

Background: α-l-Arabinofuranosidases hydrolyze arabinofuranosyl side chains from xylans.
Results: The first crystal structures of two fungal α-l-arabinofuranosidases representing two distinct subfamilies from the glycoside hydrolase GH62 family are presented. The examination of these unveils specificity determinants.
Conclusion: The structures of complexes with arabinose and cellotriose provide preliminary insight into substrate recognition and catalysis.
Significance: This work provides the first structural description members of the GH62 family.
Keywords: Biofuel, Biotechnology, Enzyme Structure, Fungi, Glycoside Hydrolases, Oligosaccharide, Arabinofuranosidase, CAZyme, GH62, Hemicellulase
Abstract
α-l-Arabinofuranosidases are glycoside hydrolases that specifically hydrolyze non-reducing residues from arabinose-containing polysaccharides. In the case of arabinoxylans, which are the main components of hemicellulose, they are part of microbial xylanolytic systems and are necessary for complete breakdown of arabinoxylans. Glycoside hydrolase family 62 (GH62) is currently a small family of α-l-arabinofuranosidases that contains only bacterial and fungal members. Little is known about the GH62 mechanism of action, because only a few members have been biochemically characterized and no three-dimensional structure is available. Here, we present the first crystal structures of two fungal GH62 α-l-arabinofuranosidases from the basidiomycete Ustilago maydis (UmAbf62A) and ascomycete Podospora anserina (PaAbf62A). Both enzymes are able to efficiently remove the α-l-arabinosyl substituents from arabinoxylan. The overall three-dimensional structure of UmAbf62A and PaAbf62A reveals a five-bladed β-propeller fold that confirms their predicted classification into clan GH-F together with GH43 α-l-arabinofuranosidases. Crystallographic structures of the complexes with arabinose and cellotriose reveal the important role of subsites +1 and +2 for sugar binding. Intriguingly, we observed that PaAbf62A was inhibited by cello-oligosaccharides and displayed binding affinity to cellulose although no activity was observed on a range of cellulosic substrates. Bioinformatic analyses showed that UmAbf62A and PaAbf62A belong to two distinct subfamilies within the GH62 family. The results presented here provide a framework to better investigate the structure-function relationships within the GH62 family.
Introduction
The increasing push toward biorefining of plant biomass is providing new impetus to the study of enzyme-mediated deconstruction of plant cell walls. However, the rather complex, refractory nature of plant cell walls renders economically viable deconstruction difficult to achieve, especially when using low specificity chemical catalysis. Therefore, a lot of emphasis is being put on the development of biotechnological tools, such as glycoside hydrolases and microorganisms displaying the ability to hydrolyze plant cell wall components.
The plant cell wall contains a large number of complex polysaccharides exemplified by arabinoxylans, which are extensively decorated at the O2 and/or O3 positions with α-l-arabinofuranosyl units (1). Therefore, the complete deconstruction of plant cell wall polysaccharides requires numerous enzymatic activities, including a complex arsenal of so-called accessory enzymes that hydrolyze arabinoxylans and produce monomeric pentose sugars (2–3). Among the hemicellulases that depolymerize arabinoxylans, α-l-arabinofuranosidases (EC 3.2.1.55) are exo-acting enzymes that remove l-arabinofuranosyl side chain moieties from the main chain xylan, thus enhancing the action of other enzymes, such as β-d-xylosidase and β-d-xylanases (4). α-l-Arabinofuranosidases are mainly grouped in glycoside hydrolase (GH)6 families GH43, GH51, and GH62 of the CAZy classification (5). Among these families, the GH62 family is the smallest with 121 members (as of 12th December 2013) and has so far received little attention. GH62 α-l-arabinofuranosidases are secreted by microorganisms and proteomic studies have identified them among the most abundant CAZymes in several fungal secretomes (6–10). To date, all of the biochemically characterized GH62 are arabinoxylan α-l-arabinofuranohydrolases, in as much that they specifically cleave either α-1,2- or α-1,3-l-arabinofuranosidic linkages in arabinoxylans (11). In the case of GH62 α-l-arabinofuranosidases from Penicillium chrysogenum and Penicillium funiculosum, 1H NMR and hydrolytic fingerprinting revealed that these enzymes cleave α-1,2- or α-1,3-bonds that specifically link arabinofuranosyl moieties to single-substituted d-xylosyl residues in arabinoxylan (12–14).
Although the catalytic mechanism of the GH62 family has not yet been elucidated, GH62 members have been predicted by the CAZy team to belong to clan GH-F (15), which is also the clan assignation of GH43 enzymes. Accordingly, it is thought that GH62 will probably operate using a single displacement or inverting mechanism, rather like enzymes in the GH43 family (16). Similarly, using sequence homologies that exist between GH43 and GH62, the identity of the catalytic residues has been predicted (16). However, in the absence of structural information and further biochemical characterization, it is impossible to be more affirmative at this stage.
In this study, we undertook the structural characterization of two GH62 α-l-arabinofuranosidases from the fungi Podospora anserina (PaAbf62A) and Ustilago maydis (UmAbf62A). PaAbf62A was mined from the genome of the ascomycete P. anserina, a fungus that displays an impressive set of hemicellulose-acting enzymes (17, 18). UmAbf62A was identified in the secretome of the phytopathogen U. maydis, a secretome that can beneficially complete that of Trichoderma reesei CL847 for the saccharification of wheat straw (7). Here, we present the crystal structures of PaAbf62A and UmAbf62A in their apo form and in complex with arabinose and cellotriose (C3). Enzyme characterization of PaAbf62A and UmAbf62A revealed differences in specificities and bioinformatics analysis of the GH62 family revealed that they belong to different subfamilies.
EXPERIMENTAL PROCEDURES
Recombinant Production of PaAbf62A and UmAbf62A
Recombinant PaAbf62A was produced as previously described (18). The U. maydis strain used in this study was obtained from the FGSC (Fungal Genetics Stock Center, Kansas City, KS) fungal collection. The strain was verified by internal transcribed spacer sequencing and archived under the number BRFM 1093 at the “Centre International de Resources Microbiennes,” which is dedicated to filamentous fungi of biotechnological interest (CIRMCF), at the National Institute of Agricultural Research (INRA), Marseille, France. The U. maydis BRFM 1093 strain was grown on MA2 (malt extract at 2% w/v) medium. Genomic DNA was purified from the mycelium using the NucleoSpin® Plant II kit (Macherey-Nagel GmbH and Co., Düren, Germany). The NW_101116 sequence encoding the UM04309 protein (GenBankTM EAK85571) was amplified by PCR from genomic DNA using Pwo Super yield DNA polymerase (GE Healthcare) and the following primers: EcoRI, forward 5′-GAATTCAACCCCGAAACTGAACG-3′ and XbaI, reverse 5′-TCTAGACCCAGACCGTACAAGGTG-3′. The amplified fragment was subcloned into the pCRII-TOPO vector (Invitrogen) and subjected to sequencing to check the integrity of the sequence. The full-length ORF corresponding to UmAbf62A was inserted at the corresponding sites (EcoRI and XbaI) into the pPICZαA vector in-frame with both the yeast α-secretion factor and C-term-(His)6 tag encoding sequences. The resulting recombinant expression plasmid was transformed into Pichia pastoris X33 and the best-producing transformant was selected for recombinant UmAbf62A production and purification as previously described (18).
Enzymatic Characterization of PaAbf62A and UmAbf62A
Arabinofuranosidase activity was determined by measuring the release of 4-nitrophenol from p-nitrophenyl-α-l-arabinofuranoside (pNP-Ara; 10 mm; Sigma) in 50 mm sodium phosphate buffer (pH 5.0) at 37 °C in a reaction volume of 100 μl. The reaction was stopped by adding 900 μl of 1 m sodium carbonate (pH 9.0). The release of 4-nitrophenol was quantified at 410 nm using a molar extinction coefficient for 4-nitrophenol of 18,300 m−1 cm−1. One unit of enzyme activity was defined as the amount of protein that released 1 μmol of arabinose per min. The apparent optimal pH was estimated using pNP-Ara in the pH range 2.6 to 7.5. The apparent optimal temperature was estimated in the 40 to 70 °C range under the conditions described above. For determination of Michaelis-Menten constants, the initial velocities of the enzymes were measured in 50 mm sodium phosphate buffer (pH 5.0) at 37 °C, with pNP-Ara concentrations ranging from 0.1 to 20 mm. The kinetic parameters were estimated by using weighted nonlinear least squares regression analysis with Grafit software (Erithacus Software, Horley, United Kingdom).
The hydrolysis of 0.1% (w/v) of wheat arabinoxylan low viscosity, sugar beet arabinan, debranched arabinan, and larch arabinogalactan (Megazyme, Wicklow, Ireland) at 37 °C, in 50 mm sodium phosphate buffer (pH 5.0), was monitored by analyzing the amount of free arabinose, using high performance anion exchange chromatography coupled with amperometric detection (ICS 3000; Dionex, Sunnyvale, CA) equipped with a Carbo-PacPA-1 analytical column (250 × 4 mm). Enzymatic reactions were stopped by the addition of 50 mm NaOH before injection (10 μl) onto the high performance anion exchange chromatography system. Elution (1 ml min−1) was carried out in 18 mm NaOH. A calibration curve was plotted using arabinose as the standard (Sigma) and used to calculate the amount of product released. All assays were carried out in triplicate. Results are expressed in μmol of arabinose released per min per mg of enzyme.
Cellulose Binding Studies
Enzyme affinity for soluble 2-HEC (Sigma) was investigated using affinity gel electrophoresis. Native polyacrylamide gels, prepared by dissolving acrylamide (10% w/v) in 25 mm Tris, 250 mm glycine buffer (pH 8.3), were polymerized in the absence and presence of ligand (0.001 to 0.1% (w/v) 2-HEC). Afterward, for the analysis, ligand-containing and ligand-free native gels were migrated simultaneously in the same tank. Five μg of target proteins and BSA (as a negative control) were loaded onto the gels and subjected to electrophoresis at 10 mA/gel for 2 h at room temperature in running buffer containing 15 g liter−1 of Tris-HCl and 72 g liter−1 of glycine (pH 8.3). A KD value was determined by plotting the values 1/(R-r) versus 1/[L], where R represents the ratio between GH62 and BSA migration in the absence of 2-HEC, r, the same ratio in the presence of 2-HEC, and [L], the 2-HEC concentration expressed in mg ml−1. −1/Kd corresponds to the y value when x = 0 (19).
Inhibition Studies
Inhibition was assayed by measuring the arabinofuranosidase activity of the enzymes using pNP-Ara as substrate in the presence of cello-oligosaccharides. Cello-oligosaccharides were added to the reaction mixture at concentrations ranging from 0 to 300 mm (for cellobiose) and from 0 to 8 mm (for C3, C4, C5, and C6) at pH 5.0 and 37 °C. The extent of inhibition was the relative enzyme activity (ratio of activity with inhibitor versus activity without inhibitor). Following the same procedure, the inhibition constant (Ki) was determined using C6 with 6 different concentrations ranging from 0.5 to 8 mm. Data were fitted to the Michaelis-Menten equation to generate an estimate of Ki, using the Grafit data analysis software.
Glycosylation Studies
15 μg of PaAbf62A were loaded onto a 10% SDS-polyacrylamide gel prior to SDS-PAGE (Tris Glycine Precast Gel, Bio-Rad). After migration, the gel was incubated 30 min in ethanol (v/v), 1 h in 0.7% (w/v) periodic acid, 5% (v/v) acetic acid, and washed three times in 0.2% (v/v) sodium metabisulfite, 5% (v/v) acetic acid. Staining was carried out using the Schiff's reagent (Sigma).
Glycosylation Site Prediction
Putative N-glycosylation sites were predicted from the amino sequence using NetNGlyc and verified by mass spectrometry.
N-Deglycosylation
Endo-Hf and amylose resin high flow were obtained from New England Labs (Beverly, MA). 0.5 milliunits of Endo-Hf was used to deglycosylate 1 mg of PaAbf62A at 30 °C during 24 h in G5 NEBuffer. Endo-Hf was eliminated by adding amylose resin. Incubation was performed for 2 h at 4 °C with gentle shaking in MBP buffer (20 mm Tris-HCl, pH 7.4, 200 mm NaCl, and 1 mm EDTA). Deglycosylated PaAbf62A was recovered in the supernatant after centrifugation.
Mass Spectrometry Analyses
Protein samples were reduced in Laemmli buffer (final composition 25 mm DTT, 2% SDS, 10% glycerol, 40 mm Tris, pH 6.8), 5 min at 95 °C, and alkylated with iodoacetamide. 5 μg of protein was subjected to 12% SDS-PAGE (Mini-Protean, Bio-Rad). Protein in the gel was digested by incubating the piece of gel with two enzymes: 25 μl of 20 ng μl−1 of modified sequencing grade trypsin (Promega) and 25 μl of 20 ng μl−1 of Endoproteinase Asp-N Sequencing grade (Roche Applied Science), in 50 mm ammonium bicarbonate, overnight at 37 °C.
These peptides were analyzed by nanoLC-MS/MS using an Ultimate3000 system (Dionex, Amsterdam, The Netherlands) coupled to an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) as previously described (20), except that peptides were eluted using a 5 to 50% gradient of solvent B during 60 min at 300 nl min−1 flow rate. The Mascot Daemon software (version 2.2.0, Matrix Science) was used to perform database searches against all entries in a homemade database containing the PaAbf62A protein sequence. Carbamidomethylation of cysteines, oxidation of methionines, acetylation on N-terminal protein, and HexNAc of asparagine were set as variable modifications for Mascot searches, without enzyme cleavage site. The mass tolerances in MS and MS/MS were set to 10 ppm and 0.8 Da, respectively, and the instrument setting was specified as “ESI-Trap.”
Size Exclusion Chromatography
200 μl of 2 mg ml−1 of proteins in 50 mm sodium acetate buffer (pH 5.0), 50 mm NaCl were loaded onto a Superdex 75 column (GE Healthcare, Life Sciences) and eluted with the same buffer at a flow rate of 0.7 ml min−1. Calibration of the column was performed using the Gel Filtration LMW Calibration Kit (GE Healthcare, Life Sciences).
Crystallization
Purified proteins were concentrated using polyethersulfone Vivaspin concentrators with a cut-off of 10 kDa (Vivascience, Sartorius, Goettingten, Germany). The final concentration was determined by measuring the absorbance at 280 nm using a Nanodrop instrument (Wilmington, DE) and theoretical extinction coefficients of 99,350 and 67,840 m−1 cm−1 for PaAbf62A and UmAbf62A, respectively.
All crystallization experiments were carried out at 12 °C. Initial crystallization conditions were screened using the commercially available JSCG I, II, III, and IV and PEG kits (Qiagen, Courtaboeuf, France) and a robotic sitting-drop method, with drops formed by mixing equal volumes (0.2 μl) of protein (12, 18, 25, or 38 mg ml−1 in 50 mm sodium acetate, pH 5.0) and precipitant solution, which were equilibrated against 80 μl of precipitant solution. Conditions were further optimized manually using the hanging-drop vapor-diffusion method in 24-well plates. Best crystals of PaAbf62A were obtained with a 1:1 (v/v) ratio of protein (18 mg ml−1 in 50 mm sodium acetate (pH 5.0) and 50 mm NaCl) to precipitant solution (25% (w/v) PEG 4000, 0.2 m calcium chloride, and 0.1 m Tris pH 8.5). Crystals appeared within a week and grew to an approximated size of 0.25 × 0.2 × 0.05 mm3.
Crystals of UmAbf62A were obtained by mixing a equal volume of protein (18 mg ml−1 in 50 mm sodium acetate (pH 5.0) and 50 mm NaCl) and reservoir solution (20% (w/v) PEG 3350 and 0.2 m sodium phosphate). Crystals of ∼0.30 × 0.30 × 0.20 mm3 appeared within 6 days.
Data Collection and Phasing
X-ray experiments were carried out at 100 K. Prior to flash cooling, optimized native crystals of UmAbf62A and PaAbf62A were soaked for a few seconds in the reservoir solution supplemented with 10% (v/v) glycerol to avoid ice formation. Diffraction data were collected in-house (Xcalibur Nova, Agilent Technologies) for PaAbf62A in complex with cellotriose, on beamlines ID23-eh1 (PaAbf62a native structure) and ID14eh1 (UmAbf62a-l-arabinofuranose complex) at the European Synchrotron Radiation Facility (Grenoble, France) and on beamline PROXIMA 1 at SOLEIL (Gif sur Yvette, France) for the native UmAbf62A structure. The diffraction data were processed scaled and merged using the XDS package (21).
The native structure of UmAbf62A was solved by the sulfur-SAD phasing method using data collected in-house at 1.7-Å resolution from a single crystal (Table 1). A highly redundant data set was collected at the Cu-Kα wavelength covering 1425° of total rotation with 1° oscillation width per frame. A native dataset was collected to 1.0 Å on beamline PROXIMA 1 at SOLEIL.
TABLE 1.
Data collection and refinement statistics
Values in parentheses indicate the outer resolution shell.
|
UmAbf62A |
PaAbf62A |
||||
|---|---|---|---|---|---|
| S-SAD | Native | Arabinose | Native | Cellotriose | |
| Space group | P 212121 | P 212121 | P 212121 | C2 | C2 |
| Unit cell parameters | a = 60.22 Å; b = 65.93 Å; c = 68.22 Å | a = 60.28 Å; b = 65.90 Å; c = 68.37 Å | a = 60.08 Å; b = 66.07 Å; c = 68.10 Å | a = 102,41 Å; b = 66.83 Å; c = 60.57 Å | a = 102,14 Å; b = 66.79 Å; c = 60.36 Å |
| α = β = γ = 90° | α = β = γ = 90° | α = β = γ = 90° | α = β = 90°; γ = 117.4° | α = β = 90°; γ = 117.3° | |
| No. of molecule/AU | 1 | 1 | 1 | 1 | 1 |
| Matthews coefficient (Å3/Da) | 1.97 | 1.97 | 1.97 | 2.47 | 2.47 |
| Solvent content (%) | 37.6 | 37.6 | 37.6 | 50.3 | 50.3 |
| Wavelength (Å) | 1,540562 | 0.82656 | 0.93340 | 0.97626 | 1.5418 |
| Resolution range (Å) | 14.84–1.71 (1.75–1.71) | 37.28–1.0 (1.03–1.00) | 47.42–1.20 (1.23–1.20) | 44.61–1.44 (1.48–1.44) | 14.84–1.80 (1.85–1.80) |
| No. of unique reflections | 55,590 (3,255) | 145,655 (10,217) | 81,763 (5,597) | 60,688 (3,880) | 64,256 (4,209) |
| No. of observed reflections | 1,179,809 (8,181) | 795,636 (40,484) | 353,317 (23,640) | 276,924 (6,384) | 40,5585 (10,712) |
| Completeness (%) | 98.0 (76.8) | 99.4 (95.4) | 95.9 (89.9) | 92.3 (80.3) | 97.6 (87.2) |
| Multiplicity | 21.2 (2.5) | 5.5 (4.0) | 4.3 (4.2) | 4.6 (1.6) | 6.3 (2.5) |
| I/σ(I) | 62.8 (4.2) | 10.1 (2.0) | 35.0 (9.3) | 21.9 (8.3) | 22.0 (2.5) |
| Rmerge (%) | 4.3 (20.8) | 9.8 (64.2) | 2.6 (14.3) | 4.8 (9.3) | 7.1 (38.1) |
| Refinement | |||||
| Rwork/Rfree | 0.13/0.15 | 0.12/0.14 | 0.14/0.16 | 0.15/0.18 | |
| Root mean square deviations | |||||
| Bond lengths (Å) | 0.022 | 0.022 | 0.027 | 0.021 | |
| Bond angles (°) | 2.293 | 2.117 | 2.52 | 2.078 | |
| Ramachandran plot | |||||
| Favored (%) | 93.57 | 93.23 | 94.77 | 94.43 | |
| Allowed (%) | 4.82 | 5.18 | 4.58 | 4.59 | |
| Mean B-factor | 10.0 | 11.0 | 15.0 | 11.1 | |
| Wilson B (Å2) | 5.6 | 7.8 | 12.5 | 13.0 | |
| Main chain (Å2) | 6.3 | 8.3 | 12.3 | 11.0 | |
| Side chain (Å2) | 8.6 | 10.9 | 14.8 | 12.7 | |
| Ligand/water (Å2) | 14.2/25.3 | 16.8/25.7 | 26.8/27.5 | 30.7/23.4 | |
| Protein Data Bank codes | 4N1I | 4N2R | 4N4B | 4N2Z | |
The anomalous signal of sulfur atoms from 6 methionine residues and 2 cysteine residues was evaluated and the eight sulfur atom positions and occupancies were determined and refined with SHELX C/D (22, 23). The initial phase calculation and phase improvement were performed using SHELX E (23). The quality of the phases was sufficient (figure of merit = 0.60 for all reflections) to obtain an interpretable map at 1.7-Å resolution after solvent flattening. An initial model containing about 90% of the whole protein sequence was automatically generated using the ARP/wARP web service (24). The missing parts were then manually built in σ-A weighted electron density maps using COOT (28) and followed by refinement steps using Refmac5 (25–27). The final R and Rfree were 0.13 and 0.15, respectively.
The crystal structure of PaAbf62A was solved by molecular replacement using UmAbf62A as the model. All protein-ligand structures were directly refined using Refmac5 and the apoenzyme structures as starting models. The coordinates and structure factors were deposited in the Protein Data Bank at RCSB under the references PDB identifiers: 4N1I, 4N2R, 4N4B, and 4N2Z for UmAbf62A, UmAbf62A in complex with arabinose, and PaAbf62A and PaAbf62A in complex with cellotriose, respectively.
Soaking Experiments
C3 was obtained from Megazyme, arabinose from Sigma, and arabino-xylo-oligosaccharides (XA3XX) were prepared as described previously (29). A PaAbf62A-cellotriose complex was obtained by soaking a PaAbf62A crystal for 35 min in the reservoir solution supplemented by C3 at saturating concentration. To form a UmAbf62A·XA3XX complex, a crystal of UmAbf62A was soaked for 2 h in solution containing 20% (w/v) PEG 3350, 0.2 m sodium phosphate, 0.1 m sodium acetate (pH 3.0), 3 mm XA3XX, and 50 mm arabinose as cryoprotectant. After soaking, crystals were picked up using a nylon loop and flash-cooled in a stream of nitrogen gas.
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) experiments were conducted at 25 °C in Tris-HCl buffer (pH 6.0) on a Microcal ITC200 instrument (GE Healthcare). To ensure minimal buffer mismatch, protein was dialyzed against 20 mm Tris-HCl buffer (pH 6.0) and the ligands were solubilized in the same buffer. Experiments consisted of a series of 20 × 2- or 40 × 1-μl injections of ligand (1.6–5 mm) into the protein-containing (PaAbf62A 114 μm, UmAbf62A 488 μm) thermostatic cell (initial delay of 60 s, duration of 4 s and spacing of 100 s). ITC experiments were systematically performed in triplicate. The corrected binding isotherms were fitted for a single binding-site model using non-linear least squares analysis to obtain values of equilibrium binding constant (Ka), stoichiometry, and enthalpy changes (ΔH) associated with ligand binding. Other thermodynamic parameters were derived from the equation: −RT ln Ka = ΔG = ΔH − TΔS.
Phylogenetic Clustering
A total of 106 sequences corresponding to individual GH62 modules were extracted from the CAZy database (5). They were aligned using the Muscle version 3.7 software (30), operating with default parameters. A matrix of maximum likelihood distances of the resulting aligned sequences was determined using Jones, Taylor, and Thornton (JTT) distances (31) and subsequently subjected to hierarchical clustering method of Ward (32).
RESULTS
Phylogenetic Analysis of GH62 Family
To better characterize and compare the two fungal arabinofuranosidases presented in this work, a phylogenetic analysis of the GH62 family was carried out using publically available bacterial and fungal sequences. Of the 106 sequences extracted from the CAZy database (12th September 2013), only 17 enzymes (including PaAbf62A and UmAbf62A) have been characterized (EC 3.2.1.55). The bioinformatics analysis revealed two distinct subfamilies that did not correspond to taxonomical segregation (Fig. 1). PaAbf62A and UmAbf62A, which share 38% identity, belong to two distinct subfamilies termed GH62-1 and GH62-2, respectively. Subfamily GH62-1 includes 30 members that are clustered into two distinct subgroups, the larger one having three biochemically characterized members, whereas none of the five members of the smaller subgroup has been characterized at present. On the other hand, subfamily GH62-2 is more homogeneous and contains 76 members, with over a dozen characterized members, which appear to display shorter loop structures than their GH62-1 counterparts.
FIGURE 1.

Phylogenetic tree of the GH62 family. Cladogram highlighting the relative position of protein sequences identified by an abbreviation of the species name and a reference public database accession number. Characterized enzymes (EC 3.2.1.55) are highlighted in bold with an asterisk. The alignment used to construct the phylogenetic tree as shown in supplemental Fig. S1 (45) deployed the maximum likelihood method using PhyML (43). The reliability of the tree was analyzed by bootstrap analysis of 100 resamplings of the data set. The phylogenetic tree was prepared using Dendroscope. un, uncultured microorganism.
Comparative Activity
After cloning and heterologous expression in P. pastoris, the specificity of UmAbf62A was investigated and compared with that of PaAbf62A (18). The capacity of UmAbf62A and PaAbf62A to hydrolyze pNP-Ara, as well as arabinose-containing polysaccharides was evaluated. Although it is generally claimed that GH62 enzymes are not able to hydrolyze pNP-Ara (33), enzyme activity was detected using pNP-Ara as substrate for both enzymes and the Michaelis-Menten parameters were determined at their pH and temperature optima (pH 5.0, 37 °C). Both PaAbf62A and UmAbf62A displayed relatively weak recognition of pNP-Ara with Km values of 6.1 and 7.5 mm, respectively. In terms of catalytic efficiency, PaAbf62A was ∼12-fold more active than UmAbf62A against pNP-Ara (Table 2). The investigation of polysaccharide hydrolysis by PaAbf62A and UmAbf62A using wheat arabinoxylan, sugar beet arabinan, and debranched or linear arabinan revealed that both enzymes released arabinose as the sole product, irrespective of the substrate. Moreover, on wheat arabinoxylan, both enzymes displayed similar catalytic rates (174 and 173 min−1 for PaAbf62A and UmAbf62A, respectively), but UmAbf62A showed an ∼2-fold higher catalytic rate on sugar beet arabinan (460 min−1 compared with 192 min−1 for PaAbf62A). Neither PaAbf62A, nor UmAbf62A displayed activity on debranched arabinan (data not shown), indicating that the hydrolytic action of these enzymes was exclusively directed toward the α-1,2 and/or the α-1,3 bonds that link side chain l-arabinofuranosyl units to the main chains of the polymers under study. Finally, determination of kcat for the different reactions clearly revealed that both enzymes displayed much higher catalytic rates on natural polysaccharide substrates than on the artificial substrate pNP-Ara.
TABLE 2.
Kinetic parameters of PaAbf62A and UmAbf62A
Michaelis constant were measured using pNP-Ara as substrate. Inhibition by C6 was measured using pNP-Ara as substrate as described under “Experimental Procedures.”
| PaAbf62A | UmAbf62A | |
|---|---|---|
| pNP-Ara | ||
| Vmaxa | 0.61 ± 0.02b | 0.06 ± 0.00 |
| kcat (min−1) | 22.6 ± 0.0 | 2.32 ± 0.02 |
| Km (mm) | 6.1 ± 0.3b | 7.5 ± 0.8 |
| kcat/Km (min−1 mm−1) | 3.70 ± 0.06b | 0.31 ± 0.03 |
| C6 | ||
| Ki (mm) | 3.40 ± 0.25 | NDc |
a Vmax is defined as the μmol of product formed per mg of enzyme per min.
b Result from Couturier et al. (18).
c ND, no inhibition detected.
Three-dimensional Structures
Crystallization trials using recombinant UmAbf62A yielded orthorhombic crystals that diffracted up to 1.7 Å using an in-house x-ray diffractometer. Sulfur SAD phasing experiment provided the means to solve the phase of the crystallographic structure of the apoenzyme.
Initial crystallization experiments using PaAbf62A all resulted in crystal twinning, which was attributed to the presence of protein glycosylation. The presence of N-glycosylation sites in the protein sequence was predicted using the NetNGlyc 1.0 server, and indeed MS/MS analysis on N-deglycosylated PaAbf62A confirmed this and unambiguously identified the Asn56 residue of the peptide 49DPTIILIN(HexNAc)GTHHVFASTAQ67 to bear the residual HexNAc sugar moiety. Similarly, MS/MS analysis of peptide 83DAPN(HexNAc)ATFYYLDQAPLGTGYR102 localized the HexNAc moiety on the Asn86 residue. In addition, the N-terminal peptide DVSIVQLSNQPPS (which is not observed in the x-ray structure) was found to bear a hexose moiety but the amino acid bearing the modification could not be determined. PaAbf62A was thus enzymatically deglycosylated, a procedure that ultimately provided the means to obtain monoclinic crystals that diffracted up to 1.44 Å. The three-dimensional structure was solved using the apo-structure of UmAbf62A as the template for molecular replacement. The statistics of data collection and processing for both proteins are summarized in Table 1.
The refined structure of UmAbf62A was solved at a resolution of 1.0 Å and comprised 312 of 336 residues of the recombinant protein. Final crystallographic Rwork and Rfree values were 0.13 and 0.15, respectively, and more than 98% of the residues were in the allowed regions of the Ramachandran plot. One molecule was present in the asymmetric unit. The overall structural fold is composed of a single domain, displaying a five-bladed β-propeller fold (Fig. 2), which is consistent with previous predictions and the fact that GH62 belongs to clan GH-F of the glycoside hydrolase classification. Each of the five radially oriented blades comprises four antiparallel β-strands with the first blade forming a so-called “molecular Velcro” (34, 35). The latter is composed of the first three inner strands composed of the C-terminal end of the polypeptide chain, and the fourth outer strand by the N terminus (blade I: residues 18–20, 301–308, 281–287, and 260–264) (Fig. 2). One α-helix is observed in the C-terminal region, which is formed by an extra amino acid resulting from the cloning of the protein. Additionally, a disulfide between Cys10 and Cys278 constrains the solvent exposed N-terminal part of the polypeptide chain in the vicinity of the loop connecting strands 18 (residues 260–264) and 19 (residues 281–287). This loop obstrudes the bottom of the tunnel delineated by the five blades of the β-propeller.
FIGURE 2.

Three-dimensional structure overview. A, wall-eyed stereo representation of the overall structure of UmAbf62A and PaAbf62A superimposed. Proteins are shown as a schematic, color ramped from the N terminus (blue) to C terminus (red) for UmAbf62A and PaAbf62A is in gray, the disulfide bridge is colored pink. B, the overall structure of UmAbf62A and PaAbf62A superimposed with indications of the five blades. C, schematic representation of β-strand organization with the molecular Velcro identified by a red circle. D, surface representation of UmAbf62A showing the catalytic pocket with the predicted catalytic amino acid (surface in red). E, surface representation of PaAbf62A showing the catalytic pocket with the predicted catalytic amino acid (surface in red).
The structure of PaAbf62A was solved at a resolution of 1.44 Å with one molecule per asymmetric unit. It comprises 319 amino acid residues of 335 for the mature polypeptide chain with final crystallographic Rwork and Rfree of 0.14 and 0.16, respectively. More than 99% of the residues are in allowed regions of the Ramachandran plot. The apo-structures of PaAbf62A and UmAbf62A are very similar, with a root mean square deviation of 1.4 Å based on the superposition of 288 Cα atoms (Fig. 2). The main differences are observed in loop regions and are due to insertions in the primary structure of PaAbf62A, which is longer than that of UmAbf62A, as evidenced by sequence alignment of the GH62 family (supplemental Fig. S1). The biggest loop extension concerns the loop that connects strand β9 from blade 3 to strand β10 from blade 4. Moreover, PaAbf62A contains two disulfide bridges, formed between Cys170 and Cys175, and between Cys303 and Cys337, respectively, whereas UmAbf62A only contains one, formed between Cys10 and Cys278. The spatial location of this single disulfide bridge in UmAbf62A is similar to that of Cys10-Cys278 in PaAbf62A.
The Active Site
Based on homology with other members of clan GH-F whose catalytic residues have been identified (36, 16), it is highly likely that the catalytic residues of UmAbf62A/PaAbf62A are Glu195/Glu216 (general acid) and Asp36/Asp49 (general base), respectively, with Asp143/Asp165 acting as the pKa modulator. Importantly, the distance measured between the putative general acid and general base amino acid is 8.0 and 8.1 Å for PaAbf62A and UmAbf62A, respectively, which is consistent with an inverting mechanism (37). It is noteworthy that the inverting mechanism has been established for the A. nidulans GH62 member.7 These amino acids are all conserved within the GH62 family, and form part of a pocket, the bottom of which is lined by a histidine residue, His261/His285, in UmAbf62A/PaAbf62A, respectively. This histidine residue is part of a SHG motif, which is also conserved within the GH62 family (supplemental Fig. S1), and is stabilized by interactions with a calcium ion that, in both enzymes, is located in the center of the five-bladed β-propeller and coordinates His261/His285 in UmAbf62A/PaAbf62A, respectively (Fig. 3). Nevertheless, the coordination of the calcium ion in UmAbf62A and PaAbf62A structures is not identical because in the former the calcium is hepta-coordinated with a pentagonal bipyramidal ligation involving His261-Nϵ2, Gln198-Nϵ2, and five water molecules, whereas in PaAbf62A, the calcium ion is hexa-coordinated with a squared bipyramidal ligation involving His285-Nϵ2 and five water molecules (Fig. 7). It is noteworthy that the structures of some GH43 members (e.g. the structure of BT2895 Bacteroides thetaiotaomicron, Protein Data Bank code 3KST) also contain a calcium ion that displays the same coordination as that observed in PaAbf62A. Moreover, the chelation of calcium by EDTA or Chelex resin had no apparent effect on enzyme activity (data not shown). Coupled to the fact that the calcium ion has no direct interaction with the active site, this observation is consistent with a structural rather than functional role.
FIGURE 3.

The active site pocket of UmAbf62A and PaAbf62A. A and B, calcium ion coordination for PaAbf62A and UmAbf62A, respectively. C, superimposition of PaAbf62A and UmAbf62A active sites. PaAbf62A is represented in cyan and UmAbf62A in gold. The residues shown in the stick calcium ions are shown in spheres. Electron density map was contoured at 2σ. This figure and other figure have been drawn with PyMOL (Delano Scientific, pymol.sourceforge.net/).
FIGURE 7.
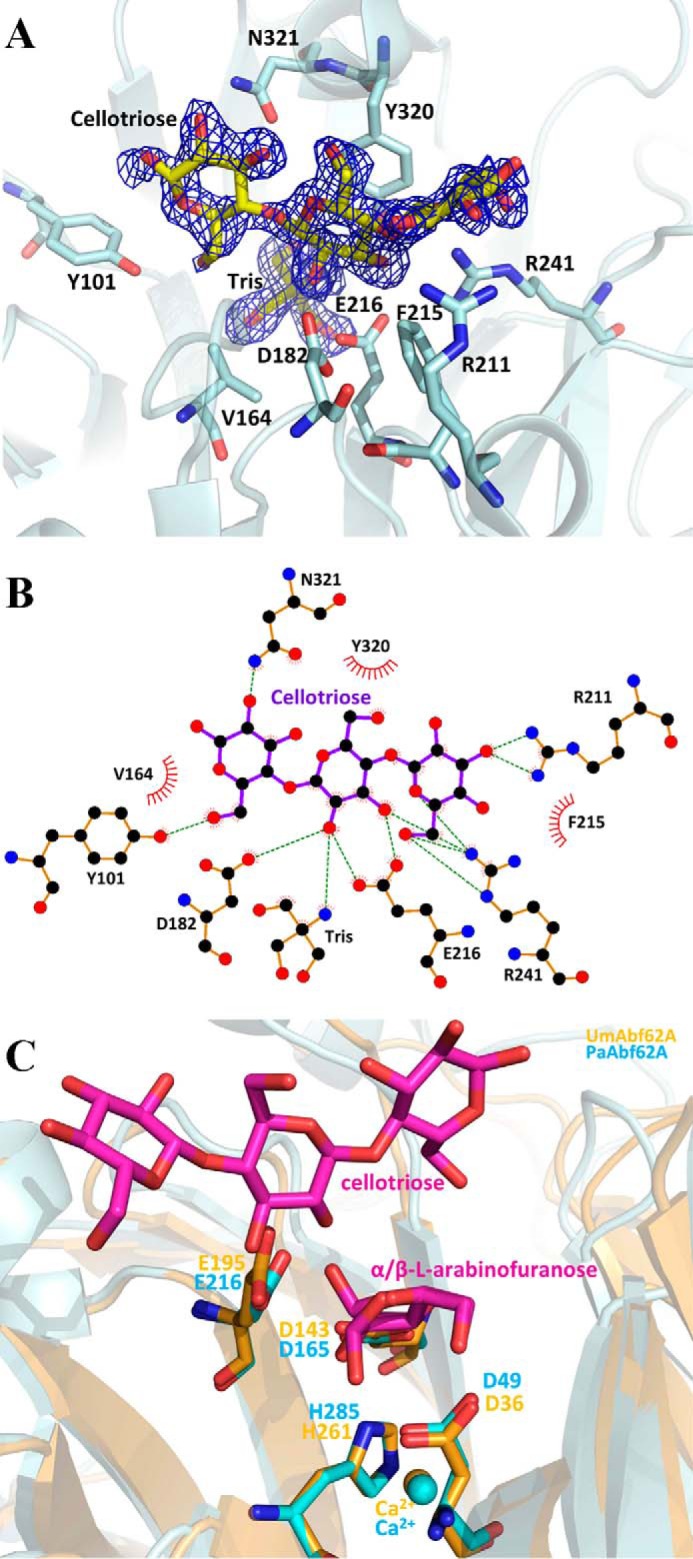
PaAbf62A in complex with cellotriose. A, structure of cellotriose in the active site pocket of PaAbf62A. Sugar is displayed as stick and protein as a schematic. Catalytic amino acids are named using the one-letter code of the amino acid followed by the position in the sequence. The electron density is displayed in dark blue and has been contoured to 1σ. B, schematic diagram showing hydrogen-bonding, water-bridged, and hydrophobic interactions between PaAbf62A and cellotriose. Amino acid residues of PaAbf62A that have hydrophobic interactions with cellotriose are shown as spiked spheres (with distances of less than 3.5 Å). Direct and water-bridged hydrogen-bonding interactions are indicated by dashed lines. C, superimposition of complex structures of PaAbf62A and UmAbf62A sugar are colored in magenta, UmAbf62A is colored in gold, and PaAbf62A is colored in cyan.
Interaction with Arabinose
To trap UmAbf62A complexed with a l-arabinofuranosyl moiety, crystals were soaked with XA3XX (29) at pH 3.0 to inactivate the enzyme (38) and l-arabinose was added to the soaking solution as a cryoprotectant. Under these experimental conditions, we hoped to observe the pentasaccharide, but in fact the electron density was too poor to reliably discern the xylosyl moieties. Consequently, only l-arabinofuranose could be built into the electron density maps (Fig. 4). The arabinofuranose is housed in the pocket described above, which constitutes the −1 subsite. The C1 hydroxyl group was observed in both α- and β-anomeric configuration. Thus, the C1-O1 covalent bond of the α-l-arabinofuranose could be considered as the scissile bond position of the substrate (α-l-arabinofuranosyl moiety), or as the anomeric oxygen of the product (β-l-arabinofuranose). In the β-configuration, O1 replaces a water molecule (Wat894) present in the apo-structure (Fig. 4). This water molecule is hydrogen bonded to the Oδ1 of the catalytic general base, Asp36, and is likely to play the role of the solvent nucleophile used by inverting enzymes. The arabinose ring is stacked against Tyr58 and hydrogen bonded to Tyr296. The O2 of the arabinose is within hydrogen bonding distance of Oδ2 of the pKa modulator Asp143, and Nδ1 of His261. Moreover, O4 of the arabinose appears to be hydrogen bonded to Lys35 (Fig. 4). Unlike UmAbf62A, no complex of PaAbf62A with arabinose could be obtained, although a complex with C3 was obtained. The details and implications of this are discussed hereafter.
FIGURE 4.
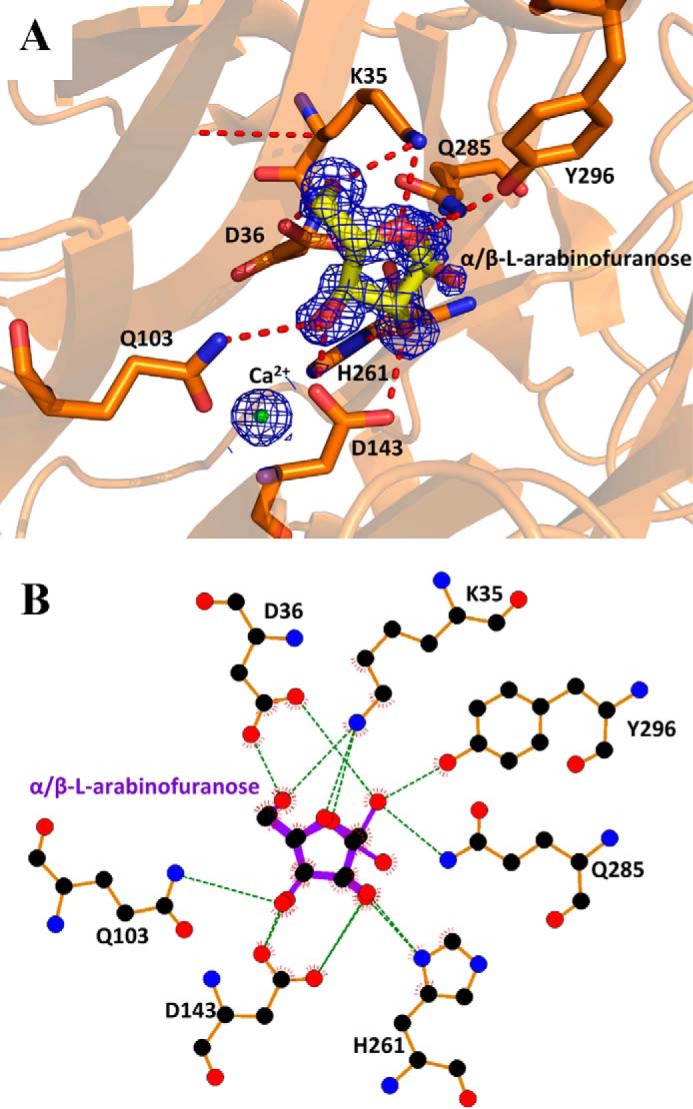
UmAbf62A in complex with arabinose. A, structure of arabinose in the active site pocket of UmAbf62A. Sugar is displayed as stick and protein as a schematic. Catalytic amino acids are named using the one-letter code of the amino acid followed by the position in the sequence. The electron density has been contoured to 1σ. B, schematic diagram showing hydrogen-bonding, water-bridged, and hydrophobic interactions between UmAbf62A and arabinose. Amino acid residues of UmAbf62A that have hydrophobic interactions with arabinose are shown as spiked spheres (with distances of less than 3.5 Å). Direct and water-bridged hydrogen-bonding interactions are indicated by dashed lines. This diagram and other have been obtained using the LIGPLOT program (44).
Interaction of PaAbf62A with Cellulose Derivatives
Hydrolysis tests clearly demonstrated that PaAbf62A was unable to hydrolyze cellulose-based substrates, such as 2-HEC, carboxymethylcellulose, Avicel, filter paper, and cello-oligosaccharides (data not shown). However, this enzyme was able to bind to Avicel with extremely weak affinity (data not shown) and to 2-HEC, with a dissociation constant (Kd) for 2-HEC being 0.77 mg ml−1 (Fig. 5). To evaluate the affinity of PaAbf62A for cello-oligosaccharides, ITC was performed using C3 and C6. Accordingly, when using C6 as the ligand, a Kd value in the range of 80 μm was determined compared with 12 μm for X6 (Fig. 6). However, no binding to C3 was detected. Likewise, in the similar assays, UmAbf62A failed to display any binding to either of the two cello-oligosaccharides or to xylohexaose (Fig. 6) in addition, binding to 2-HEC was not possible to determine because UmAbf62A did not penetrate within the native gels. This result emphasizes the unexpectedness of PaAbf62A behavior toward C6 and 2-HEC, especially considering that the enzyme does not possess a carbohydrate binding module. Taken together, these results imply that cello-oligosaccharide/2-HEC binding by PaAbf62A might be due either to direct binding in the active site cleft, or at secondary binding sites. Therefore, to further investigate this binding phenomenon, the inhibition of PaAbf62A-mediated hydrolysis of pNP-Ara by cello-oligosaccharides was studied. This revealed that, unlike UmAbf62A, the activity of PaAbf62A was inhibited by cello-oligosaccharides or cellulose derivatives and that the strength of this inhibition (expressed as an IC50) was correlated to the chain length of the cello-oligosaccharides (i.e. IC50 C3, 20 mm < C4, 8 mm < C5, 2.2 mm < C6, 0.8 mm). C6 acted as an inhibitor of PaAbf62A with a Ki of 3.4 mm (Table 2), which is roughly 40-fold lower than the Kd value determined using ITC for C6. The explanation for this unexpected result is not apparent, but clearly it is highly unlikely that the non-reducing terminal glucosyl moiety of C6 actually binds in the place of the l-arabinosyl moiety. Therefore, although the model used to calculate the Ki was that of competitive inhibition, the actual inhibition mode was probably more complex, with some binding configurations failing to (fully) inhibit the hydrolysis of pNP-Ara.
FIGURE 5.
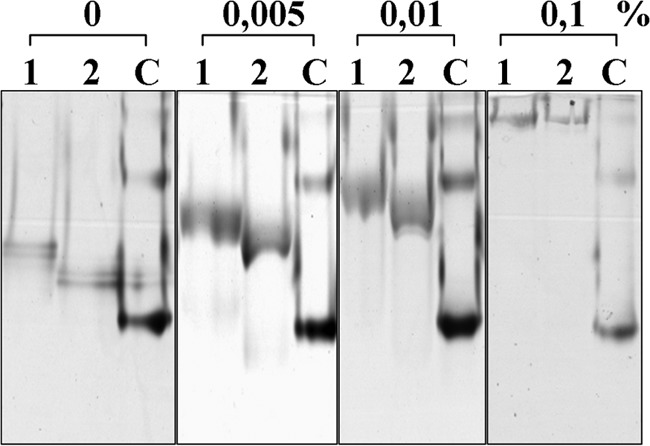
PaAbf62A binding to cellulose. Affinity gel electrophoresis for 2-hydroxyethyl cellulose. Concentrations are displayed at the top of the gel, from 0 to 0.1%. Lane 1, PaAbf62A; lane 2, deglycosylated PaAbf62A; C, control BSA.
FIGURE 6.

Representative ITC data of PaAbf62A and UmAbf62A binding to xylo- and cello-oligosaccharides. A–D, the upper parts of each panel show the raw binding heats, the lower parts show the integrated binding heats minus the dilution control heats fitted to a single-site binding model. Experiments were performed at 25 °C. The concentration of PaAbf62A was 114 μm; and UmAbf62A was 488 and 95 μm. A, PaAbf62A titration with X6 (1.6 mm); B, PaAbf62A titration with C6 (2.2 mm); C, UmAbf62A (488 μm) titration with X6 (5 mm); and D, UmAbf62A (95 μm) titration with C6 (3 mm). In panels A and B, n is the number of binding sites and Ka is the affinity constant both determined by ITC, the values are the mean of three experiments.
Positive Subsite Interactions in PaAbf62A
In light of previous results, it is somewhat surprising that C3 is the only ligand that was found to make a crystallographic complex with PaAbf62A. In this complex, the trisaccharide is located in front of the catalytic pocket, which houses a Tris molecule, in the central region of the roughly straight surface channel, confirming the identity of this topological feature as the substrate binding cleft, which can accommodate polymer backbones. According to the protein-ligand complex described in this work, topology of the PaAbf62A substrate binding cleft is formed from at least four subsites, with the scissile bond being located between subsites −1 and +1, the latter being occupied by the central glucosyl moiety of cellotriose (39). Likewise, the adjacent subsites +2NR and +2R are occupied by non-reducing and reducing glucosyl moieties, respectively (Fig. 7). Further subsites might exist either at the reducing (i.e. +3R) or non-reducing sides (i.e. +3NR), but these are not evidenced by our data, although they are implied by the fact that C6 displays the highest inhibitory potential of the cellooligosaccharides tested. In the case that subsites +3R and +3NR do exist, these would be highly solvent exposed.
The glucosyl residue at the +2R subsite stacks with the Tyr162 residue, which is present in half of the GH62-1 subfamily, and is stabilized by two strong hydrogen bonds between its O6 and the hydroxyl of Tyr101 and Nϵ2 of Glu121, respectively. Moreover, the O2 of this glucosyl moiety interacts with Nδ2 of Asn321. Interestingly, Tyr101 is only present in the GH62-1 subfamily, where it is well conserved. Significantly, in UmAbf62 the equivalent of Tyr101 is Trp104, with a tryptophan being strictly conserved at this position in the subfamily GH62-2. Finally, it is noteworthy that in the few cases where Tyr101 is absent from subfamily GH62-1, it is replaced by a tryptophan residue, like Trp104 in UmAbf62A (Fig. 6 and supplemental Fig. S1).
At the +1 subsite, Tyr320 displays a stacking interaction with the glucose, whereas the O2 atom of the sugar points toward the catalytic pocket, making a hydrogen bond with the Tris molecule. The Oϵ1 atom of Glu216, the predicted catalytic general acid, also interacts with the O2 of the glucose at the +1 subsite. This is consistent with the fact that the O2 of the sugar moiety in subsite +1 (i.e. a xylosyl or arabinosyl group if the substrate is an arabinoxylan) is engaged in the scissile glycosidic bond that links to the arabinosyl moiety bound in subsite −1. Moreover, the O3 of the glucose bound in the +1 subsite displays strong interactions with the Oϵ2 of Glu216 and Nη2 of Arg241, respectively, suggesting that the enzyme might hydrolyze both α-1,2 and α-1,3 linkages.
In addition to its interactions with the glucosyl moiety in subsite −1, the Nη2 of Arg241 also makes a bidentate hydrogen bond to O5 and O6 of the +2NR, suggesting that this residue might play an important role. This observation is consistent with the fact that Arg241 is highly conserved in GH62. Overall, the glucosyl moiety present in +2NR interacts strongly with Arg241 through its O6, and with Arg211 via its O2 and O3. Unlike Arg241, Arg211 is not conserved in the GH62 family and the corresponding residue in UmAbf62A is Glu190. Finally, a water molecule (Wat819) is located at the same position as the water molecule Wat894 in UmAbf62A implying it could take the role of the solvent nucleophile in PaAbf62A.
DISCUSSION
Structural Characterization of GH62 α-l-Arabinofuranosidases
This study has revealed the first structures of two GH62 family members in their apo forms, and in complex with either arabinose or C3. The data presented demonstrate that GH62 arabinofuranosidases share common structural features with GH43 arabinofuranosidases, because both families are characterized by a five-bladed β-propeller fold. The catalytic apparatus is localized in a pocket, which can accommodate the arabinose sugar constituting the −1 subsite. The three predicted catalytic residues interact either with the arabinose in the −1 subsite in the complexed form or with a water molecule in the apo form. This water molecule is at the same position as the anomeric hydroxyl group in the complex with arabinose. Furthermore, the water molecule is absent when arabinose is in an inverted conformation compared with the substrate, suggesting that, not only water molecules Wat894 and Wat819 in UmAbf62A and PaAbf62A, respectively, could play the role of the catalytic water molecule, but also that hydrolysis would occur with an inverting mechanism. Taken together, these data strongly implies that the GH62 family belongs to the same clan as the GH43 family (i.e. clan GH-F), which is consistent with the predictions made by Pons et al. (16). Furthermore, it is interesting to note that His161/His285 in UmAbf62A/PaAbf62A, respectively, which coordinates the calcium ion and binds the arabinose in the −1 subsite, is also conserved in the GH43 arabinofuranosidases and arabinanases from Bacillus subtilis (PDB code 3C7E) and Cellvibrio japonicus (PDB code 3QED), respectively. Consistent with this observation, we suggest that this histidine plays an important functional role in arabinose hydrolysis within the clan GH-F.
In this work, tentative identification of the positive subsites involved in PaAbf62A xylan binding have been performed using the PaAbf62A-C3 complex. We believe that this extrapolation is valid, because as well as binding X6, PaAbf62A also binds C6. Moreover, taking into account the fact that the conformation of β-glucans and xylan main chains are similar, it is noteworthy that PaAbf62A is more specific for xylans than arabinans, the latter displaying a quite different main chain conformation. Nevertheless, caution is required because examination of the complex also highlighted specific glucan-binding features, such as the stabilizing hydrogen bonds that are formed with O6 of the primary alcohol of glucosyl moieties that occupy the +2R and +2NR subsites (Fig. 7). Obviously in the case of arabinoxylans these interactions do not occur, because the 6-hydroxyl group of glucopyranose is absent in xylopyranose. This might explain why no xylose could be attributed using XA3XX (data not shown) whatever the soaking condition tested and it suggests that xylan binding involves other interactions that account for the specific backbone conformation of xylooligosaccharides and xylans.
Comparison of PaAbf62A and UmAbf62A
The activities of the two GH-62 enzymes on pNP-Ara and arabinoxylan are comparable, but UmAbf62A is more active on sugar beet arabinan. In this respect, it is noteworthy that mono-arabinose substitutions in wheat arabinoxylan are α-linked to the main chain via α-1,3 linkages (40), whereas sugar beet arabinan displays both α-1,2- and α1,3-arabinofuranosyl substitutions (41). Therefore, both enzymes are able to hydrolyze α-1,2 and α-1,3 linkages, a conclusion that is supported by the crystallographic data. When superimposing both holoenzyme structures, and looking at the ligands, the arabinose in α conformation in UmAbf62A is orientated toward the position occupied by the O2 atom of the +1 glucopyranosyl moiety of the C3 in the PaAbf62A-C3 complex (Fig. 7). Because xylan chains are pseudosymetrical and arabinose side chains can rotate around the glycosidic linkage, xylan might bind to the active site cleft in either orientation, leading to hydrolysis of either α-1,2 or α-1,3 linkages.
Regarding binding of PaAbf62A to C3 and C6, it is noteworthy that Arg241 is strictly conserved in the GH62 family. According to our results this residue interacts with the sugar moiety in subsites +2NR and +1, whereas Arg211 makes strong interactions with sugars in the +2NR subsite. However, absence of Arg211 in UmAbf62A might explain why this enzyme apparently does not bind C3. This difference in the C3 binding ability might be further explained by the fact that Tyr101 in PaAbf62A makes a hydrogen bond with the O1 of the glucosyl moiety positioned at the reducing end of C3, whereas, the equivalent residue in UmAbf62A, Trp104, appears to be too close to the glucosyl and thus would make a steric clash. Binding to cellulose derivatives can probably be explained in terms of mimicry of the xylose backbone. Nevertheless, further studies will be needed to investigate this property within the different GH62 subfamilies.
This report constitutes the first determination of crystal structures for the GH62 family and, gratifyingly, reveals structures of members of two distinct subfamilies. Moreover, thanks to the isolation of enzyme-ligand complexes, our study provides some insights into the finer active site details. Consistent with previous predictions, this study confirms that GH62 enzymes are structurally and functionally related to GH43 enzymes, and as such belong to the same GH-F clan. Surprisingly, cellulose binding was observed with the P. anserina arabinofuranosidase, even though this enzyme does not possess a carbohydrate binding module. Altogether, the data presented are a first step to a better understanding of GH62 enzymes, and constitutes an essential step for the further investigation of structure-function relationships, and for the future design of tailored plant cell wall degrading enzymes for industrial applications.
Supplementary Material
Acknowledgments
The equipment used for the crystallization experiments and x-ray crystallography as well as isothermal titration calorimetry is part of the Integrated Screening Platform of Toulouse (PICT, AuthorIBiSA), we are grateful to Dr. V. Gervais for helpful assistance in the ITC experiment. We thank the European Synchrotron Radiation Facility (ESRF) at Grenoble (France) in particular the beamline ID29 staff and French synchrotron (SOLEIL) at Saint-Aubin (France) and the beamline Proxima1 staff for their assistance.
This work was supported in part by grants from the region Midi-Pyrénées (to B. S.) and the National Institute of Agricultural Research (INRA) CEPIA research department.

This article contains supplemental Fig. S1.
The atomic coordinates and structure factors (codes 4N4B, 4N2Z, 4N1I, and 4N2R) have been deposited in the Protein Data Bank (http://wwpdb.org/).
C. Wilkens, B. O. Petersen, and B. Svensson, personal communication.
- GH
- glycoside hydrolase
- PaAbf62A
- Podospora anserina GH62 α-l-arabinofuranosidase A
- UmAbf62A
- Ustilago maydis GH62 α-l-arabinofuranosidase A
- pNP-Ara
- para-nitrophenyl-α-l-arabinofuranoside
- C3-C6
- cello-oligosaccharides containing 3, 4, 5, or 6 glucosyl subunits
- 2-HEC
- 2-hydroxyethylcellulose
- ITC
- isothermal titration calorimetry
- PDB
- Protein Data Bank
- X6
- xylohexaose
- XA3XX
- an AXOS:arabino-xylo-oligosaccharide named according to the nomenclature used in Ref. 42.
REFERENCES
- 1. Kormelink F. J., Gruppen H., Viëtor R. J., Voragen A. G. (1993) Mode of action of the xylan-degrading enzymes from Aspergillus awamori on alkali-extractable cereal arabinoxylans. Carbohydr. Res. 249, 355–367 [DOI] [PubMed] [Google Scholar]
- 2. Dumon C., Song L., Bozonnet S., Fauré R., O'Donohue M. J. (2012) Progress and future prospects for pentose-specific biocatalysts in biorefining. Process Biochem. 47, 346–357 [Google Scholar]
- 3. Couturier M., Berrin J. G. (2013) The Saccharification Step: The Main Enzymatic Components. In Lignocellulose Conversion (Faraco V., ed) pp. 93–110, Springer, Berlin Heidelberg [Google Scholar]
- 4. Vincent P., Shareck F., Dupont C., Morosoli R., Kluepfel D. (1997) New α-l-arabinofuranosidase produced by Streptomyces lividans. Cloning and DNA sequence of the abfB gene and characterization of the enzyme. Biochem. J. 322, 845–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., Henrissat B. (2009) The Carbohydrate-Active EnZymes database (CAZy). An expert resource for glycogenomics. Nucleic Acids Res. 37, D233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Herpoël-Gimbert I., Margeot A., Dolla A., Jan G., Mollé D., Lignon S., Mathis H., Sigoillot J. C., Monot F., Asther M. (2008) Comparative secretome analyses of two Trichoderma reesei RUT-C30 and CL847 hypersecretory strains. Biotechnol. Biofuels 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Couturier M., Navarro D., Olivé C., Chevret D., Haon M., Favel A., Lesage-Meessen L., Henrissat B., Coutinho P. M., Berrin J. G. (2012) Post-genomic analyses of fungal lignocellulosic biomass degradation reveal the unexpected potential of the plant pathogen Ustilago maydis. BMC Genomics 13, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ravalason H., Grisel S., Chevret D., Favel A., Berrin J. G., Sigoillot J. C., Herpoël-Gimbert I. (2012) Fusarium verticillioides secretome as a source of auxiliary enzymes to enhance saccharification of wheat straw. Bioresour. Technol. 114, 589–596 [DOI] [PubMed] [Google Scholar]
- 9. Chen B., Gui F., Xie B., Deng Y., Sun X., Lin M., Tao Y., Li S. (2013) Composition and expression of genes encoding carbohydrate-active enzymes in the straw-degrading mushroom Volvariella volvacea. PLoS One 8, e58780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ribeiro D. A., Cota J., Alvarez T. M., Brüchli F., Bragato J., Pereira B. M., Pauletti B. A., Jackson G., Pimenta M. T., Murakami M. T., Camassola M., Ruller R., Dillon A. J., Pradella J. G., Paes Leme A. F., Squina F. M. (2012) The Penicillium echinulatum secretome on sugar cane bagasse. PLoS One 7, e50571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kellett L. E., Poole D. M., Ferreira L. M., Durrant A. J., Hazlewood G. P., Gilbert H. J. (1990) Xylanase B and an arabinofuranosidase from Pseudomonas fluorescens subsp. cellulosa contain identical cellulose-binding domains and are encoded by adjacent genes. Biochem. J. 272, 369–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sakamoto T., Ogura A., Inui M., Tokuda S., Hosokawa S., Ihara H., Kasai N. (2011) Identification of a GH62 α-l-arabinofuranosidase specific for arabinoxylan produced by Penicillium chrysogenum. Appl. Microbiol. Biotechnol. 90, 137–146 [DOI] [PubMed] [Google Scholar]
- 13. De La Mare M., Guais O., Bonnin E., Weber J., Francois J. M. (2013) Molecular and biochemical characterization of three GH62 α-l-arabinofuranosidases from the soil deuteromycete Penicillium funiculosum. Enzyme Microb. Technol. 53, 351–358 [DOI] [PubMed] [Google Scholar]
- 14. Hashimoto K., Yoshida M., Hasumi K. (2011) Isolation and characterization of CcAbf62A, a GH62 α-l-arabinofuranosidase, from the basidiomycete Coprinopsis cinerea. Biosci. Biotechnol. Biochem. 75, 342–345 [DOI] [PubMed] [Google Scholar]
- 15. Henrissat B., Bairoch A. (1996) Updating the sequence-based classification of glycosyl hydrolases. Biochem. J. 316, 695–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pons T., Naumoff D. G., Martínez-Fleites C., Hernández L. (2004) Three acidic residues are at the active site of a β-propeller architecture in glycoside hydrolase families 32, 43, 62, and 68. Proteins 54, 424–432 [DOI] [PubMed] [Google Scholar]
- 17. Espagne E., Lespinet O., Malagnac F., Da Silva C., Jaillon O., Porcel B. M., Couloux A., Aury J. M., Ségurens B., Poulain J., Anthouard V., Grossetete S., Khalili H., Coppin E., Déquard-Chablat M., Picard M., Contamine V., Arnaise S., Bourdais A., Berteaux-Lecellier V., Gautheret D., de Vries R. P., Battaglia E., Coutinho P. M., Danchin E. G., Henrissat B., Khoury R. E., Sainsard-Chanet A., Boivin A., Pinan-Lucarré B., Sellem C. H., Debuchy R., Wincker P., Weissenbach J., Silar P. (2008) The genome sequence of the model ascomycete fungus Podospora anserina. Genome Biol. 9, R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Couturier M., Haon M., Coutinho P. M., Henrissat B., Lesage-Meessen L., Berrin J.-G. (2011) Podospora anserina hemicellulases potentiate the Trichoderma reesei secretome for saccharification of lignocellulosic biomass. Appl. Environ. Microbiol. 77, 237–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bolam D. N., Xie H., Pell G., Hogg D., Galbraith G., Henrissat B., Gilbert H. J. (2004) X4 modules represent a new family of carbohydrate-binding modules that display novel properties. J. Biol. Chem. 279, 22953–22963 [DOI] [PubMed] [Google Scholar]
- 20. Gautier V., Mouton-Barbosa E., Bouyssié D., Delcourt N., Beau M., Girard J.-P., Cayrol C., Burlet-Schiltz O., Monsarrat B., Gonzalez de Peredo A. (2012) Label-free quantification and shotgun analysis of complex proteomes by one-dimensional SDS-PAGE/NanoLC-MS evaluation for the large scale analysis of inflammatory human endothelial cells. Mol. Cell Proteomics 11, 527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schneider T. R., Sheldrick G. M. (2002) Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 58, 1772–1779 [DOI] [PubMed] [Google Scholar]
- 23. Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E. Combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Collaborative Computational Project, Number 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 27. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 29. Paës G., Skov L. K., O'Donohue M. J., Rémond C., Kastrup J. S., Gajhede M., Mirza O. (2008) The structure of the complex between a branched pentasaccharide and Thermobacillus xylanilyticus GH-51 arabinofuranosidase reveals xylan-binding determinants and induced fit. Biochemistry 47, 7441–7451 [DOI] [PubMed] [Google Scholar]
- 30. Edgar R. C. (2004) MUSCLE. Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jones D. T., Taylor W. R., Thornton J. M. (1992) The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282 [DOI] [PubMed] [Google Scholar]
- 32. Ward J. H. (1963) Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 58, 236–244 [Google Scholar]
- 33. Gilbert H. J. (2013) Glycoside hydrolase family 62, in CAZypedia. [Google Scholar]
- 34. Neer E. J., Smith T. F. (1996) Protein heterodimers. New structures propel new questions. Cell 84, 175–178 [DOI] [PubMed] [Google Scholar]
- 35. Paoli M. (2001) An elusive propeller-like fold. Nat. Struct. Biol. 8, 744–745 [DOI] [PubMed] [Google Scholar]
- 36. Nurizzo D., Turkenburg J. P., Charnock S. J., Roberts S. M., Dodson E. J., McKie V. A., Taylor E. J., Gilbert H. J., Davies G. J. (2002) Cellvibrio japonicus α-l-arabinanase 43A has a novel five-blade β-propeller fold. Nat. Struct. Biol. 9, 665–668 [DOI] [PubMed] [Google Scholar]
- 37. McCarter J. D., Withers S. G. (1994) Mechanisms of enzymatic glycoside hydrolysis. Curr. Opin. Struct. Biol. 4, 885–892 [DOI] [PubMed] [Google Scholar]
- 38. Davies G. J., Mackenzie L., Varrot A., Dauter M., Brzozowski A. M., Schülein M., Withers S. G. (1998) Snapshots along an enzymatic reaction coordinate. Analysis of a retaining β-glycoside hydrolase. Biochemistry 37, 11707–11713 [DOI] [PubMed] [Google Scholar]
- 39. Davies G. J., Wilson K. S., Henrissat B. (1997) Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 321, 557–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saulnier L., Sado P.-E., Branlard G., Charmet G., Guillon F. (2007) Wheat arabinoxylans. Exploiting variation in amount and composition to develop enhanced varieties. J. Cereal Sci. 46, 261–281 [Google Scholar]
- 41. Leijdekkers A. G., Bink J. P., Geutjes S., Schols H. A., Gruppen H. (2013) Enzymatic saccharification of sugar beet pulp for the production of galacturonic acid and arabinose. A study on the impact of the formation of recalcitrant oligosaccharides. Bioresour. Technol. 128, 518–525 [DOI] [PubMed] [Google Scholar]
- 42. Faure R., Courtin C. M., Delcour J. A., Dumon C., Faulds C. B., Fincher G. B., Fort S., Fry S. C., Halila S., Kabel M. A., Pouvreau L., Quemener B., Rivet A., Saulnier L., Schols H. A., Driguez H., O'Donohue M. J. (2009) A brief and informationally rich naming system for oligosaccharide motifs of heteroxylans found in plant cell walls. Aust. J. Chem. 62, 533–537 [Google Scholar]
- 43. Guindon S., Gascuel O. (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 [DOI] [PubMed] [Google Scholar]
- 44. Wallace A. C., Laskowski R. A., Thornton J. M. (1995) LIGPLOT. A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8, 127–134 [DOI] [PubMed] [Google Scholar]
- 45. Gouet P., Courcelle E., Stuart D. I., Métoz F. (1999) ESPript. Multiple sequence alignments in PostScript. Bioinformatics. 15, 305–308 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.