

Abstract
Asbestos exposure results in pulmonary fibrosis (asbestosis) and malignancies (bronchogenic lung cancer and mesothelioma) by mechanisms that are not fully understood. Alveolar epithelial cell (AEC) apoptosis is important in the development of pulmonary fibrosis after exposure to an array of toxins, including asbestos. An endoplasmic reticulum (ER) stress response and mitochondria-regulated (intrinsic) apoptosis occur in AECs of patients with idiopathic pulmonary fibrosis, a disease with similarities to asbestosis. Asbestos induces AEC intrinsic apoptosis, but the role of the ER is unclear. The objective of this study was to determine whether asbestos causes an AEC ER stress response that promotes apoptosis. Using human A549 and rat primary isolated alveolar type II cells, amosite asbestos fibers increased AEC mRNA and protein expression of ER stress proteins involved in the unfolded protein response, such as inositol-requiring kinase (IRE) 1 and X-box–binding protein-1, as well as ER Ca²2+ release ,as assessed by a FURA-2 assay. Eukarion-134, a superoxide dismutase/catalase mimetic, as well as overexpression of Bcl-XL in A549 cells each attenuate asbestos-induced AEC ER stress (IRE-1 and X-box–binding protein-1 protein expression; ER Ca²2+ release) and apoptosis. Thapsigargin, a known ER stress inducer, augments AEC apoptosis, and eukarion-134 or Bcl-XL overexpression are protective. Finally, 4-phenylbutyric acid, a chemical chaperone that attenuates ER stress, blocks asbestos- and thapsigargin-induced AEC IRE-1 protein expression, but does not reduce ER Ca²2+ release or apoptosis. These results show that asbestos triggers an AEC ER stress response and subsequent intrinsic apoptosis that is mediated in part by ER Ca²2+ release.
Keywords: alveolar epithelium, asbestos, mitochondria, endoplasmic reticulum, apoptosis
Clinical Relevance
Because the mechanisms underlying asbestos-induced pulmonary toxicity are not fully established, this work examines how asbestos fibers activate the endoplasmic reticulum (ER) stress response to trigger mitochondria-regulated alveolar epithelial cell (AEC) apoptosis. Our findings show that important crosstalk between the ER and the mitochondria in AEC exposed to oxidative stress is important in the pathophysiologic events leading to oxidant-induced toxicity as seen in various degenerative disorders, respiratory diseases (e.g., asbestosis, pulmonary fibrosis), tumors, and aging.
Asbestos fibers are a naturally occurring group of mineral silicates (amphiboles and chrysotile) in which environmental and occupational exposure causes pulmonary and pleural fibrosis, lung cancer, and mesothelioma by mechanisms that are not fully established (see Refs. 1–3 for review). Alveolar epithelial cell (AEC) apoptosis is one important early event implicated in the pathogenesis of pulmonary fibrosis after exposure to various toxins, including asbestos (3, 4). Asbestos fibers are internalized by AECs soon after exposure, resulting in the production of iron-derived reactive oxygen species (ROS), DNA damage, and apoptosis (1–3). The mitochondria (intrinsic) apoptotic death pathway is mediated by proapoptotic Bcl-2 family members (e.g., Bax, Bak, and others) after activation by diverse stimuli, such as ROS, DNA damage, ceramide, and calcium, while antiapoptotic Bcl-2 family members (e.g., Bcl-2, Bcl-XL, etc.) are protective (5, 6). Apoptotic stimuli subsequently result in permeabilization of the outer mitochondrial membrane, reductions in mitochondrial membrane potential and apoptosome formation that activates caspase-9 and downstream caspase-3. We previously showed that iron-derived ROS from the mitochondria mediate asbestos-induced AEC DNA damage and apoptosis via the mitochondria-regulated death pathway, and that overexpression of Bcl-XL is protective (7, 8). Endoplasmic reticulum (ER) stress can also lead to intrinsic apoptosis, but its role after asbestos exposure has not been studied. The ER is responsible for both intracellular Ca2+ storage and for the folding, maturation, and transport of nascent proteins. Conditions that disrupt these processes, including oxidative stress, perturbation of Ca²2+, and/or accumulation of unfolded and/or misfolded proteins, result in ER stress (see Refs. 3, 4, 6 for review).
Accumulating evidence convincingly show that ER stress occurs in AECs undergoing apoptosis in patients with idiopathic pulmonary fibrosis (IPF), and may contribute to epithelial–mesenchymal transition, but the pathophysiologic significance of this finding is unknown (4, 9–12). Overexpression of mutant surfactant proteins in AECs results in misfolded proteins in the ER that causes ER stress and apoptosis, as well as enhanced susceptibility to bleomycin-induced pulmonary fibrosis (11, 13, 14). Given the radiographic and histopathologic similarities between IPF and asbestosis, ER stress may be important in asbestosis. A rodent model of asbestosis documented abnormal AEC ER morphology as assessed by electron microscopy (15). However, it is unknown whether asbestos fibers induce an AEC ER stress response and, if so, whether ER stress is important for activating intrinsic AEC apoptosis.
The ER and mitochondria are interconnected physically and functionally, thereby regulating mitochondrial metabolism, intracellular Ca2+ levels and complex cell survival/death signals (see Refs. 3, 5, 6 for review). Bcl-2 family members have an important role in regulating ER/mitochondrial cross-talk. Transient ER Ca2+ release activates prosurvival signaling (adaptive response), whereas intrinsic apoptotic agents require sustained ER Ca2+ release along with mitochondrial Bax/Bak binding. Bax and Bak are required to maintain homeostatic concentrations of ER Ca2+ necessary for regulating intrinsic apoptosis, although mitochondrial localization of Bax/Bak is sufficient for triggering BH3-only induced cell death (5, 6, 16–18). ER stress can trigger intrinsic apoptosis by activating ER transmembrane proteins involved in the unfolded protein response (UPR), including inositol-requiring kinase (IRE) 1, protein kinase R–like ER kinase (PERK) and activating transcription factor 6, which activate downstream UPR genes, including X-box–binding protein 1 (XBP-1) and C/EBP homologous protein (CHOP), as well as proapoptotic Bcl-2 family members (see Refs. 3–5 for review). Proapoptotic factors, such as Bax and Bak, modulate ER Ca2+ homeostasis, whereas Bcl-XL interacts directly with the inositol 1,4,5-triphosphate receptor (IP3R) to enhance spontaneous Ca2+ signaling (5, 6, 16–18). Overexpression of sarcoplasmic ER Ca2+ATP (SERCA) in Bax/Bak double-knockout murine embryonic fibroblasts restores ER Ca2+ levels and intrinsic apoptotic cell death in response to oxidative stress, suggesting that ER-localized Bax/Bak acts as an apoptotic gateway by inducing ER Ca2+ release (16–18). ER Ca2+ release is necessary, but not sufficient, for inducing intrinsic apoptosis by coordinating the ER stress survival signaling through IRE-1α/TNF receptor–associated factor 2 that results in activation of apoptosis signal–regulating kinase 1 and c-Jun N-terminal kinase (6, 19). Collectively, these data show important cross-talk between the ER and mitochondria in regulating intrinsic apoptosis, but the relevance of this cross-talk to AECs exposed to asbestos is unknown.
We reasoned that asbestos causes an AEC ER stress response that results in ER Ca2+ release important in augmenting mitochondria-regulated apoptosis. We show that amosite asbestos fibers induce AEC ER stress, as evidenced by increased mRNA and protein expression of ER stress proteins (IRE-1, XBP-1 spliced, and CHOP), as well as ER Ca²2+ release, in human A549 and rat alveolar epithelial type (AT) 2 cells. Asbestos-induced AEC ER stress and apoptosis were reduced by eukarion (Euk) 134, a superoxide dismutase (SOD)/catalase mimetic that attenuates mitochondrial ROS production (20), as well as in A549 cells overexpressing Bcl-XL. Thapsigargin, a known ER stress inducer, also augments AEC apoptosis, and both Euk-134 or Bcl-XL overexpression are protective. 4-Phenylbutyric acid (4-PBA), a small chemical chaperone known to block the UPR, reduces asbestos- and thapsigargin-induced AEC IRE-1 protein expression, but does not attenuate ER Ca2+ release or apoptosis. These findings demonstrate that asbestos stimulates an AEC ER stress response, and suggest an important role for ER Ca²2+ release in mediating AEC intrinsic apoptosis.
Materials and Methods
Reagents
Amosite asbestos fibers used in this study were Union International Centere le Cancer reference standard samples kindly supplied by Drs. V. Timbrell (21) and Andy Ghio (U.S. Environmental Protection Agency), and were handled as described in the online supplement.
Cell Culture
A549 and primary isolated rat AT2 cells were plated in six-well plates and grown to confluence before adding asbestos, H2O2, or thapsigargin for various time periods (1, 4, and 24 h) as described in the online supplement. A549 cells that stably overexpress Bcl-XL were used as described elsewhere (7). Primary isolated rat AT2 cells were isolated from the lungs of Sprague-Dawley rats, as previously described and approved by the Animal Care and Use Committee for these studies (7, 8).
Real-Time RT-PCR
Real-time RT-PCR analysis of ER-UPR mRNA was performed by isolating total RNA from treated wells, synthesizing cDNA from 2 μg of RNA using oligo-d(T) primers by reverse transcriptase Superscript III (Invitrogen, Carlsbad, CA), and cDNA production was performed with specific TaqMan probes that were commercially available (Taqman Assays; Life Technologies/Applied Biosystems, Carlsbad, CA) to detect human IRE-1, CHOP, XBP-1, and glucose-regulated protein (GRP) 78 by real-time RT-PCR, as described in the online supplement. The relative expression of each was determined from a cDNA standard curve and normalized by the expression value of a control TaqMan probe.
Western Analysis
Cell lysates were collected and immunoblotting was performed as previously described (22) using antibodies that included monoclonal antibodies directed against XBP-1 (1:500; ABCam, Cambridge, MA; detects both the nonspliced [29 kD] and active, spliced [40 kD] components), IRE-1 (1:500; Cell Signaling Technologies, Danvers, MA), CHOP (1:500; Cell Signaling Technologies), and actin (1:200; Santa Cruz Biotechnologies, Santa Cruz, CA). The protein bands were quantified by densitometry, as described in the online supplement.
Measurement of Intracellular Ca2+
A549 and rat AT2 cells were loaded with FURA-2/AM, which was used as a fluorescence indicator of intracellular free Ca2+ levels, as previously described (23). As described in the online supplement, changes in intracellular calcium concentration were expressed using conventional F340:F380 ratio (the ratio of the Fura-2 fluorescence intensities measured at wavelengths of 340 and 380 nm).
Apoptosis Assay
Apoptosis was assessed using a histone-associated DNA fragmentation (mono and oligo nucleosomes) ELISA assay (Roche Diagnostics, Indianapolis, IN), as previously described (7, 8, 22). Asbestos, H2O2, or thapsigargin were added to the cultured A549 cells 24 hours before performing the assay.
Immunofluorescence Microscopy
As described in the online supplement, A549 cells in the presence or absence of amosite asbestos (5 or 25 μg/cm2) were immunostained for COX IV (mitochondria; Cell Signaling Technology, Danvers, MA), calnexin (ER; BD Transduction Laboratories, San Jose, CA), and Hoechst 34580 (nucleus; Invitrogen, Grand Island, NY). Individual cells were stratified into three categories: (1) cells expressing COX IV and calnexin with low colocalization (arbitrarily defined six or fewer colocalized areas per cell); (2) cells expressing COX IV and calnexin with high colocalization (arbitrarily defined > 6 colocalized areas per cell); and (3) cells expressing calnexin, but negligible COX IV (mitochondrial dysfunction).
Statistical Analysis
Data are expressed as the means (±SEM; n = 6 unless otherwise stated) and analyzed as detailed in the online supplement.
Results
Asbestos Causes AEC ER Stress Response
To determine whether asbestos induces an ER stress response in AECs, we assessed A549 cell mRNA expression of IRE-1, XBP-1, GRP78/BIP, and CHOP by real-time RT-PCR after exposure to amosite asbestos (5–25 μg/cm2) for various periods of time (0.5, 4, and 24 h). As compared to control, asbestos (5 μg/cm2) significantly increased mRNA expression of IRE-1, XBP-1, CHOP, and GRP78/Ig heavy chain binding protein (BiP) as early as 30 minutes and, in the case of XBP-1 and CHOP, the increases persisted over 24 hours (Figure 1A). Similar changes in XBP-1 and CHOP mRNA were noted after exposure of A549 cells to a higher dose of asbestos (25 μg/cm2) that causes apoptosis (7, 8, 22), but the increases were less pronounced at 24 hours when apoptosis was evident (Figure 1B; see also Figure E1A in the online supplement). After asbestos (25 μg/cm2) exposure for 30 minutes or 4 hours, time points where we see little cell death (7), asbestos significantly increased XBP-1 and CHOP mRNA by nearly twofold (Figure 1B and Figure E1A). As shown in Figure 1C, we confirmed by RT-PCR and gel electrophoresis that asbestos increased the spliced isoform of XBP-1, as did H2O2 (100 μM) and thapsigargin (80 μM).
Figure 1.

Asbestos fibers trigger endoplasmic reticulum (ER) stress mRNA expression. (A) A549 cells were exposed to amosite asbestos (5 μg/cm2) for various periods, and then mRNA for inositol-requiring kinase (IRE) 1, C/EBP homologous protein (CHOP), glucose-regulated protein (GRP) 78/BIP, and X-box–binding protein (XBP) 1 were assessed by real-time RT-PCR, and data expressed as fold control normalized to 18S. (B) A549 cells were exposed to various doses of amosite asbestos (5–25 μg/cm2) for various periods, and then mRNA for XBP-1 was assessed by real-time RT-PCR. *P < 0.05 versus control (n = 5). (C) Using RT-PCR and gel electrophoresis, asbestos (25 μg/cm2) was found to increase the spliced isoform of XBP-1 in A549 cells after a 4-hour exposure period, as did H2O2 (100 μM) and thapsigargin (80 μM); blot is representative of three separate experiments.
Given the findings previously described here, plus work by other groups implicating a role for IRE-1 and downstream signaling by XBP-1 in modulating ER stress–induced intrinsic apoptosis and pulmonary fibrosis (9–11, 14, 19), we focused on this arm of ER stress activation in AECs. Amosite asbestos (5–25 μg/cm2) augmented IRE-1 protein expression in both human A549 (Figure 2A) and primary isolated rat AT2 cells (Figure 2B). Furthermore, asbestos-induced IRE-1 expression was comparable to thapsigargin, a known ER stress inducer. Asbestos also increased XBP-1 spliced protein (active version) expression after a 1- to 24-hour exposure period (Figure 2C). In general, 1.5- to twofold increases in IRE-1 and XBP-1 spliced protein expression were evident as early as 1 hour, and persisted over 24 hours. H2O2, an endogenous oxidative stress that acts similarly to asbestos in triggering DNA damage, p53 activation, and mitochondria-regulated apoptosis (8, 22), also stimulated AEC IRE-1 and XBP-1 spliced protein expression. Furthermore, asbestos and H2O2 each augmented A549 cell BiP protein expression over 1 to 4 hours to levels that were comparable to thapsigargin (Figure E2). Taken together, these findings suggest that AEC ER stress activation occurs after exposure to both exogenous (asbestos fibers) and endogenous (H2O2) oxidative stress (Figure 2 and Figure E2).
Figure 2.

Oxidative stress from asbestos or H2O2 induces alveolar epithelial cell (AEC) ER stress as assessed by protein expression of IRE-1 and XBP-1 (spliced). (A) A549 cells were exposed to amosite asbestos (5–25 μg/cm2) or H2O2 (20–50 μM) for 1 hour, and then IRE-1 protein expression was assessed by Western blot analysis (blot is representative of four separate experiments). (B) Rat alveolar epithelial type (AT) 2 cells were exposed to amosite asbestos (25 μg/cm2), H2O2 (20 μM), or thapsigargin (Thap; 80 μM) for 1 hour, and then IRE-1 protein expression was assessed by Western blot analysis (representative blot of eight separate experiments). (C) A549 cells were exposed to amosite asbestos (5–25 μg/cm2) or H2O2 (20 μM) for various periods of time, and then XBP-1 spliced protein expression was assessed by Western blot analysis (blot is representative of six separate experiments). *P < 0.05 versus control.
Asbestos Stimulates AEC ER Ca2+ Release and Apoptosis
Calcium release from the ER to the mitochondria is triggered by several intrinsic apoptotic stimuli (6, 24, 25). However, ER Ca2+ release to the mitochondria appears necessary, but not sufficient, for inducing intrinsic apoptosis in some cells (19, 24, 25). Crocidolite asbestos–induced release of intracellular Ca2+ stores is important in mediating DNA damage in human white blood cells (26). We reasoned that oxidative stress (e.g., asbestos fibers and H2O2) augments AEC ER Ca2+ release, which may lead to intrinsic apoptosis. To address AEC ER Ca2+ release, we used FURA-2–loaded AECs exposed to amosite asbestos, H2O2, or thapsigargin. As shown in Figure 3, amosite asbestos (25 μg/cm2) and H2O2 (20 μM) rapidly augmented Ca2+ release from A549 and rat AT2 cells in a manner comparable to thapsigargin (80 μM). In general, Ca2+ release occurred within 5 to 10 minutes, and remained slightly elevated for over 12 minutes of monitoring. As shown in Figure 3D, asbestos-induced Ca2+ release occurred in Ca2+-free media, implicating an important role for the ER as the source of the Ca2+. In contrast, control AT2 cells not exposed to asbestos demonstrated negligible Ca2+ release for over 15 minutes of monitoring (Figure 3F). These findings demonstrate that AEC oxidative stress after exposure to asbestos fibers or H2O2 results in intracellular ER Ca2+ release that is similar to thapsigargin.
Figure 3.
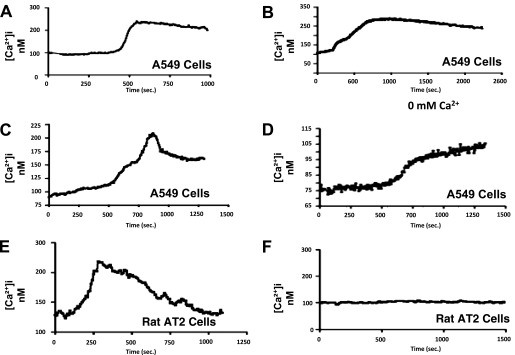
Oxidative stress from asbestos or H2O2 induces AEC Ca2+ signaling that is similar to a known ER stressor, thapsigargin. A549 cells were exposed to (A) thapsigargin (80 μM), (B) H2O2 (50 μM), or (C) amosite asbestos (25 μg/cm2) for 15 minutes, and Ca2+ signaling assessed by a Fura-2 assay was evident under all conditions. (D) A549 cells exposed to amosite asbestos (25 μg/cm2) for 15 minutes in a Ca2+-free media still showed evidence of Ca2+ release, suggesting that the ER is a source of Ca2+. (E) Rat AT2 cells were exposed to amosite asbestos (25 μg/cm2) or control media without asbestos (F) for 15 minutes; Ca2+ release was evident only in the asbestos-exposed AT2 cells. Representative data from at least three separate experiments are shown.
Previous studies have established that agents inducing ER stress, such as thapsigargin and tunicamycin, can trigger apoptosis in various cells (27, 28). To confirm whether thapsigargin alone causes AEC apoptosis, we used a highly sensitive DNA fragmentation assay. Thapsigargin (30–200 nM) augmented A549 cell apoptosis in a dose-dependent manner after a 24-hour exposure period (Figure E3A). To assess whether blocking ER Ca2+ release attenuates oxidant-induced AEC apoptosis, A549 cells were exposed to either amosite asbestos (25 μg/cm2) or H2O2 (100 μM) for 24 hours in the presence or absence of BAPTA (10 μM), an intracellular Ca2+ chelator known to inhibit ER stress–induced intrinsic apoptosis in A549 cells exposed to the antitumor agent, rhein (29). As expected, asbestos and H2O2 each induced apoptosis by two- to 2.5-fold, respectively (Figure E3B). BAPTA modestly reduced asbestos- and H2O2-induced apoptosis, although these differences did not reach statistical significance (Figure E3B). Collectively, these data show that oxidative stress after asbestos or H2O2 exposure induces intracellular AEC Ca2+ release, and suggest that ER Ca2+ release may be important in mediating AEC apoptosis.
Euk-134 Attenuates Asbestos-Induced ER Stress and Apoptosis
We previously demonstrated that mitochondria-derived ROS are important for mediating asbestos-induced AEC intrinsic apoptosis (7, 8, 22). Furthermore, our group showed that Euk-134, a combined SOD and catalase mimetic, prevents particulate matter–induced mitochondrial ROS production assessed using a mitochondria-targeted ratiometric sensor, as well as intrinsic apoptosis of human A549 and primary isolated rat AT2 cells (20). Because antioxidants, including those targeted to the mitochondria, attenuate ER stress and apoptosis (30–33), we reasoned that Euk-134 would be protective in our model. To determine whether Euk-134 attenuates asbestos-induced AEC ER Ca2+ release, we used FURA-2–loaded A549 cells that had been pretreated with Euk-134 (20 μM) or control media and then exposed to amosite asbestos (25 μg/cm2). As compared to untreated asbestos-exposed A549 cells, Euk-134 greatly diminished asbestos-induced Ca2+ release over the 15 minutes of monitoring (Figures 4A and 4B). To address whether Euk-134 alters AEC ER stress and downstream apoptosis, we examined the protective effects of Euk-134 (20 μM) against amosite asbestos- (25–50 μg/cm2) and thapsigragin (80 μM) -induced IRE-1 protein expression and apoptosis. Euk-134 prevented asbestos- and thapsigragin-induced IRE-1 protein expression after a 4-hour exposure period (Figure 4C), as well as apoptosis assessed by DNA fragmentation at 24 hours (Figure 4D). Thus, these findings suggest an important upstream role for mitochondrial-derived ROS in mediating asbestos-induced AEC ER stress response and subsequent intrinsic apoptosis.
Figure 4.

Eukarion (Euk) 134 attenuates asbestos-induced ER stress and apoptosis. A549 cells were pretreated with either control media or Euk-134 (20 μM) for 2 hours and then exposed to amosite asbestos (25 μg/cm2) in the absence (A) or presence (B) of Euk-134 for 15 minutes to assess Ca2+ release by a Fura-2 assay. Euk-134 reduces asbestos-induced Ca2+ release (representative data depicted from three separate experiments). (C) A549 cells were pretreated as described previously here, then exposed to either control media, amosite asbestos (5–25 μg/cm2), or thapsigargin (80 μM) in the absence or presence of Euk-134 for 1 hour, and then IRE-1 protein expression was assessed. A densitometric analysis of IRE-1 expression corrected for protein loading from three separate experiments is shown above the blot. *P < 0.05 versus control, †P < 0.05 versus asbestos or TG in the absence of Euk-134 (n = 3). (D) A549 cells were pretreated as described previously here, then exposed to control media, amosite asbestos (5–25 μg/cm2), or thapsigargin (TG; 80 μM) in the absence (blue bars) or presence (red bars) of Euk-134 for 24 hours, and then apoptosis was assessed by DNA fragmentation expressed as fold control. *P < 0.05 versus control, †P < 0.05 versus asbestos or TG in the absence of Euk-134 (n = 6).
Bcl-XL Overexpression Blocks Asbestos-Induced AEC ER Stress and Apoptosis
Others, as well as our group, have shown that overexpression of antiapoptotic Bcl-2 family members, such as Bcl-XL, prevent the reductions in mitochondrial membrane potential and intrinsic apoptosis after exposure to various apoptogenic agents, including asbestos (5, 7). Antiapoptotic Bcl-2 family members, which act at both the mitochondria and the ER, modulate ER Ca2+ release to the mitochondria in part by controlling the phosphorylation state of IP3R located in the mitochondria-associated membrane via its BH4 domain (5, 6, 17, 24, 25). We reasoned that the protective effects of Bcl-XL overexpression in AECs occurs by attenuating asbestos-induced ER stress response and Ca2+ release. To address this possibility, we used A549 cells that stably overexpress Bcl-XL as previously characterized (7). As compared to lentivirus empty vector–transfected A549 cells, Bcl-XL overexpression greatly diminished amosite asbestos (25 μg/cm2)–induced Ca2+ release over the 15 minutes of monitoring (Figures 5A and 5B). To determine whether Bcl-XL overexpression reduces AEC ER stress, we examined the effects of Bcl-XL overexpression against asbestos- (5–25 μg/cm2) and thapsigragin (80 μM)-induced IRE-1 and XBP-1 spliced protein expression over 4 hours. Bcl-XL–overexpressing A549 cells had negligible increases in asbestos- and thapsigragin-induced ER stress response (IRE-1 and XBP-1 spliced protein expression) as compared to lentivirus empty vector–transfected controls (Figure 5C). Finally, we determined whether Bcl-XL–overexpressing A549 cells have diminished thapsigargin-induced apoptosis similar to what we previously reported in asbestos-exposed Bcl-XL–overexpressing A549 cells (7). As compared to lentivirus empty vector–transfected controls, Bcl-XL–overexpressing A549 cells have significantly reduced amosite asbestos- (25–50 μg/cm2) and thapsigargin (80 μM)-induced apoptosis (Figure 5D). Taken together, these findings show an important role for Bcl-XL in regulating asbestos-induced AEC ER stress response, Ca2+ release, and subsequent intrinsic apoptosis.
Figure 5.

Bcl-XL reduces asbestos-induced ER stress and apoptosis. As compared to lentivirus empty vector–transfected A549 cells (A), A549 cells that overexpress Bcl-XL (B) have reduced baseline Ca2+ levels and less asbestos-induced Ca2+ release as assessed by a Fura-2 assay over 15 minutes (representative data depicted from three separate experiments). (C) Lentivirus empty vector–transfected A549 cells and A549 cells that overexpress Bcl-XL were exposed to either control media, amosite asbestos (5–25 μg/cm2), or thapsigargin (80 μM) for 1 hour, and then IRE-1, XBP-1 spliced, and Bcl-XL protein expression was assessed. A densitometric analysis of IRE-1 and XBP-1 spliced expression corrected for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) from at least three separate experiments shows negligible asbestos- or thapsigargin-induced protein expression. (D) As compared to lentivirus empty vector–transfected A549 cells (blue bars), A549 cells that overexpress Bcl-XL (red bars) have reduced amosite asbestos– (25–50 μg/cm2) or thapsigargin (TG; 80 μM)-induced apoptosis, as assessed by DNA fragmentation at 24 hours, expressed as fold control. *P < 0.05 versus control, †P < 0.05 versus asbestos or TG in lentivirus empty vector–transfected A549 cells (n = 3–6).
Asbestos Does Not Alter Mitochondria–ER Colocalization
The mitochondria and the ER are intimately associated, and this connection can be increased by apoptotic signals (34, 35). However, the association between the mitochondria and the ER in AECs, and whether this association is altered in the setting of intrinsic apoptosis after asbestos exposure, are unknown. To determine whether asbestos alters AEC mitochondria–ER colocalization, we used immunofluorescence microscopy to label A549 cell mitochondria (COX IV) and ER (calnexin) after a 4-hour exposure period in the presence or absence of amosite asbestos. As shown in Figure 6, semiquantitative analysis revealed that, under control conditions, nearly 90% of the cells had evidence of colocalization of the mitochondria and the ER, with roughly 45% of the cells with low-level colocalization (arbitrarily defined as six or fewer colocalized areas per cell) and another 45% with high-level colocalization (defined as over six colocalized areas per cell). As compared to controls, amosite asbestos (5–25 μg/cm2) afforded negligible changes in the levels of mitochondria–ER colocalization: low- and high-level colocalization was approximately 38 and 45%, respectively, for over 200 cells assessed for each condition. Consistent with our previous reports (7, 8), asbestos induced AEC mitochondrial dysfunction, as assessed by loss of COX IV immunostaining (Figure 6D).
Figure 6.

Asbestos causes AEC mitochondrial dysfunction without altering colocalization of the mitochondria and the ER. A549 cells were exposed to control media (A and B) or 25 μg/cm2 amosite asbestos (C and D) for 4 hours, and then immunofluorescence with semiquantitative analysis of the mitochondria (COXIV; punctate red), the ER (calnexin; punctate green), colocalization (punctate yellow–orange; arrows), and mitochondrial dysfunction (loss of mitochondrial red staining) (D) was performed as described in the Materials and Methods. The graph depicts the semiquantitative analysis for control and asbestos (Asb-5, 5 μg/cm2; Asb-25, 25 μg/cm2)-exposed cells; x-axis (percentage of 200 cells analyzed for each condition). A representative cell with low mitochondria–ER colocalization (A), high mitochondria–ER colocalization (B and C), and mitochondrial dysfunction (D) are shown. Data are from one of two separate experiments with similar results.
4-PBA Attenuates Asbestos- and Thapsigargin-Induced AEC IRE-1 Expression, but Affords Negligible Protection against ER Ca2+ Release and Apoptosis
Small molecular chaperones, such as 4-PBA, can improve ER protein folding and mitigate the ER–UPR (36). The role of 4-PBA is being investigated in the treatment of diverse ER-related diseases involving misfolded proteins, such as cystic fibrosis, diabetes, Alzheimer’s disease, cardiac fibrosis, and myofibroblast differentiation of lung fibroblasts important in pulmonary fibrosis (36–40). To determine whether 4-PBA is protective in our model, we treated AECs with 4-PBA (2.5 mM) for 4 hours and then assessed amosite asbestos–, H2O2-, and thapsigargin-induced IRE protein expression at 4 hours, ER Ca2+ release over 15 minutes, and apoptosis at 24 hours. As expected, 4-PBA reduced asbestos-, H2O2-, and thapsigargin-induced increases in A549 cell IRE-1 protein expression (Figure 7A). Notably, 4-PBA afforded negligible protection against asbestos-induced AT2 cell ER Ca2+ release (Figure 7B), thapsigargin-induced A549 cell ER Ca2+ release (Figure 7C), and asbestos-induced apoptosis in human A549 or rat AT2 cells (Figure 7D).
Figure 7.

4-Phenylbutyric acid (4-PBA) prevents oxidant- (asbestos and H2O2) and thapsigargin-induced AEC IRE-1 expression, but does not attenuate Ca2+ release or apoptosis. (A) A549 cells were pretreated with either control media (blue bars) or 4-PBA (red bars) for 4 hours, then exposed to amosite asbestos (25 μg/cm2), H2O2 (20 μM), or thapsigargin (80 μM) for 4 hours, and IRE-1 protein expression was assessed. The densitometric analysis of IRE-1 expression for each condition corrected for actin from at least three separate experiments is shown. (B) Rat AT2 cells or (C) A549 cells were pretreated with 4-PBA, as described previously, and then exposed to either (B) amosite asbestos (25 μg/cm2) or (C) thapsigargin (80 μM) in the absence (blue lines) or presence (red lines) of 4-PBA for 15 minutes while assessing Ca2+ release by a Fura-2 assay (representative data depicted from two separate experiments). (D) A549 (blue bars) and rat AT2 (red bars) cells were pretreated as described previously, then exposed to either control media (Con), amosite asbestos (Asb; 25 μg/cm2), 4-PBA (2.5 mM), or asbestos plus 4-PBA for 24 hours, and then apoptosis was assessed by DNA fragmentation. Data are expressed as fold control. *P < 0.05 versus control (n = 6); †P < 0.05 versus asbestos, H2O2, or thapsigargin in the absence of 4-PBA.
Discussion
AEC apoptosis is an important early event implicated in the pathogenesis of pulmonary fibrosis, including that due to asbestos exposure. The major findings in this study are that exposure of AECs to oxidative stress (e.g., amosite asbestos fibers or H2O2) induces an ER stress response consisting of ER Ca2+ release and increased mRNA and protein expression of IRE-1 and XBP-1 spliced that are comparable to thapsigargin, a known ER stress inducer. Furthermore, we show that Euk-134, a SOD/catalase mimetic, as well as Bcl-XL overexpression each attenuate asbestos- and thapsigargin-induced ER stress response (IRE-1 expression and ER Ca2+ release) and apoptosis. Finally, we show that 4-PBA reduced asbestos- and thapsigargin-induced AEC IRE-1 expression, but did not attenuate ER Ca²+ release or apoptosis. Collectively, our results demonstrate that asbestos and H2O2 activate an AEC ER stress response, and that oxidant-induced ER Ca²+ release has an important role in promoting AEC apoptosis.
An important observation in this study is that asbestos fibers induce an ER stress response in cultured AECs—both human A549 and primary isolated rat AT2 cells. Asbestos-induced AEC ER stress is supported by several lines of evidence that includes the following: (1) increased mRNA expression of molecules involved in the ER UPR (e.g. IRE-1, XBP-1, BiP, and CHOP); (2) increased ER UPR protein expression (e.g. IRE-1, XBP-1 spliced, BiP, and, to a lesser extent, CHOP); and (3) augmented ER Ca²2+ release, even in the presence of a Ca²2+-free media (Figures 1–3; Figures E1 and E2). These findings of oxidative stress–induced ER stress in AECs parallel our earlier studies showing that asbestos fibers and H2O2 induce comparable levels of mitochondrial dysfunction, p53 activation, and intrinsic apoptosis in A549 and rat AT2 cells (7, 8, 22). Our observation that asbestos activates an ER stress response in AECs concurs with prior in vitro studies showing that asbestos stimulates ER stress in breast cancer and white blood cells (26, 41), as well as a rat model of asbestosis demonstrating abnormalities in the ER of AECs (15). Although we chose to focus on AEC ER stress pathway involving IRE-1 expression and ER Ca²2+ release given their implicated roles in apoptosis, it will be of interest determining whether other arms of the ER stress pathway contribute to AEC apoptosis after asbestos exposure (e.g. PERK and ATF6).
Similar to various groups working in other cell types (27, 28), we show that a well known ER stressor (thapsigargin) induces AEC apoptosis (Figure E3). Collectively, our data support a model in which oxidative stress from asbestos or H2O2 induce AEC ER stress that can lead to apoptosis (Figure E4). This model is consistent with the observations of several groups showing that the alveolar epithelium in patients with IPF demonstrates colocalization of ER stress response in apoptotic AECs (4, 9, 10). Interestingly, using a novel murine model of AEC-specific mutant surfactant protein expression, Lawson and colleagues (14) demonstrated that AEC ER stress occurs in vivo, but that this does not cause pulmonary fibrosis unless the mice were also exposed to a low dose of a fibrogenic agent (e.g., bleomycin). Notably, augmented pulmonary fibrosis in the mice with AEC-specific mutant surfactant protein expression exposed to low-dose bleomycin was associated with increased AEC apoptosis. Future in vivo studies are warranted to assess the causal role of AEC ER stress in the pathogenesis of asbestosis.
A novel finding in this study is that Euk-134, a SOD/catalase mimetic that we have previously shown blocks particulate matter–induced mitochondrial ROS production and intrinsic AEC apoptosis, as well as Bcl-XL overexpression each attenuated asbestos- and thapsigargin-induced ER stress response (IRE-1 expression and ER Ca2+ release) as well as apoptosis (Figures 5 and 6). We also confirm that the ER and mitochondria are closely associated in AECs, as assessed by immunofluorescence microscopy with semiquantitative analysis (Figure 6). Moreover, we showed that asbestos exposure does not alter this close association, despite evidence of inducing mitochondrial dysfunction. These findings implicating mitochondrial ROS production as the principal source of asbestos-induced free radicals in our model are in accord with our prior studies that include the following: (1) p0-A549 cells lacking mitochondrial DNA and incapable of mitochondrial ROS production, are protected against asbestos-induced DNA damage, p53 activation, and apoptosis (7, 8, 22); (2) using highly sensitive reduction–oxidation–sensitive green fluorescent proteins targeted to the mitochondria or cytoplasm to detect ROS production, we have previously reported that the mitochondria are the primary source of ROS generation (42); (3) the mitochondria-regulated (intrinsic) death pathway is the primary pathway mediating AEC apoptosis in vitro (7); and (4) a mitochondria-targeted DNA repair protein (8-oxoguanine DNA glycosylase) or mitochondrial aconitase 2 overexpression each prevent oxidant-induced AEC apoptosis, despite high levels of mitochondrial ROS production (42). Our data with asbestos-exposed AECs implicating mitochondrial ROS in triggering ER stress also concur with studies showing that antioxidants, including those targeted to the mitochondria, attenuate oxidative stress–induced ER stress and apoptosis in other cell types (30–33). Mitochondria-targeted antioxidants protect against oxidant-induced ER stress and intrinsic apoptosis of pancreatic beta cells (31, 33). Mitochondrial ROS activate the UPR in cancer cells, thereby promoting cell survival under glucose deprivation conditions (43). Although not examined in our study, there is likely a spectrum of mitochondrial ROS production whereby low levels of ROS support cell survival, but higher levels promote cell death. We acknowledge that mitochondria-independent ROS derived from the plasma membrane and/or cytoplasmic sources may contribute to the protective effects of Euk-134. However, work by others (44–48), as well as our group (20), using a variety of experimental systems has established that Euk-134 primarily functions by limiting mitochondrial ROS production. Taken together, these observations implicate important cross-talk between the ER and the mitochondria in AECs exposed to oxidative stress that determines whether cells will survive or undergo apoptosis.
Numerous studies show that Ca2+ transfer from the ER to the mitochondria, which acts in conjunction with a variety of apoptotic signals, is a crucial trigger for opening the permeability transition pore that irreversibly commits cells to intrinsic apoptosis (5, 6). Several lines of evidence presented herein implicate an important role for ER Ca2+ release in mediating oxidant-induced AEC apoptosis. First, similar to thapsigargin, asbestos and H2O2 each trigger AEC ER Ca2+ release within minutes after exposure. Second, Euk-134 and overexpression of Bxl-XL each reduce AEC ER Ca2+ release as well as apoptosis after exposure to oxidative stress or thapsigargin. Finally, 4-PBA, which facilitates ER protein folding and reduces the ER UPR (36–40), attenuates oxidative stress–induced IRE-1 protein expression, as expected, but does not prevent AEC ER Ca2+ release or apoptosis (Figure 7). This suggests that ER Ca2+ release, rather than activation of the IRE-1 pathway, is important in mediating oxidative stress–induced AEC apoptosis. The detailed molecular mechanism(s) underlying ER-mitochondria cross-talk in AECs exposed to asbestos that promotes apoptosis is unclear. Some possible mechanisms involved in our model are suggested by the work of several groups demonstrating that the antiapoptotic molecule, Bcl-XL, interacts directly with the IP3R located on the ER to empty ER Ca2+ stores, thereby preventing mitochondrial Ca2+ loading, whereas proapoptotic molecules (e.g., BAX) enhance ER Ca2+ release to the mitochondria in part by binding to Bcl-XL to counteract its effect on IP3R (6, 16, 17). Bax/Bak double-knockout murine embryonic fibroblasts have reduced ER Ca2+ levels, and are resistant to intrinsic apoptotic cell death, an effect that can be overcome by overexpression of sarcoplasmic ER Ca2+ATP, suggesting that ER-localized Bax/Bak acts as an apoptotic gateway by inducing ER Ca2+ release (16–18). Furthermore, recent studies have established that close ER–mitochondria juxtapositioning at mitochondria-associated membranes enables ER Ca2+ transfer to the mitochondria by protein–protein interactions between the IP3R and the voltage-dependent anion channel located on the outer mitochondrial membrane, followed by Ca2+ transfer via the mitochondrial Ca2+ uniporter located in the inner mitochondrial membrane (6, 24, 25, 34, 35). Another possibility is that the inability of 4-PBA to prevent asbestos-induced AEC apoptosis in our model may be mediated by a recently described effect of 4-PBA in preventing prosurvival signaling via NF-κB in lung epithelial cells (49). Although the precise molecular mechanism(s) that account for asbestos-induced AEC mitochondria–ER cross-talk that drives intrinsic apoptosis, as well as the in vivo relevance of our in vitro findings, await further study, our data suggest an important role for mitochondrial ROS production, ER Ca2+ release, and modulation by Bcl-2 family members.
In conclusion, our data establish an innovative role for ER Ca2+ release in mediating oxidant-induced AEC mitochondrial dysfunction and apoptosis. We demonstrate that Euk-134, a SOD/catalase mimetic, as well as Bcl-XL overexpression, each reduce asbestos- and thapsigargin-induced ER stress response (IRE-1 expression and ER Ca2+ release) and apoptosis. Asbestos-induced AEC ER stress and apoptosis occur despite not altering the close association between the ER and mitochondria. Finally, 4-PBA, a small molecular chaperone known to inhibit the ER UPR, reduces asbestos- and thapsigargin-induced AEC IRE-1 expression, but does not attenuate ER Ca²2+ release or apoptosis. A hypothetical model illustrating how these events might be coordinated is shown in Figure E3. The coupling of mitochondrial ROS production to an ER stress response, especially ER Ca²2+ release, is crucial for mediating intrinsic AEC apoptosis after exposure to oxidative stress, such as asbestos fibers. We reason that these findings, showing cross-talk between the ER and the mitochondria in AECs exposed to oxidative stress, are important in the pathophysiologic events leading to oxidant-induced toxicity, as seen in various degenerative disorders, respiratory diseases (e.g. asbestosis and pulmonary fibrosis), tumors, and aging.
Footnotes
This work was supported by a Department of Veterans Affairs Merit Review grant (D.W.K.) and National Institutes of Health grants RO1ES020357 (D.W.K.), P30 HL101292 (A.P.L.), and P01HL071643 (K.R. and N.S.C.).
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2013-0053OC on July 25, 2013
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Mossman BT, Lippmann M, Hesterberg TW, Kelsey KT, Barchowsky A, Bonner JC. Pulmonary endpoints (lung carcinoma and asbestosis) following inhalation exposure to asbestos. J Toxicol Environ Health. 2011;14:76–121. doi: 10.1080/10937404.2011.556047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang SXL, Jaurand M-C, Kamp DW, Whysner J, Hei TK. Role of mutagenicity in asbestos fiber-induced carcinogenicity and other diseases. J Toxicol Environ Health. 2011;14:179–245. doi: 10.1080/10937404.2011.556051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu G, Cheresh P, Kamp DW. Molecular basis of asbestos-induced lung disease. Annu Rev Pathol Mech Dis. 2013;8:161–187. doi: 10.1146/annurev-pathol-020712-163942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302:L721–L729. doi: 10.1152/ajplung.00410.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szegezdi E, MacDonald DC, Chonghaile TN, Gupta S, Samali A. Bcl-2 family on guard at the ER. Am J Physiol Cell Physiol. 2009;296:C941–C953. doi: 10.1152/ajpcell.00612.2008. [DOI] [PubMed] [Google Scholar]
- 6.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signaling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 7.Panduri V, Weitzman SA, Chandel N, Kamp DW. The mitochondria-regulated death pathway mediates asbestos-induced alveolar epithelial cell apoptosis. Am J Respir Cell Mol Biol. 2003;28:241–248. doi: 10.1165/rcmb.4903. [DOI] [PubMed] [Google Scholar]
- 8.Panduri V, Weitzman SA, Chandel NS, Kamp DW. Mitochondrial-derived free radicals mediate asbestos-induced alveolar epithelial cell apoptosis. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1220–L1227. doi: 10.1152/ajplung.00371.2003. [DOI] [PubMed] [Google Scholar]
- 9.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1119–L1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 10.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle R-M, Seeger W, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong Q, Zhou B, Ann DK, Minoo P, Liu Y, Banfalvi A, Krishnaveni MS, Dubourd M, Demaio L, Willis BC, et al. Role of endoplasmic reticulum stress in epithelial-mesenchymal transition of alveolar epithelial cells: effects of misfolded surfactant protein. Am J Respir Cell Mol Biol. 2011;45:498–509. doi: 10.1165/rcmb.2010-0347OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanjore H, Cheng D-S, Degryse AL, Zoz DF, Abdolrasulnia R, Lawson WE, Blackwell TS. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. 2011;286:30972–30980. doi: 10.1074/jbc.M110.181164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maguire JA, Mulugeta S, Beers MF. Multiple ways to die: delineation of the unfolded protein response and apoptosis induced by Surfactant Protein C BRICHOS mutants. Int J Biochem Cell Biol. 2012;44:101–112. doi: 10.1016/j.biocel.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Newcomb DC, Jones BR, Roldan J, Lane KB, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA. 2011;108:10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pinkerton KE, Young SL, Fram EK, Crapo JD. Alveolar type II cell responses to chronic inhalation of chrysotile asbestos in rats. Am J Respir Cell Mol Biol. 1990;3:543–552. doi: 10.1165/ajrcmb/3.6.543. [DOI] [PubMed] [Google Scholar]
- 16.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 17.White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol. 2005;7:1021–1028. doi: 10.1038/ncb1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, Korsmeyer SJ. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102:105–110. doi: 10.1073/pnas.0408352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klee M, Pallauf K, Alcala S, Fleischer A, Pimentel-Muinos FX. Mitochondrial apoptosis induced by BH3-only molecules in the exclusive presence of endoplasmic reticular Bak. EMBO J. 2009;28:1757–1768. doi: 10.1038/emboj.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soberanes S, Urich D, Baker CM, Burgess Z, Chiarella SE, Bell EL, Ghio AJ, De Vizcaya-Ruiz A, Liu J, Ridge KM, et al. Mitochondrial complex III-generated oxidants activate ASK1 and JNK to induce alveolar epithelial cell death following exposure to particulate matter air pollution. J Biol Chem. 2009;284:2176–2186. doi: 10.1074/jbc.M808844200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Timbrell V.Characteristics of International Union Against Cancer standard reference samples of asbestos. Shapiro HA, editor. New York: Oxford University Press; 1970. 28–36. [Google Scholar]
- 22.Panduri V, Surapureddi S, Soberanes S, Weitzman SA, Chandel N, Kamp DW. P53 mediates amosite asbestos-induced alveolar epithelial cell mitochondria-regulated apoptosis. Am J Respir Cell Mol Biol. 2006;34:443–452. doi: 10.1165/rcmb.2005-0352OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vadász I, Dada LA, Briva A, Trejo HE, Welch LC, Chen J, Tóth PT, Lecuona E, Witters LA, Schumacker PT, et al. AMP-activated protein kinase regulates CO2-induced alveoalar epithelial dysfunction in rats and human cells by promoting Na,K-ATPase endocytosis. J Clin Invest. 2008;118:752–762. doi: 10.1172/JCI29723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, Rizzuto R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012;19:267–273. doi: 10.1038/cdd.2011.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, Pinton P. Selective regulation of subtype III IP3R by AKT regulates ER Ca2+ release and apoptosis. Cell Death and Disease. 2012;3:e304. doi: 10.1038/cddis.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faux SP, Michelangeli F, Levy LS. Calcium chelator Quin-2 prevents crocidolite-induced DNA strand breakage in human white blood cells. Mutat Res. 1994;311:209–215. doi: 10.1016/0027-5107(94)90178-3. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and Noxa by p53. J Biol Chem. 2006;281:7260–7270. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- 28.Cha S-I, Ryerson CJ, Lee JS, Kukreja J, Barry SS, Jones KD, Elicker BM, Kim DS, Papa FR, Collard HR, et al. Cleaved cytokeratin-18 is mechanistically informative biomarker in idiopathic pulmonary fibrosis. Respir Res2012. 13:105. [DOI] [PMC free article] [PubMed]
- 29.Hsia TC, Yang JS, Chen GW, Chiu TH, Lu HF, Yang MD, Yu FS, Liu KC, Lai KC, Lin CC, et al. The roles of endoplasmic reticulum stress and Ca2+ on rhein-induced apoptosis in A549 human lung cancer cells. Anticancer Res. 2009;29:309–318. [PubMed] [Google Scholar]
- 30.Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, Kaufman RJ. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci USA. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin N, Chen H, Zhang H, Wan X, Su Q. Mitochondrial reactive oxygen species (ROS) inhibition ameliorates palmitate-induced INS-1 beta cell death. Endocrine. 2012;42:107–117. doi: 10.1007/s12020-012-9633-z. [DOI] [PubMed] [Google Scholar]
- 32.Liang S, Wang G, Briazova T, Zhang C, Wang A, Zheng Z, Gow A, Chen AF, Rajagopalan S, Chen LC, et al. Airborne particulate matter selectively activates endoplasmic reticulum stress response in the lung and liver tissues. Am J Physiol Cell Physiol. 2010;299:C736–C749. doi: 10.1152/ajpcell.00529.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lim S, Rashid MA, Jang M, Kim Y, Won H, Lee J, Woo J-T, Kim YS, Murphy MP, Ali L, et al. Mitochondria-targeted antoxidants protect pancreatic beta cells against oxidative stress and improve insulin secretion in glucotoxicity and glucolipotoxicity. Cell Physiol Biochem. 2011;28:873–886. doi: 10.1159/000335802. [DOI] [PubMed] [Google Scholar]
- 34.Giorgi C, De Stefani D, Bononi A, Rizzuto R, Piinton P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int J Biochem Cell Biol. 2009;41:1817–1827. doi: 10.1016/j.biocel.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raturi A, Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM) Biochim Biophys Acta. 2013;1833:213–224. doi: 10.1016/j.bbamcr.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 36.de Almeida SF, Picarote G, Fleming JV, Carmo-Fonseca M, Azvedo JE, de Sousa M. Chemical chaperones reduce endoplasmic reticulum stress and prevent mutant HFE aggregate formation. J Biol Chem. 2007;282:27905–27912. doi: 10.1074/jbc.M702672200. [DOI] [PubMed] [Google Scholar]
- 37.Baek HA, Kim DS, Park HS, Jang KY, Kang MJ, Lee DG, Moon WS, Chae HJ, Chung MJ. Involvement of endoplasmic reticulum stress in myofibroblast differentiation of lung fibroblasts. Am J Respir Cell Mol Biol. 2012;46:731–739. doi: 10.1165/rcmb.2011-0121OC. [DOI] [PubMed] [Google Scholar]
- 38.Ayala P, Montenegro J, Vivar R, Letelier A, Urroz PA, Copaja M, Pivet D, Humeres C, Troncoso R, Vicencio JM, et al. Attenuation of endoplasmic reticulum stress using the chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis indued by isoproterenol. Exp Mol Pathol. 2012;92:97–104. doi: 10.1016/j.yexmp.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 39.Zeitlin PL, Diener-West M, Rubenstein RC, Boyle MP, Lee CK, Brass-Ernst L. Evidence of CFTR function in cystic fibrosis after systemic administration of 4-phenylbutyrate. Mol Ther. 2002;6:119–126. doi: 10.1006/mthe.2002.0639. [DOI] [PubMed] [Google Scholar]
- 40.Wang WJ, Mulugeta S, Russo SJ, Beers MF. Deletion of exon 4 from human surfactant protein C results in aggresome formation and generation of a dominant negative. J Cell Sci. 2003;116:683–692. doi: 10.1242/jcs.00267. [DOI] [PubMed] [Google Scholar]
- 41.Nozaki S, Sledge GW, Jr, Nakshatri H. Repression of GADD153/CHOP by NF-kB: a possible cellular defense against endoplasmic stress-induced cell death. Oncogene. 2001;20:2178–2185. doi: 10.1038/sj.onc.1204292. [DOI] [PubMed] [Google Scholar]
- 42.Panduri V, Liu G, Surapureddi S, Kondapalli J, Soberanes S, de Souza-Pinto NC, Bohr VA, Budinger GRS, Schumacker PT, Weitzman SA, et al. Role of mitochondrial hOGG1 and aconitase in oxidant-induced lung epithelial cell apoptosis. Free Radic Biol Med. 2010;47:750–759. doi: 10.1016/j.freeradbiomed.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haga N, Saito S, Tsukumo Y, Sakurai J, Furuno A, Tsuruo T, Tomida A. Mitochondria regulate the unfolded protein response leading to cancer cell survival under glucose deprivation conditions. Cancer Sci. 2010;101:1125–1132. doi: 10.1111/j.1349-7006.2010.01525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Melov S, Ravenscroft J, Malik S, Gill MS, Walker DW, Clayton PE, Wallace DC, Malfroy B, Doctrow SR, Lithgow GJ. Extension of life-span with superoxide dismutase/catalase mimetics. Science. 2000;289:1567–1569. doi: 10.1126/science.289.5484.1567. [DOI] [PubMed] [Google Scholar]
- 45.Melov S, Doctrow SR, Schneider JA, Haberson J, Patel M, Coskun PE, Huffman K, Wallace DC, Malfroy B. Lifespan extension and rescue of spongiform encephalpathy in SOD2 nullizygous mice treated with SOD-catalase mimetics. J Neurosci. 2001;21:8348–8353. doi: 10.1523/JNEUROSCI.21-21-08348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Na N, Chandel NS, Litvan J, Ridge KM. Mitochondrial reactive oxygen species are required for hypoxia-induced degradation of keratin intermediate filaments. FASEB J. 2010;24:799–809. doi: 10.1096/fj.08-128967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pramanik KC, Boreddy SR, Srivastava SK. Role of mitochondrial electron transport chain complexes in capsaicin mediated oxidative stress leading to apoptosis in pancreatic cancer cells. PLoS ONE. 2011;6:e20151. doi: 10.1371/journal.pone.0020151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamanaka RB, Glasauer A, Hoover P, Yang S, Blatt H, Mullen AR, Getsios S, Gottardi CJ, DeBerardinis RJ, Lavker RM, et al. Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development. Sci Signal. 2013;6:1–9. doi: 10.1126/scisignal.2003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maguire JA, Mulugeta S, Beers MF. Endoplasmic reticulum stress induced by surfactant protein C BRISCHOS mutants promotes proinflammatory signaling by epithelial cells. Am J Respir Cell Mol Biol. 2010;44:404–414. doi: 10.1165/rcmb.2009-0382OC. [DOI] [PMC free article] [PubMed] [Google Scholar]