

Background: Calcium channels control membrane excitability. The mechanisms underlying Cav2.1/Cav2.2 targeting are not well understood.
Results: Ankyrin-B associates with Cav2.1/Cav2.2. Loss of ankyrin-B results in reduced expression of Cav2.1/Cav2.2 in select brain regions.
Conclusion: Ankyrin-B plays a role in the expression of Cav2.1/Cav2.2.
Significance: Results identify pathway for membrane targeting of calcium channels and regulation of membrane excitability.
Keywords: Adaptor Proteins, Cytoskeleton, Ion Channels, Membrane Trafficking, Protein Sorting, Protein Targeting
Abstract
N-type and P/Q-type calcium channels are documented players in the regulation of synaptic function; however, the mechanisms underlying their expression and cellular targeting are poorly understood. Ankyrin polypeptides are essential for normal integral membrane protein expression in a number of cell types, including neurons, cardiomyocytes, epithelia, secretory cells, and erythrocytes. Ankyrin dysfunction has been linked to defects in integral protein expression, abnormal cellular function, and disease. Here, we demonstrate that ankyrin-B associates with Cav2.1 and Cav2.2 in cortex, cerebellum, and brain stem. Additionally, using in vitro and in vivo techniques, we demonstrate that ankyrin-B, via its membrane-binding domain, associates with a highly conserved motif in the DII/III loop domain of Cav2.1 and Cav2.2. Further, we demonstrate that this domain is necessary for proper targeting of Cav2.1 and Cav2.2 in a heterologous system. Finally, we demonstrate that mutation of a single conserved tyrosine residue in the ankyrin-binding motif of both Cav2.1 (Y797E) and Cav2.2 (Y788E) results in loss of association with ankyrin-B in vitro and in vivo. Collectively, our findings identify an interaction between ankyrin-B and both Cav2.1 and Cav2.2 at the amino acid level that is necessary for proper Cav2.1 and Cav2.2 targeting in vivo.
Introduction
Neurons are highly polarized cells, where structural, functional, and molecular differences within the neuronal physiology underlie the ability to receive, process, and transmit information. The specific expression profile of ion channels, receptors, and cytoskeletal proteins present within select neuronal membrane domains plays a primary role in defining local function of the entire cell. Recently, studies focused on the role of cytoskeletal proteins in the genesis and maintenance of local membrane domains in excitable cells have revealed a new and exciting avenue of study for these proteins in physiology and disease (1, 2).
Calcium ions are critical second messengers involved in the regulation of multiple neuronal cellular processes, but primarily synaptic transmission (3–7). Ca2+ entry across the plasma membrane occurs via voltage-dependent calcium channels (VDCCs).2 To maintain the tight spatio-temporal distribution of intracellular Ca2+ necessary to mediate a diverse array of functions, VDCCs have evolved with respect to their biophysical properties, regulation, and subcellular localization (8). The physiological importance of VDCCs is further exemplified by the variety of mutations in VDCC channel components linked to neurological and motor diseases (9). VDCCs have been classified into five groups, identified as T, L, N, P/Q, and R, based on electrophysiological and pharmacological properties (9–12). N- and P/Q-type currents are observed primarily in neurons and co-localize with a subset of docked vesicles at the synapse, where they control exocytosis (13–15) and initiate synaptic neurotransmission (3, 4, 16). VDCCs are heteromultimers composed of the pore-forming α1 subunit and associated auxiliary subunits, Cavβ and α2δ (for review, see Ref. 4). Although auxiliary subunits have been demonstrated to enhance α1B expression (17, 18), the mechanisms underlying Cav channel membrane expression and localization are not clearly defined.
Ankyrins are a class of membrane adaptor proteins necessary for the targeting and regulation of select membrane and cytoplasmic proteins (19). In the brain, ankyrin-G is a necessary component for the functional organization of initial segments and nodes of Ranvier. Targeted disruption of cerebellar ankyrin-G results in severe ataxia, loss of ability to fire action potentials, and loss of Nav1.6 from the initial segment (20, 21). Additionally, there is loss of nodal βIV spectrin and KCNQ2/3 channels with indiscriminate localization of the cell adhesion molecule neurofascin (21, 22). Loss of neurofascin expression at the initial segment of Purkinje neurons results in disruption of interneuron synapses connecting Purkinje neurons in the cerebellum (23). Likewise, ankyrin-B has been demonstrated to play a critical role in the establishment and maintenance of excitable domains in brain (24). Loss of ankyrin-B in mice results in significant nervous system defects, including hypoplasia of the corpus callosum and pyramidal tracts, dilation of the lateral ventricles, and deterioration of long axon tracts (25). Notably, recent work outside of the brain has defined new roles for ankyrin-B for targeting select voltage-gated Ca2+ channels in the pacemaking cells of the cardiac sinoatrial node (26, 27). We therefore tested the role of ankyrin-B for targeting select calcium channels in the brain.
Our findings define a role for ankyrin-B in neuronal cell biology by demonstrating ankyrin-B-dependent interactions that are required for proper Cav2.1 and Cav2.2 expression in brain. We observed a significant decrease in Cav2.1 and Cav2.2 expression in cortex, cerebellum, and brain stem tissues of ankyrin-B-deficient mice. In defining the structural requirement for this interaction, we demonstrate that the ankyrin-B membrane-binding domain associates directly with Cav2.1 and Cav2.2. Specifically, this interaction occurs in the DII/III loop region of both VDCCs. Finally, a single amino acid mutation at residue Tyr-797 in Cav2.1 and Tyr-788 in Cav2.2 is sufficient to disrupt association of ankyrin-B with Cav2.1 or Cav2.2. Collectively, our data demonstrate a necessary role for ankyrin-B in the expression of Cav2.1 and Cav2.2 in brain.
EXPERIMENTAL PROCEDURES
Animals
Mice were age-matched wild-type, ankyrin-B+/−, and ankyrin-G cerebellar-specific knock-out littermates. Animals were backcrossed at least 20 generations (99.8% pure) into the C57BL/6 background (Jackson Laboratories). Ankyrin-G cerebellar-specific knock-out mice were generated as described (25). Animals were handled according to approved protocols and animal welfare regulations of the Institutional Animal Care and Use Committee at Ohio State University. All mice were housed and handled identically.
Antibodies
The following primary antibodies were used for immunoblotting and/or immunostaining: anti-ankyrin-B (polyclonal), anti-ankyrin-G (polyclonal), anti-Cav2.2 (polyclonal; Thermo Scientific and Alomone Labs), anti-Cav2.1 (polyclonal; Alomone Labs); anti-GAPDH (monoclonal; Fitzgerald Industries International); anti-GFP (NeuroMabs). Secondary antibodies used for immunoblotting were HRP-conjugated donkey anti-rabbit (Jackson Laboratories) and HRP-conjugated donkey anti-mouse (Jackson Laboratories). Secondary antibody used for immunostaining were Alexa Fluor 568 goat anti-rabbit Ig (Invitrogen). Cav2.1 and Cav2.2 antibodies were validated using immunoblots from HEK293 cells transfected with either GFP-Cav2.1 or GFP-Cav2.2. Cav2.1 Ig specifically recognized GFP-Cav2.1, but not GFP-Cav2.2. Conversely, Cav2.2 Ig specifically recognized GFP-Cav2.2, but not GFP-Cav2.1.
Cav2.1 and Cav2.2 Constructs
Primers were designed to clone amino acids 708–1194 (Cav2.1 DII/DIII loop), 788–1194, and 808–1194 from rat full-length Cav2.1 cDNA based on the sequence obtained from GenBank (NM_012918.3). To facilitate subcloning into pcDNA3.1+, primers were designed to incorporate 5′ EcoRI and 3′ XhoI restriction sites. To facilitate subcloning into pEGFP-C3, primers were designed to incorporate 5′ EcoRI and 3′ BamHI restriction sites. Full-length Cav2.1 constructs were generated from rat Cav2.1. To facilitate subcloning into pAcGFP1-N2, primers were designed to incorporate 5′ HindIII and 3′ SalI restriction sites. Primers were designed to clone amino acids 710–1139 (Cav2.2 DII/III loop), 779–1139, and 799–1139 from a murine brain cDNA library based on the sequence obtained from GenBank (NM_001042528). To facilitate subcloning into pcDNA3.1+ (Invitrogen), primers were designed to incorporate 5′ EcoRI and 3′ XhoI restriction sites. To facilitate subcloning into pEGFP-C3 (Clontech), primers were designed to incorporate 5′ EcoRI and 3′ BamHI restriction sites. Full-length Cav2.2 constructs were generated from rat Cav2.2. To facilitate subcloning into pAcGFP1-N2, primers were designed to incorporate 5′ SalI and 3′ KpnI restriction sites. All sequences were verified prior to experiments.
Site-directed Mutagenesis
Primers designed to insert the Y788E mutation into the Cav2.2 DII/III loop in pcDNA3.1+ and pEGFP-C3 and the full-length Cav2.2 in pAcGFP1-N2 or the Y797E in Cav2.1 DII/III loop in pcDNA3.1+ and pEGFPC3 and the full-length Cav2.1 in pAcGFP1-N2 were used in concert with the Stratagene QuikChange Site-directed Mutagenesis kit and manufacturer's instructions. All sequences were verified prior to experiments.
In Vitro Translation/Binding
Cav2.1 and Cav2.2 constructs were in vitro translated using rabbit reticulocyte lysate, [35S]methionine (20 μCi of Redivue l-[35S]methionine; GE Healthcare), T7 polymerase, and 0.63 ng of plasmid DNA (TNT Coupled Rabbit Reticulocyte Lysate System; Promega). For binding experiments, 20 μg of purified GST, ankyrin-B membrane-binding domain (MBD)-GST, ankyrin-B spectrin-binding domain (SBD)-GST, or ankyrin-B C-terminal domain (CTD)-GST was coupled to glutathione-Sepharose for 2 h at 4 °C in IV Binding Buffer (50 mm Tris-HCl (pH 7.35), 1 mm EDTA, 1 mm EGTA, 150 mm NaCl, 0.1% Triton X-100). Following extensive washing in IV Wash Buffer (50 mm Tris-HCl (pH 7.35), 1 mm EDTA, 1 mm EGTA, 350 mm NaCl, 0.1% Triton X-100), conjugated beads were incubated with Cav2.1 or Cav2.2 in vitro translated products overnight at 4 °C in IV Wash Buffer. The beads were washed five times in IV Wash Buffer, eluted, and separated by SDS-PAGE. Gels were stained with Coomassie Blue to show the presence of GST proteins before drying the gel (Bio-Rad Laboratories). Radiolabeled proteins were detected by phosphorimaging or standard autoradiography.
Tissue Preparation and Homogenization
For immunoblot and co-immunoprecipitation (co-IP) analysis, brain tissues (cortex, cerebellum, and brain stem) were flash-frozen in liquid nitrogen and ground into a fine powder. The powder was resuspended in 2 volumes of ice-cold homogenization buffer (50 mm Tris-HCl (pH 7.35), 10 mm NaCl, 0.32 m sucrose, 5 mm EDTA, 2.5 mm EGTA, 1 mm PMSF, 1 mm AEBSF, 10 μg/ml leupeptin, and 10 μg/ml pepstatin) and homogenized using a hand-held homogenizer (27, 28). The homogenate was centrifuged at 1000 × g at 4 °C to remove nuclei. Triton X-100 and deoxycholate were added to the postnuclear supernatant for final concentrations of 0.75% Triton X-100 and 1% deoxycholate. The lysate was pelleted at high speed for 15 min at 4 °C. The resulting supernatant was quantitated by bicinchoninic acid assay prior to analysis.
Immunoblots
Immunoblots from anti-ankyrin-B, anti-Cav2.1, anti-Cav2.2, and anti-ankyrin-G blots were evaluated by densitometry and expression normalized to anti-GAPDH blots (29, 30). Histograms represent expression as a percentage of wild-type expression (wild-type expression normalized to 100%).
Tissue Preparation for Immunostaining
Freshly extracted tissues from wild-type and ankyrin-B+/− mice were fixed in 4% paraformaldehyde for 12 h and the tissues transferred to 10, 20, and 30% sucrose solutions for 12 h each. Tissues were cryosectioned to 10-μm thickness. Cryosections were rehydrated in PBS prior to preblocking in 3 mg/ml BSA in PBS. Primary antibodies were made in a vehicle of 3 mg/ml BSA with 0.1% Triton X-100 in PBS and incubated on sections overnight at 4 °C. The slides were washed three times in vehicle and incubated with Alexa Fluor donkey anti-rabbit 568 secondary antibodies for 3 h at 4 °C. Following three washes in vehicle, the slides were mounted with VectaShield (Vector Laboratories) and no. 1 coverslips. Images were collected on a Zeiss 510 Meta confocal microscopy using Carl Zeiss software.
Co-IP from Brain Lysates
Protein A-conjugated agarose beads (AffiGel; Bio-Rad) were incubated with either control Ig or anti-Cav2.2 Ig, anti-Cav2.1 Ig, or anti-ankyrin-B Ig in co-IP binding buffer (PBS with 0.1% Triton X-100 and protease inhibitor mixture (Sigma)) for 12 h at 4 °C. Beads were centrifuged and washed three times in ice-cold PBS. Wild-type cortex, cerebellum, or brain stem tissue lysate were added to the washed beads, along with protease inhibitor mixture and co-IP binding buffer, and incubated for 12 h at 4 °C. The reactions were washed three times in ice-cold co-IP buffer. The samples were eluted and the proteins separated by SDS-PAGE prior to immunoblots with ankyrin-B, Cav2.1, or Cav2.2 Ig. Experiments were performed multiple times with similar results. Due to the low copy number of Cav2.1 and Cav2.2 in brain regions, lysate inputs were scaled up. For experiments where Cav2.1 or Cav2.2 Igs were immobilized on beads, 1 mg of cortex and cerebellum lysate was used, whereas 2 mg of brain stem lysate was used. Input lanes represent 10% of total lysate used. For experiments where ankyrin-B Ig was immobilized on beads, 1 mg of lysate for each brain region was utilized. Input lanes represent 5% of total lysate for cortex and cerebellum and 10% of total lysate for brain stem.
Co-IP from Transfected Cells
Protein A-conjugated agarose beads were incubated with either control IgG or affinity-purified anti-ankyrin-B Ig for 12 h at 4 °C. Beads were centrifuged and washed three times in ice-cold PBS. 100 μg of transfected HEK lysate was added to the washed beads, along with protease inhibitor mixture and IP buffer, and incubated for 12 h at 4 °C. The reactions were washed three times in ice-cold IP buffer. The samples were eluted and the proteins separated by SDS-PAGE prior to immunoblots with anti-GFP Ig. For co-IP using full-length calcium channel-transfected cells, input lanes represent 20% of total lysate input (400 μg). For GFP-Cav channel loop transfections, input lanes represent 10% of total lysate input (200 μg).
Pulldowns
One hundred micrograms of wild-type cortex, cerebellum, or brain stem lysate was added to 10 μg of GST, GST-ankyrin-B MBD, GST-ankyrin-B SBD, or GST-ankyrin-B CTD-conjugated beads, along with 500 μl of Binding Buffer, protease inhibitor mixture (Sigma), and PMSF. The reactions incubated for 12 h at 4 °C. The beads were centrifuged and washed three times in binding buffer. The proteins were eluted, separated by SDS-PAGE, and immunoblotted using anti-Cav2.1 Ig and anti-Cav2.2 Ig.
HEK293 Cell Culture and Transfection of Cav2.1 and Cav2.2 Fragments
HEK293 cells were maintained in Dulbecco's modified essential medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS; HyClone) and 0.1% penicillin/streptomycin. Cells were cultured at 37 °C in 5% CO2. Cultured cells were split 24 h prior to transfection and plated to obtain 30% confluence at time of transfection. Effectene reagent (Qiagen) was used to transfect cells with 0.2 μg Cav2.2 DII/III pEGFP-C3, Cav2.2 DII/III Y788E pEGFP-C3, or pEGFP-C3 alone. Transfection was carried out for 7 h at 37 °C and the cells recovered overnight. After 48 h, the cells were fixed with 2% paraformaldehyde, stained with ToPro (to discriminate nuclear material), and evaluated by confocal microscopy. Likewise, transfections using Cav2.1 DII/III pEGFP-C3, Cav2.1 DII/III Y797E pEGFP-C3, and pEGFP-C3 alone were also performed using the same protocol.
HEK293 Cell Culture and Transfection for Confocal Microscopy
HEK293 cells were maintained in Dulbecco's modified essential medium supplemented with 10% FBS and 0.1% penicillin/streptomycin. Cells were cultured at 37 °C in 5% CO2. Cultured cells were split 24 h prior to transfection and plated on MatTek plates to obtain 90% confluence at time of transfection. Lipofectamine (Invitrogen) was used to transfect cells with 1 μg of Cav2.2-GFP, 1 μg of Cav2.2 Y788E-GFP, 1 μg of Cav2.1-GFP, 1 μg of Cav2.1 Y797E-GFP, or pAcGFP1-N2 alone. Transfection was carried out for 5 h at 37 °C and the cells recovered overnight. After 24 h, the cells were fixed with 2% paraformaldehyde, mounted with VectaShield (with DAPI), and evaluated by confocal microscopy.
HEK293 Cell Culture and Transfection for Co-immunoprecipitations
HEK293 cells were maintained in Dulbecco's modified essential medium supplemented with 10% FBS and 0.1% penicillin/streptomycin. Cells were cultured at 37 °C in 5% CO2. Cultured cells were split 24 h prior to transfection and plated on 100-mm2 plates to obtain 90% confluence at time of transfection. Lipofectamine (Invitrogen) was used to transfect cells with 1 μg of Cav2.2-GFP, 1 μg of Cav2.2 Y788E-GFP, 1 μg of Cav2.1-GFP, 1 μg of Cav2.1 Y797E-GFP, or pAcGFP1-N2 alone. Untransfected cells were used as a negative control. Transfection was carried out for 5 h at 37 °C and the cells recovered overnight. After 24 h, the cells were lysed in IP buffer (25 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 5% glycerol, protease inhibitor mixture (Sigma)) and centrifuged to remove cellular debris, and the protein was quantitated by bicinchoninic acid assay (Pierce).
Statistics
Data are presented as a mean ± S.D., and statistical significance was assessed using Student's t test. The null hypothesis was rejected for p < 0.05.
RESULTS
Cav2.1 and Cav2.2 Expression Is Reduced in Ankyrin-B+/− Brain
Ankyrins are key players for targeting ion channels in excitable cells. Recent work defined new roles for ankyrin-B for targeting select voltage-gated Ca2+ channels in the pacemaking cells of the cardiac sinoatrial node (26, 27). We therefore tested the role of ankyrin-B for targeting select calcium channels in the nervous system. Specifically, we investigated Cav2.1 and Cav2.2 subunit expression in ankyrin-B+/− cortex, cerebellum, and brain stem. As expected, ankyrin-B expression was significantly reduced in ankyrin-B+/− cortex, cerebellum, and brain stem lysates (Fig. 1A; decreased 53, 52, and 51%, respectively; n = 4, p < 0.0001), whereas the expression levels of the related ankyrin gene product, ankyrin-G (Fig. 1B), and GAPDH (loading control; Fig. 1E) were unchanged. We observed decreased Cav2.1 expression in cortex (41%), cerebellum (20%), and brain stem (35%) in ankyrin-B+/−-derived tissues compared with wild-type lysates (Fig. 1C; n = 4, p < 0.05). Cav2.2 expression was also reduced in ankyrin-B+/− cortex (49%), cerebellar (40%), and brain stem tissue (50%) compared with lysates from wild-type littermates (Fig. 1D; n = 4, p < 0.05).
FIGURE 1.

Cav2.1 and Cav2.2 expression levels are decreased in ankyrin-B+/− brain lysates. Ankyrin-B, Cav2.1, and Cav2.2 expression levels are significantly decreased in cortex, cerebellum, and brain stem in ankyrin-B+/− mice compared with wild-type mice (A, C, and D, respectively). There are no significant differences in expression between wild-type and ankyrin-B+/− lysates of ankyrin-G (B) or GAPDH (E, loading control). For each panel, n = 4 for each genotype (p < 0.05).
Based on our immunoblot findings, we performed confocal analysis for the localization of Cav2.1 and Cav2.2 in wild-type and ankyrin-B+/− mouse cerebellum. In the cerebellum, Cav2.1 and Cav2.2 are expressed in Purkinje neurons and localized in the cell body (31, 32). Ankyrin-B is also expressed in the cerebellar Purkinje neuron cell body, as well as throughout the molecular layer (33). As shown in Fig. 2, whereas ankyrin-B, Cav2.1, and Cav2.2 are robustly expressed in Purkinje neurons from wild-type cerebellum, we observed a significant decrease of both Cav2.1 and Cav2.2 expression in response to the reduction in ankyrin-B expression (Fig. 2).
FIGURE 2.
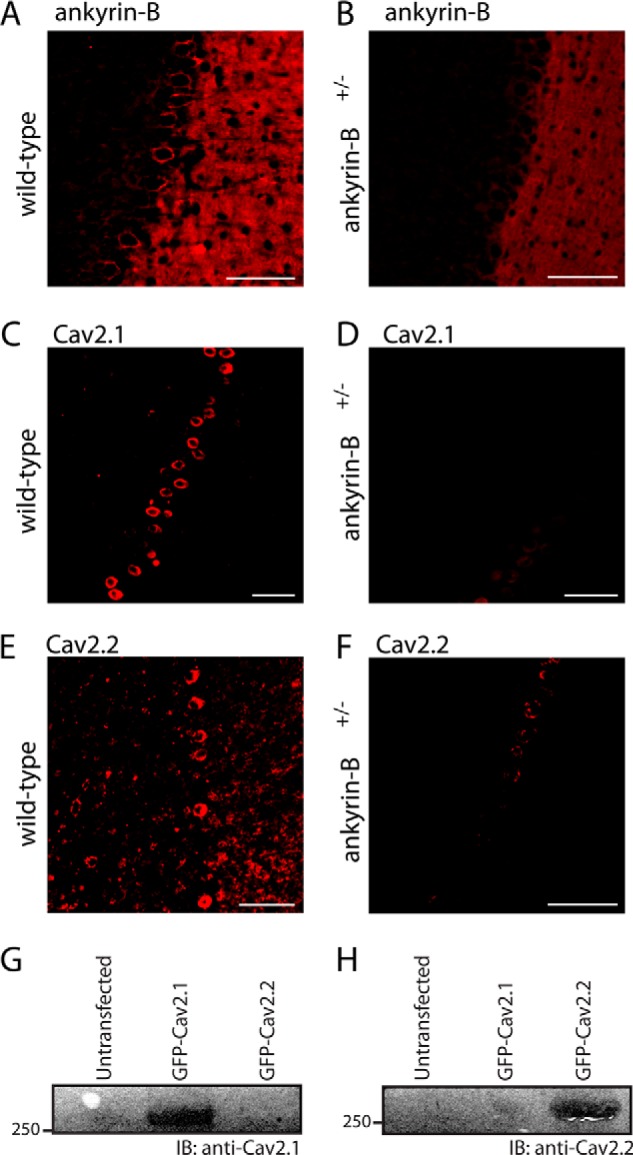
Ankyrin-B+/− mice display a significant decrease in ankyrin-B, Cav2. 1, and Cav2.2 expression in cerebellar Purkinje neurons. A and B, ankyrin-B expression is significantly reduced in ankyrin-B+/− Purkinje neurons. Note that ankyrin-B is expressed in both Purkinje cell layer and molecular layer. C–F, Cav2.1 (C and D) and Cav2.2 (E and F) expression is significantly reduced in the Purkinje neurons of ankyrin-B+/− mice. Scale bar denotes 20 μm. G and H, Cav2.1 and Cav2.2 antibodies are validated. Untransfected HEK293 cells and HEK293 cells transfected with either full-length GFP-Cav2.1 or GFP-Cav2.2 were analyzed by co-immunoprecipitation or immunoblotting (IB). In G, Cav2.1 Ig recognizes GFP-Cav2.1 but not GFP-Cav2.2. In H, Cav2.2 Ig recognizes GFP-Cav2.2 but not GFP-Cav2.1.
Ankyrin-G Is Not Required for Cav2.1 or Cav2.2 Expression
Although encoded from different genes, ankyrin-B and ankyrin-G display similar domain organization, and both target ion channels and transporters in diverse excitable cell types (2). As both ankyrin-B and ankyrin-G are co-expressed in cell types harboring Cav2.1 and Cav2.2, we evaluated whether ankyrin-G is required for Cav2.1 and Cav2.2 expression as shown for ankyrin-B (Figs. 1 and 2). In contrast to findings for ankyrin-B-deficient tissue, we observed no difference in Cav2.1 or Cav2.2 expression in cerebellar lysates generated from a well characterized mouse model homozygous for a null mutation in cerebellar ankyrin-G (Fig. 3 and Ref. 20). These data demonstrate that ankyrin-B, but not ankyrin-G, is required for regulating Cav2.1 and Cav2.2 expression in the cerebellum. Moreover, these data suggest that ankyrins-G and -B play nonredundant roles in calcium channel targeting in brain.
FIGURE 3.
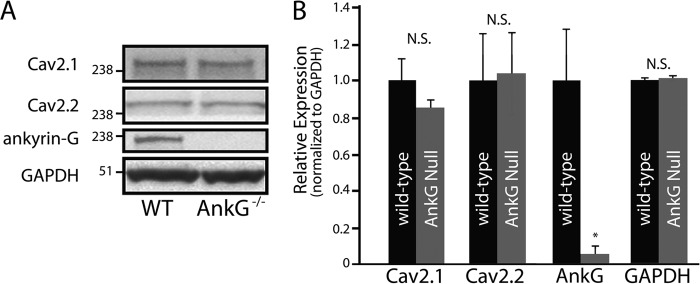
Ankyrin-G is not required for Cav2.1 or Cav2.2 expression. Although ankyrin-G cerebellar-specific knock-out mice display a significant reduction in the expression of ankyrin-G (A), there are no significant differences in the expression of Cav2.1 or Cav2.2 between wild-type and ankyrin-G cerebellar-specific knock-out cerebellar lysates (A and B, n = 4; N.S.). Ankyrin-G levels by immunoblotting in ankyrin-G cerebellar-specific knock-out mice were significantly reduced and similar to background.
Ankyrin-B Associates with Cav2.1 and Cav2.2 in Brain
Considering that Cav2.1 and Cav2.2 expression levels were significantly reduced in ankyrin-B+/− brain tissue (Figs. 1 and 2), we evaluated the ability of ankyrin-B to associate with Cav2.1 and Cav2.2 in brain. Co-IP analysis using detergent-soluble lysates from adult mouse cortex, cerebellum, and brain stem demonstrated that affinity-purified anti-Cav2.1 Ig co-immunoprecipitated ankyrin-B from cortex (Fig. 4A), cerebellum (Fig. 4B), and brain stem (Fig. 4C). Likewise, anti-Cav2.2 Ig co-immunoprecipitated ankyrin-B from cortex, cerebellum, and brain stem lysates (Fig. 4, D–F). We observed no interaction between ankyrin-B and control Ig in any tissue lysate (Fig. 4, A–F). To confirm this interaction, we performed reciprocal co-immunoprecipitations utilizing affinity-purified ankyrin-B Ig. Ankyrin-B Ig co-immunoprecipitated Cav2.1 and Cav2.2 from detergent-soluble lysates from cortex, cerebellum, and brain stem, whereas we observed no interaction with a control Ig (Fig. 4, G–L). Collectively, these data support an in vivo interaction between ankyrin-B and Cav2.1, as well as an in vivo interaction between ankyrin-B and Cav2.2, in mouse cortex, cerebellum, and brain stem.
FIGURE 4.
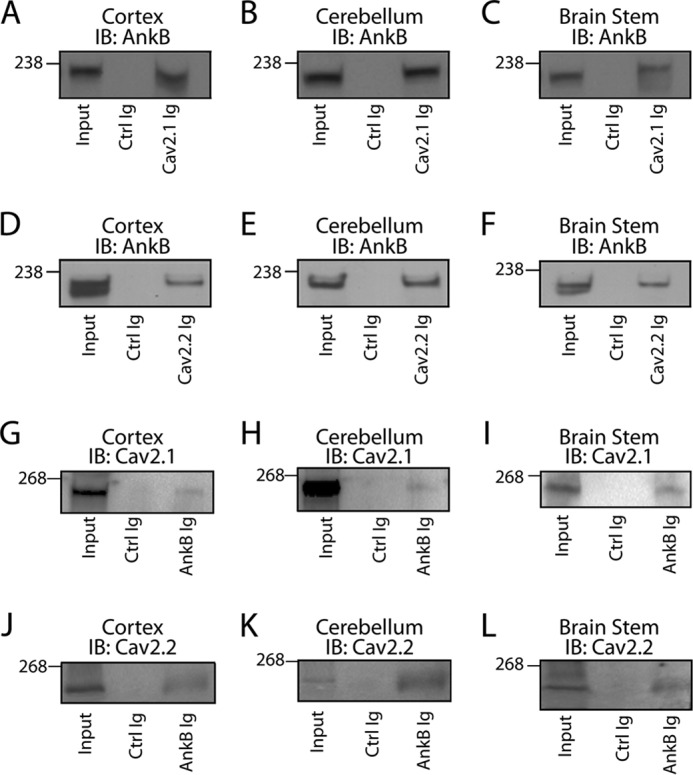
Ankyrin-B co-immunoprecipitates with Cav2.1 and Cav2.2. A–F, Cav2.1 Ig and Cav2.2 Ig co-immunoprecipitate ankyrin-B from detergent-soluble lysates of cortex (A and D), cerebellum (B and E), and brain stem (C and F), respectively. No significant interaction was observed with control (Ctrl) Ig. G–L, ankyrin-G Ig co-immunoprecipitates Cav2.1 and Cav2.2 from detergent-soluble lysates of cortex (G and J), cerebellum (H and K), and brain stem (I and L), respectively. No significant interaction was observed with control Ig. IB, immunoblotting.
We further evaluated the neuronal ankyrin-B/Cav2.1 and ankyrin-B/Cav2.2 interactions using pulldown experiments from mouse detergent-soluble cortex, cerebellum, and brain stem lysates. Ankyrin-B comprises three distinct structural domains: MBD, SBD, and CTD (Fig. 5A). Of note, GST-ankyrin-B MBD, but neither GST-ankyrin-B SBD or GST-ankyrin-B CTD interacted with Cav2.1 nor Cav2.2 from cortex (Fig. 5B), cerebellum (Fig. 5C), and brain stem (Fig. 5D; fusion proteins are shown in Fig. 5E). Collectively, these data demonstrate an interaction between Cav2.1 and Cav2.2 and ankyrin-B within multiple regions of the brain. These data further identify the structural requirements on ankyrin-B for Cav channel binding as the ANK repeats of the membrane-binding domain.
FIGURE 5.

Ankyrin-B associates with Cav2.1 and Cav2.2. A, ankyrin-B comprises a membrane-binding domain, spectrin-binding domain, and a C-terminal regulatory domain. B–D, GST-ankyrin-B MBD associates with both Cav2.1 and Cav2.2 from detergent-soluble lysates of cortex (B), cerebellum (C), and brain stem (D). No interaction was observed between lysates and GST-ankyrin-B SBD, GST-ankyrin-B CTD, or GST alone. E, GST fusion proteins were utilized for pull-down experiments. Fusion proteins were purified from bacteria and analyzed by Coomassie Blue staining. Whereas GST migrates at 29 kDa, GST-ankyrin-B MBD migrates at 94 kDa. GST-ankyrin-B SBD migrates at 62 kDa, and GST-ankyrin-B CTD migrates at 79 kDa.
Ankyrin-B Interacts with the Cav2.1 and Cav2.2 DII/III Loops
Next, we evaluated the requirements on Cav2.1 and Cav2.2 for neuronal ankyrin-B binding. Previous studies have suggested that a region in the DII/III loop region of Cav2.2 contains a region postulated to play a role in the targeting of calcium channels (34) (Fig. 6, A and B). This DII/III region contains a 20-amino acid motif that is highly conserved among species (Fig. 6, C and D). Three constructs based on Cav2.1 and Cav2.2 sequences were designed to evaluate the role of this motif in ankyrin-B/Cav2.1 and ankyrin-B/Cav2.2 association (Fig. 6, E and F). The first construct encompassed the entire DII/III loop (Cav2.1 amino acids 708–1194; Cav2.2 amino acids 710–1139); the second initiated proximal to the putative ankyrin-binding motif (ABM; Cav2.1 amino acids 788–1194; Cav2.2 amino acids 779–1139); and the third began distal to the ABM (Cav2.1 amino acids 808–1194; Cav2.2 amino acids 799–1139). In vitro binding with immobilized GST-ankyrin-B MBD revealed that the full-length Cav2.1 DII/III loop and the Cav2.1 construct containing residues 788–1194 displayed binding activity for GST-ankyrin-B MBD (Fig. 6G). In contrast, the Cav2.1 construct lacking residues 788–808 (construct 808–1194) had binding activity similar to GST alone (Fig. 6G). Similarly, full-length Cav2.2 DII/III loop and the Cav2.2 construct containing residues 779–799 (construct 779–1139) displayed binding activity for GST-ankyrin-B MBD (Fig. 6H). The Cav2.2 construct lacking residues 779–799 (construct 799–1139) had binding activity similar to GST alone (Fig. 6H). These data identify a 20-amino acid motif in both the Cav2.1 and Cav2.2 DII/III loops that is necessary for interaction with ankyrin-B.
FIGURE 6.

Cav2.1 and Cav2.2 DII/DIII motif associates with ankyrin-B. A and B, location of putative ABM in the DII/DIII cytoplasmic loop of Cav2.1 and Cav2.2 is shown. C and D, the putative ABM in Cav2.1 and Cav2.2 is conserved across species and harbors a central conserved tyrosine (bolded). E and F, three constructs were designed to evaluate the role of this amino acid sequence in ankyrin-B binding. These constructs include the entire DII/III loop, a portion of the loop that begins just prior to the putative ABM, and a portion of the loop that begins just after the putative ABM. G and H, in vitro binding assays demonstrate that, whereas the full-length DII/III loops of Cav2.1 and Cav2.2 and the smaller loop portion that contains the putative ABM are able to associate with GST-ankyrin-B MBD, the smaller loop portion that lacks this domain demonstrates no binding with GST-ankyrin-B MBD. I and J, Coomassie Blue staining demonstrating that equal loading of GST constructs was performed prior to imaging (denoted by asterisks).
A Conserved Tyrosine Residue Regulates Association with Ankyrin-B
Although inhibition of calcium channels is voltage-dependent, it is mediated by direct interaction of G protein βγ subunits with the α1 pore-forming subunit of the channel (35, 36). Additionally, phosphorylation by tyrosine kinases has been shown to inhibit calcium channels (37) with G protein-dependent inhibition of calcium current due in part to a decrease in the open probability of the channel, resulting in reduced current density (38–40). We had previously defined a 20-amino acid motif in the DII/III loops of Cav2.1 and Cav2.2 that associates with ankyrin-B (Fig. 6). Notably, this motif contains a conserved tyrosine residue. To evaluate the role of this tyrosine in the regulation of Cav2.1 and Cav2.2 association with ankyrin-B, we incorporated a Y797E mutation in the Cav2.1 DII/III loop and evaluated the effect on in vitro binding (Fig. 7A). Mutagenesis of the Tyr-797 residue to a glutamic acid resulted in loss of association with GST-ankyrin-B MBD by in vitro binding analysis (Fig. 7B). Mutation of the analogous residue, Tyr-788, in the Cav2.2 DII/III to a glutamic acid (Fig. 7D) demonstrated a similar loss of in vitro binding with GST-ankyrin-B MBD (Fig. 7E).
FIGURE 7.
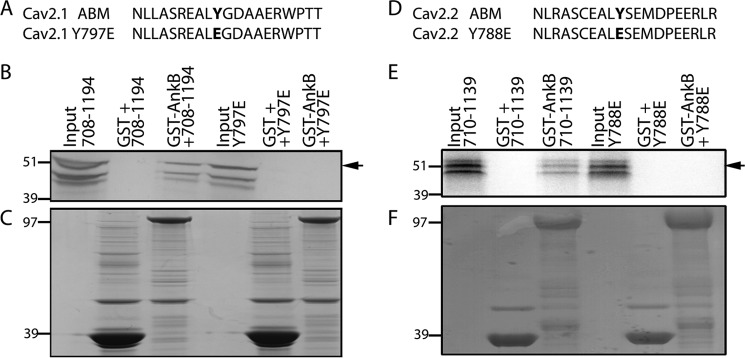
A conserved tyrosine residue in the Cav2. 1 and Cav2.2 DII/III loop is necessary for ankyrin-B interaction in vitro. A and D, Cav2.1 and Cav2.2 DII/III loop domains contain a conserved tyrosine residue that was experimentally mutated to a glutamic acid to assess ankyrin-B binding. B, GST-ankyrin-B MBD was able to associate with the Cav2.1 DII/III loop domain. In contrast, there was a loss of interaction with the domain containing the Y797E mutation. There was no appreciable binding with GST alone. E, Cav2.2 DII/III loop domain associated with GST-ankyrin-B MBD is shown. However, there was no appreciable association between GST-ankyrin-B MBD and the Cav2.2 DII/III Y788E. There was no detectable binding to GST alone. C and F, Coomassie Blue stains demonstrate that equal loading of GST constructs was performed prior to imaging.
DII/DIII Tyrosine Residues Are Critical for Cav2.1/Cav2.2 Targeting
Ankyrin proteins are critical for targeting ion channels and transporters in diverse cell types including neurons. In fact, ion channel or transporter ankyrin-binding motifs alone are sufficient to target GFP and other markers in heterologous cells and primary cultures (41). We therefore tested (i) whether the DII/DIII loops of Cav2.1 and Cav2.2 (that harbor ankyrin-binding motifs) were sufficient to behave as a dominant target signal in HEK293 cells, and (ii) whether the single tyrosine mutation in each channel altered this trafficking pattern. DII/DIII loops from Cav2.1 and Cav2.2 were subcloned in-frame into the pEGFP-C3 expression vector and transfected into HEK293 cells (express endogenous ankyrin-B). Confocal analysis revealed that unlike GFP that was exclusively found in the HEK293 cell nucleus (Fig. 8, A and D), GFP-Cav2.1 DII/III loop and GFP-Cav2.2 DII/III were in fact targeted outside of the nucleus (versus GFP) and had a diffuse pattern of expression throughout the cell (Fig. 8, B and E). Consistent with these findings, both GFP-Cav2.1 DII/III loop and GFP-Cav2.2 DII/III loop interacted with ankyrin-B in parallel co-IP experiments from transfected cells (Fig. 8, G–I). In contrast, GFP-Cav2.1 DII/III Y797E and GFP-Cav2.2 DII/III Y788E lacked ankyrin-B-binding activity (Fig. 8, G–I) and were restricted in localization to a tight and consistent perinuclear distribution (Fig. 8, C and F). These data demonstrate that the Cav2.1 and Cav2.2 DII/DIII loops harbor dominant targeting motifs that divert them from the nucleus. Further, these data implicate the Tyr-797 in the Cav2.1 and the Tyr-788 residue in the Cav2.2 as critical components in the regulation of calcium channel targeting.
FIGURE 8.

Cav2.1/Cav2.2 tyrosine residues in ABM are critical for cellular targeting. HEK293 cells transfected with pEGFP-C3 demonstrated a primarily nuclear localization pattern (A and D), whereas HEK293 cells transfected with Cav2.1 DII/III-GFP (B) or Cav2.2 DII/III-GFP (E) displayed a diffuse cellular pattern of expression unique from the nucleus. C and F, unlike WT constructs, both Cav2.1 Y797E-GFP and Cav2.2 Y788E-GFP were distributed in a tight perinuclear pattern. Scale bars denote 8 μm. G–I, a conserved tyrosine residue in the Cav2.1 and Cav2.2 DII/III loop is necessary for ankyrin-B interaction in vitro. G, in control experiments, ankyrin-B Ig did not interact with GFP alone expressed in HEK293 cells. H and I, ankyrin-B Ig co-immunoprecipitates GFP fusion protein containing the Cav2.1 or Cav2.2 DII/DII loop from transfected HEK293 cells. In contrast, ankyrin-B Ig does not co-immunoprecipitate GFP fusion proteins of Cav2.1 DII/DIII Y797E or Cav2.2 DII/III Y788E in parallel experiments. IB, immunoblotting.
We confirmed these findings in the context of full-length Cav2.1 and Cav2.2. We observed both GFP-Cav2.1 and GFP-Cav2.2 with diffuse expression across transfected HEK293 cells, whereas GFP-Cav2.1 Y797E and full-length GFP-Cav2.2 Y788E displayed abnormal targeting with a clustered perinuclear distribution (Fig. 9, A–E). Consistent with these findings, whereas GFP-Cav2.1 and GFP-Cav2.2 associated with ankyrin-B in co-IP experiments, ankyrin-B Ig was unable to co-immunoprecipitate either full-length GFP-Cav2.1 Y797E or full-length GFP-Cav2.2 Y788E from HEK293 cell lysates (Fig. 9, F–H). These results further support a critical role for ankyrin-B-mediated Cav2.1 and Cav2.2 expression and targeting.
FIGURE 9.
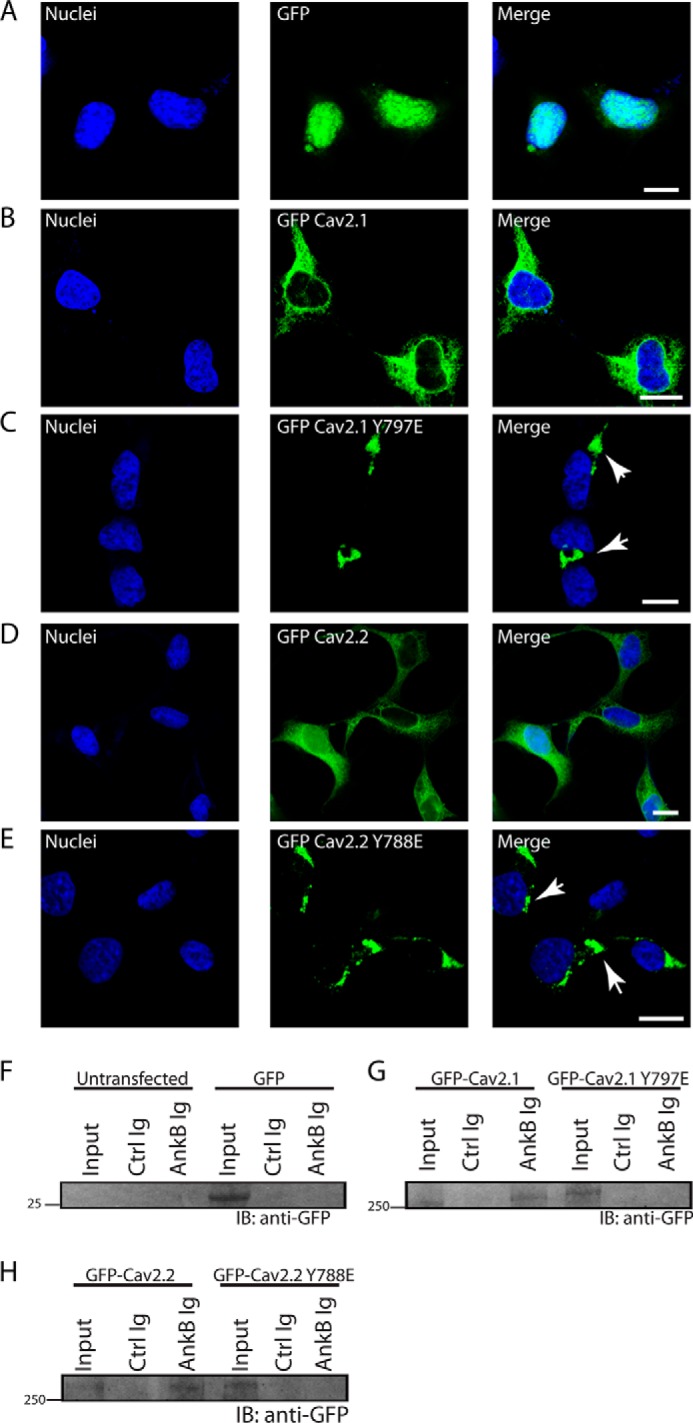
Cav2. 1/Cav2.2 tyrosine residues in ABM are critical for cellular targeting and ankyrin interaction. A, HEK293 cells transfected with pEGFP-C3 demonstrated a primarily nuclear localization pattern. B and D, in contrast, HEK293 cells transfected with full-length GFP-Cav2.1 (B) or GFP-Cav2.2 (D) displayed a diffuse cellular pattern of expression unique from the nucleus or perinuclear region. C and E, unlike WT Cav2.1 and Cav2.2 constructs, both full-length GFP-Cav2.1 Y797E and full-length GFP-Cav2.2 Y788E-GFP were distributed in a tight perinuclear pattern (white arrowheads). Scale bars denote 8 μm. F, control experiments demonstrate that ankyrin-B Ig does not associate with GFP alone. G, ankyrin-B co-immunoprecipitates full-length GFP-Cav2.1, but not full-length GFP-Cav2.1 Y797E from transfected HEK293 lysates. H, ankyrin-B co-immunoprecipitates full-length GFP-Cav2.2, but not full-length GFP-Cav2.2 Y788E from transfected HEK293 lysates.
DISCUSSION
Our findings demonstrate a role of ankyrin-B for Cav2.1 and Cav2.2 channel expression and targeting in brain. Loss of ankyrin-B results in decreased expression of Cav2.1 and Cav2.2 in cortex, cerebellum, and brain stem. Moreover, mutation of a single residue in the Cav2.1 and Cav2.2 DII/III loop results in loss of association with ankyrin-B and abnormal cellular targeting. Ankyrin-B has been demonstrated to play a critical role in the establishment and maintenance of excitable domains in brain. Loss of ankyrin-B in mice results in significant nervous system defects, including hypoplasia of the corpus callosum and pyramidal tracts, dilation of the lateral ventricles, and deterioration of long axon tracts (25). Specifically, these phenotypes are associated with loss of L1CAM proteins throughout the brain. Interestingly, mutations in the human L1CAM gene cause mental retardation as well as a collection of neurological symptoms referred to as CRASH syndrome (corpus callosum hypoplasia, retardation, aphasia, spastic paraplegia, and hydrocephalus) (42). The ankyrin-B−/− mouse model, although lethal at post-natal day 1, exhibits loss of L1CAM from axons and exhibits many of the CRASH symptoms, although with greater severity (25). No humans with null mutations in the ankyrin-B gene (ANK2) have been identified; however, given that heterozygote individuals with loss-of-function mutations in the ANK2 gene are present in the population (43) and are found in patients with cardiac arrhythmia phenotypes (26, 44–47), it is possible that rare homozygous or mixed heterozygotes may exhibit CRASH symptoms. S1224L and Y1229H mutations in the human L1CAM gene are associated with CRASH syndrome (48). Moreover, these mutations are located within the highly conserved ankyrin-binding site and abolish association of L1 with ankyrin-B (48).
Cav2.1 and Cav2.2 channel activity in brain is necessary for proper initiation of synaptic transmission. Cav2.2−/− animals demonstrate normal motor coordination; however, these mice exhibit reduced responses to pain stimuli (49) and elevated aggression (50). Cav2.1−/− animals demonstrate dystonia at postnatal day 10–12 and rarely survive past weaning (51). Likewise, Cav2.1−/− mice exhibit reduced responses to pain stimuli (52). As noted above, ankyrin-B−/− die at birth, therefore we currently lack data on the in vivo phenotypes of ankyrin-B deficiency in an adult mouse. Future studies utilizing inducible ankyrin-B knock-out strategies in the postnatal mouse will be important for understanding the role of this cytoskeletal protein in synaptic function.
Finally, although our data link ankyrin-B with regulation of targeting via the DII/DIII loop of both Cav2.1 and Cav2.2 via specific tyrosine residues, it is possible that other protein interactions in this loop may also play critical roles in ion channel processing and membrane regulation. For example, the Cav2.2 DII/DIII loop maintains a SNARE binding or synprint region that has been linked with Cav channel targeting (34). It will be important in future work to understand the potential dual regulation of Cav channel targeting by ankyrin and other proteins.
This work was supported, in whole or in part, by National Institutes of Health Grants HL084583, HL083422, and HL114383 (to P. J. M.) and HL096805 and HL114893 (to T. J. H.). This work was also supported by the American Heart Association (to P. J. M.), the Saving Tiny Hearts Society (to P. J. M.), and The Ohio State University William D. and Jacquelyn L. Wells Fund for Cardiovascular Research (to P. J. M.).
- VDCC
- voltage-dependent calcium channel
- ABM
- ankyrin-binding motif
- CTD
- C-terminal domain
- IP
- immunoprecipitation
- MBD
- membrane-binding domain
- SBD
- spectrin-binding domain.
REFERENCES
- 1. Cheever T. R., Ervasti J. M. (2013) Actin isoforms in neuronal development and function. Int. Rev. Cell Mol. Biol. 301, 157–213 [DOI] [PubMed] [Google Scholar]
- 2. Bennett V., Healy J. (2008) Organizing the fluid membrane bilayer: diseases linked to spectrin and ankyrin. Trends Mol. Med. 14, 28–36 [DOI] [PubMed] [Google Scholar]
- 3. Dunlap K., Luebke J. I., Turner T. J. (1995) Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 18, 89–98 [PubMed] [Google Scholar]
- 4. Catterall W. A. (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16, 521–555 [DOI] [PubMed] [Google Scholar]
- 5. Clapham D. E. (1995) Calcium signaling. Cell 80, 259–268 [DOI] [PubMed] [Google Scholar]
- 6. Snutch T. P., Reiner P. B. (1992) Ca2+ channels: diversity of form and function. Curr. Opin. Neurobiol. 2, 247–253 [DOI] [PubMed] [Google Scholar]
- 7. Tedford H. W., Zamponi G. W. (2006) Direct G protein modulation of Cav2 calcium channels. Pharmacol. Rev. 58, 837–862 [DOI] [PubMed] [Google Scholar]
- 8. Tsien R. W., Lipscombe D., Madison D. V., Bley K. R., Fox A. P. (1988) Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 11, 431–438 [DOI] [PubMed] [Google Scholar]
- 9. Miller R. J. (1997) Calcium channels prove to be a real headache. Trends Neurosci. 20, 189–192 [DOI] [PubMed] [Google Scholar]
- 10. Ellinor P. T., Zhang J. F., Randall A. D., Zhou M., Schwarz T. L., Tsien R. W., Horne W. A. (1993) Functional expression of a rapidly inactivating neuronal calcium channel. Nature 363, 455–458 [DOI] [PubMed] [Google Scholar]
- 11. Fox A. P., Nowycky M. C., Tsien R. W. (1987) Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurones. J. Physiol. 394, 149–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Usowicz M. M., Sugimori M., Cherksey B., Llinás R. (1992) P-type calcium channels in the somata and dendrites of adult cerebellar Purkinje cells. Neuron 9, 1185–1199 [DOI] [PubMed] [Google Scholar]
- 13. Wheeler D. B., Randall A., Tsien R. W. (1994) Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science 264, 107–111 [DOI] [PubMed] [Google Scholar]
- 14. Takahashi T., Momiyama A. (1993) Different types of calcium channels mediate central synaptic transmission. Nature 366, 156–158 [DOI] [PubMed] [Google Scholar]
- 15. Nooney J. M., Lodge D. (1996) The use of invertebrate peptide toxins to establish Ca2+ channel identity of CA3-CA1 neurotransmission in rat hippocampal slices. Eur. J. Pharmacol. 306, 41–50 [DOI] [PubMed] [Google Scholar]
- 16. Olivera B. M., Miljanich G. P., Ramachandran J., Adams M. E. (1994) Calcium channel diversity and neurotransmitter release: the ω-conotoxins and ω-agatoxins. Annu. Rev. Biochem. 63, 823–867 [DOI] [PubMed] [Google Scholar]
- 17. Leroy J., Richards M. S., Butcher A. J., Nieto-Rostro M., Pratt W. S., Davies A., Dolphin A. C. (2005) Interaction via a key tryptophan in the I-II linker of N-type calcium channels is required for β1 but not for palmitoylated β2, implicating an additional binding site in the regulation of channel voltage-dependent properties. J. Neurosci. 25, 6984–6996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Davies A., Hendrich J., Van Minh A. T., Wratten J., Douglas L., Dolphin A. C. (2007) Functional biology of the α2δ subunits of voltage-gated calcium channels. Trends Pharmacol. Sci. 28, 220–228 [DOI] [PubMed] [Google Scholar]
- 19. Bennett V., Baines A. J. (2001) Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol. Rev. 81, 1353–1392 [DOI] [PubMed] [Google Scholar]
- 20. Zhou D., Lambert S., Malen P. L., Carpenter S., Boland L. M., Bennett V. (1998) Ankyrin-G is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J. Cell Biol. 143, 1295–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jenkins S. M., Bennett V. (2001) Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J. Cell Biol. 155, 739–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pan Z., Kao T., Horvath Z., Lemos J., Sul J. Y., Cranstoun S. D., Bennett V., Scherer S. S., Cooper E. C. (2006) A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 26, 2599–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ango F., di Cristo G., Higashiyama H., Bennett V., Wu P., Huang Z. J. (2004) Ankyrin-based subcellular gradient of neurofascin, an immunoglobulin family protein, directs GABAergic innervation at Purkinje axon initial segment. Cell 119, 257–272 [DOI] [PubMed] [Google Scholar]
- 24. Galiano M. R., Jha S., Ho T. S., Zhang C., Ogawa Y., Chang K. J., Stankewich M. C., Mohler P. J., Rasband M. N. (2012) A distal axonal cytoskeleton forms an intra-axonal boundary that controls axon initial segment assembly. Cell 149, 1125–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scotland P., Zhou D., Benveniste H., Bennett V. (1998) Nervous system defects of ankyrin-B−/− mice suggest functional overlap between the cell adhesion molecule L1 and 440-kD ankyrin-B in premyelinated axons. J. Cell Biol. 143, 1305–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Le Scouarnec S., Bhasin N., Vieyres C., Hund T. J., Cunha S. R., Koval O., Marionneau C., Chen B., Wu Y., Demolombe S., Song L. S., Le Marec H., Probst V., Schott J. J., Anderson M. E., Mohler P. J. (2008) Dysfunction in ankyrin-B-dependent ion channel and transporter targeting causes human sinus node disease. Proc. Natl. Acad. Sci. U.S.A. 105, 15617–15622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cunha S. R., Hund T. J., Hashemi S., Voigt N., Li N., Wright P., Koval O., Li J., Gudmundsson H., Gumina R. J., Karck M., Schott J. J., Probst V., Le Marec H., Anderson M. E., Dobrev D., Wehrens X. H., Mohler P. J. (2011) Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation 124, 1212–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abdi K. M., Mohler P. J., Davis J. Q., Bennett V. (2006) Isoform specificity of ankyrin-B: a site in the divergent C-terminal domain is required for intramolecular association. J. Biol. Chem. 281, 5741–5749 [DOI] [PubMed] [Google Scholar]
- 29. Cunha S. R., Bhasin N., Mohler P. J. (2007) Targeting and stability of Na/Ca exchanger 1 in cardiomyocytes requires direct interaction with the membrane adaptor ankyrin-B. J. Biol. Chem. 282, 4875–4883 [DOI] [PubMed] [Google Scholar]
- 30. Cunha S. R., Mohler P. J. (2008) Obscurin targets ankyrin-B and protein phosphatase 2A to the cardiac M-line. J. Biol. Chem. 283, 31968–31980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Westenbroek R. E., Sakurai T., Elliott E. M., Hell J. W., Starr T. V., Snutch T. P., Catterall W. A. (1995) Immunochemical identification and subcellular distribution of the α1A subunits of brain calcium channels. J. Neurosci. 15, 6403–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Indriati D. W., Kamasawa N., Matsui K., Meredith A. L., Watanabe M., Shigemoto R. (2013) Quantitative localization of Cav2.1 (P/Q-type) voltage-dependent calcium channels in Purkinje cells: somatodendritic gradient and distinct somatic coclustering with calcium-activated potassium channels. J. Neurosci. 33, 3668–3678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kordeli E., Davis J., Trapp B., Bennett V. (1990) An isoform of ankyrin is localized at nodes of Ranvier in myelinated axons of central and peripheral nerves. J. Cell Biol. 110, 1341–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mochida S., Westenbroek R. E., Yokoyama C. T., Zhong H., Myers S. J., Scheuer T., Itoh K., Catterall W. A. (2003) Requirement for the synaptic protein interaction site for reconstitution of synaptic transmission by P/Q-type calcium channels. Proc. Natl. Acad. Sci. U.S.A. 100, 2819–2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ikeda S. R. (1996) Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature 380, 255–258 [DOI] [PubMed] [Google Scholar]
- 36. Herlitze S., Garcia D. E., Mackie K., Hille B., Scheuer T., Catterall W. A. (1996) Modulation of Ca2+ channels by G-protein βγ subunits. Nature 380, 258–262 [DOI] [PubMed] [Google Scholar]
- 37. Dolphin A. C. (2003) G protein modulation of voltage-gated calcium channels. Pharmacol. Rev. 55, 607–627 [DOI] [PubMed] [Google Scholar]
- 38. Yue D. T., Backx P. H., Imredy J. P. (1990) Calcium-sensitive inactivation in the gating of single calcium channels. Science 250, 1735–1738 [DOI] [PubMed] [Google Scholar]
- 39. Delcour A. H., Tsien R. W. (1993) Altered prevalence of gating modes in neurotransmitter inhibition of N-type calcium channels. Science 259, 980–984 [DOI] [PubMed] [Google Scholar]
- 40. Patil P. G., de Leon M., Reed R. R., Dubel S., Snutch T. P., Yue D. T. (1996) Elementary events underlying voltage-dependent G-protein inhibition of N-type calcium channels. Biophys. J. 71, 2509–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Garrido J. J., Giraud P., Carlier E., Fernandes F., Moussif A., Fache M. P., Debanne D., Dargent B. (2003) A targeting motif involved in sodium channel clustering at the axonal initial segment. Science 300, 2091–2094 [DOI] [PubMed] [Google Scholar]
- 42. Fransen E., Van Camp G., Vits L., Willems P. J. (1997) L1-associated diseases: clinical geneticists divide, molecular geneticists unite. Hum. Mol. Genet. 6, 1625–1632 [DOI] [PubMed] [Google Scholar]
- 43. Mohler P. J., Healy J. A., Xue H., Puca A. A., Kline C. F., Allingham R. R., Kranias E. G., Rockman H. A., Bennett V. (2007) Ankyrin-B syndrome: enhanced cardiac function balanced by risk of cardiac death and premature senescence. PLoS ONE 2, e1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mohler P. J., Le Scouarnec S., Denjoy I., Lowe J. S., Guicheney P., Caron L., Driskell I. M., Schott J. J., Norris K., Leenhardt A., Kim R. B., Escande D., Roden D. M. (2007) Defining the cellular phenotype of “ankyrin-B syndrome” variants: human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation 115, 432–441 [DOI] [PubMed] [Google Scholar]
- 45. Mohler P. J., Schott J. J., Gramolini A. O., Dilly K. W., Guatimosim S., duBell W. H., Song L. S., Haurogné K., Kyndt F., Ali M. E., Rogers T. B., Lederer W. J., Escande D., Le Marec H., Bennett V. (2003) Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421, 634–639 [DOI] [PubMed] [Google Scholar]
- 46. Mohler P. J., Trafford A. W. (2011) The devil is in the details: methodological reviews, a new JMCC initiative. J. Mol. Cell. Cardiol. 50, 939. [DOI] [PubMed] [Google Scholar]
- 47. Sedlacek K., Stark K., Cunha S. R., Pfeufer A., Weber S., Berger I., Perz S., Kääb S., Wichmann H. E., Mohler P. J., Hengstenberg C., Jeron A. (2008) Common genetic variants in ANK2 modulate QT interval: results from the KORA study. Circ. Cardiovasc. Genet. 1, 93–99 [DOI] [PubMed] [Google Scholar]
- 48. Needham L. K., Thelen K., Maness P. F. (2001) Cytoplasmic domain mutations of the L1 cell adhesion molecule reduce L1-ankyrin interactions. J. Neurosci. 21, 1490–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim C., Jun K., Lee T., Kim S. S., McEnery M. W., Chin H., Kim H. L., Park J. M., Kim D. K., Jung S. J., Kim J., Shin H. S. (2001) Altered nociceptive response in mice deficient in the α1B subunit of the voltage-dependent calcium channel. Mol. Cell. Neurosci. 18, 235–245 [DOI] [PubMed] [Google Scholar]
- 50. Kim C., Jeon D., Kim Y. H., Lee C. J., Kim H., Shin H. S. (2009) Deletion of N-type Ca2+ channel Cav2.2 results in hyperaggressive behaviors in mice. J. Biol. Chem. 284, 2738–2745 [DOI] [PubMed] [Google Scholar]
- 51. Jun K., Piedras-Rentería E. S., Smith S. M., Wheeler D. B., Lee S. B., Lee T. G., Chin H., Adams M. E., Scheller R. H., Tsien R. W., Shin H. S. (1999) Ablation of P/Q-type Ca2+ channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the α1A-subunit. Proc. Natl. Acad. Sci. U.S.A. 96, 15245–15250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Luvisetto S., Marinelli S., Panasiti M. S., D'Amato F. R., Fletcher C. F., Pavone F., Pietrobon D. (2006) Pain sensitivity in mice lacking the Cav2.1α1 subunit of P/Q-type Ca2+ channels. Neuroscience 142, 823–832 [DOI] [PubMed] [Google Scholar]