

Background: QSOX enzymes are medically important catalysts of disulfide bond generation.
Results: Mechanistic studies of the three redox centers in QSOX revealed an unexpected thermodynamic mismatch in the reaction coordinate.
Conclusion: QSOX circumvents this thermodynamic barrier using a mixed disulfide intermediate that links distal redox centers.
Significance: The strategy of coupling disulfide exchanges appears to be exploited by other enzymes of oxidative protein folding.
Keywords: Disulfide, Enzyme Mechanisms, Oxidase, Oxidation-Reduction, Protein Folding, Redox, Sulfhydryl, QSOX, Quiescin Sulfhydryl Oxidase
Abstract
The quiescin sulfhydryl oxidase (QSOX) family of enzymes generates disulfide bonds in peptides and proteins with the reduction of oxygen to hydrogen peroxide. Determination of the potentials of the redox centers in Trypanosoma brucei QSOX provides a context for understanding catalysis by this facile oxidant of protein thiols. The CXXC motif of the thioredoxin domain is comparatively oxidizing (E′0 of −144 mV), consistent with an ability to transfer disulfide bonds to a broad range of thiol substrates. In contrast, the proximal CXXC disulfide in the ERV (essential for respiration and vegetative growth) domain of TbQSOX is strongly reducing (E′0 of −273 mV), representing a major apparent thermodynamic barrier to overall catalysis. Reduction of the oxidizing FAD cofactor (E′0 of −153 mV) is followed by the strongly favorable reduction of molecular oxygen. The role of a mixed disulfide intermediate between thioredoxin and ERV domains was highlighted by rapid reaction studies in which the wild-type CGAC motif in the thioredoxin domain of TbQSOX was replaced by the more oxidizing CPHC or more reducing CGPC sequence. Mixed disulfide bond formation is accompanied by the generation of a charge transfer complex with the flavin cofactor. This provides thermodynamic coupling among the three redox centers of QSOX and avoids the strongly uphill mismatch between the formal potentials of the thioredoxin and ERV disulfides. This work identifies intriguing mechanistic parallels between the eukaryotic QSOX enzymes and the DsbA/B system catalyzing disulfide bond generation in the bacterial periplasm and suggests that the strategy of linked disulfide exchanges may be exploited in other catalysts of oxidative protein folding.
Introduction
Several pathways have been described for the introduction of disulfide bonds in the secretory pathway of higher eukaryotes. In protein-disulfide isomerase-first models of oxidative protein folding (1–3), a direct interaction between the oxidized forms of one or more of the protein-disulfide isomerases leads to the net oxidation of protein clients. The resulting reduced protein-disulfide isomerases may then be reoxidized by one of several enzymes including the flavin-linked Ero1 oxidases utilizing molecular oxygen (4–6) and peroxiredoxin 4 (7–9) or glutathione peroxidase 7/8 (10) using hydrogen peroxide as a co-substrate. Other reduced thioredoxin-like resident proteins of the endoplasmic reticulum are believed to deliver reducing equivalents to vitamin-K-epoxide reductase (11), thereby providing an additional pathway for oxidative protein folding. In contrast, oxidases of the QSOX3 family (1, 12–17) are capable of the direct oxidation of client proteins. In vitro studies with avian and recombinant human and protozoan QSOXs showed that oxidation of protein thiols is facile when substrates are unfolded or at least conformationally flexible (3, 18). The inclusion of micromolar levels of reduced protein-disulfide isomerase with low nanomolar levels of QSOX leads to the efficient oxidative refolding of a client protein with nine disulfide bonds and correspondingly more than 34 million potential disulfide pairings (2). Of the two human paralogs (14, 19), QSOX1 is better understood on a mechanistic and biological level (1, 15, 16). QSOX1 is highly expressed in tissues with a heavy secretory load (1, 14–16) and is markedly up-regulated in certain cancers including those of prostate (20, 21), pancreas (22), and breast (23, 24). Recent studies have revealed that QSOX peptides from plasma serve as a biomarker for acute decompensated heart failure (25) and pancreatic cancer (22, 26), and investigations probing the biological function of QSOX demonstrate that overexpression provides cancer cells with enhanced invasive potential (22, 23, 27).
A wide range of non-fungal unicellular eukaryotes (from the smallest free living eukaryote to a number of pathogenic protists) contain a single QSOX enzyme (1, 14–16, 19). This study deals with the QSOX found in Trypanosoma brucei, the causative agent of one form of African sleeping sickness. TbQSOX proves especially tractable to express in Escherichia coli (28), and its three-domain structure represents the minimal architecture for this family of multidomain sulfhydryl oxidases (16, 19, 28, 29). Fig. 1A shows the domain organization of TbQSOX together with key catalytic steps deduced from studies of both metazoan (30–32) and protist (3, 28, 29) QSOXs. Crystal structures of TbQSOX in open and closed conformations are shown in Fig. 1, B and C (29). Catalysis is initiated with the transfer of reducing equivalents from the client protein to the CIXXCII motif of the thioredoxin (TRX) domain (Fig. 1A, step 1). The TRX domain appears to be capable of rapidly sampling multiple open conformations while tethered to the relatively rigid helix-rich region (HRR)-ERV domains (29) via a flexible linker (shown as a dashed line in Fig. 1, B and C). The crystal structures of TbQSOX show no obvious binding site for protein or peptide substrates (3, 29) in accord with the biochemical evidence for a “hit and run” mode of catalysis (3). After the reduced TRX domain has disengaged from its protein substrate, it docks at the interface of the HRR and ERV domains where it forms a CysI-CysIII mixed disulfide with the redox-active CIIIXXCIV disulfide proximal to the flavin cofactor within the ERV domain (Fig. 1C). Step 2 is completed by resolution of this mixed disulfide with dissociation of the oxidized TRX domain, allowing reducing equivalents to migrate to the flavin and thence to molecular oxygen (steps 3 and 4, respectively (1, 16, 32)).
FIGURE 1.
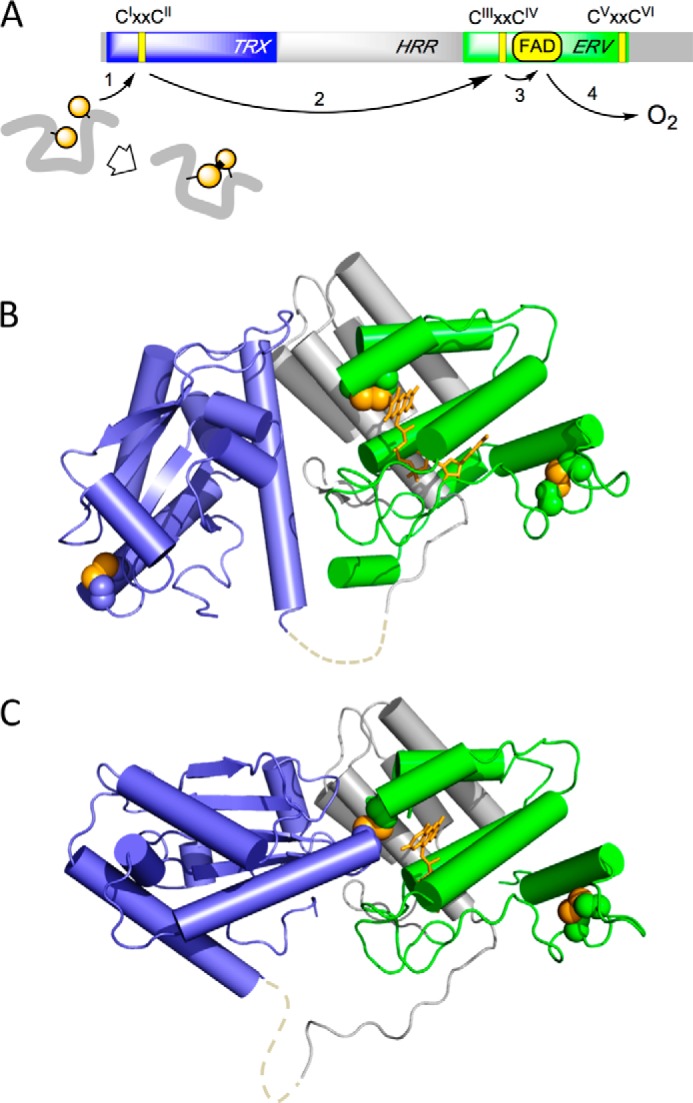
Domain organization and crystal structures of TbQSOX. A represents the domain structure of TbQSOX: the three CXXC motifs (CIXXCII, CIIIXXCIV, and CVXXCVI) together with the TRX domain, HRR, and ERV domain shown in blue, gray, and green, respectively. The flow of reducing equivalents from reduced protein substrate to molecular oxygen is schematically depicted with arrows 1–4. The third conserved CXXC disulfide (CVXXCVI) is not required for the oxidation of small molecule or protein substrates in vitro and is not considered further here. B shows the crystal structure of an open form of the enzyme (Protein Data Bank code 3QCP) using the same domain coloring and orientation as in A. The CXXC sulfur atoms are depicted as yellow spheres, and the FAD is depicted using yellow sticks. Cysteine to alanine mutations at CysII and CysIV allowed the oxidative capture of the interdomain CysI-CysIII mixed disulfide shown in the closed form of TbQSOX in C (Protein Data Bank code 3QD9). The dashed lines in B and C depict a mobile loop not resolved in the crystal structures.
This study probes the mechanism of this proficient stand-alone catalyst of disulfide bond formation. Here, we determined the reduction potentials of the three redox centers in TbQSOX catalysis and investigated the consequence of modulating the redox potential of the TRX domain on catalytic proficiency using rapid reaction and steady state kinetics. These studies provide the first thermodynamic context for a QSOX family member, revealing a surprisingly unfavorable thermodynamic barrier in the catalytic mechanism. Finally, our analysis identified intriguing parallels with the DsbA/B proteins catalyzing oxidative protein folding in the bacterial periplasm and provides a rationalization for this striking example of mechanistic convergence.
EXPERIMENTAL PROCEDURES
Materials and Reagents
Unless noted, chemical reagents and bacterial culture components were obtained as described previously (28). Primers were from Integrated DNA Technologies. Reduced glutathione (GSH), oxidized glutathione (GSSG), and oxidized DTT were from Sigma. Reduced DTT and isopropyl d-thiogalactopyranoside were from GoldBio. Methyl-PEG24-maleimide (MM(PEG)24) was from Thermo Fisher Scientific. 5-Deaza-FAD was obtained as described previously (33).
Spectrophotometry, Assays, and Kinetics
Routine aerobic and anaerobic UV-visible spectrophotometric experiments, QSOX enzyme assays, and rapid reaction studies with an SF-61 DX2 double mixing stopped-flow spectrophotometer (Hi-Tech Scientific) were performed as described earlier (3, 28, 34).
TbQSOX Constructs and Mutants
A truncated form containing only the thioredoxin domain (TbQSOX-TRX; corresponding to residues 20–199 in the full-length protein) was subcloned using the TbQSOX-FL-Fwd (28) and Trx-Rev (GTCCTCGAGTTAGACAGAACGTTTGACCAG) primers. Mutants of the intervening XX dipeptide of the CIXXCIITRX motif in the context of the TbQSOX-TRX or full-length TbQSOX constructs were obtained using the QuikChange II Mutagenesis kit (Stratagene) with appropriate primers. All final expression constructs in the pET-28a(+) vector contained N-terminal His tags and were sequenced to verify integrity.
Expression and Purification of TbQSOX
Full-length TbQSOX and truncated constructs were expressed and purified as before (3, 28) with the following minor modifications. The hydrophobic interaction chromatography step for full-length proteins utilized 35%, not 40%, saturated ammonium sulfate. This step was omitted for the purification of TbQSOX-TRX construct. Protein yields for all constructs were similar to that of wild-type TbQSOX (∼7 mg/liter of cell culture (28)).
Redox Potential Determinations
All redox potential experiments were carried out at 25 °C in 50 mm potassium phosphate buffer, pH 7.0 containing 1 mm EDTA. Stock solutions of reduced and oxidized glutathione were adjusted to pH 7 using potassium hydroxide. Thiol-containing solutions were standardized using 5,5-dithiobis(2-nitrobenzoate). The fraction of protein containing a particular reduced redox center was plotted against the composition of the corresponding redox couple ([GSH]2/[GSSG] or [DTTred]/[DTTox]). Redox data were analyzed using non-linear curve fitting with the Hill equation in GraphPad Prism 6 (GraphPad Software) to obtain the equilibrium constant, Kox. Standard redox potentials were obtained via the Nernst equation using E′0 values of −240 mV for GSH/GSSG (35) and −327 mV for DTTred/DTTox (36).
Redox Potentials of Thioredoxin Domain CIXXCII
Approximately 5 μm protein was incubated at 25 °C for 2 h in 1.5-ml centrifuge tubes containing a range of glutathione redox buffers comprising 5 mm GSSG and 0.05–75 mm GSH. The mixtures were then rapidly quenched by the addition of 100% (w/v) ice-cold trichloroacetic acid (TCA) while vigorously vortexing to give a final concentration of 20% TCA. The precipitated protein samples were recovered by centrifugation (6000 relative centrifugal force for 30 min at 4 °C), and the pellets were washed twice with ice-cold acetone and allowed to air dry for 30 min. The denatured samples were then resuspended in 10 mm MM(PEG)24 in 2× non-reducing Laemmli buffer before being analyzed by SDS-PAGE using 12% gels. Gels were imaged, and bands were quantitated using a FluorChem Q imaging system and the FluorChem Q software (Protein Simple).
Redox Potential of the CIIIXXCIV Proximal Disulfide
The 5-deaza-FAD-substituted enzyme was first generated by reconstituting the apoprotein with the flavin analog. Wild-type TbQSOX (45 nmol in 50 mm phosphate buffer, pH 7.5) was bound to 0.5 ml of ProBond nickel-chelating resin (Invitrogen) retained in a small plastic capped column. Flavin release was initiated by the addition of 1.5 ml of 6 m guanidine hydrochloride in 50 mm phosphate buffer adjusted to pH 7.5. The column was recapped and incubated with rocking for 15 min at room temperature. The column was then allowed to drain and was washed with 4 column volumes of the denaturant followed by 3 column volumes of phosphate buffer without guanidine hydrochloride. Washes were evaluated spectrophotometrically to follow loss of FAD from the resin. The resin was then rocked overnight at 4 °C with a 1.7-fold molar excess of 5-deaza-FAD in 1 ml of phosphate buffer, pH 7.5. 5-Deaza-FAD-TbQSOX was eluted from the column with 500 mm imidazole and exchanged into 50 mm phosphate buffer, pH 7.0 containing 1 mm EDTA using Amicon Ultra centrifugal filters (Millipore). Redox experiments were conducted in open cuvettes including 50 nm glucose oxidase and 10 mm glucose as a precaution to maintain dissolved oxygen concentrations to low levels. Aliquots of reduced DTT were added to 15 μm 5-deaza-FAD-enzyme in the presence of 20 mm oxidized DTT. Spectra were recorded immediately and found to be unchanged 30 s after mixing. Spectra were scatter-corrected over 490–800 nm and corrected for dilution. The maximum absorbance change was centered around 445 nm (see Fig. 3A, inset). However, the absorbance change over five consecutive wavelengths (443–447 nm) was averaged to minimize instrumental noise prior to calculation of the fraction of enzyme carrying the reduced CIIIXXCIV motif.
FIGURE 3.
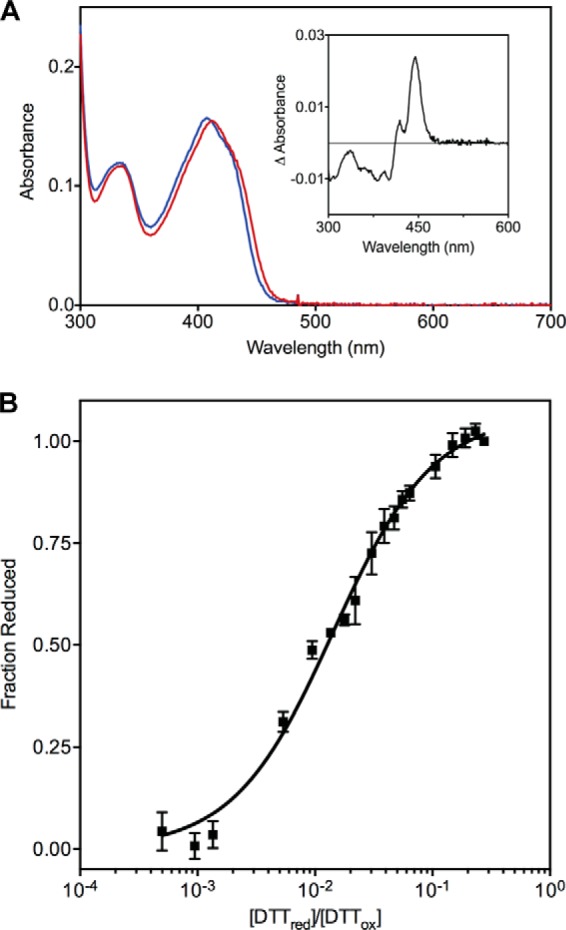
Determination of the redox potential of the proximal CXXC disulfide in TbQSOX. 5-Deaza-FAD-substituted TbQSOX was prepared as described under “Experimental Procedures.” A, the spectrum of the protein was recorded before and after reduction with 5 mm DTT (red and blue lines, respectively). The ∼5-nm blue shift upon reduction of the proximal disulfide resulted in maximum differences in absorbance between oxidized and reduced forms centered around 445 nm (inset), which was used to calculate the fraction of reduced enzyme with increasing ratios of reduced to oxidized DTT (see text). B, the fractions of reduced enzyme were plotted, and a non-linear fit to the data from three independent experiments gave a redox potential of −273 ± 3 mV for the proximal disulfide. Error bars represent S.E.
TbQSOX-bound FAD Redox Potential
Wild-type TbQSOX (30 μm in 1 ml of 50 mm phosphate buffer, pH 7.0 containing 1 mm EDTA, 10 mm GSSG, and 10 mm glucose) was deoxygenated by cycles of vacuum and nitrogen in an anaerobic cuvette (34) prior to the addition of 50 nm glucose oxidase to ensure continued anaerobiosis. A stock solution of GSH was delivered via a Hamilton gas-tight syringe, and UV-visible spectra were recorded immediately and 60 s after each addition. The 60-s spectra were scatter-corrected (over 700–850 nm), and the fraction of reduced enzyme FAD was calculated from the dilution-corrected absorbance at 456 nm.
RESULTS
Overview of Redox Potential Measurements
We started this work by determining the redox potentials of the centers participating in the oxidation of protein substrates (Fig. 1A). Because the TRX domain of TbQSOX is linked by a mobile and disordered loop region to the HRR/ERV pseudodimer (Fig. 1B and Refs. 29 and 37), truncations of the protein allow them to be studied separately. It should be noted that there is an additional CXXC motif toward the C terminus of the ERV domain. Although this CVXXCVI motif is conserved in all QSOX sequences examined to date (1, 19), single or double mutations at this locus have an insignificant impact on the turnover of protein substrates in vitro using either human QSOX1 or TbQSOX (28, 32). Furthermore, the recent crystal structures of open and closed forms of TbQSOX (Fig. 1, B and C) do not provide an obvious rationale for a catalytic role of the CVXXCVI disulfide. Hence, this third CXXC motif will not be considered further in the present study.
Redox Measurement of CIXXCII Motif
Determining the redox potential of the CIXXCII motif requires isolating the redox-active TRX domain from the HRR-ERV domains to prevent transfer of reducing equivalents from the CIXXCII dithiol to the CIIIXXCIV and FAD centers of TbQSOX (Fig. 1A). The thioredoxin domain with an additional embedded α-helix (colored in blue in Fig. 1B) is tethered to the rigid and structurally independent HRR-ERV domains in TbQSOX by a flexible linker. This TbQSOX-TRX construct was expressed independently as described under “Experimental Procedures” and was found to contain no free thiols on 5,5-dithiobis(2-nitrobenzoate) titration. The small size of this construct (201 residues) allows the use of a gel shift procedure commonly used to determine the redox potentials of thioredoxin family members (38, 39). TbQSOX-TRX was equilibrated at pH 7.0 in redox buffers containing varying ratios of reduced and oxidized glutathione, the mixtures were quenched by TCA precipitation, and the reduced component was labeled with a small PEG-maleimide derivative (MM(PEG)24; see “Experimental Procedures”), adding 1.24 kDa per thiol. Non-reducing SDS-PAGE revealed the expected gel shift for the introduction of two maleimide labels for the reduced protein. As observed by others (40), an intermediate band appeared (denoted with * in Fig. 2A), likely reflecting the formation of a mixed disulfide intermediate with glutathione. Under conditions where very high concentrations of reduced glutathione were present, a portion of the protein becomes over-reduced as one of the three structural disulfides present in this construct is reduced (Fig. 2, A and B). This over-reduced protein was included with the reduced fraction of protein, and the mixed glutathione-disulfide bands were excluded from band analysis. Quantitation of the reduced and oxidized bands by densitometry yielded the data in Fig. 2C. Four independent experiments gave a redox potential of −144 ± 1 mV. This relatively oxidizing potential is consistent with a role of this redox center as the initial oxidant for reduced, conformationally mobile protein substrates (see later text). Thus, the observed redox potential is intermediate between the highly oxidizing DsbA protein of the bacterial periplasm (∼−120 mV (41, 42)) and that of the relatively oxidizing two CXXC motifs of mammalian protein-disulfide isomerase (∼−170 mV (43–45)) from the endoplasmic reticulum.
FIGURE 2.
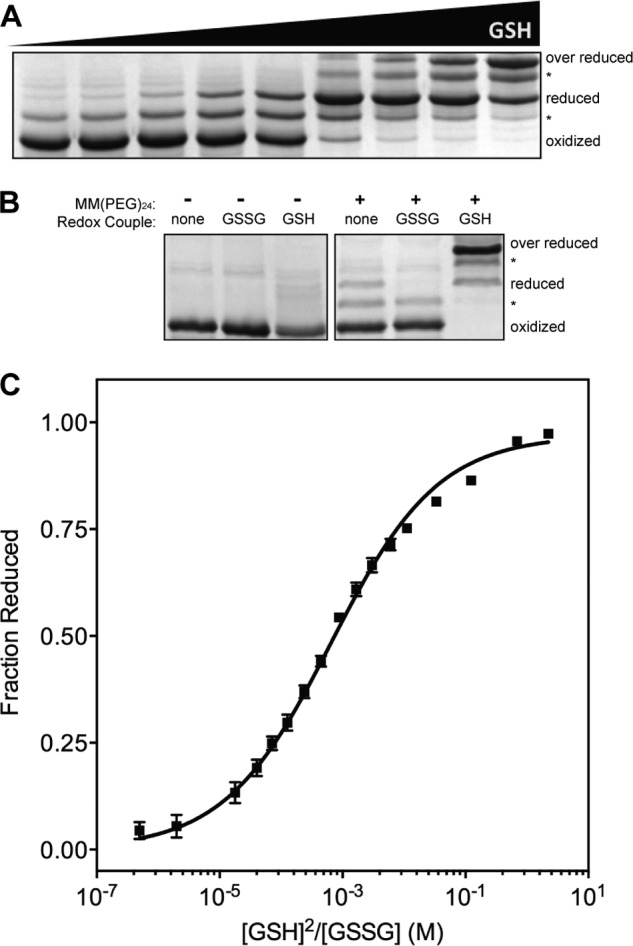
Redox potential determination of the thioredoxin domain CXXC motif in TbQSOX. A, SDS-PAGE showing the redox equilibration of TbQSOX-TRX incubated with glutathione redox buffers where the proportion of reduced glutathione increases from left to right. The portion of protein containing reduced CXXC reacts with MM(PEG)24 and thus runs as a higher molecular weight band than the oxidized protein. Minor intermediate bands corresponding to protein containing a mixed disulfide species with glutathione (*) were excluded from analysis. A portion of the protein becomes over-reduced at high GSH concentrations. B, gel shift controls using 20 mm GSSG or 20 mm GSH in the presence or absence of MM(PEG)24. High levels of reduced GSH cause the majority of the protein to become over-reduced. C, the band intensities of reduced forms and oxidized protein in A were quantified by densitometry, and the fractions of reduced protein from four independent experiments were plotted, and a non-linear fit of the data yielded a redox potential of −144 ± 1 mV (see “Experimental Procedures”). Error bars represent S.E.
Redox Measurement of CIIIXXCIV Motif
Upon net transfer of reducing equivalents from protein clients to the CIXXCII motif of QSOX family members, the mobile TRX domain then reduces the CIIIXXCIV proximal disulfide adjacent to the oxidized flavin cofactor within the ERV domain (Fig. 1A) (29, 31, 32). Several lines of evidence suggest that this proximal disulfide is significantly more reducing than the adjacent flavin. For example, dithionite titrations of full-length TbQSOX in which the strongly oxidizing CIXXCII disulfide in the TRX domain is disabled with a CIS mutation showed that the FAD cofactor is stoichiometrically reduced after the addition of 2 electrons before any reduction of the proximal disulfide occurs (28). Comparable results were obtained in an HRR-ERV truncation construct of TbQSOX in which the TRX domain is removed entirely (28). This behavior is not unique to TbQSOX; it was previously encountered in an HRR-ERV fragment of avian QSOX1 that was generated by partial proteolysis (31) and is found in the ERV domains of several small stand-alone sulfhydryl oxidases including augmenter of liver regeneration and ERV2p (46, 47).
Because we wanted to determine the redox potential of the proximal disulfide in the context of an oxidized flavin, we explored the use of the highly reducing flavin analog 5-deaza-FAD (48, 49). Substitution of the flavin prosthetic group in full-length TbQSOX with 5-deaza-FAD (see “Experimental Procedures”) yields an enzyme with undetectable activity (data not shown). This is to be expected because 5-deazaflavins are unreactive in their reduced forms toward oxygen (48). Although incubation of substituted TbQSOX with 5 mm DTT leads to insignificant reduction of the bound deazaflavin, the oxidized flavin envelope is blue shifted by ∼5 nm (from 412 to 407 nm; Fig. 3A). Precedent for such blue shifts upon removal of a disulfide bond proximal to the flavin was previously observed in lipoamide dehydrogenase (50); augmenter of liver regeneration (51); and avian (52), human (32), and trypanosomal (28) QSOXs. Reduction of the proximal disulfide in 5-deaza-FAD-TbQSOX is then conveniently followed by changes in absorbance centered around 445 nm (see Fig. 3A, inset, and “Experimental Procedures”) upon the incremental addition of reduced DTT in the presence of 20 mm oxidized DTT. A plot of the percentage of reduction as a function of the composition of the ratio DTTred/DTTox yields a redox potential of −273 ± 3 mV for the proximal disulfide (Fig. 3B). Thus, the CIIIXXCIV proximal disulfide is much more reducing than the TRX domain; the ∼130-mV mismatch implies a more than ∼10,000-fold equilibrium bias in favor of reduction of the thioredoxin domain in TbQSOX. A further discussion of the redox imbalance among the three redox centers in QSOX will be presented after determination of the redox potential of the flavin prosthetic group.
Redox Measurement of the FAD Prosthetic Group
To evaluate the third redox center, the bound FAD of TbQSOX, we utilized the full-length protein and delivered reducing equivalents using the relatively weak thermodynamic reductant glutathione. Although this monothiol is a poor substrate of both protist and mammalian QSOX enzymes (28, 53, 54), communication between GSH and the CIXXCII disulfide is rapid enough to ensure equilibration with the FAD after several seconds under rigorously anaerobic conditions (see “Experimental Procedures”). The corresponding spectra (Fig. 4A) show a progressive decline in absorbance at 456 nm when increasing concentrations of GSH are added to a solution of 10 mm GSSG and allowed calculation of a redox potential for the bound flavin of −153 ± 1 mV. Thus, the flavin and the TRX CIXXCII motif are almost equipotential (ΔE′0 of 9 mV). This is consistent with the outcome of dithionite titrations of TbQSOX: 4 electrons are required to completely reduce the flavin with an almost linear decrease in absorbance at 456 nm (28), implying that both the FAD and the CIXXCII motif are of comparable redox potential.
FIGURE 4.
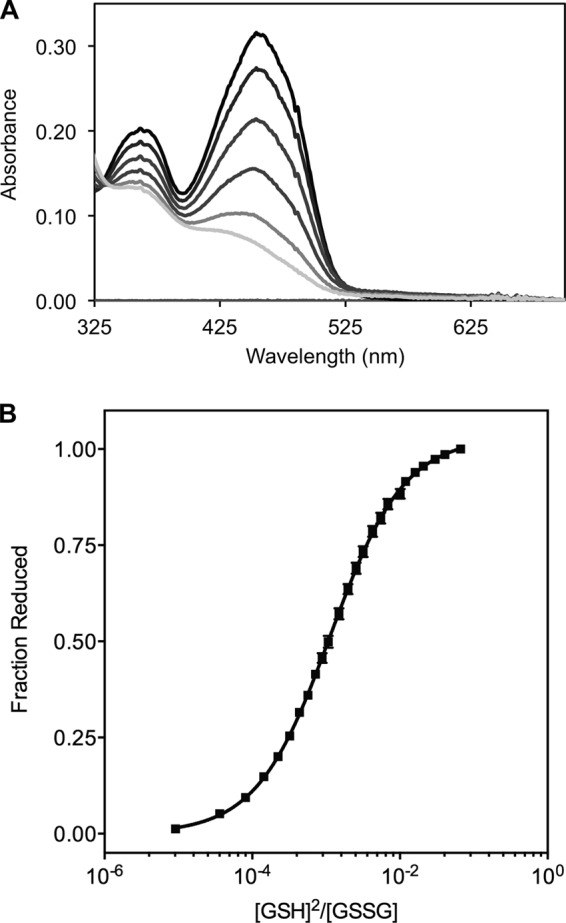
The redox potential of the FAD cofactor in TbQSOX. Full-length TbQSOX was equilibrated with reduced and oxidized glutathione under rigorously anaerobic conditions. A, reduction of the FAD was quantified by the decline in absorbance at 456 nm with increasing GSH additions. B, a non-linear fit of the fraction of reduced FAD yields a redox potential of −153 ± 1 mV. Error bars represent S.E.
Modulating Catalysis by Mutating the CIXXCII Motif
Multiple studies have shown that the intervening XX dipeptide in CXXC motifs in thioredoxin superfamily members can modulate the redox potential in a predictable way (41, 55–59). Thus, when the dipeptide of E. coli thioredoxin is changed from GP to PH (a sequence found in the highly oxidizing DsbA protein), the redox potential becomes about 60 mV more positive (56, 57). Conversely, substitution of the PH sequence to a GP in the context of the DsbA protein effects a change of redox potential from the highly oxidizing −122 mV to the more reducing −214 mV (60, 61). One might expect that the potentials for each couple in an enzyme containing multiple redox centers would be optimized for its physiological roles. Here, we explore the catalytic impact of modulating the redox potential of the TRX CGAC motif in TbQSOX using CGPC and CPHC sequences.
Redox Potentials of Mutant TRX Domains and Steady State Kinetics of Corresponding Mutant TbQSOX Enzymes
We first determined the redox potentials of the TbQSOX TRX domain containing the CGPC and CPHC sequences following the gel shift procedure outlined earlier (see “Experimental Procedures”). As expected, the GP sequence is more reducing than the wild-type sequence (by 41 mV; Table 1). Again, in accord with the trends observed with previous thioredoxin family members, replacement of the CGAC sequence by CPHC generates a mutant that is 31 mV more oxidizing than the wild-type domain (Table 1).
TABLE 1.
Redox potentials of wild-type TbQSOX and CIXXCII mutants
| Redox center | Redox couple | Kox | E′0a |
|---|---|---|---|
| mV | |||
| CIGACIIWT | GSH/GSSG | 6.05 × 10−4 m | −144 ± 1 |
| CIIIKECIVWT | DTTred/DTTox | 1.23 × 10−2 | −273 ± 3 |
| FAD | GSH/GSSG | 1.11 × 10−3 m | −153 ± 1 |
| CIGPCII | GSH/GSSG | 1.35 × 10−2 m | −185 ± 4 |
| CIPHCII | GSH/GSSG | 4.58 × 10−5 m | −113 ± 3 |
a Redox potentials were calculated from the Kox determined from the mean of two to four replicates. Uncertainty represents the 95% confidence limits.
Full-length TbQSOX constructs incorporating the CIGPCII and CIPHCII sequences in the TRX domain were purified for kinetics characterization. The mutant oxidases showed flavin spectra, purities, and stabilities comparable with those of the wild-type protein (data not shown). Both proteins were assayed with the model substrate DTT and using reduced RNase as a tractable unfolded reduced protein substrate (3, 18). A comparison of steady state kinetic parameters is shown in Table 2. The GP enzyme shows very modest decreases in kcat/Km for DTT and rRNase (4.2- and 1.7-fold, respectively) dominated by a small decrease in kcat (Table 2). In contrast, the more oxidizing TbQSOX mutant shows correspondingly larger decreases in kcat values (of 50- and 28-fold, respectively), but these values are substantially offset by a 5.7- and 9-fold decrease in the Km term, again leading to a very small overall decrease in catalytic efficiency when compared with the wild-type enzyme. Although Michaelis-Menten behavior was observed for both DTT and rRNase using the three mutant enzymes, the behavior of the CIPHCII enzyme is distinctly anomalous at higher DTT concentrations (Fig. 5). The rates show a saturable initial phase giving the Km estimate in Table 2 followed by a linear dependence from 1 to 15 mm DTT. Strikingly, inactivation of the TRX domain using the CIS mutation in TbQSOX shows a linear dependence on DTT concentration but without the initial phase (28). Hence, the first phase involves saturation of the TRX domain by DTT, and the limiting rate constant of 0.9 s−1 reflects an internal rate-limiting step that is more than 40-fold slower than observed with the WT enzyme (28). The subsequent linear phase reflects the ability of DTT to short circuit catalysis by reducing the CysIII-CysIV proximal disulfide without the obligatory participation of the thioredoxin domain. The protein substrate rRNase does not show this secondary phase because it cannot efficiently communicate with the ERV domain directly (28).
TABLE 2.
Kinetics of wild-type TbQSOX and CIXXCII mutants
Values for the wild-type protein were adapted from Kodali and Thorpe (28). Turnover numbers are expressed as thiols oxidized per second with substrate concentrations denoted in terms of total thiols. Uncertainty represents S.E.
| Mutant | Kinetics with DTT |
Kinetics with rRNase |
||||
|---|---|---|---|---|---|---|
| kcat | Km | kcat/Km | kcat | Km | kcat/Km | |
| s−1 | mm | m−1 s−1 | s−1 | mm | m−1 s−1 | |
| CIGACIIWT1 | 45.1 ± 0.7 | 0.17 ± 0.01 | 2.6 × 105 | 21.9 ± 1.8 | 0.36 ± 0.06 | 6.0 × 104 |
| CIGPCII | 13.7 ± 0.5 | 0.22 ± 0.02 | 6.2 × 104 | 11.0 ± 1.1 | 0.32 ± 0.08 | 3.5 × 104 |
| CIPHCII | 0.9 ± 0.1 | 0.03 ± 0.01 | 7.2 × 104 | 0.79 ± 0.01 | 0.04 ± 0.01 | 1.8 × 104 |
FIGURE 5.
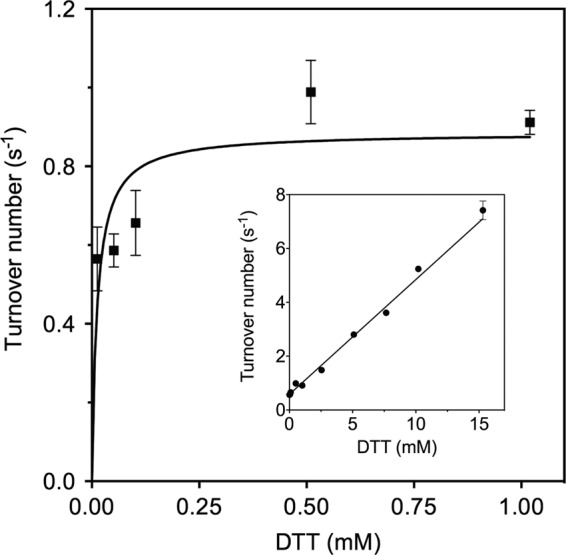
Turnover number of the CIPHCII mutation of TbQSOX with DTT. The main panel represents turnover numbers up to 1 mm DTT. The inset shows the linear dependence of the turnover number of the mutant enzyme at concentrations above 1 mm DTT. Error bars represent S.E.
Monitoring Turnover with DTT in the Stopped-flow Spectrophotometer
Although the mutant TbQSOX enzymes show relatively modest decreases in catalytic efficiency, they demonstrate marked differences in the ratio of species populating the steady state during aerobic turnover with DTT. TbQSOX shows only modest levels of a charge transfer species in the steady state (28) as shown in Fig. 6A with the time course at 456 and 580 nm observed when the oxidase is mixed with 5 mm DTT in aerobic phosphate buffer, pH 7.5 depicted in Fig. 6B. The steady state is maintained for ∼0.5 s before depletion of oxygen leads to the accumulation of the reduced enzyme. The two mutants show strongly contrasting behavior. The CIPHCII enzyme is almost completely oxidized in the steady state with no detectable charge transfer band at 580 nm (Fig. 6C). Significant reduction of this enzyme only occurs after a sharp transition at 5 s. During the subsequent accumulation of reduced enzyme, no long-wavelength spectral features are evident as oxygen is depleted from the solution.
FIGURE 6.
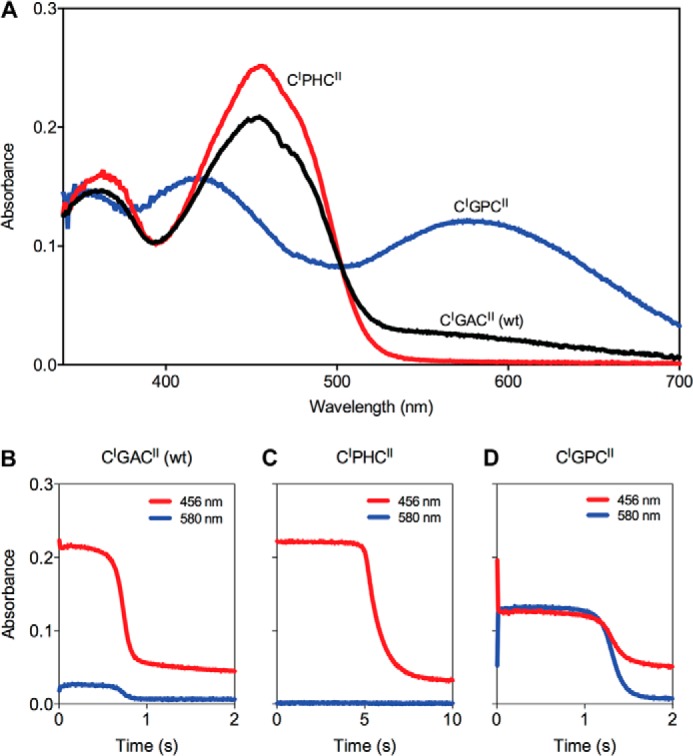
Steady state spectra of wild-type, CIPHCII, and CIGPCII mutations of TbQSOX during the oxidation of DTT in air-saturated buffer. A, wild-type TbQSOX shows a small amount of charge transfer absorbance in the steady state observed when the enzyme is mixed aerobically with 5 mm DTT in 50 mm phosphate buffer, pH 7.5, 25 °C. By contrast, the CIPHCII mutant (red) lacks any detectable charge transfer species, and the CIGPCII mutant (blue) shows a strong charge transfer absorbance. B–D, the corresponding time courses for the absorbance changes at 456 and 580 nm, respectively, are shown.
In marked contrast, the CIGPCII mutant shows an intense blue color during turnover; this strong charge transfer band (centered at 580 nm) remains almost unchanged for 1 s before reduced flavin begins to accumulate (Fig. 6D). This prominent species is also observed under anaerobic conditions when the CIGPCII mutant is mixed with DTT in the stopped-flow spectrophotometer (Fig. 7). Here, the formation of the charge transfer band shows a limiting apparent rate constant of 936 ± 40 s−1, which is some 3.3-fold faster than the 280 s−1 observed for the wild-type protein (28). In contrast, conversion of the charge transfer species to yield reduced flavin is 7.1 ± 0.3 s−1 for the GP mutant, which is about 2.6-fold slower than the wild-type protein. It is important to note that thiolate to flavin charge transfer species are undetectable in the absence of the TRX domain in both static and rapid reaction experiments (28). Thus, the charge transfer species observed during earlier rapid reaction studies of both the T. brucei and avian QSOXs (28, 30) is almost certainly dominated by the formation of a mixed disulfide intermediate in which the TRX domain is docked against the HRR-ERV fragment (Fig. 8A). The crystal structure of the closed conformation of TbQSOX (Fig. 1C and Ref. 29) provides a structural approximation for this critical link between the two catalytic modules of QSOX catalysis. Although this work shows that the formation of this species is highly responsive to the sequence of the CXXC motifs within the TRX domain, it is clearly unwarranted to justify the differences in behavior in terms of redox potentials alone. Obviously, the insertion of a proline at the first or second position within the intervening XX dipeptide motif (from CGAC of the wild-type enzyme to CPHC and CGPC) may perturb the ability either of the CysI cysteinyl sulfur to serve as a nucleophile during the formation of the CysI-CysIII mixed disulfide intermediate or of the CysII sulfur to achieve the in-line geometry needed to resolve this interdomain disulfide (1, 16).
FIGURE 7.
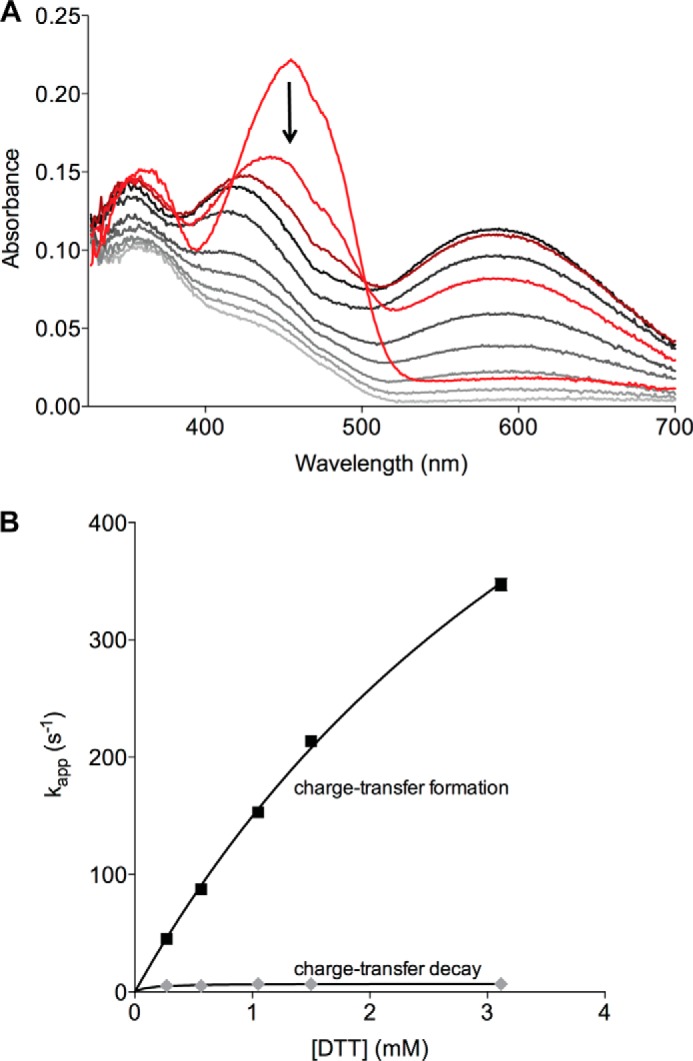
Reduction of the CIGPCII mutant under anaerobic conditions and analysis of the dependence of the apparent rate constants for formation and decay of the charge transfer complex on DTT concentration. A, anaerobic stopped-flow spectra demonstrating the formation of a long-wavelength charge transfer intermediate and subsequent disappearance of this species as flavin is reduced. For clarity, only select spectra are shown (starting with red, dark red, and brown traces). B, apparent rate constants for the formation (solid squares) and decay (gray diamonds) of the long-wavelength charge transfer species characterized at 580 nm.
FIGURE 8.

Schematic of free energy coordinates and equilibria depicting overall catalysis by TbQSOX and DsbA/B. A shows a schematic free energy coordinate for TbQSOX. The transfer of electrons from reduced substrate to CIXXCII of TbQSOX is energetically favorable, whereas the subsequent intramolecular transfer of electrons to CIIIXXCIV is strongly unfavorable based on free energy coordinates alone. The reduction of the FAD prosthetic group and the final transfer from dihydroflavin to molecular oxygen (73) are both favored energetically. A also depicts the interconversion of selected 2-electron reduced forms in TbQSOX. B presents the corresponding schemes for DsbA/B. The redox potentials for DsbB disulfides in B are the average of those of Regeimbal and Bardwell (74) and Inaba and Ito (61). Cysteine residues are labeled by their position in the sequence of DsbA and DsbB.
DISCUSSION
TbQSOX in Thermodynamic Context: Redox Potentials of Thiol Substrates
The trypanosomal enzyme (28), like other QSOXs (3, 18, 32, 54), is capable of oxidizing a very wide range of mono-, di-, and multithiol substrates. Conformationally flexible peptides and proteins containing two or more cysteine residues appear to be excellent substrates of the enzyme, although there is comparatively little data regarding their redox potentials. A series of dithiol-containing peptides and unfolded proteins show redox potentials of −190 to −220 mV (60, 62); these values are considerably more reducing than the −144 mV observed for the thioredoxin domain of TbQSOX. In terms of potential protein clients of QSOX, many structural proteins contain multiple disulfides, greatly complicating the determination of redox potentials. Thus, values for a particular disulfide bridge would likely depend on the number of disulfides that were already introduced and the degree to which those predecessors were correctly paired. Gilbert (63) has tabulated a wide range of redox potentials for intramolecular protein structural disulfides spanning −185 to −450 mV. In summary, the CIXXCII redox disulfide of the TRX domain appears to be much more oxidizing than the majority of dithiol substrates that QSOX is likely to encounter (1, 16, 27, 64). However, this strongly oxidizing couple must now serve as the reductant of the proximal disulfide in a reaction that appears to constitute a significant barrier to the overall reaction (Fig. 8A). Thereafter, transfer of reducing equivalents to the flavin and then to molecular oxygen is very energetically favorable.
Coupled Disulfide Exchange Reactions Promote Efficient Catalysis
Fig. 8 shows, somewhat surprisingly, that two evolutionarily unrelated catalysts of oxidative protein folding, QSOX and the bacterial DsbA/DsbB oxidoreductase enzyme system, appear to share a common mechanistic strategy. First, the oxidation of client proteins in both systems is initiated by a strongly oxidizing thioredoxin domain or subunit. After reduction, the soluble and highly oxidizing periplasmic DsbA (Fig. 8B) must dock with membrane-bound DsbB at the bacterial plasma membrane (65–67). In contrast, QSOX incorporates a tethered, and oxidizing, N-terminal thioredoxin domain (Fig. 1A). Second, both systems apparently show a strongly uphill mismatch associated with reoxidation of their cognate thioredoxin partners. Third, in both systems, a charge transfer interaction between a thiolate and the oxidized cofactor (flavin in QSOX and a quinone in DsbB) is the first observable intermediate in rapid reaction studies (28, 30, 68). Fourth, decomposition of this charge transfer intermediate to yield dihydroflavin or hydroquinone is rate-limiting in overall catalysis in both QSOX and DsbB, respectively (25, 27, 54). Finally, accumulation of these reduced enzyme species is believed to occur in both instances via thiol-cofactor covalent adducts (1, 69, 70).
In terms of charge transfer complex formation, one or two disulfide exchanges are required to form a path of communication from the reduced thioredoxin domain to the organic cofactor in QSOX and DsbA/B (Fig. 8). Thus, in TbQSOX, a CysI-CysIII mixed disulfide traps the closed conformation, thereby allowing CysIV to form a charge transfer complex with the flavin (Fig. 8A). Such thiolate to flavin charge transfer complexes can provide an additional thermodynamic stabilization favoring product formation (70). In the case of DsbB, a recent rapid reaction study showed that Cys30 of reduced DsbA efficiently captures Cys104 of DsbB in an interprotein mixed disulfide (68). The liberated cysteine Cys130 of DsbB can facilely attack Cys41 located on the second mobile periplasmic peptide loop of DsbB, thereby releasing the charge transfer thiolate to interact with the highly oxidizing quinone cofactor (68, 71).
It is important to note that an uphill mismatch in redox potential between dithiol/disulfide centers is not a disqualification for the efficient formation of mixed disulfide intermediates; differences in redox potential cannot be used to predict the thermodynamic stability of a mixed disulfide that may form between them. Hence, in the case of DsbA/B, a series of facile disulfide exchange reactions (66, 68, 72) provides a covalent pathway for thermodynamic coupling that can link the oxidation of a comparatively distal DsbA to a highly oxidizing quinone center in DsbB provided that DsbA does not dissociate prior to completion of this series of stepwise reactions. Subsequently, the Cys44 thiolate of DsbB likely forms a covalent adduct with the quinone (69) in analogy to the C4a adducts believed to intervene in the final step of transfer of reducing equivalents to the flavin cofactor in QSOX (1). Indeed, both cofactor adducts may provide an additional thermodynamic drive toward the accumulation of the reduced cofactor, allowing electrons to be ultimately delivered to the respiratory chain or to molecular oxygen directly.
Inaba et al. (69) suggest that the relatively negative potentials of the two loop disulfides in DsbB (Cys41-Cys44 and Cys103-Cys130) may restrict reactivity with non-cognate potential reductants within the periplasmic space. The same argument may be advanced for QSOX within its physiological locales: the proximal disulfide shows a redox potential of −273 mV, and this likely contributes to restricting direct communication of potential thiol substrates with the ERV domain. Such short circuiting only seems to be evident under forcing conditions with a small non-physiological substrate when communication between thioredoxin and ERV domains is significantly impaired as observed with the CIPHCII mutant of TbQSOX described earlier (Fig. 5).
In summary, the evolutionarily unrelated bacterial DsbA/B and eukaryotic QSOX systems have apparently adopted a common general strategy for disulfide bond formation. Both systems initially receive electrons from thiol-containing peptides via a strongly oxidizing CXXC motif in the context of a thioredoxin fold. Reducing equivalents are then transferred through a second redox-active disulfide motif in what appears to be a prohibitive thermodynamic mismatch, a strategy that likely evolved to restrict nonspecific oxidation of extraneous thiols. Rather than the full expression of this thermodynamic mismatch, a series of disulfide exchange reactions provides an alternate pathway that couples oxidation of the cognate thioredoxin donor with reduction of a distal oxidizing cofactor. It seems likely that examination of additional oxidoreductases of oxidative protein folding may provide further examples of this strategy to thermodynamically couple distal centers with a series of linked thiol-disulfide exchange reactions.
Acknowledgments
We thank Dr. Bruce Palfey for an insightful discussion of thiol-disulfide exchange reactions. Shawn Gannon provided help with the interpretation of redox titrations.
This work was supported, in whole or in part, by National Institutes of Health Grants GM26643 (to C. T.) and 1-T32-GM08550, a United States Public Health Service training grant (to B. A. I.).
- QSOX
- quiescin sulfhydryl oxidase
- ERV
- essential for respiration and vegetative growth
- GSH
- reduced glutathione
- GSSG
- oxidized glutathione
- MM(PEG)24
- methyl-PEG24-maleimide
- rRNase
- reduced bovine pancreatic ribonuclease A
- TRX
- thioredoxin
- HRR
- helix-rich region
- Tb
- T. brucei.
REFERENCES
- 1. Heckler E. J., Rancy P. C., Kodali V. K., Thorpe C. (2008) Generating disulfides with the quiescin-sulfhydryl oxidases. Biochim. Biophys. Acta 1783, 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rancy P. C., Thorpe C. (2008) Oxidative protein folding in vitro: a study of the cooperation between quiescin-sulfhydryl oxidase and protein disulfide isomerase. Biochemistry 47, 12047–12056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Codding J. A., Israel B. A., Thorpe C. (2012) Protein substrate discrimination in the quiescin sulfhydryl oxidase (QSOX) family. Biochemistry 51, 4226–4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cabibbo A., Pagani M., Fabbri M., Rocchi M., Farmery M. R., Bulleid N. J., Sitia R. (2000) ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J. Biol. Chem. 275, 4827–4833 [DOI] [PubMed] [Google Scholar]
- 5. Pagani M., Fabbri M., Benedetti C., Fassio A., Pilati S., Bulleid N. J., Cabibbo A., Sitia R. (2000) Endoplasmic reticulum oxidoreductin 1-lβ (ERO1-Lβ), a human gene induced in the course of the unfolded protein response. J. Biol. Chem. 275, 23685–23692 [DOI] [PubMed] [Google Scholar]
- 6. Araki K., Inaba K. (2012) Structure, mechanism, and evolution of Ero1 family enzymes. Antioxid. Redox Signal. 16, 790–799 [DOI] [PubMed] [Google Scholar]
- 7. Zito E., Melo E. P., Yang Y., Wahlander Å., Neubert T. A., Ron D. (2010) Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol. Cell 40, 787–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tavender T. J., Springate J. J., Bulleid N. J. (2010) Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 29, 4185–4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sato Y., Kojima R., Okumura M., Hagiwara M., Masui S., Maegawa K., Saiki M., Horibe T., Suzuki M., Inaba K. (2013) Synergistic cooperation of PDI family members in peroxiredoxin 4-driven oxidative protein folding. Sci. Rep. 3, 2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nguyen V. D., Saaranen M. J., Karala A. R., Lappi A. K., Wang L., Raykhel I. B., Alanen H. I., Salo K. E., Wang C. C., Ruddock L. W. (2011) Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bond formation. J. Mol. Biol. 406, 503–515 [DOI] [PubMed] [Google Scholar]
- 11. Schulman S., Wang B., Li W., Rapoport T. A. (2010) Vitamin K epoxide reductase prefers ER membrane-anchored thioredoxin-like redox partners. Proc. Natl. Acad. Sci. U.S.A. 107, 15027–15032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hoober K. L., Glynn N. M., Burnside J., Coppock D. L., Thorpe C. (1999) Homology between egg white sulfhydryl oxidase and quiescin Q6 defines a new class of flavin-linked sulfhydryl oxidases. J. Biol. Chem. 274, 31759–31762 [DOI] [PubMed] [Google Scholar]
- 13. Benayoun B., Esnard-Fève A., Castella S., Courty Y., Esnard F. (2001) Rat seminal vesicle FAD-dependent sulfhydryl oxidase. Biochemical characterization and molecular cloning of a member of the new sulfhydryl oxidase/quiescin Q6 gene family. J. Biol. Chem. 276, 13830–13837 [DOI] [PubMed] [Google Scholar]
- 14. Thorpe C., Hoober K. L., Raje S., Glynn N. M., Burnside J., Turi G. K., Coppock D. L. (2002) Sulfhydryl oxidases: emerging catalysts of protein disulfide bond formation in eukaryotes. Arch. Biochem. Biophys. 405, 1–12 [DOI] [PubMed] [Google Scholar]
- 15. Coppock D. L., Thorpe C. (2006) Multidomain flavin-dependent sulfhydryl oxidases. Antioxid. Redox Signal. 8, 300–311 [DOI] [PubMed] [Google Scholar]
- 16. Kodali V. K., Thorpe C. (2010) Oxidative protein folding and the quiescin-sulfhydryl oxidase family of flavoproteins. Antioxid. Redox Signal. 13, 1217–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sevier C. (2012) Erv2 and quiescin sulfhydryl oxidases: Erv-domain enzymes associated with the secretory pathway. Antioxid. Redox Signal. 16, 800–808 [DOI] [PubMed] [Google Scholar]
- 18. Hoober K. L., Sheasley S. L., Gilbert H. F., Thorpe C. (1999) Sulfhydryl oxidase from egg white: a facile catalyst for disulfide bond formation in proteins and peptides. J. Biol. Chem. 274, 22147–22150 [DOI] [PubMed] [Google Scholar]
- 19. Limor-Waisberg K., Ben-Dor S., Fass D. (2013) Diversification of quiescin sulfhydryl oxidase in a preserved framework for redox relay. BMC Evol. Biol. 13, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ouyang X., DeWeese T. L., Nelson W. G., Abate-Shen C. (2005) Loss-of-function of Nkx3.1 promotes increased oxidative damage in prostate carcinogenesis. Cancer Res. 65, 6773–6779 [DOI] [PubMed] [Google Scholar]
- 21. Song H., Zhang B., Watson M. A., Humphrey P. A., Lim H., Milbrandt J. (2009) Loss of Nkx3.1 leads to the activation of discrete downstream target genes during prostate tumorigenesis. Oncogene 28, 3307–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katchman B. A., Antwi K., Hostetter G., Demeure M. J., Watanabe A., Decker G. A., Miller L. J., Von Hoff D. D., Lake D. F. (2011) Quiescin sulfhydryl oxidase 1 promotes invasion of pancreatic tumor cells mediated by matrix metalloproteinases. Mol. Cancer Res. 9, 1621–1631 [DOI] [PubMed] [Google Scholar]
- 23. Soloviev M., Esteves M. P., Amiri F., Crompton M. R., Rider C. C. (2013) Elevated transcription of the gene QSOX1 encoding quiescin Q6 sulfhydryl oxidase 1 in breast cancer. PLoS One 8, e57327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Katchman B. A., Ocal I. T., Cunliffe H. E., Chang Y.-H., Hostetter G., Watanabe A., Lobello J., Lake D. F. (2013) Expression of quiescin sulfhydryl oxidase 1 is associated with a highly invasive phenotype and correlates with a poor prognosis in Luminal B breast cancer. Breast Cancer Res. 15, R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mebazaa A., Vanpoucke G., Thomas G., Verleysen K., Cohen-Solal A., Vanderheyden M., Bartunek J., Mueller C., Launay J. M., Van Landuyt N., D'Hondt F., Verschuere E., Vanhaute C., Tuytten R., Vanneste L., De Cremer K., Wuyts J., Davies H., Moerman P., Logeart D., Collet C., Lortat-Jacob B., Tavares M., Laroy W., Januzzi J. L., Samuel J. L., Kas K. (2012) Unbiased plasma proteomics for novel diagnostic biomarkers in cardiovascular disease: identification of quiescin Q6 as a candidate biomarker of acutely decompensated heart failure. Eur. Heart J. 33, 2317–2324 [DOI] [PubMed] [Google Scholar]
- 26. Antwi K., Hostetter G., Demeure M. J., Katchman B. A., Decker G. A., Ruiz Y., Sielaff T. D., Koep L. J., Lake D. F. (2009) Analysis of the plasma peptidome from pancreas cancer patients connects a peptide in plasma to overexpression of the parent protein in tumors. J. Proteome Res. 8, 4722–4731 [DOI] [PubMed] [Google Scholar]
- 27. Ilani T., Alon A., Grossman I., Horowitz B., Kartvelishvily E., Cohen S. R., Fass D. (2013) A secreted disulfide catalyst controls extracellular matrix composition and function. Science 341, 74–76 [DOI] [PubMed] [Google Scholar]
- 28. Kodali V. K., Thorpe C. (2010) Quiescin sulfhydryl oxidase from Trypanosoma brucei: catalytic activity and mechanism of a QSOX family member with a single thioredoxin domain. Biochemistry 49, 2075–2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alon A., Grossman I., Gat Y., Kodali V. K., DiMaio F., Mehlman T., Haran G., Baker D., Thorpe C., Fass D. (2012) The dynamic disulphide relay of quiescin sulphydryl oxidase. Nature 488, 414–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hoober K. L., Thorpe C. (1999) Egg white sulfhydryl oxidase: kinetic mechanism of the catalysis of disulfide bond formation. Biochemistry 38, 3211–3217 [DOI] [PubMed] [Google Scholar]
- 31. Raje S., Thorpe C. (2003) Inter-domain redox communication in flavoenzymes of the quiescin/sulfhydryl oxidase family: role of a thioredoxin domain in disulfide bond formation. Biochemistry 42, 4560–4568 [DOI] [PubMed] [Google Scholar]
- 32. Heckler E. J., Alon A., Fass D., Thorpe C. (2008) Human quiescin-sulfhydryl oxidase, QSOX1: probing internal redox steps by mutagenesis. Biochemistry 47, 4955–4963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gorelick R. J., Thorpe C. (1986) Electron-transferring flavoprotein from pig kidney: flavin analogue studies. Biochemistry 25, 7092–7098 [DOI] [PubMed] [Google Scholar]
- 34. Williams C. H., Jr., Arscott L. D., Matthews R. G., Thorpe C., Wilkinson K. D. (1979) Methodology employed for anaerobic spectrophotometric titrations and for computer-assisted data analysis. Methods Enzymol. 62, 185–198 [DOI] [PubMed] [Google Scholar]
- 35. Williams C. H., Jr. (1992) in Chemistry and Biochemistry of Flavoenzymes (Müller F., ed) pp. 121–211, CRC Press, Boca Raton, FL [Google Scholar]
- 36. Lees W. J., Whitesides G. M. (1993) Equilibrium constants for thiol-disulfide interchange reactions—a coherent, corrected set. J. Org. Chem. 58, 642–647 [Google Scholar]
- 37. Alon A., Heckler E. J., Thorpe C., Fass D. (2010) QSOX contains a pseudo-dimer of functional and degenerate sulfhydryl oxidase domains. FEBS Lett. 584, 1521–1525 [DOI] [PubMed] [Google Scholar]
- 38. Appenzeller-Herzog C., Ellgaard L. (2008) In vivo reduction-oxidation state of protein disulfide isomerase: the two active sites independently occur in the reduced and oxidized forms. Antioxid. Redox Signal. 10, 55–64 [DOI] [PubMed] [Google Scholar]
- 39. Jessop C. E., Bulleid N. J. (2004) Glutathione directly reduces an oxidoreductase in the endoplasmic reticulum of mammalian cells. J. Biol. Chem. 279, 55341–55347 [DOI] [PubMed] [Google Scholar]
- 40. Hagiwara T., Inaba H., Yoshida S., Nagaizumi K., Arai M., Hanabusa H., Fukutake K. (1996) A novel mutation Gly1672→Arg in type 2A and a homozygous mutation in type 2B von Willebrand disease. Thromb. Haemost. 76, 253–257 [PubMed] [Google Scholar]
- 41. Huber-Wunderlich M., Glockshuber R. (1998) A single dipeptide sequence modulates the redox properties of a whole enzyme family. Fold. Des. 3, 161–171 [DOI] [PubMed] [Google Scholar]
- 42. Zapun A., Bardwell J. C., Creighton T. E. (1993) The reactive and destabilizing disulfide bond of DsbA, a protein required for protein disulfide bond formation in vivo. Biochemistry 32, 5083–5092 [DOI] [PubMed] [Google Scholar]
- 43. Chambers J. E., Tavender T. J., Oka O. B., Warwood S., Knight D., Bulleid N. J. (2010) The reduction potential of the active site disulfides of human protein disulfide isomerase limits oxidation of the enzyme by Ero1α. J. Biol. Chem. 285, 29200–29207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hatahet F., Ruddock L. W. (2009) Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 11, 2807–2850 [DOI] [PubMed] [Google Scholar]
- 45. Lundström J., Holmgren A. (1993) Determination of the reduction-oxidation potential of the thioredoxin-like domains of protein disulfide-isomerase from the equilibrium with glutathione and thioredoxin. Biochemistry 32, 6649–6655 [DOI] [PubMed] [Google Scholar]
- 46. Farrell S. R., Thorpe C. (2005) Augmenter of liver regeneration: a flavin dependent sulfhydryl oxidase with cytochrome C reductase activity. Biochemistry 44, 1532–1541 [DOI] [PubMed] [Google Scholar]
- 47. Wang W., Winther J. R., Thorpe C. (2007) Erv2p: characterization of the redox behavior of a yeast sulfhydryl oxidase. Biochemistry 46, 3246–3254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Walsh C., Fisher J., Spencer R., Graham D. W., Ashton W. T., Brown J. E., Brown R. D., Rogers E. F. (1978) Chemical and enzymatic properties of riboflavin analogues. Biochemistry 17, 1942–1951 [DOI] [PubMed] [Google Scholar]
- 49. Stankovich M. T., Massey V. (1976) Determination of the redox potential of deazariboflavin by equilibration with flavins. Biochim. Biophys. Acta 452, 335–344 [DOI] [PubMed] [Google Scholar]
- 50. Thorpe C., Williams C. H., Jr. (1976) Differential reactivity of the two active site cysteine residues generated on reduction of pig heart lipoamide dehydrogenase. J. Biol. Chem. 251, 3553–3557 [PubMed] [Google Scholar]
- 51. Daithankar V. N., Farrell S. R., Thorpe C. (2009) Augmenter of liver regeneration: substrate specificity of a flavin-dependent oxidoreductase from the mitochondrial intermembrane space. Biochemistry 48, 4828–4837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brohawn S. G., Miksa I. R., Thorpe C. (2003) Avian sulfhydryl oxidase is not a metalloenzyme: adventitious binding of divalent metal ions to the enzyme. Biochemistry 42, 11074–11082 [DOI] [PubMed] [Google Scholar]
- 53. Hoober K. L., Joneja B., White H. B., 3rd, Thorpe C. (1996) A sulfhydryl oxidase from chicken egg white. J. Biol. Chem. 271, 30510–30516 [DOI] [PubMed] [Google Scholar]
- 54. Jaje J., Wolcott H. N., Fadugba O., Cripps D., Yang A. J., Mather I. H., Thorpe C. (2007) A flavin-dependent sulfhydryl oxidase in bovine milk. Biochemistry 46, 13031–13040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chivers P. T., Prehoda K. E., Raines R. T. (1997) The CXXC motif: a rheostat in the active site. Biochemistry 36, 4061–4066 [DOI] [PubMed] [Google Scholar]
- 56. Jonda S., Huber-Wunderlich M., Glockshuber R., Mössner E. (1999) Complementation of DsbA deficiency with secreted thioredoxin variants reveals the crucial role of an efficient dithiol oxidant for catalyzed protein folding in the bacterial periplasm. EMBO J. 18, 3271–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Quan S., Schneider I., Pan J., Von Hacht A., Bardwell J. C. (2007) The CXXC motif is more than a redox rheostat. J. Biol. Chem. 282, 28823–28833 [DOI] [PubMed] [Google Scholar]
- 58. Lewin A., Crow A., Hodson C. T., Hederstedt L., Le Brun N. E. (2008) Effects of substitutions in the CXXC active-site motif of the extracytoplasmic thioredoxin ResA. Biochem. J. 414, 81–91 [DOI] [PubMed] [Google Scholar]
- 59. Wunderlich M., Glockshuber R. (1993) Redox properties of protein disulfide isomerase (DsbA) from Escherichia coli. Protein Sci. 2, 717–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Aslund F., Berndt K. D., Holmgren A. (1997) Redox potentials of glutaredoxins and other thiol-disulfide oxidoreductases of the thioredoxin superfamily determined by direct protein-protein redox equilibria. J. Biol. Chem. 272, 30780–30786 [DOI] [PubMed] [Google Scholar]
- 61. Inaba K., Ito K. (2002) Paradoxical redox properties of DsbB and DsbA in the protein disulfide-introducing reaction cascade. EMBO J. 21, 2646–2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Siedler F., Rudolph-Böhner S., Doi M., Musiol H. J., Moroder L. (1993) Redox potentials of active-site bis(cysteinyl) fragments of thiol-protein oxidoreductases. Biochemistry 32, 7488–7495 [DOI] [PubMed] [Google Scholar]
- 63. Gilbert H. F. (1995) Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 251, 8–28 [DOI] [PubMed] [Google Scholar]
- 64. Rudolf J., Pringle M. A., Bulleid N. J. (2013) Proteolytic processing of QSOX1A ensures efficient secretion of a potent disulfide catalyst. Biochem. J. 454, 181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kadokura H., Katzen F., Beckwith J. (2003) Protein disulfide bond formation in prokaryotes. Annu. Rev. Biochem. 72, 111–135 [DOI] [PubMed] [Google Scholar]
- 66. Inaba K., Ito K. (2008) Structure and mechanisms of the DsbB-DsbA disulfide bond generation machine. Biochim. Biophys. Acta 1783, 520–529 [DOI] [PubMed] [Google Scholar]
- 67. Nakamoto H., Bardwell J. C. (2004) Catalysis of disulfide bond formation and isomerization in the Escherichia coli periplasm. Biochim. Biophys. Acta 1694, 111–119 [DOI] [PubMed] [Google Scholar]
- 68. Tapley T. L., Eichner T., Gleiter S., Ballou D. P., Bardwell J. C. (2007) Kinetic characterization of the disulfide bond-forming enzyme DsbB. J. Biol. Chem. 282, 10263–10271 [DOI] [PubMed] [Google Scholar]
- 69. Inaba K., Takahashi Y. H., Ito K., Hayashi S. (2006) Critical role of a thiolate-quinone charge transfer complex and its adduct form in de novo disulfide bond generation by DsbB. Proc. Natl. Acad. Sci. U.S.A. 103, 287–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dmitrenko O., Thorpe C. (2008) A computational analysis of the interaction between flavin and thiol(ate) groups. Implications for flavoenzyme catalysis. J. Sulfur Chem. 29, 415–421 [Google Scholar]
- 71. Inaba K., Murakami S., Suzuki M., Nakagawa A., Yamashita E., Okada K., Ito K. (2006) Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell 127, 789–801 [DOI] [PubMed] [Google Scholar]
- 72. Inaba K., Murakami S., Nakagawa A., Iida H., Kinjo M., Ito K., Suzuki M. (2009) Dynamic nature of disulphide bond formation catalysts revealed by crystal structures of DsbB. EMBO J. 28, 779–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wood P. M. (1988) The potential diagram for oxygen at pH 7. Biochem. J. 253, 287–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Regeimbal J., Bardwell J. C. (2002) DsbB catalyzes disulfide bond formation de novo. J. Biol. Chem. 277, 32706–32713 [DOI] [PubMed] [Google Scholar]