRP and 20 μm Amplex® red. H2O2-dependent Amplex® red oxidation was measured using a Fluostar BMG Lab plate fluorescence reader (λex = 515 nm, λem = 590 nm). H2O2 concentration of each sample was determined according to appropriate calibration curves.
NADPH-dependent Peroxidase Activity in the Presence of MtMrx1 and MtAhpE
NADPH oxidation during MtAhpE-mediated H2O2 reduction was determined using an enzyme-coupled assay. Briefly, reaction mixtures containing 100 μm NADPH, 20 μm reduced MtAhpE, 5 μm reduced wt or CXXA MtMrx1, were incubated in 50 mm HEPES, 0.5 mm EDTA, pH 7.8 at 25 °C, followed by sequential addition of 0.13 μm CgMR, 30 μm MSSM, and 20 μm H2O2. NADPH reduction was monitored at 340 nm (ϵ340 = 6,220 m−1cm−1) using a thermostated Shimadzu UV-2450 spectrophotometer. Reaction mixtures lacking MtAhpE or MtMrx1 were used as negative controls.
RESULTS
Reaction of AhpE-SOH with MSH
When reduced MtAhpE was incubated with an excess of PEG-maleimide and then analyzed by SDS-PAGE, a protein molecular weight shift of 5 kDa was observed, in agreement with the addition of one molecule of PEG/protein and the presence of one thiol per MtAhpE monomer. As expected, this increase in the molecular weight was not observed when the enzyme was first oxidized to its sulfenic acid derivative by addition of equimolar H2O2, in agreement with specific alkylation of reduced cysteine residues (Fig. 1A). When oxidized MtAhpE was incubated with reduced MSH, no thiol alkylation was observed, indicating that no reduction of AhpE-SOH by MSH took place (Fig. 1A). However, incubation with MSH did protect MtAhpE from oxidation-dependent dimerization, a slow process that takes place after initial sulfenic acid formation (22). Considering that AhpE-SOH reacts with glutathione and N-acetylcysteine to form mixed disulfides, we hypothesized that protection was due to the formation of an MtAhpE-SS-M adduct according to Equation 5.
MS studies demonstrated the S-thiolation of MtAhpE on cysteine 45 (Fig. 1B): the mass of the Cys-45 containing peptide was found to be 484 Da higher, consistent with the covalent attachment of MSH. Upon fragmentation of the precursor ion of m/z 1026.5 (z = 3), a predominant neutral loss of 180 Da was observed, corresponding to inositol and consistent with previous results (15). The LC-MS/MS spectrum also allowed exact localization of the mixed disulfide between mycothiol and the cysteine residue.
FIGURE 1.

Oxidized MtAhpE reacts with MSH forming a mixed disulfide that is reduced by MtMrx1. A, alkylation of MtAhpE with PEG-maleimide. Reduced or oxidized MtAhpE (20 μm) was incubated with or without MSH (200 μm). Proteins were precipitated with TCA, treated with PEG-maleimide (5 mm), and evaluated on a CBB-stained 15% SDS-PAGE. B, identification of S-mycothiolation on cysteine 45 of MtAhpE. Sample was obtained by adding MSH (60 μm) to oxidized MtAhpE (20 μm). The LC-MS/MS spectrum shows data obtained from a 3+ parent ion with m/z = 1026.5. The spectrum displays one major daughter ion at m/z 966.4 corresponding to the neutral loss of inositol (180 Da) after fragmentation at a C-O bond. The y- and b- series of ions allowed exact localization of the mixed disulfide between mycothiol and the cysteine residue. C, kinetics of reaction of oxidized MtAhpE with MSH. Time-dependent decrease (oxidation) and increase (overoxidation) in the intrinsic fluorescence intensity (λex = 295 nm, λem = 340 nm) of MtAhpE (2 μm) in the absence (A) or presence (B) of MSH (18 μm) upon addition of H2O2 (150 μm) in 100 mm sodium phosphate buffer plus 0.1 mm DTPA. D, effect of the MSH concentration on the observed rate constants of MtAhpE intrinsic fluorescence change caused by overoxidation. E, MtAhpE-SS-M reduction by MtMrx1. Reduced (lanes 1 and 2) or oxidized (lanes 3 and 4) MtAhpE (20 μm) was incubated with MSSM (lane 2) or MSH (lanes 3 and 4)(20 μm) for 30 min, followed by incubation without (lane 3) or with MtMrx1 (lane 4) (20 μm) for 15 min. Samples were treated with PEG-maleimide (5 mm) and evaluated by CBB-stained 15% SDS-PAGE.
Kinetics of MtAhpE-SOH Reaction with Mycothiol
Incubation of reduced MtAhpE with H2O2 in excess caused enzyme overoxidation to sulfinic acid, followed by the accompanying change in Trp fluorescence as previously described (22). After the addition of H2O2 in the presence of excess mycothiol, we observed a decrease in the amplitude and an increase in the observed rate constants (kobs) of MtAhpE overoxidation (Fig. 1C). The kobs of the process linearly depended on the MSH concentration (Fig. 1D). The slope results in a second-order rate constant (k2) of 237 ± 30 m−1 s−1 for the reaction of MtAhpE-SOH with MSH at pH 7.4 and 25 °C. The offset corresponds to k1 × [H2O2] and perfectly agrees with the previously determined rate constant of enzyme overoxidation (22).
Reduction of MtAhpE-SS-M by MtMrx1
When reduced MtAhpE (20 μm) was incubated with mycothiol disulfide (MSSM, 20 μm) alkylation with PEG-maleimide was not abolished, indicating that at least under these conditions the reduced enzyme is not oxidized by the disulfide form of mycothiol. When MtMrx1 was added to pre-formed MtAhpE-SS-M and further treated with PEG-maleimide, the protein was alkylated (Fig. 1E), indicating that MtMrx1 reduces MtAhpE-SS-M.
Reduction of MtAhpE by MtMrx1
When oxidized MtAhpE (10 μm) was incubated with reduced MtMrx1 (16 μm) for 0.2–4 min and further treated with PEG-maleimide, the enzyme was alkylated (Fig. 2A), indicating that MtAhpE is reduced by MtMrx1 according to Equation 6.
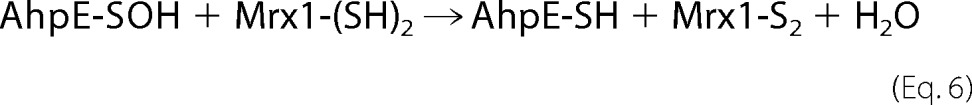 |
MtMrx1-dependent reduction of MtAhpE was ∼50% complete after 0.2–0.5 min, suggesting a relatively fast reaction. Interestingly, the addition of MtMrx1 partially inhibits the dimerization of MtAhpE, which slowly occurs after oxidation (Fig. 2A).
FIGURE 2.

MtAhpE is reduced by wild type MtMrx1 by a dithiolic mechanism. A, reduced (lane 1) or oxidized (lanes 2–6) MtAhpE (10 μm) incubated in the absence (lanes 1–3) or presence of reduced MtMrx1 (16 μm) for indicated times (lanes 4–6) and treated with 5 mm PEG-maleimide were evaluated on a Coomassie Brilliant Blue (CBB) stained 15% SDS-PAGE. B, reduced and oxidized MtAhpE alone (10 μm), or oxidized MtAhpE incubated with MtMrx1 wt or MtMrx1 CXXA (30 μm) for 15 min were evaluated on a CBB-stained 15% SDS-PAGE in the absence (lanes 1–4, respectively) or presence (lanes 4–8, respectively) of β-ME. A novel band with a molecular mass compatible with a mixed disulfide formation between MtAhpE and MtMrx1CXXA is indicated as MtAhpE-SS-MtMrx1. C, mass spectrometric analysis of the MtAhpE-SS-MtMrx1 complex is shown in Fig. 2B. A quadruply charged parent ion of [M+4H]4+ = 1183.7 Da shows fragmentation characteristics of a disulfide linkage between Cys17 of MtMrx1 and Cys45 of MtAhpE, as determined by the DBond software (29). P*, one strand of a dipeptide; p*, the other strand of a dipeptide; capital letters, fragment ions from peptide P*; lowercase letters, fragment ions from peptide p*. The loss of 34 atomic mass units represents formation of dehydroalanine from C-S bond fragmentation. D, reaction of MtAhpE-SS-M with MtMrx1CXXA does not form a protein-protein intermolecular mixed disulfide. Reduced (lane 1) and oxidized MtAhpE (lanes 2–7) alone (lanes 2, 4, and 6), or incubated with MSH during 30 min (lanes 3, 5, and 7), were incubated in the absence (lanes 2 and 3) or presence of MtMrx1 wt (lanes 4 and 5) or MtMrx1CXXA (lanes 6 and 7) for 15 min. Reaction was stopped by addition of 5 mm NEM, and samples were evaluated on a CBB-stained 15% SDS-PAGE under non-reducing conditions. A novel band with a molecular mass compatible with a mixed disulfide formation between MtAhpE and MtMrx1CXXA is indicated as MtAhpE-SS-MtMrx1.
Di- versus mono-thiolic Reduction of MtAhpE by MtMrx1
In C. glutamicum Mrx1, only the N-terminal cysteine residue of the CXXC active site sequence motif was found to be essential for the reduction of the arsenate-mycothiol adduct intermediate (16, 33). For the MtMrx1, however, the C-terminal cysteine mutated to alanine (MtMrx1 CXXA) was not able to reduce MtAhpE. It formed a mixed disulfide (MtAhpE-MtMrx1 CXXA), which was reducible by β-ME (Fig. 2B), indicating that both cysteine residues of MtMrx1 are essential for the reduction of MtAhpE under these conditions, according to the following mechanism in Equations 7 and 8.
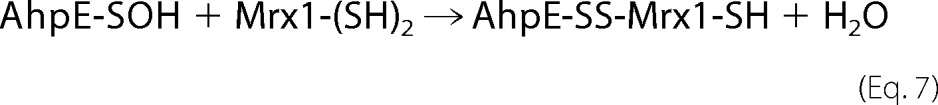 |
Mass spectrometrical studies confirmed the mixed disulfide formation. A tryptic digest of the proteins present in the band corresponding to MtAhpE-MtMrx1 CXXA in Fig. 2B revealed a mixed disulfide between two cysteine-containing tryptic peptides from each individual protein (Fig. 2C).
As above indicated, wild type MtMrx1 could directly reduce MtAhpE-SOH (Fig. 2, A and B). Interestingly, when MtMrx1CXXA was added to MtAhpE-SS-M, the AhpE-SS-Mrx1 mixed disulfide was not observed (Fig. 2D), indicating that the cysteine in MtMrx1CXXA reacts with the sulfur atom of MSH, yielding reduced MtAhpE through the following reaction in Equation 9,
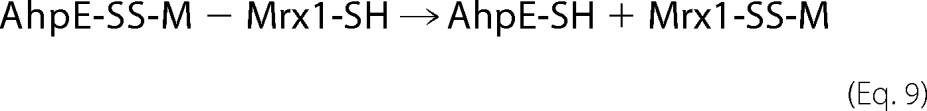 |
which is the reaction for a monothiolic mechanism of reduction.
Kinetics of MtAhpE-SOH Reaction with MtMrx1CXXA
When we oxidized MtAhpE to its sulfenic acid derivative in the presence of excess MtMrx1CXXA, a time-dependent increase of the mixed disulfide between MtAhpE and MtMrx1CXXA was observed (Fig. 3A). Time courses of formation of this mixed disulfide fitted to exponential curves (Fig. 3B), and the observed rate constants were dependent on the MtMrx1CXXA concentration (Fig. 3C). From the slope of the plot shown in Fig. 3C, a second-order rate constant, for the reaction between oxidized MtAhpE and the N-terminal thiol in MtMrx1CXXA to form a mixed disulfide, of (1.6 ± 0.3) × 103 m−1 s−1 at pH 7.4 and 25 °C was determined.
FIGURE 3.

Kinetics of the reaction of MtAhpE-SOH with the nucleophilic thiol in MtMrx1CXXA. A, oxidized MtAhpE (1 μm) was incubated with MtMrx1CXXA (25 μm), aliquots were taken at different incubation times and the reaction was stopped by addition of 10% TCA. Samples were evaluated on a CBB-stained 15% SDS-PAGE. B, time-dependent increase of the relative band intensity of the mixed disulfide shown in A, expressed as MtAhpE-SS-MtMrx1/MtMrx1CXXA. MtMrx1CXXA in concentrations of more than 10 times excess remain constant, and was used as protein load control. The continuous line shows the best fit to an exponential curve. C, effect of increasing MtMrx1CXXA concentrations on the observed rate constants of intermolecular disulfide formation.
pKa Values of the Nucleophylic Cysteines in MtMrx1wt and MtMrx1CXXA
In previous work, we had determined the pKa value of the nucleophilic cysteine residue of MtMrx1wt as 6.8 (15). To investigate the possible influence of the CXXA mutation on the reactivity of the nucleophilic cysteine, we determined the pKa value of the nucleophilic cysteine of MtMrx1CXXA. We obtained a midpoint value of 7.6 (Fig. 4A).
FIGURE 4.

MtMrx1 thiol pKa determinations and kinetics of oxidation by H2O2. A, pKa titration curves for wild type (circles) (15) and the CXXA mutant (triangles). The specific absorption of the thiolate ion at 240 nm as a function of the pH is shown. Aexp is determined as described (31). Data were fitted with the Henderson-Hasselbach equation. B, time-dependent decrease in the total intrinsic fluorescence intensity (λex = 295 nm, λem = 335 nm) of MtMrx1wt (10 μm) upon oxidation by H2O2 in 100 mm sodium phosphate buffer plus 0.1 mm DTPA, at pH 7.4 and room temperature. The first arrow indicates the addition of excess H2O2 (1.5 mm) and the second, the addition of DTT (1.5 mm). C, effect of H2O2 concentration on the observed rate constants of wtMtMrx1 intrinsic fluorescence change.
Kinetics of MtMrx1 Thiol Oxidation by H2O2
When MtMrx1 (10 μm) was oxidized with H2O2 (1 mm), a time-dependent decrease in protein intrinsic fluorescence was observed in a way that was fully reversible by DTT treatment, indicating specificity for protein thiol oxidation (Fig. 4B). Time courses of fluorescence decay fitted to exponential curves, and observed rate constants were dependent on H2O2 concentrations (Fig. 4C). From the slope of the plot, a second-order rate constant for MtMrx1 oxidation by H2O2 of (6.6 ± 0.6) m−1 s−1 at pH 7.4 and 25 °C was obtained.
MtAhpE-catalyzed H2O2 Reduction via MtMrx1
We tested whether MtAhpE was able to catalytically consume H2O2 in the presence of reduced MtMrx1. When a flux of H2O2 (1 μm min−1) was infused to reduced MtMrx1 (50 μm), a time-dependent increase of H2O2 was observed (Fig. 5). After 15 min, nearly 14 μm H2O2 was accumulated, indicating that <10% of the H2O2 was consumed. This is consistent with a low reactivity of MtMrx1 toward H2O2 (6.6 ± 0.6 m−1 s−1 at pH 7.4, Fig. 4C). Reduced MtAhpE (2 μm) alone was not able to catalytically consume H2O2. However, when the same flux of H2O2 was infused to a mixture containing both reduced MtMrx1 and MtAhpE, a much slower increase in H2O2 concentration was observed: 2.8 μm H2O2 accumulated after 15 min, indicating that 81% of the infused H2O2 has been consumed (Fig. 5). Importantly, while MtAhpE catalytically consumed H2O2 in the presence of wt MtMrx1, the peroxidase activity of MtAhpE abolished when the MtMrx1CXXA variant was used, once again indicating that both cysteine residues of MtMrx1 are essential for direct MtAhpE reduction (Fig. 5).
FIGURE 5.

MtAhpE catalyzes H2O2 reduction in the presence of MtMrx1. H2O2 was infused (J = 1 μm min−1) to reaction mixtures containing 50 μm reduced MtMrx1 wt (triangles), 50 μm reduced MtMrx1 CXXA + 2 μm MtAhpE (circles), or 50 μm reduced MtMrx1 wt + 2 μm MtAhpE (squares). Remaining H2O2 was measured at different time points, using Amplex®red oxidation assay. Data points represent an average of n = 3 ± S.D.
MtAhpE Catalyzes the H2O2-dependent NADPH Oxidation
The addition of MSSM (30 μm) to a reaction mixture containing NADPH (100 μm), reduced MtAhpE (20 μm), reduced MtMrx1 wt (5 μm), and CgMR (0.13 μm) caused a rapid decrease in NADPH concentration, coupled to the reduction of MSSM, after which the absorbance remained constant. Subsequent addition of H2O2 (20 μm) led to an acceleration of NADPH consumption (1.6 ± 0.1 μm/min) indicating that the complete system supports a NADPH-dependent peroxidase activity (Fig. 6, A and B). This acceleration was not seen in the absence of MtMrx-1 or MtAhpE. Notably, even when the CXXA mutant instead of wt MtMrx-1 was used, a H2O2-dependent acceleration of NADPH consumption was observed, although to a lower extent (∼65% with respect to wt MtMrx-1)(Fig. 6B).
FIGURE 6.

NADPH consumption during MtAhpE-mediated H2O2 reduction. A, time-dependent consumption of NADPH (100 μm) in a coupled assay containing 20 μm MtAhpE, 5 μm MtMrx1wt, 0.13 μm CgMR, 30 μm MSSM, and 20 μm H2O2 in 50 mm HEPES, 0.5 mm EDTA, pH 7.8 at 25 °C. The arrows indicate the addition of the last three mentioned components to the mixture. B, NADPH consumption upon addition of H2O2 (arrow) in mixtures as in A (Mrx1 wt); with CXXA instead of wt MtMrx1 (Mrx1 CXXA); in the absence of MtMrx1 (w/o Mrx1) or in the absence of MtAhpE (w/o AhpE). Representative traces that were repeated in independent days with the same results are shown.
DISCUSSION
In previous work, we demonstrated the peroxidase activity of the one-cysteine peroxiredoxin from M. tuberculosis, MtAhpE, using different peroxides and artificial reducing substrates. We found peroxynitrite and fatty acid hydroperoxides as preferential oxidizing substrates for this enzyme (22, 23), which interestingly was found associated to the membrane fraction of the bacterium (34). We have also proposed and used MtAhpE as a model to study the mechanisms of cysteine residues oxidation (from thiol to sulfenic acid) and overoxidation (from sulfenic to sulfinic acid) in proteins (23). However, the identification of a biologically relevant reducing pathway to complete its catalytic cycle was lacking so far.
In the present work, we demonstrate that MSH is not able to reduce the sulfenic derivative of MtAhpE, but forms a mixed disulfide with the enzyme, as confirmed by mass spectrometry (Fig. 1, A and B), with a rate constant of 237 m−1s−1 at pH 7.4 and 25 °C (Fig. 1, C and D). Mycoredoxin-1 was able to reduce the mixed disulfide formed between the enzyme and mycothiol (Fig. 1E). The second thiol moiety of MtMrx1 was not needed for MtAhpE-SS-M reduction, as indicated by the lack of detection of MtAhpE-S-S-MtMrx1 adduct upon incubation with the MtMrx1CXXA mutant (Fig. 2D). Moreover the catalytic consumption of NADPH by a coupled assay consisting of NADPH/CgMR/MSH/MtMrx1/MtAhpE/H2O2 was also functional when MtMrx1 was substituted for MtMrx1 CXXA (Fig. 6B). These results are consistent with the reported data for CgMrx1-dependent reduction of arsenate by CgArsC1 and CgArsC2. In these enzymes, the proposed mechanism of reaction involves the initial formation of an arseno (V)-sulfur complex, followed by a nucleophilic attack by MSH resulting in a arseno mycothiol (As(V)-MSH) complex. Mrx1 reacts with the latter, releasing As(III) and forming a mixed disulfide with MSH which is then reduced by a second molecule of MSH yielding reduced Mrx1 and mycothiol disulfide. For MtAhpE-SS-M reduction by MtMrx1, we propose an analogous monothiolic mechanism (16, 33), as illustrated in Fig. 7 described below.
FIGURE 7.

Mechanisms proposed for MSH/Mrx1-dependent MtAhpE reduction. MtAhpE is oxidized by the peroxide to form a sulfenic acid (reaction 1). Sulfenic acid is then directly reduced by MtMrx1 (reactions 2 and 3), or through an intermediate disulfide formation with mycothiol (reaction 4), followed by reduction by MtMrx1 (reaction 5). The leaving Mrx1-S2 and Mrx1-SS-M disulfide species are then reduced by a second mycothiol molecule forming mycothiol disulfide (MSSM) and reduced MtMrx1 as reported (7, 15). The formed MSSM is in turn reduced by the NADPH dependent flavoenzyme, mycothiol disulfide reductase (MR), as previously reported (8, 15). Both reducing pathways may compete with enzyme oxidative inactivation (overoxidation) to a sulfinic acid (reaction 6). The lower pathway (reactions 4–5) would predominate in the cytosol while the upper one (reactions 2–3) could be favored in membrane-associated compartments due to the hydrophilic nature of MSH.
We also showed that MtMrx1 reduces oxidized MtAhpE as shown by thiol alkylation assays (Fig. 2A). However, a mutant form of MtMrx1 where the C-terminal cysteine was substituted for alanine (MtMrx1CXXA) forms a mixed disulfide with oxidized MtAhpE, which was evidenced on SDS-PAGE (Fig. 2B) and peptide identification by mass spectrometry (Fig. 2C). Thus, the sulfenic acid at the peroxidatic cysteine in oxidized MtAhpE reacts with the N-terminal Cys residue in MtMrx1, which is subsequently reduced by the second thiol moiety present in wild type MtMrx1, regenerating reduced MtAhpE. Accordingly, wild type but not CXXA MtMrx1 supports the catalytic consumption of fluxes of H2O2 by MtAhpE (Fig. 5). To note, reduced MtMrx1 caused a marginal H2O2 consumption in the absence of MtAhpE, in agreement with the lack of peroxidase activity of MtMrx1 previously reported (15), and consistent with the rate constant of the reaction determined herein (6.6 ± 0.6 m−1s−1 at pH 7.4, Fig. 4). This value agrees with the quite low nucleophilic cysteine pKa value (6.8) previously reported (15), and pH-independent rate constants of thiolate oxidation by H2O2 in the 18–26 m−1s−1 range (35).
The MtMrx1CXXA mutant was used to estimate the second-order rate constant of the reaction between the sulfenic acid of MtAhpE and the nucleophilic cysteine in MtMrx1 as (1.6 ± 0.3) × 103 m−1s−1 at pH 7.4 (Fig. 3). This value is similar to that determined for the reaction between the sulfenic acid of MtAhpE and the aromatic thiolate thionitrobenzoate ((1.5 ± 0.3) × 103 m−1s−1 at pH 7.4 (22)) and higher than that determined for enzyme reduction by DTT (90 m−1s−1, at pH 7.4 (23)). The rate constant determined this way is that of the first step leading to MtAhpE reduction (Equation 8) and relies on a mutant with potentially altered properties. In this respect, the pKa value of the nucleophilic cysteine in MtMrx1CXXA is 7.6, which is 0.8 pH units higher than that of MtMrx1wt (Fig. 4A). Thus, the nucleophilic thiol is 80% deprotonated at pH 7.4 for MtMrx1wt and only 40% for its CXXA mutant, which could result in a ∼50% lower reactivity at physiological pH. The rate constant obtained using this mutant is most probably a lower limit for the rate-limiting step during the overall process (Equation 7), since experiments using wild type MtMrx1 (16 μm) showed an important fraction of MtAhpE (10 μm) reduction (≥50%) in only 0.2–0.5 min (Fig. 2A), which is consistent with a global rate constant of reduction in the ∼103-104 m−1s−1 range according to computer-assisted simulations using Gepasy 3 software. When comparing with reduction of other MtPrxs by thioredoxins, MtMrx-1-catalyzed MtAhpE reduction seems to approach the reported rate constant of thioredoxin-mediated reduction of other Prxs (MtTPx or MtAhpC), which occur with rate constants in the 104 m−1s−1 range (13, 36). The rate constant of MtAhpE reduction by MtMrx1 is ∼10-fold higher than that of mixed disulfide formation with mycothiol (Fig. 1, C and D). However, as mentioned above, the concentration of mycothiol is in the millimolar range in the M. tuberculosis cytosolic fraction (6) and therefore, to effectively compete with mycothiol for oxidized MtAhpE, the MtMrx1concentration (which is unknown so far) should be ≥50 μm. As indicated, proteomic analysis found MtAhpE in the membrane-associated fraction. Direct MtMrx1-dependent MtAhpE reduction might be favored in these hydrophobic compartments that are difficult to reach for a polar molecule, such as MSH. In any case, both direct reduction by MtMrx1 and mixed disulfide formation with mycothiol are fast enough to compete with H2O2-mediated enzyme oxidative inactivation (rate constant of 40 m−1s−1) (Figs. 3C and 1D, respectively). Whether MtAhpE oxidative inactivation can compete with enzyme reduction by MSH/MtMrx1 in vivo will not only depend on the rate constants of reactions, but also on the steady-state concentrations of reducing as well as oxidizing substrates.
MtAhpE reduction by the MSH/Mrx-1 system led to mycothiol oxidation that was reduced by CgMR as indicated by the NADPH consumption observed using a coupled assay shown in Fig. 6. No NADPH-dependent peroxidase activity occurred in the absence of MtAhpE. This was also true in the absence of MtMrx-1, indicating that oxidized MtAhpE or the mixed disulfide MtAhpE-SM adduct could not be reduced directly by MR/NADPH. Moreover, MtMrx1CXXA had 65% of the activity measured using MtMrx1wt, indicating that in the presence of mycothiol most MtAhpE reduction occurs through a monothiolic mechanism. These results are in agreement with the lack of AhpE-Mrx1CXXA adduct detection by SDS-PAGE in the presence of mycothiol (Fig. 2D, lane 7). To our knowledge, this is the first identification of a biologically relevant reducing enzyme for this one-cysteine peroxiredoxin from M. tuberculosis or any other member of the AhpE family of Prxs. It is also the first report on the reduction of a mycothiol-containing protein mixed disulfide by Mrx1 from Mycobacteria. Moreover, the data reported herein specifies a molecular link between a peroxidase system and the mycothiol/mycoredoxin-1 pathway in Mycobacteria. We propose a mechanism of peroxide sensing and/or detoxification (Fig. 7) where MtAhpE is oxidized by the peroxide to form a sulfenic acid (Fig. 7, reaction 1). The sulfenic acid is then directly reduced by MtMrx1 (Fig. 7, reactions 2 and 3), by a dithiolic mechanism involving a MtAhpE-SS-MtMrx1 intermediate. Alternatively, disulfide formation with MSH followed by reduction by MtMrx1 occurs (Fig. 7, reactions 4 and 5). In any case, the leaving MtMrx1-S2 and MtMrx1-SS-M disulfide are then reduced by mycothiol (7, 15) which in turn is maintained in its reduced state by mycothiol disulfide reductase (MR) at the expense of NADPH (8). Both reducing pathways compete with enzyme overoxidation to sulfinic acid (Fig. 7, reaction 6). To note, glutaredoxins from different cellular sources have been reported to be involved in homologous Prxs reduction and both monothiolic and dithiolic mechanisms have been proposed (37–41).
A recent report using thiol redox proteomic and mass spectrometry to identify S-mycothiolated proteins in C. glutamicum during NaOCl stress revealed that TPx was one of the proteins that is modified on its thiols. S-Mycothiolation affected CgTPx activity that was restored by incubation with CgMrx1 (17). MSH/Mrx1-dependent reduction has not been described in other Prxs from Mycobacteria (TPx and AhpC) studied so far, where NADPH/thioredoxin reductase is directly used to reduce different isoforms of thioredoxin, the reducing substrates for most of the bacterial Prxs (13, 18). For MtAhpC, an alternative reducing system, based on dihydrolipoamide linked to metabolic enzymes, has been demonstrated (42). Mrx-1 was not evaluated as reducing substrate for other peroxiredoxins from M. tuberculosis (Bcp and BcpB) so far.
Thioredoxins, tryparedoxins, as well as different glutaredoxins and NrdH-redoxins, are known to reduce many other protein substrates besides thiol-dependent peroxidases, such as ribonucleotide reductases (43–45). Similarly, we propose that reduction of protein oxidized cysteine residues, either at the sulfenic acid or MSH-mixed disulfide state, by MtMrx1, may be not specific for MtAhpE. Ongoing structural and functional studies on the MtAhpE-Mrx1 adduct will help to understand the bases of the interaction, which could predict protein molecular features required for a dithiolic mechanism of Mrx1-dependent protein reduction to operate. Similarly, structural data regarding the MtAhpE-SSM mixed disulfide could provide the basis for the identification of proteins susceptible to this modification, which, as glutathionylation in other cellular systems, may importantly affect protein function.
MtMrx1-dependent MtAhpE reduction provides an explanation for the increased susceptibility to oxidative stress in strains of Mycobacteria lacking functional Mrx1 or with a low MSH content8 (7–9, 11). Moreover, Prxs and other thiol-dependent peroxidases have recently been proposed as key mediators in peroxide signaling (46–48). The results shown herein, taken together with our previous reports establishing peroxynitrite and fatty acid hydroperoxides as preferential substrates for MtAhpE (23), provide a possible route for sensing these peroxides in Mycobacteria (12). The importance of this metabolic pathway in vivo and its possible consequences on infectivity and pathogenesis will be the subject of future investigations.
Acknowledgments
We thank Dr. E. Paek (Hanyang University, Korea) for help with the use of DBond software and Gaetan Herinckx for experimental assistance.
This work was supported by grants from the National Institutes of Health (NIH) and Universidad de la República (to R. R.), agentschap voor innovatie door Wetenschap en technologie (IWT) (to K. V. L.), the Vlaams Instituut voor Biotechnologie (VIB) and the FWO (Project Grant G.0305.12) (to J. M.), Agencia Nacional de Investigación e Innovación (ANII, Uruguay, FCE_2011_1_5706) (to M. T.). We thank EMBO for the Visiting Grant 439-2011 (to M. H.).
List of Gen Accession numbers (TubercuList) of peroxidases and mycoredoxin 1 from M. tuberculosis: catalase peroxidase, Rv1908; alkyl hydroperoxide reductase C, Rv2428; thioredoxin peroxidase, Rv1932; bacterioferritin comigratory protein, Rv2521; bacterioferritin comigratory protein B, Rv1608c; alkyl hydroperoxide reductase E, Rv2238c; mycoredoxin 1, Rv3198A.
Mrx1-deletion studies were performed in M. smegmatis, which contains an AhpE with 71% protein identity to MtAhpE according to PeroxiBase (20).
- TB
- tuberculosis disease
- AhpE
- alkyhydroperoxide reductase E
- β-ME
- β-mercaptoethanol
- DTPA
- diethylene triamine pentaacetic acid
- DTT
- dithiothreitol
- NEM
- N-ethylmaleimide
- H2O2
- hydrogen peroxide
- HRP
- horseradish peroxidase
- Mrx1
- mycoredoxin-1 from Mycobacterium tuberculosis
- MSH
- mycothiol
- MR
- mycothiol disulfide reductase
- PEG-maleimide
- methoxypolyethylene glycol maleimide
- TCA
- trichloroacetic acid.
REFERENCES
- 1. World Health Organization (2012) Global Tuberculosis Report [Google Scholar]
- 2. Nathan C. (2009) Taming tuberculosis: a challenge for science and society. Cell Host Microbe. 5, 220–224 [DOI] [PubMed] [Google Scholar]
- 3. Alvarez M. N., Peluffo G., Piacenza L., Radi R. (2011) Intraphagosomal peroxynitrite as a macrophage-derived cytotoxin against internalized Trypanosoma cruzi: consequences for oxidative killing and role of microbial peroxiredoxins in infectivity. J. Biol. Chem. 286, 6627–6640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fang F. C. (2004) Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2, 820–832 [DOI] [PubMed] [Google Scholar]
- 5. Nathan C., Shiloh M. U. (2000) Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. U.S.A. 97, 8841–8848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Newton G. L., Fahey R. C. (2002) Mycothiol biochemistry. Arch. Microbiol. 178, 388–394 [DOI] [PubMed] [Google Scholar]
- 7. Van Laer K., Hamilton C. J., Messens J. (2013) Low-molecular-weight thiols in thiol-disulfide exchange. Antioxid. Redox Signal. 18, 1642–1653 [DOI] [PubMed] [Google Scholar]
- 8. Patel M. P., Blanchard J. S. (1999) Expression, purification, and characterization of Mycobacterium tuberculosis mycothione reductase. Biochemistry 38, 11827–11833 [DOI] [PubMed] [Google Scholar]
- 9. Rawat M., Johnson C., Cadiz V., Av-Gay Y. (2007) Comparative analysis of mutants in the mycothiol biosynthesis pathway in Mycobacterium smegmatis. Biochem. Biophys. Res. Commun. 363, 71–76 [DOI] [PubMed] [Google Scholar]
- 10. Rawat M., Kovacevic S., Billman-Jacobe H., Av-Gay Y. (2003) Inactivation of mshB, a key gene in the mycothiol biosynthesis pathway in Mycobacterium smegmatis. Microbiology 149, 1341–1349 [DOI] [PubMed] [Google Scholar]
- 11. Rawat M., Newton G. L., Ko M., Martinez G. J., Fahey R. C., Av-Gay Y. (2002) Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob Agents Chemother 46, 3348–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ta P., Buchmeier N., Newton G. L., Rawat M., Fahey R. C. (2011) Organic hydroperoxide resistance protein and ergothioneine compensate for loss of mycothiol in Mycobacterium smegmatis mutants. J. Bacteriol. 193, 1981–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jaeger T., Budde H., Flohé L., Menge U., Singh M., Trujillo M., Radi R. (2004) Multiple thioredoxin-mediated routes to detoxify hydroperoxides in Mycobacterium tuberculosis. Arch. Biochem. Biophys. 423, 182–191 [DOI] [PubMed] [Google Scholar]
- 14. Cole S. T., Barrell B. G. (1998) Analysis of the genome of Mycobacterium tuberculosis H37Rv. Novartis Found Symp. 217, 160–172; discussion 172–167 [PubMed] [Google Scholar]
- 15. Van Laer K., Buts L., Foloppe N., Vertommen D., Van Belle K., Wahni K., Roos G., Nilsson L., Mateos L. M., Rawat M., van Nuland N. A., Messens J. (2012) Mycoredoxin-1 is one of the missing links in the oxidative stress defense mechanism of Mycobacteria. Mol. Microbiol. 86, 787–804 [DOI] [PubMed] [Google Scholar]
- 16. Ordoñez E., Van Belle K., Roos G., De Galan S., Letek M., Gil J. A., Wyns L., Mateos L. M., Messens J. (2009) Arsenate reductase, mycothiol, and mycoredoxin concert thiol/disulfide exchange. J. Biol. Chem. 284, 15107–15116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chi B. K., Busche T., Laer K. V., Basell K., Becher D., Clermont L., Seibold G. M., Persicke M., Kalinowski J., Messens J., Antelmann H. (2013) Protein S-Mycothiolation Functions as Redox-Switch and Thiol Protection Mechanism in Corynebacterium glutamicum Under Hypochlorite Stress. Antioxid. Redox Signal., In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hugo M., Radi R., Trujillo M. (2012) Thiol dependent peroxidases in Mycobacterium tuberculosis in Understanding Tuberculosis: Deciphering the Secret Life of the Bacilli (Cardona P.-J., ed.), InTech; pp. 293–316 [Google Scholar]
- 19. Soito L., Williamson C., Knutson S. T., Fetrow J. S., Poole L. B., Nelson K. J. (2011) PREX: PeroxiRedoxin classification indEX, a database of subfamily assignments across the diverse peroxiredoxin family. Nucleic Acids Res. 39, D332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oliva M., Theiler G., Zamocky M., Koua D., Margis-Pinheiro M., Passardi F., Dunand C. (2009) PeroxiBase: a powerful tool to collect and analyse peroxidase sequences from Viridiplantae. J. Exp. Bot. 60, 453–459 [DOI] [PubMed] [Google Scholar]
- 21. Li S., Peterson N. A., Kim M. Y., Kim C. Y., Hung L. W., Yu M., Lekin T., Segelke B. W., Lott J. S., Baker E. N. (2005) Crystal Structure of AhpE from Mycobacterium tuberculosis, a 1-Cys peroxiredoxin. J. Mol. Biol. 346, 1035–1046 [DOI] [PubMed] [Google Scholar]
- 22. Hugo M., Turell L., Manta B., Botti H., Monteiro G., Netto L. E., Alvarez B., Radi R., Trujillo M. (2009) Thiol and sulfenic acid oxidation of AhpE, the one-cysteine peroxiredoxin from Mycobacterium tuberculosis: kinetics, acidity constants, and conformational dynamics. Biochemistry 48, 9416–9426 [DOI] [PubMed] [Google Scholar]
- 23. Reyes A. M., Hugo M., Trostchansky A., Capece L., Radi R., Trujillo M. (2011) Oxidizing substrate specificity of Mycobacterium tuberculosis alkyl hydroperoxide reductase E: kinetics and mechanisms of oxidation and overoxidation. Free Radic. Biol. Med. 51, 464–473 [DOI] [PubMed] [Google Scholar]
- 24. Hildebraundt A. G., Roots I. (1975) Reduced nicotinamide adenine dinucleotide phosphate (NADPH) dependent formation and breakdown of hydrogen peroxide during mixed function oxidation reactions in liver microsomes. Arch. Biochem. Biophys. 171, 385–397 [DOI] [PubMed] [Google Scholar]
- 25. Schonbaum G. R., Lo S. (1972) Interaction of peroxidases with aromatic peracids and alkyl peroxides. Product analysis. J. Biol. Chem. 247, 3353–3360 [PubMed] [Google Scholar]
- 26. Ellman G. L. (1959) Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82, 70–77 [DOI] [PubMed] [Google Scholar]
- 27. Riddles P. W., Blakeley R. L., Zerner B. (1979) Ellman's reagent: 5,5′-dithiobis(2-nitrobenzoic acid)–a reexamination. Anal. Biochem. 94, 75–81 [DOI] [PubMed] [Google Scholar]
- 28. Pyr Dit Ruys S., Wang X., Smith E. M., Herinckx G., Hussain N., Rider M. H., Vertommen D., Proud C. G. (2012) Identification of autophosphorylation sites in eukaryotic elongation factor-2 kinase. Biochem. J. 442, 681–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choi S., Jeong J., Na S., Lee H. S., Kim H. Y., Lee K. J., Paek E. (2010) New algorithm for the identification of intact disulfide linkages based on fragmentation characteristics in tandem mass spectra. J. Proteome Res. 9, 626–635 [DOI] [PubMed] [Google Scholar]
- 30. Mendes P. (1993) GEPASI: a software package for modelling the dynamics, steady states and control of biochemical and other systems. Comput. Appl. Biosci. 9, 563–571 [DOI] [PubMed] [Google Scholar]
- 31. Roos G., Garcia-Pino A., Van Belle K., Brosens E., Wahni K., Vandenbussche G., Wyns L., Loris R., Messens J. (2007) The conserved active site proline determines the reducing power of Staphylococcus aureus thioredoxin. J. Mol. Biol. 368, 800–811 [DOI] [PubMed] [Google Scholar]
- 32. Piñeyro M. D., Arcari T., Robello C., Radi R., Trujillo M. (2011) Tryparedoxin peroxidases from Trypanosoma cruzi: high efficiency in the catalytic elimination of hydrogen peroxide and peroxynitrite. Arch. Biochem. Biophys. 507, 287–295 [DOI] [PubMed] [Google Scholar]
- 33. Villadangos A. F., Van Belle K., Wahni K., Dufe V. T., Freitas S., Nur H., De Galan S., Gil J. A., Collet J. F., Mateos L. M., Messens J. (2011) Corynebacterium glutamicum survives arsenic stress with arsenate reductases coupled to two distinct redox mechanisms. Mol. Microbiol. 82, 998–1014 [DOI] [PubMed] [Google Scholar]
- 34. Gu S., Chen J., Dobos K. M., Bradbury E. M., Belisle J. T., Chen X. (2003) Comprehensive proteomic profiling of the membrane constituents of a Mycobacterium tuberculosis strain. Mol. Cell Proteomics 2, 1284–1296 [DOI] [PubMed] [Google Scholar]
- 35. Winterbourn C. C., Metodiewa D. (1999) Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol. Med. 27, 322–328 [DOI] [PubMed] [Google Scholar]
- 36. Trujillo M., Mauri P., Benazzi L., Comini M., De Palma A., Flohé L., Radi R., Stehr M., Singh M., Ursini F., Jaeger T. (2006) The mycobacterial thioredoxin peroxidase can act as a one-cysteine peroxiredoxin. J. Biol. Chem. 281, 20555–20566 [DOI] [PubMed] [Google Scholar]
- 37. Rouhier N., Gelhaye E., Jacquot J. P. (2002) Glutaredoxin-dependent peroxiredoxin from poplar: protein-protein interaction and catalytic mechanism. J. Biol. Chem. 277, 13609–13614 [DOI] [PubMed] [Google Scholar]
- 38. Pedrajas J. R., Padilla C. A., McDonagh B., Bárcena J. A. (2010) Glutaredoxin participates in the reduction of peroxides by the mitochondrial 1-CYS peroxiredoxin in Saccharomyces cerevisiae. Antioxid Redox Signal 13, 249–258 [DOI] [PubMed] [Google Scholar]
- 39. Hanschmann E. M., Lönn M. E., Schütte L. D., Funke M., Godoy J. R., Eitner S., Hudemann C., Lillig C. H. (2010) Both thioredoxin 2 and glutaredoxin 2 contribute to the reduction of the mitochondrial 2-Cys peroxiredoxin Prx3. J. Biol. Chem. 285, 40699–40705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reeves S. A., Parsonage D., Nelson K. J., Poole L. B. (2011) Kinetic and thermodynamic features reveal that Escherichia coli BCP is an unusually versatile peroxiredoxin. Biochemistry 50, 8970–8981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reynolds C. M., Meyer J., Poole L. B. (2002) An NADH-dependent bacterial thioredoxin reductase-like protein in conjunction with a glutaredoxin homologue form a unique peroxiredoxin (AhpC) reducing system in Clostridium pasteurianum. Biochemistry 41, 1990–2001 [DOI] [PubMed] [Google Scholar]
- 42. Bryk R., Lima C. D., Erdjument-Bromage H., Tempst P., Nathan C. (2002) Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science 295, 1073–1077 [DOI] [PubMed] [Google Scholar]
- 43. Dormeyer M., Reckenfelderbäumer N., Ludemann H., Krauth-Siegel R. L. (2001) Trypanothione-dependent synthesis of deoxyribonucleotides by Trypanosoma brucei ribonucleotide reductase. J. Biol. Chem. 276, 10602–10606 [DOI] [PubMed] [Google Scholar]
- 44. Holmgren A. (1979) Glutathione-dependent synthesis of deoxyribonucleotides. Characterization of the enzymatic mechanism of Escherichia coli glutaredoxin. J. Biol. Chem. 254, 3672–3678 [PubMed] [Google Scholar]
- 45. Van Laer K., Dziewulska A. M., Fislage M., Wahni K., Hbeddou A., Collet J. F., Versées W., Mateos L. M., Tamu Dufe V., Messens J. (2013) NrdH-redoxin of Mycobacterium tuberculosis and Corynebacterium glutamicum dimerizes at high protein concentration and exclusively receives electrons from thioredoxin reductase. J. Biol. Chem. 288, 7942–7955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Delaunay A., Pflieger D., Barrault M. B., Vinh J., Toledano M. B. (2002) A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell 111, 471–481 [DOI] [PubMed] [Google Scholar]
- 47. Fomenko D. E., Koc A., Agisheva N., Jacobsen M., Kaya A., Malinouski M., Rutherford J. C., Siu K. L., Jin D. Y., Winge D. R., Gladyshev V. N. (2011) Thiol peroxidases mediate specific genome-wide regulation of gene expression in response to hydrogen peroxide. Proc. Natl. Acad. Sci. U.S.A. 108, 2729–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Azevedo D., Tacnet F., Delaunay A., Rodrigues-Pousada C., Toledano M. B. (2003) Two redox centers within Yap1 for H2O2 and thiol-reactive chemicals signaling. Free Radic Biol. Med. 35, 889–900 [DOI] [PubMed] [Google Scholar]