

Background: IFN-λ gene induction in hepatitis C virus infected hepatocytes is not well defined.
Results: IRF-3, IRF-7, and NF-κB transcribe type III IFNs. Treatment with IFN-λ reduced HCV RNA and miR122.
Conclusion: IFN-λ gene regulation in HCV-infected hepatocytes is similar to immune cells. IFN-λ may exert antiviral activity by down-regulating miR122.
Significance: Hepatocytes can produce IFN-λ, which may be effective against hepatitis C virus.
Keywords: Cytokines induction, Hepatitis c virus, Interferon, Transcription factors, Transcription promoter, Interferon regulatory factor, NF-κB
Abstract
Hepatitis C virus (HCV) infection in hepatocytes stimulates innate antiviral responses including the production of type III interferons (IFN-λ), including IL-28A, IL-28B, and IL-29. However, the molecular mechanism(s) regulating the expression of IFN-λ genes in HCV-infected hepatocytes remains undefined. In this study, we examined regulatory elements involved in the induction of IFN-λ genes following HCV infection in hepatocytes and further determined the binding of specific transcription factor(s) to promoter regions of IFN-λ genes. Our studies reveal that the regulatory portion for IL-28A, IL-28B, and IL-29 genes is localized to a 1-kb region in their respective promoters. Notably, interferon regulatory factor (IRF)-3 and -7 are the key transcriptional factors for the induction of IL-28A and IL-28B genes, whereas NF-κB is an additional requirement for the induction of the IL-29 gene. Ligation of Toll-like receptors (TLR) 3, 7, 8, and 9, which also activate IRFs and NF-κB, resulted in more robust production of IFN-λ than that observed with HCV infection, verifying the importance of TLR pathways in IFN-λ production. Furthermore, the addition of IFN-λ to HCV-infected hepatocytes decreased viral replication and produced a concurrent reduction in microRNA-122 (miR-122). The decrease in viral replication was enhanced by the co-administration of IFN-λ and miR-122 inhibitor (miRIDIAN), suggesting that such combinatorial therapies may be beneficial for the treatment of chronic HCV infection.
Introduction
Hepatitis C virus (HCV)2 is a serious worldwide health problem, with more than 170 million people infected globally. HCV establishes persistent infection in 70% of infected individuals, leading to chronic liver inflammation, fibrosis, and cirrhosis (1). Combination therapy of type I IFN with ribavirin is currently used for treating chronic HCV patients. Although the addition of viral protease inhibitors to this regimen has significantly improved treatment outcomes, their use is presently limited to one of several HCV genotypes, indicating the continued need for the development of antiviral agents. Type III IFNs (also known as IFN-λ) are the newest members of the IFN family and include IFN-λ1 (IL-29), IFN-λ2 (IL-28A), IFN-λ3 (IL-28B), and the most recent addition, IFN-λ4 (3, 4). Notably, single nucleotide polymorphisms (SNPs) in the 3-kb promoter region of IL-28B strongly predict the sustained virological response to pegylated IFN-α/ribavirin treatment in HCV patients. Therefore, IFN-λ might influence the responsiveness of patients to IFN-α therapy.
The IFN-λ receptor is a heterodimer composed of the ligand binding chain, IFN-λR, and the accessory chain, IL-10R. The binding of IFN-λ to its receptor induces interferon-stimulated genes such as IFIT1, MX1, CXCL10, and CXCL11, similar to the effect of type I IFN (5). In contrast to widespread expression of the type I IFN receptor on multiple cell types, expression of the IFN-λ receptor is largely limited to epithelial cells, including hepatocytes, but is surprisingly absent from most hematopoietic cells (3). This restricted expression of the IFN-λ receptor suggests that IFN-λ-based treatment may be less toxic when compared with IFN-α/β therapy.
Recently, HCV infection in hepatocytes has been reported to induce IFN-λ production (6–8). Moreover, production of IL-28 by HCV-infected primary human hepatocytes resulted in the expression of interferon-stimulated genes, presumably through autocrine signaling via the IFN-λ receptor (8). However, the molecular mechanisms for HCV-mediated induction of IFN-λ genes have not been well defined in hepatocytes. To this end, we found that HCV or poly(I:C), which mimics intracellular viral RNA, induced transcription of IFN-λ genes in human hepatoma cells and primary human hepatocytes. Expression of IL-28A and IL-28B genes in hepatocytes required IRF-3 and IRF-7, whereas expression of IL-29 gene was dependent upon IRF-3, IRF-7, and NF-κB. Mutation of the binding sites for these transcription factors abolished IFN-λ gene expression, as did knockdown of IRF-3, IRF-7, and NF-κB. Production of IFN-λ was increased following exposure to ligands of TLR3, -7, -8, and -9 when compared with HCV, confirming the importance of IRF- and NF-κB-dependent pathways in IFN-λ induction. We also demonstrated that treatment of HCV-infected hepatocytes with IFN-λ suppresses miR-122 and HCV replication. In addition, co-treatment with IFN-λ and miR-122 inhibitor (miRIDIAN) resulted in a more pronounced suppression of HCV when compared with treatment with IFN-λ alone. These observations affirm the antiviral effects of IFN-λ and miR-122 inhibitors and suggest their use as potential alternatives to the current treatment for HCV infection.
EXPERIMENTAL PROCEDURES
Reagents
Antibodies to human IgG, IRF-3, IRF-7, NF-κB p50 or p65 were purchased from Cell Signaling. siRNA to target IRF-3, IRF-7, RELA(NF-κB p65), or control siRNA and miRIDIAN miR122 hairpin inhibitor and negative control #1 were purchased from Dharmacon. Toll-like receptor (TLR) agonists were purchased from InvivoGen. Recombinant human IL-28 and IL-29 were purchased from PeproTech.
Cells and Viruses
Cryopreserved primary human hepatocytes were purchased from Invitrogen. Primary human hepatocytes were cultured on collagen I-coated 24-well plates in Williams' E medium supplemented with 1% GlutaMAX and 1% penicillin/streptomycin and maintained with Williams' E medium supplemented with 1% GlutaMAX, dexamethasone, 1% penicillin/streptomycin, insulin, transferrin, selenium complex, BSA, and linoleic acid. Liver biopsies from chronic HCV or control patients were generously provided by Dr. Hugo Rosen (University of Colorado), and patient information regarding the age, gender, and HCV genotype was as described previously (9). The human hepatoma cell lines PH5CH8 and Huh 7.5.1 were maintained in DMEM with 10% FBS, penicillin/streptomycin (100 μg/ml), l-glutamine (2 mm), and 1× nonessential amino acids. The HCV JFH-1 strain was kindly provided by Dr. Wakita (Tokyo Metropolitan institute) and grown as described previously (10).
Plasmid Construction
Promoter fragments were amplified by PCR, using primers generated from genomic sequences of the respective genes. Promoter length was determined from the translational start site. The PCR product was then cloned into pGL3-basic vector. Mutant constructs were generated using the QuikChange site-directed mutagenesis kit. Site-directed mutagenesis was introduced as follows (lowercase letters indicate mutated nucleotides): IL-28A·AP-1 (GCAGTTAGTCA changed to GCgcTTAacCA); IL-28A·IRF-1 (TTTCTCTTTC changed to cgTCTCcgTC); IL-28A·NF-κB-1 (TGGGCTTTCCCA changed to TGttCTTTaaCA); IL-28A·IRF-2 (TTTCACTTTTC changed to cccCACcccTC); IL-28A·NF-κB-2 (GGGACTGCCC changed to GttACTGaaC); IL-28B·IRF-1 (GTTTCTCTTT changed to GcccCTCccc); IL-28B·NF-κB-1 (TGGGCTTTCCCAG changed to TGcGCTaTggCAG); IL-28B·IRF-2 (GTTTTCACTTTTC changed to GTcTcCACcTcTC); IL-28B·NF-κB-2 (GGGACTGCCC changed to GcGACTGggC); IL-29·GATA-1 (CCTAATCTCAG changed to CagAATagCAG); IL-29·IRF (AGTTTCTCTTTCCT changed to AGcccCTCcccCCT); IL-29·NF-κB (TGGAAATTCT changed to TccAtATTgT). IRF-3 and IRF-7 plasmids were kindly provided by Dr. Shizuo Akira (Osaka University). NF-κB and dominant negative IκB constructs were designed as described previously (11).
Transfection and Luciferase Assay
Cells (5 × 105) were plated in 6-well plates and transfected with 1 μg of luciferase reporter plasmid driven by the IL-28A, IL-28B, or IL-29 promoter and 10 ng of phRL-TK plasmid driven by HSV thymidine kinase promoter using Mirus transfection reagent (Mirus Bio Corp.) according to the manufacturer's protocol. Cells were cultured for 24 h and then stimulated for 6 h with poly(I:C) or infected for 12 h with JFH-1. In the cDNA expression plasmid experiment, a mixture containing 1 μg of luciferase plasmid, 0.2 μg of cDNA expression plasmid, and 10 ng of phRL-TK plasmid was transfected into the cells, which were then cultured for 24 h. Cells were harvested and lysed in 100 μl of lysis buffer (Promega). For siRNA experiments, PH5CH8 cells were transfected according to the DharmaFECT Duo transfection reagent with 10 ng of phRL-TK plasmid and 1 μg of luciferase constructs containing IFN-λ promoter plasmid in combination with 100 nM of IRF-3, IRF-7, RELA(NF-κB p65), or control siRNA, respectively. At 36 h after transfection, cells were stimulated with poly(I:C) (50 μg/ml) for 6 h or infected with JFH-1 for 12 h. Firefly and Renilla luciferase activities were measured by GLOMAX multidetection system. Firefly luciferase activity from the luciferase reporter vector was normalized to the Renilla luciferase activity from the phRL-TK vector. Data are the ratio of relative light units measured for luciferase activity to relative light units measured for Renilla luciferase activity. For miR-122 suppression activity, Huh 7.5.1 cells were transfected with 100 nM of negative control or miRIDIAN (miR-122 hairpin inhibitor) using DharmaFECT Duo transfection reagent. After 48 h of transfection, cells were infected with JFH-1 for 5 days. HCV-infected cells were stimulated with 100 ng of recombinant IL-28 and/or IL-29 for the indicated time course. Cells were harvested and analyzed for HCV mRNA. In each experiment, samples were analyzed in triplicate, and each experiment was repeated at least three times.
Real-time Quantitative PCR
RNA was extracted using the RNeasy kit (Qiagen) according to the manufacturer's protocol. Real-time RT-PCR was performed on an ABI Prism sequence detection system (Applied Biosystems). The primers used were as follows: IL-28A/B, 5′-AGTCGCTTCTGCTGAAGGAC-3′ and 5′-TCCAGAACCTTCAGCGTCAG-3′; IL-29, 5′-CTGCCACATTGGCAGGTTCA-3′ and 5′-AGACAGGAGAGCTGCAACTC-3′; GAPDH, 5′-ATGGCACCGTCAAGGCTGAG-3′ and 5′-GCTAAGCAGTTGGTGGTGCA-3′. The results were normalized to GAPDH. For microRNA real-time PCR, microRNAs were reverse-transcribed using the TaqMan microRNA reverse transcription kit (Invitrogen) according to the manufacturer's protocol, with reverse transcriptase primers from hsa-miR-122 and U18 TaqMan microRNA assay kits. MicroRNA quantitative PCR reactions were assembled according to the TaqMan small RNA assay protocol with 2× TaqMan master mix.
ELISA
IL-28 and IL-29 secretion (R&D Systems) in culture supernatants and HCV NS3 (BioFront Tec.) in cell lysates were analyzed by ELISA according to the manufacturer's protocol.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assay was performed as described previously (12). Anti-IRF-3, anti-IRF-7, anti-NF-κB, or normal rabbit IgG (Millipore) antibodies were used in immunoprecipitation experiments. Purified ChIP DNA was measured by real-time quantitative PCR using Platinum SYBR Green quantitative PCR SuperMix-UDG (Invitrogen). PCR conditions were 95 °C for 10 min followed by 40 cycles consisting of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s. The level of ChIP DNA was normalized with that of input DNA. In each experiment, samples were analyzed in triplicate. The primers used were as follows: IL-28A/B, 5′-TGCTCATCTGGCTCACTAGC-3′ and 5′-GAGCTCTGTCTGGGATGTAA-3′; IL-29, 5′-CTTCCTCTCTGCCACTCAGG-3′ and 5′-ACTGCTTCCCCAGCGGCATG-3′.
Statistical Analysis
Experimental results were analyzed for statistical significance (Student's t test or analysis of variance with Tukey's post test where appropriate). p values <0.01 were considered significant and are indicated in the figures.
RESULTS
Induction of IFN-λ Genes in HCV-infected Liver Tissue and Human Primary Liver Cells
To determine the production of IFN-λ at the site of HCV infection, we first examined the induction of IFN-λ genes (IL-28A/B, IL-29) in liver biopsy specimens from healthy or HCV-infected patients by real-time quantitative PCR analysis. As shown in Fig. 1, A and B, transcription of IL-28A/B and IL-29 was significantly increased in HCV-infected patients when compared with healthy controls. To identify the kinetics and magnitude of IFN-λ gene induction, we assessed mRNA levels of IL-28A/B and IL-29 genes from human primary liver cells at various time points (0, 4, 12, 24, 48, and 72 h) after JFH-1 infection. The expression of IL-28A/B and IL-29 genes was maximally induced at 12 h after infection, which was likely due to the recognition of viral pathogen-associated molecular patterns and not a direct effect of viral replication (Fig. 1, C and E). Moreover, the expression of IFN-λ genes was specific to actively replicating HCV because UV-treated JFH-1 failed to enhance transcription of IL-28A/B and IL-29 (Fig. 1, C and E). These results were confirmed at the protein level as determined by ELISA (Fig. 1, D and F). IFN-λ mRNA and protein levels were also increased in the HCV-infected human hepatocyte cell line PH5CH8 (data not shown). Taken together, these results indicate that HCV induces IFN-λ synthesis during acute and chronic infection.
FIGURE 1.
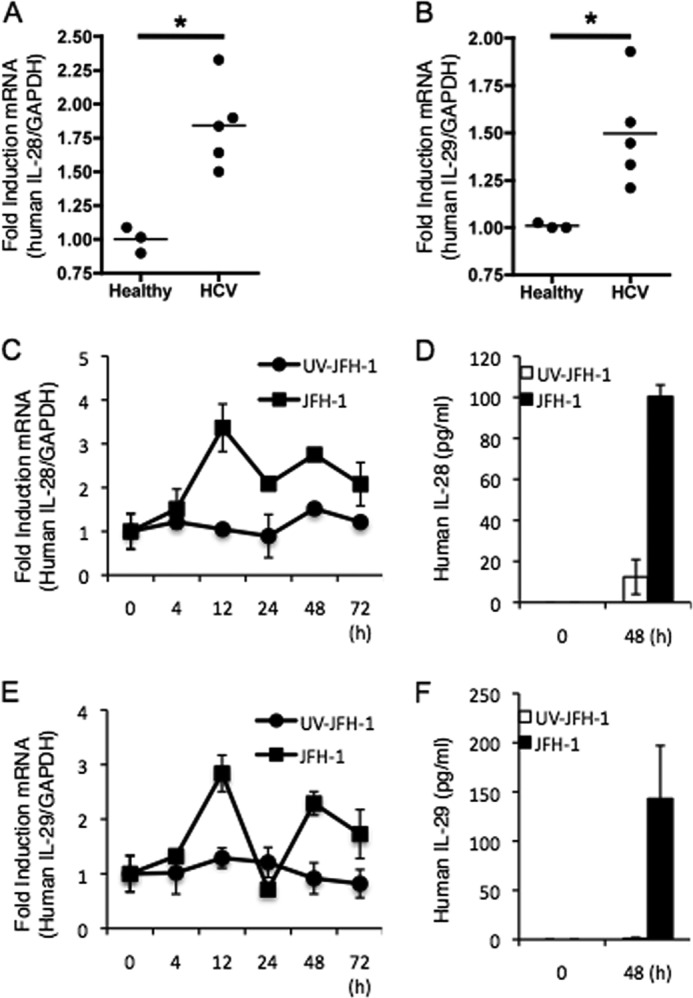
Hepatic induction of IFN-λ genes during HCV infection. A and B, total RNA was extracted from liver tissues of healthy and chronic HCV patients for IL-28A/B (A) and IL-29 (B) mRNA quantification by real-time PCR. C–F, human primary hepatocytes were inoculated with UV-irradiated JFH-1 or JFH-1 for the indicated amounts of time, and IFN-λ mRNA levels were quantified by real-time PCR (C and E), whereas protein levels were analyzed by ELISA (D and F). Data represent means ± S.D. of three independent experiments (* indicates p < 0.01).
Proximal Regulatory Elements Are Required for Transcriptional Activation at the Human IFN-λ Promoter
We next defined the critical regions upstream of the IFN-λ genes necessary for transcription by generating a nested series of deletion constructs of the human IFN-λ gene promoter. All constructs included the transcriptional start codon. These constructs were transfected into PH5CH8 cells with or without JFH-1 infection. After 12 h of infection, transcription of IL-28A, IL-28B, and IL-29 was determined by measuring luciferase activity. Although all promoter constructs were robustly responsive to JFH-1 infection, luciferase activity was noticeably increased with the addition of as little as 1.0 kb upstream of the putative start site (Fig. 2, A–C). These results indicate that IFN-λ gene transcription induced by HCV infection is dependent on sequences present within this region. In addition, similar results were observed in PH5CH8 cells following treatment with poly(I:C), which mimics intracellular viral RNA (data not shown). It also implies that shared cis-elements in the proximal region are likely involved in regulating the expression of IFN-λ genes in response to HCV infection.
FIGURE 2.
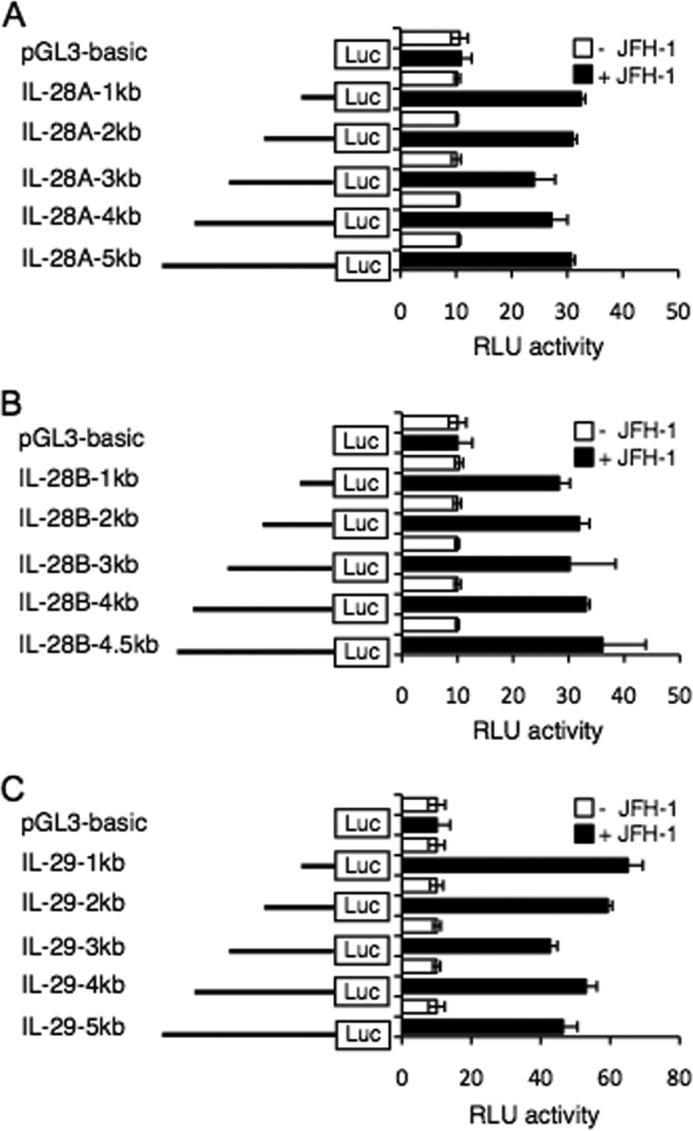
Transcriptional activity at the IL-28A, IL-28B, and IL-29 promoter by HCV infection. A–C, PH5CH8 cells were transfected with a mixture of the luciferase (Luc) constructs containing various 5′ deletions of the human IL-28A (A), IL-28B (B), or IL-29 (C) promoters and Renilla luciferase control vector. The cells were cultured for 18 h and then infected with JFH-1 for 12 h. Luciferase activity in whole cell lysates was normalized to Renilla luciferase activity. Data are the mean ± S.D. of triplicate data points from five independent experiments. RLU, relative light units.
HCV-induced IL-28A/B and IL-29 Genes Are Differentially Regulated by IRFs and NF-κB
Subsequent analysis within the 1.0-kb region of IL-28A/B and IL-29 gene regulatory domain uncovered specific transcription factor(s) involved in the induction of these genes. Although there was one putative AP-1 binding site and two putative IRF and NF-κB binding sites on the IL-28A promoter, the IL-29 promoter contained one putative binding site each for GATA-1, IRF, and NF-κB transcription factors (Fig. 3A). To determine whether these distinct motifs were required for the transcription of IFN-λ in response to HCV or poly(I:C), we performed site-directed mutagenesis at each binding site. As shown in Fig. 3B, mutations in the AP-1, IRF, or NF-κB binding sites resulted in little to no difference in IL-28A transcription. In contrast, mutation of the IRF binding motif closest to the transcription initiation site of IL-28A significantly decreased IL-28A transcription when stimulated with JFH-1 or poly(I:C) (Fig. 3B). Similar results were seen at the IL-28B promoter (Fig. 3C). Transcription of IL-29 was unaffected by mutating the GATA-3 binding site (Fig. 3D); however, mutation of the IRF and NF-κB binding motifs produced a notable decrease in IL-29 transcription, which was drastically enhanced upon mutation of both binding sites. These results indicate that the induction of IL-28A and IL-28B gene expression in response to HCV and poly(I:C) is likely mediated through IRF, whereas both IRF and NF-κB are required for transcription at the IL-29 locus.
FIGURE 3.

IRF-3, IRF-7, and NF-κB differentially determine expression of IL-28A/B and IL-29. A, nucleotide sequence from 1.0 kb upstream of the transcription start site of human IL-28A, IL-28B, and IL-29. The positions of the IRF, NF-κB, AP-1, and GATA-1 binding sites are indicated. B–D, PH5CH8 cells were transiently transfected with control plasmid pGL3-basic, and plasmids encoding luciferase constructs containing human IL-28A (pGL3-IL-28A 1 kb) (B), IL-28B (pGL3-IL-28B 1 kb) (C), or IL-29 (pGL3-IL-29 1 kb) (D) promoter regions. Various mutation constructs of the above promoter regions were also transfected into PH5CH8 cells, using a Renilla luciferase vector as a control. The cells were cultured for 24 h and then stimulated with poly(I:C) for 6 h or infected with JFH-1 for 12 h. Luciferase activity in whole cell lysates was normalized to Renilla luciferase activity. Data are the mean ± S.D. of triplicate data points from five independent experiments. RLU, relative light units.
To identify specific IRF and NF-κB proteins involved in transcriptional regulation of IFN-λ genes, we co-transfected PH5CH8 cells with reporter constructs of the IL-28A, IL-28B, or IL-29 promoter and an individual vector containing NF-κB p65, IRF-3, or IRF-7 and stimulated cells with poly(I:C). As shown in Fig. 4, the induction of IFN-λ genes depends on poly(I:C) stimulation. The overexpression of IRF-3 (Fig. 4, B and E) and IRF-7 (Fig. 4, C and F) resulted in robust transcriptional activity at the IL-28A and IL-28B promoters following poly(I:C) stimulation when compared with control cells overexpressing NF-κB with the promoter reporter construct alone (Fig. 4, A and D). These results suggest that IRF-3 and IRF-7 are crucial for IL-28A and IL-28B transcription, whereas NF-κB is dispensable for inducing IL-28A/B expression. In contrast, overexpression of the p65 subunit resulted in increased transcription at the IL-29 promoter, which was enhanced by the addition of poly(I:C) (Fig. 4G). Transfection with the dominant-negative mutant of IκB kinase (IKKβ) inhibited poly(I:C)-mediated IL-29 transcription, confirming the essential role of NF-κB in regulating IL-29 gene expression (Fig. 4H). The contributions of IRF-3 and IRF-7 to the induction of IL-29 gene were verified by the increase in luciferase activity evident upon their overexpression following poly(I:C) stimulation (Fig. 4, I and J). We also conducted parallel studies of IRF3, IRF7, and NF-κB overexpression in HCV-infected hepatocytes and found similar, but less dramatic induction of IFN-λ when compared with stimulation with poly(I:C) (data not shown).
FIGURE 4.

Overexpression of IRFs and NF-κB confirms their role in expression of IFN-λ genes. A–J, PH5CH8 cells were transiently transfected with plasmids encoding different promoter regions of human IL-28A (pGL3-IL-28A 1 kb) (A–C), IL-28B (pGL3-IL-28B 1 kb) (D–F), or IL-29 (pGL3-IL-29 1 kb) (G–J), and expression vectors encoding IRF-3, IRF-7, and NF-κB or a dominant-negative mutant of IκB kinase (DN-IκB). At 18 h after transfection, cells were stimulated with poly(I:C) for 6 h. Luciferase activity in whole cell lysates was normalized to Renilla luciferase activity. Data represent mean ± S.D. of triplicate data points of one experiment and are representative of five independent experiments (* indicates p < 0.01). RLU, relative light units.
To further verify the role of IRF and NF-κB in inducing IFN-λ genes by HCV infection, we employed siRNA gene silencing to knock down the expression of IRF-3, IRF-7, and RELA(NF-κB p65). IRF-3 and IRF-7 knockdown significantly reduced both poly(I:C)-induced and JFH-1-induced expression of IL-28A/B and IL-29 genes (Fig. 5, A–C). NF-κB knockdown reduced IL-29 gene induction following either poly(I:C) stimulation or JFH-1 infection (Fig. 5C), but did not affect the induction of IL-28A/B genes (Fig. 5, A and B).
FIGURE 5.

IRFs are core transcription factors of IFN-λ genes. A–C, PH5CH8 cells were transfected with control plasmid or promoter plasmids encoding IL-28A (A), IL-28B (B), or IL-29 (C) in combination with siRNA specific for IRF-3, IRF-7, RELA(NF-κB p65), or control. At 36 h after transfection, cells were stimulated with poly(I:C) for 6 h or infected with JFH-1 for 12 h. Luciferase activity in whole cell lysates was normalized to Renilla luciferase activity. Data represent mean ± S.D. of triplicate data points of one experiment and are representative of three independent experiments. Statistical significance is indicated by * (p < 0.01) and is relative to control + poly(I:C) or control + JFH-1. RLU, relative light units.
IL-28A/B and IL-29 Promoters Are Directly Bound by IRF-3·IRF-7 and IRF-3·IRF-7·NF-κB, Respectively
To verify the recruitment and direct binding of IRF-3, IRF-7, and NF-κB to the endogenous IFN-λ promoter, we performed chromatin immunoprecipitation (ChIP) assays on PH5CH8 cells that were stimulated with JFH-1 or poly(I:C) using antibodies directed against IRF-3, IRF-7, NF-κB, or isotype control. As expected, IRF-3 and IRF-7 were recruited to the IL-28A and IL-28B promoters in response to JFH-1 and poly(I:C), whereas recruitment of NF-κB was comparable with the isotype control (Fig. 6, A and B). However, recruitment of NF-κB to the IL-29 promoter was markedly elevated in response to JFH-1 or poly(I:C) (Fig. 6, C and D). Indeed, NF-κB bound the IL-29 promoter to the same extent as IRF-3 and IRF-7, confirming the contribution of all three transcription factors to the expression of IL-29 (Fig. 6, C and D).
FIGURE 6.
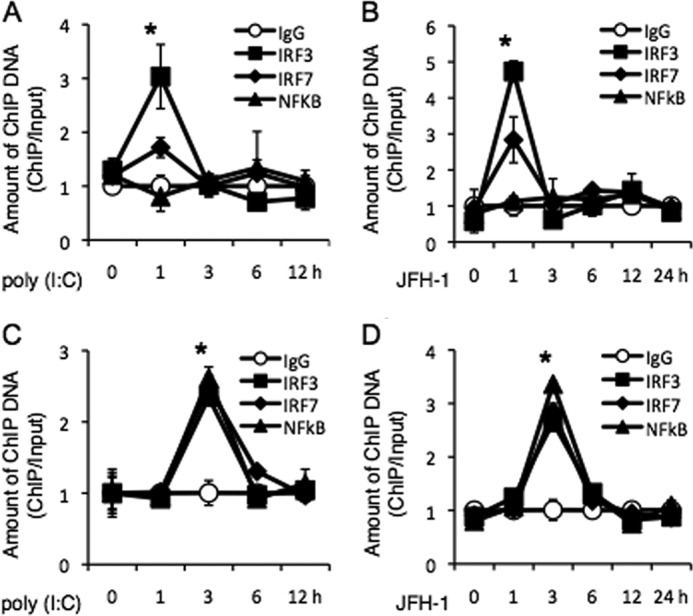
IRFs and NF-κB directly bind the promoters of IFN-λ genes. A–D, PH5CH8 cells were stimulated with poly(I:C) (A and C) or infected with JFH-1 (B and D) for the indicated lengths of time. Chromatin was immunoprecipitated with anti-IRF-3, anti-IRF-7, or anti-NF-κB antibody, or control normal rabbit IgG. Purified ChIP and input DNA were analyzed by real-time quantitative PCR. The amount of ChIP DNA was normalized to that of input DNA. The mean value of control antibody before stimulation was arbitrarily defined as 1. Data are the mean ± S.D. of triplicate data points from a representative of five independent experiments. Statistical significance is indicated by * (p < 0.01) and is relative to IgG.
IFN-λ Treatment Decreases miR-122 Expression and Has Enhanced Antiviral Activity upon Co-treatment with miR-122 Inhibitor
miR-122, the most abundant microRNA in the liver, is known to facilitate HCV replication and translation (13) and regulates type I IFN signaling via epigenetic control of SOCS3 (14). Based on previous studies demonstrating a potential role for IFN-λ in regulating responses to IFN-α therapy (27), we hypothesized that IFN-λ might exert its antiviral effects by modulating the expression of miR-122 in addition to triggering the induction of interferon-stimulated genes. To test this possibility, we first determined the time course of miR-122 and HCV RNA expression by infecting Huh 7.5.1 cells with JFH-1 for various time points (1, 3, 5, 7, 9, and 11 days). miR-122 expression increased early in infection and peaked at day 3 after infection, whereas intracellular HCV RNA accrued to high levels starting at day 5 after infection (Fig. 7A). Next, we determined whether treatment with IFN-λ might modulate the expression of miR-122 and found that IFN-λ treatment significantly decreased both miR122 and HCV RNA (Fig. 7, B and C). Consequently, transfection of the miR-122 inhibitor, miRIDIAN, into HCV-infected cells for 36 h and subsequent treatment with IFN-λ resulted in a more pronounced inhibition of HCV replication when compared with treatment with IFN-λ alone (Fig. 7D). In addition, a slight decrease in HCV NS3 protein upon treatment with miRIDIAN alone was also enhanced upon co-treatment with IFN-λ (Fig. 7E). Collectively, these results identify a correlation between IFN-λ treatment and miR-122 levels, which likely reflects the decrease in viral burden.
FIGURE 7.

miR-122-dependent replication of HCV is suppressed by IFN-λ stimulation. A, Huh 7.5.1 cells infected with JFH-1 for the indicated number of days and HCV RNA or miR-122 levels were quantified by real-time PCR. The mean value of day 1 after infection was arbitrarily defined as 1 in -fold increase in HCV or as 10 in -fold increase in miR-122. Cells were infected with JFH-1 for 5 days and then replated in 6-well plates. After 24 h, the HCV-infected cells were stimulated with recombinant IFN-λ (100 ng) for 0–48 h. HPRT, hypoxanthine-guanine phosphoribosyltransferase. B and C, the amount of HCV RNA or miR-122 was quantified by real-time PCR. D and E, similarly, HCV-infected cells were replated in 6-well plates after 5 days of infection. After 24 h, cells were transfected with miRIDIAN or negative control miRIDIAN (20 pmol) for 36 h and stimulated with or without IFN-λ (100 ng each) for 1–3 days. HCV RNA was quantified by real-time PCR (D), whereas HCV NS3 protein was measured by ELISA (E). Data shown are from one experiment and are representative of three experiment with similar results. Statistical significance is indicated by * (p < 0.01) and is relative to the control group that received no treatment.
Stimulation of Primary Human Hepatocytes with TLR Ligands Induces Robust IFN-λ Expression
Because IFN-λ elicits antiviral activity to limit HCV replication and engagement of TLRs with TLR ligands also activates IRF/NF-κB, we examined the ability of various TLR ligands to stimulate IFN-λ production in hepatocytes relative to HCV infection. Primary human hepatocytes were stimulated with a panel of TLR agonists (Fig. 8A) for 24 h, and IL-28B and IL-29 production was then determined by ELISA. Ligation of TLR3, -7, -8, and -9 produced increased IL-28B (Fig. 8B) and IL-29 (Fig. 8C) relative to JFH-1-infected hepatocytes. These results indicate that various TLR ligands are capable of triggering production of IFN-λ and may have an important role in enhancing IFN-λ production in vivo.
FIGURE 8.
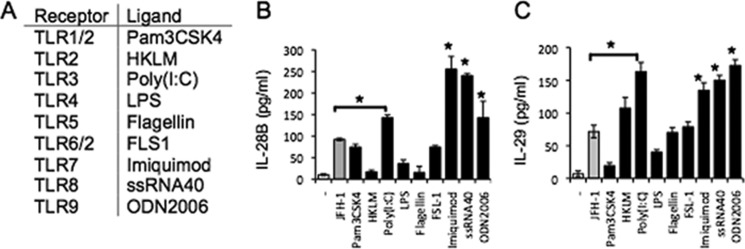
TLR3, -7, -8, and -9 induce human IFN-λ gene expression in human primary hepatocytes. Human primary hepatocytes were incubated with JFH-1, Pam3CSK4 (1 μg/ml), Heat-killed Listeria monocytogenes (HKLM) (108 cells/ml), Poly (I-C) (5 μg/ml), LPS (10 μg/ml), flagellin (1 μg/ml), FSL-1 (100 ng/ml), Imiquimod (1 μg/ml), ssRNA (1 μg/ml), or ODN2006 (5 μm) for 24 h. A, Toll-like receptors (TLR) and their respective ligands. B and C, supernatants were harvested, and IL-28B (B) and IL-29 (C) were measured by ELISA. Data are the mean ± S.D. of triplicate data points from one experiment. Results are representative of three independent experiments. Statistical significance is indicated by * (p < 0.01) and is relative to JFH-1 infection.
DISCUSSION
In this study, we identified the involvement of proximal regions of the IFN-λ promoter in HCV-induced expression of IFN-λ genes in hepatocytes and demonstrated that IRF-3·IRF-7 are required for IL-28A and IL-28B gene expression, whereas IL-29 gene induction requires NF-κB in addition to IRF-3·IRF-7. Using human primary hepatocytes and the human hepatocyte cell line PH5CH8, we first determined that HCV infection and poly(I:C) stimulation are capable of inducing IL-28A, IL-28B, and IL-29 genes and the production of their proteins. We then established that IRF-3 and IRF-7 are essential for the transcription of IL-28A/B and IL-29 genes; NF-κB was an additional requirement for the transcription of IL-29 upon HCV infection or poly(I:C) stimulation. Moreover, IFN-λ treatment in HCV-infected hepatocytes reduces miR-122 expression, and co-treatment of IFN-λ and miR-122 inhibitor results in increased suppression of HCV replication. Taken together, our results suggest that IFN-λ-based immunotherapy would be a potential alternative to treat HCV patients.
The IFN-λ genes share a high degree of homology; the IL-28B gene shares sequence homology with the IL-28A gene (96%) and IL-29 gene (81%), suggesting that these genes might have evolved from a common ancestor (15). This homology is reflected in our findings that overlapping regulatory factors dictate the expression of IFN-λ genes. However, in terms of transcriptional factors regulating the expression of IL-28A, IL-28B, and IL-29 genes, there are conflicting results between previous studies and our results: previous studies reported that IRF-7 alone was sufficient for the expression of IL-28A and IL-28B genes whereas IRF-3, IRF-7, and NF-κB are required for the transcription of IL-29 in human myeloid and epithelial cells (16–18). For instance, regulation of the IL-29 gene is similar to the IFNβ gene as they both require IRF-3 and IRF-7 in human monocyte-derived dendritic cells, whereas IRF-7 controlled IL-28A and IL-28B gene expression, which is similar to the transcription of the IFN-α gene (16). These results were further verified by another study, demonstrating that IL-29 gene expression in LPS-stimulated human monocyte-derived dendritic cells required IRF and NF-κB binding spatially separated elements in distal regions of the promoter (17), in contrast to our results, which demonstrate the involvement of proximal regions (1 kb) of the promoter in HCV-induced production of IFN-λ. Furthermore, a recent study by Ding et al. (19) indicates that both NF-κB and IRF-3 are required for transcriptional regulation of IL-28A/B genes in response to HCV genomic RNA in human alveolar basal epithelial cells (A549) infected with Sendai virus. Our results on the transcriptional regulation of IFN-λ genes in HCV-infected hepatocytes are similar to those previously identified in myeloid cells and other cells of epithelial origin (16–18). It is likely that this discrepancy in transcriptional regulation of IFN-λ genes may be due to cell-specific or stimulus-specific induction patterns of IFN-λ. It also implicates that certain cell types or inflammatory stimuli may be more efficient inducers of type III IFNs than others.
Because NF-κB is uniquely required for the transcriptional regulation of the IL-29 gene but not for IL-28A and IL-28B genes, the production and action of IL-29 in pathogenic infections may differ when compared with those of other members of the type III IFN family. We further determined whether one or more subunits of NF-κB were particularly relevant to its action at the IFN-λ locus. Using siRNA, we knocked down expression of p50 or p65 and measured the induction of the IL-29 gene. p65 knockdown significantly decreased IL-29 transcription in response to HCV and poly(I:C), whereas p50 knockdown showed no difference in IL-29 transcription in response to HCV and poly(I:C) (data not shown). These results indicate that the p65 subunit of NF-κB is essential for its activity at the IL-29 promoter. Because NF-κB was dispensable for the induction of IL-28A/B, the differential requirement for NF-κB in IFN-λ gene expression indicates that IL-28A/B can be expressed without the simultaneous induction of IL-29. Given that NF-κB is readily activated in infection, the significance of these divergent regulatory mechanisms continues to be an area of investigation and may be explained by the existence of other cis- and trans-regulatory elements that have yet to be discovered. Furthermore, it would be interesting to investigate the relevance of these regulatory mechanisms to the transcription of the most recent addition to the type III IFN family, IFN-λ4.
Because IFN-λ exerts antiviral activity to limit HCV replication and IFN-λ-based therapies could be utilized to treat HCV patients, it is important to define the specific cellular sources of IFN-λ during HCV infection. Because dendritic cells play a pivotal role in initiating innate immunity to pathogenic infections, they are robust producers of IFNs and are well equipped for this task due to their expression of a wide range of pattern recognition receptors that sense conserved microbial motifs known as pathogen-associated molecular patterns. TLRs are well studied among pattern recognition receptors and are expressed both at the cell surface and within endocytic compartments (20). Interestingly, IFN-λ was shown to enhance TLR-mediated antiviral immunity; TLR3 or TLR9 agonist treatment of IFN-λR1-deficient mice significantly reduced antiviral activity when compared with normal mice (21). Our results demonstrate that ligation of TLR3, -7, -8, and -9 results in robust production of IL-28B and IL-29 relative to JFH-1-infected hepatocytes (Fig. 8). Therefore, it is likely that HCV-activated TLR ligands also induce production of IFN-λ in hepatocytes to activate antiviral programs in infected and neighboring uninfected cells.
IFNs represent the first line of defense against viral pathogens and act both directly on viral replication and indirectly through activation of host immune response genes (22). Previous studies report that numerous viruses including herpes simplex virus 1, influenza A virus, HIV, hepatitis B virus (HBV), and HCV are sensitive to the antiviral effects of IFN-λ (23–25). Antiviral activity of IFN-λ has also been demonstrated against HCV using both HCV replicon and cell culture infectious virus model systems in hepatocellular carcinoma cells, affirming that IFN-λ is a potent antiviral cytokine against human hepatotropic viruses (22). We observed that IFN-λ gene expression is enhanced in patients with chronic HCV (Fig. 1, A and B), suggesting that the amount of endogenous IFN-λ produced is insufficient to clear virus or that the immune evasion strategies of HCV are effective in overcoming the antiviral effects of IFN-λ. However, we also demonstrate that treatment of HCV-infected hepatocytes with exogenous IFN-λ not only reduced intracellular viral content, but also suppressed expression of miR-122, which is known to enhance HCV replication and translation (Fig. 7). However, it is possible that the decrease in miR-122 may be secondary to the decrease in viral RNA rather than a direct effect of IFN-λ on inhibiting miR-122 synthesis. Moreover, given that miR-122 has been reported to block type I IFN signaling, the net decrease in miR-122 suggests that IFN-λ might trigger an antiviral state in HCV-infected hepatocytes both through classical IFN-stimulated pathways and through direct or indirect down-regulation of host factors necessary for HCV replication (26). Although a precise role of IFN-λ in regulating the expression of miR-122 needs to be determined, the enhanced inhibition of HCV replication upon the addition of IFN-λ and miRIDIAN, an miR-122 inhibitor, suggests that the two agents may act in concert to limit HCV infection.
In conclusion, we demonstrate that HCV infection induces transcription of IFN-λ genes in human hepatocytes, indicating that the contribution of hepatocytes to the antiviral state in HCV infection is regulated in a manner similar to immune cells and other epithelial cells. Despite some understanding of IFN-λ gene regulation, several questions regarding the mechanisms dictating IFN-λ production and activity remain to be addressed, including the identity of additional regulatory elements and the functional significance of differential expression of IL-28A/B and IL-29. Accordingly, further studies are necessary for better understanding of IFN-λ biology as it may have a significant impact on major chronic inflammatory diseases such as hepatitis C.
Acknowledgments
We thank Susan Landes for excellent technical assistance, Dr. Thomas J. Braciale and Dr. Taeg S. Kim for critical discussion of the manuscript prior to submission, and members of the Hahn laboratory for helpful discussions throughout the course of this work.
This work was supported, in whole or in part, by National Institutes of Health Grants AI098126 and AI066328 (to Y. S. H.).
- HCV
- hepatitis C virus
- IRF
- interferon regulatory factor
- TLR
- Toll-like receptor
- miR
- microRNA.
REFERENCES
- 1. Rosen H. R. (2011) Clinical practice. Chronic hepatitis C infection. N. Engl. J. Med. 364, 2429–2438 [DOI] [PubMed] [Google Scholar]
- 2. Deleted in proof. [Google Scholar]
- 3. Sheppard P., Kindsvogel W., Xu W., Henderson K., Schlutsmeyer S., Whitmore T. E., Kuestner R., Garrigues U., Birks C., Roraback J., Ostrander C., Dong D., Shin J., Presnell S., Fox B., Haldeman B., Cooper E., Taft D., Gilbert T., Grant F. J., Tackett M., Krivan W., McKnight G., Clegg C., Foster D., Klucher K. M. (2003) IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 4, 63–68 [DOI] [PubMed] [Google Scholar]
- 4. Kotenko S. V., Gallagher G., Baurin V. V., Lewis-Antes A., Shen M., Shah N. K., Langer J. A., Sheikh F., Dickensheets H., Donnelly R. P. (2003) IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 4, 69–77 [DOI] [PubMed] [Google Scholar]
- 5. Donnelly R. P., Sheikh F., Kotenko S. V., Dickensheets H. (2004) The expanded family of class II cytokines that share the IL-10 receptor-2 (IL-10R2) chain. J. Leukoc. Biol. 76, 314–321 [DOI] [PubMed] [Google Scholar]
- 6. Langhans B., Kupfer B., Braunschweiger I., Arndt S., Schulte W., Nischalke H. D., Nattermann J., Oldenburg J., Sauerbruch T., Spengler U. (2011) Interferon-λ serum levels in hepatitis C. J. Hepatol. 54, 859–865 [DOI] [PubMed] [Google Scholar]
- 7. Marukian S., Andrus L., Sheahan T. P., Jones C. T., Charles E. D., Ploss A., Rice C. M., Dustin L. B. (2011) Hepatitis C virus induces interferon-λ and interferon-stimulated genes in primary liver cultures. Hepatology 54, 1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thomas E., Gonzalez V. D., Li Q., Modi A. A., Chen W., Noureddin M., Rotman Y., Liang T. J. (2012) HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology 142, 978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tacke R. S., Lee H. C., Goh C., Courtney J., Polyak S. J., Rosen H. R., Hahn Y. S. (2012) Myeloid suppressor cells induced by hepatitis C virus suppress T-cell responses through the production of reactive oxygen species. Hepatology 55, 343–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee H. C., Sung S. S., Krueger P. D., Jo Y. A., Rosen H. R., Ziegler S. F., Hahn Y. S. (2013) Hepatitis C virus promotes T-helper (Th)17 responses through thymic stromal lymphopoietin production by infected hepatocytes. Hepatology 57, 1314–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee H. C., Ziegler S. F. (2007) Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NFκB. Proc. Natl. Acad. Sci. U.S.A. 104, 914–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee H. C., Headley M. B., Iseki M., Ikuta K., Ziegler S. F. (2008) Cutting edge: Inhibition of NF-κB-mediated TSLP expression by retinoid X receptor. J. Immunol. 181, 5189–5193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jopling C. L., Yi M., Lancaster A. M., Lemon S. M., Sarnow P. (2005) Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309, 1577–1581 [DOI] [PubMed] [Google Scholar]
- 14. Yoshikawa T., Takata A., Otsuka M., Kishikawa T., Kojima K., Yoshida H., Koike K. (2012) Silencing of microRNA-122 enhances interferon-α signaling in the liver through regulating SOCS3 promoter methylation. Sci. Rep. 2, 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li M., Liu X., Zhou Y., Su S. B. (2009) Interferon-λs: the modulators of antivirus, antitumor, and immune responses. J. Leukoc. Biol. 86, 23–32 [DOI] [PubMed] [Google Scholar]
- 16. Osterlund P. I., Pietilä T. E., Veckman V., Kotenko S. V., Julkunen I. (2007) IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-λ) genes. J. Immunol. 179, 3434–3442 [DOI] [PubMed] [Google Scholar]
- 17. Thomson S. J., Goh F. G., Banks H., Krausgruber T., Kotenko S. V., Foxwell B. M., Udalova I. A. (2009) The role of transposable elements in the regulation of IFN-λ1 gene expression. Proc. Natl. Acad. Sci. U.S.A. 106, 11564–11569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Siegel R., Eskdale J., Gallagher G. (2011) Regulation of IFN-λ1 promoter activity (IFN-λ1/IL-29) in human airway epithelial cells. J. Immunol. 187, 5636–5644 [DOI] [PubMed] [Google Scholar]
- 19. Ding Q., Huang B., Lu J., Liu Y. J., Zhong J. (2012) Hepatitis C virus NS3/4A protease blocks IL-28 production. Eur. J. Immunol. 42, 2374–2382 [DOI] [PubMed] [Google Scholar]
- 20. Akira S., Takeda K. (2004) Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511 [DOI] [PubMed] [Google Scholar]
- 21. Ank N., Iversen M. B., Bartholdy C., Staeheli P., Hartmann R., Jensen U. B., Dagnaes-Hansen F., Thomsen A. R., Chen Z., Haugen H., Klucher K., Paludan S. R. (2008) An important role for type III interferon (IFN-λ/IL-28) in TLR-induced antiviral activity. J. Immunol. 180, 2474–2485 [DOI] [PubMed] [Google Scholar]
- 22. Bonjardim C. A. (2005) Interferons (IFNs) are key cytokines in both innate and adaptive antiviral immune responses–and viruses counteract IFN action. Microbes Infect. 7, 569–578 [DOI] [PubMed] [Google Scholar]
- 23. Zhu H., Butera M., Nelson D. R., Liu C. (2005) Novel type I interferon IL-28A suppresses hepatitis C viral RNA replication. Virol. J. 2, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hou W., Wang X., Ye L., Zhou L., Yang Z. Q., Riedel E., Ho W. Z. (2009) λ interferon inhibits human immunodeficiency virus type 1 infection of macrophages. J. Virol. 83, 3834–3842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Robek M. D., Boyd B. S., Chisari F. V. (2005) λ interferon inhibits hepatitis B and C virus replication. J. Virol. 79, 3851–3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wick G., Grundtman C., Mayerl C., Wimpissinger T. F., Feichtinger J., Zelger B., Sgonc R., Wolfram D. (2013) The immunology of fibrosis. Annu. Rev. Immunol. 31, 107–135 [DOI] [PubMed] [Google Scholar]
- 27. Marcello T., Grakoui A., Barba-Spaeth G., Machlin E. S., Kotenko S. V., MacDonald M. R., Rice C. M. (2006) Interferons α and λ inhibit hepatitis C replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 131, 1887–1898 [DOI] [PubMed] [Google Scholar]