

Abstract
The methanol extract of Melochia odorata yielded three 4-quinolone alkaloids including waltherione A (1) and two new alkaloids, waltherione C (2) and waltherione D (3). Waltheriones A and C showed significant activities in an in vitro anti-HIV cytoprotection assay at concentrations of 56.2 and 0.84 μM, and inhibition of HIV P24 formation of more than 50% at 1.7 and 0.95 μM, respectively. The structures of the alkaloids were established by spectroscopic data interpretation.
Quinolone-based agents have demonstrated antiviral activity. A review by Ahmed and Daneshtalab1 of different quinolone-based antiviral structures contained the basic β-diketo 4- quinolones, which are active against human-immunodeficiency virus-1 (HIV-1), and the fluoroquinolones, which protect cells from HIV-mediated cytotoxicity. The review also cited several proposed mechanisms for these compounds, including inhibition of the transcription of proviral DNA into mRNA, inhibition of HIV integrase, and interference with post-integrational processes. Antiviral 2-quinolones, exemplified by efavirenz, are HIV-1 reverse transcriptase (RT) inhibitors.2,3 In the present study, a set of 4-quinolones possessing activities against HIV-1 are presented.
Under a drug discovery program targeting infectious diseases, known as the “Conservation and Sustainable Use of Biodiversity in Papua New Guinea (PNG)” International Cooperative Biodiversity Group (ICBG), a cell-based anti-HIV assay4,5 was used to screen botanical collections from PNG. A methanol extract of the stems and twigs of Melochia odorata L.f. (Sterculiaceae) was identified as active. Bioassay-guided isolation yielded quinolone alkaloids including waltherione A (1), and two new analogues that we named waltheriones C (2) and D (3).
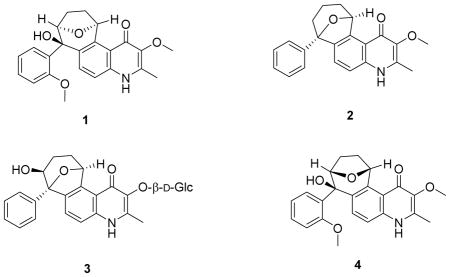
Waltherione A (1) was isolated previously from the root bark6 and stems7 of Waltheria douradinha St.-Hil., the roots of Melochia chamaedrys A. St.-Hil.,8 and the leaves of Melochia odorata L.f.9 Waltherione A was reported to possess antifungal activity against Candida albicans, Cryptococcus neoformans, and Saccharomyces cerevisiae9 and to inhibit acetylcholinesterase.10 4-Quinolone alkaloids that are structurally related to waltherione A include waltherione B (4),7 vanessine,7 antidesmone,11 chamaedrone,12 and melochinone.13 Vanessine was reported to have weak antibacterial activities against Escherichia coli, Salmonella setubal, and Klebsiella pneumoniae, 7 while antidesmone was reported to have antifungal activity.11 Chamaedrone was found to be antimicrobial against the bacteria Staphylococcus epidermidis and Escherichia coli and against the fungi Candida albicans and Sacharomyces cereviseae.12 In Papua New Guinea, chewing of Melochia odorata roots with lime and betel nut to treat painful urination has been reported in Siwai, located in the Autonomous Region of Bougainville.14
The 13C, 1H, COSY, HSQC, and HMBC NMR spectra, specific rotation, and IR data from alkaloid 1 were consistent with literature values reported for waltherione A.6 The absolute configurations of waltheriones A (1) and B (4) have been established previously by X-ray crystallography.7
Waltherione C (2) was isolated as an off-white solid. Its molecular formula, C22H22NO3, was determined by HRESIMS ([M + H]+ at m/z 348.1600, calcd 348.15942). The 13C and 1H NMR data of alkaloids 1 and 2 were very similar (Table 1). Both have the 4- quinolone moiety fused to a bicyclic ether with an attached phenyl ring. However, the methoxy group attached to C-2′ of alkaloid 1 is not present in 2 as evidenced by the presence of a monosubstituted benzene spin system (H-2′–H-6′) exhibiting the expected symmetry. The other major structural difference between alkaloids 1 and 2 is the loss of oxygenation of C-10 in 2 as evident from the loss of the signal at δH 4.73 and the presence of an additional methylene signal at δH 2.10 (H2-10). Finally, the HMBC correlation between C-9 and H-13 indicated an ether bridge connecting C-9 to C-13. The change in the coupling constant of the doublet signal of H-13 from J = 6.5 Hz in 1 to J = 2.0 Hz in 2 gave additional proof to this change from a five-membered fused ether ring encompassing C-10 to C-13 in 1 to a six-membered fused ether ring encompassing C-9 to C-13 in 2. Additionally, C-9 showed HMBC correlations with the aromatic protons H-2′/H-6′ and with H-7. Other relevant HMBC correlations are shown in Figure 1. Correlations in the COSY spectra showed the vicinal connectivities of H-10, H2-11, H2-12, and H-13 (Figure 1).
Table 1.
1H NMR (CD3OD, 500 MHz) and 13C NMR (CD3OD, 125 MHz) Data for Alkaloids 2– 3.
| 2a | 3b | |||
|---|---|---|---|---|
|
| ||||
| Position | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) |
| 2 | 144.5, C | - | 147.4, C | - |
| 3 | 142.2, C | - | 139.1, C | - |
| 4 | 174.3, C | - | 174.1, C | - |
| 4a | 121.1, C | - | 121.0, C | - |
| 5 | 142.6, C | - | 143.8, C | - |
| 6 | 141.4, C | - | 141.4, C | - |
| 7 | 124.5, CH | 7.03, d (8.0) | 127.8, CH | 7.44, d (8.0) |
| 8 | 118.3, CH | 7.35, d (8.0) | 118.7, CH | 7.49, d (8.0) |
| 8a | 139.7, C | - | 139.9, C | - |
| 9 | 86.6, C | - | 89.0, C | - |
| 10 | 32.8, CH2 | 2.12, m | 69.6, CH | 4.27, dd (9.5, 6.0) |
| 11 | 18.6, CH2 | 1.00, 1.63, m | 27.7, CH2 | 0.74, 1.88, m |
| 12 | 26.7, CH2 | 1.86, 2.02, m | 28.8, CH2 | 1.90, 2.10, m |
| 13 | 81.3, CH | 6.30, d (2.5) | 80.7, CH | 6.17, d (2.0) |
| 1′ | 143.4, C | - | 140.4, C | - |
| 2′ | 126.9, CH | 7.51, d (8.0) | 129.1, CH | 7.71, d (8.0) |
| 3′ | 129.1, CH | 7.37, t (8.0) | 129.1, CH | 7.39, t (7.5) |
| 4′ | 128.6, CH | 7.30, t (8.0) | 129.1, CH | 7.33, t (8.0) |
| 5′ | 129.1, CH | 7.37, t (8.0) | 129.1, CH | 7.39, t (7.5) |
| 6′ | 126.9, CH | 7.51, d (8.0) | 129.1, CH | 7.71, d (8.0) |
| 1″ | - | 107.6, CH | 4.68, d (7.0) | |
| 2″ | - | 75.5, CH | 3.46, m | |
| 3″ | - | 78.5, CH | 3.32c | |
| 4″ | - | 71.2, CH | 3.42, m | |
| 5″ | - | 78.6, CH | 3.28c | |
| 6″ | - | 62.6, CH2 | 3.70, dd (12.0, 5.5) 3.86, brd (12.0) |
|
| OCH3 (3) | 60.4 | 3.82, s | - | - |
| CH3 (2) | 14.6 | 2.46, s | 15.9, CH3 | 2.61, s |
Measured in CD3OD and CDCl3
Measured in CD3OD
Signal obscured by the CD3OD solvent peak
Figure 1.
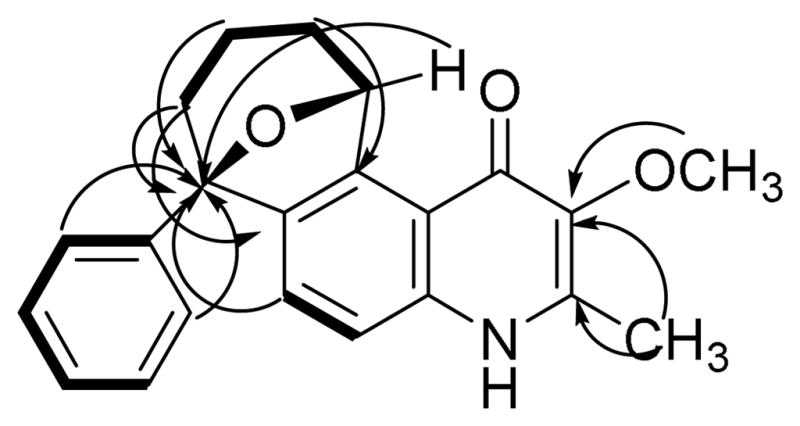
Key COSY (solid lines) and HMBC (arrows) correlations in alkaloid 2.
The bicyclic ring system in 2 is geometrically constrained to have the 4-quinolone moiety fused to the tetrahydropyran ring in a 1,3-diaxial arrangement. This places both the phenyl and hydrogen substituents at C-9 and C-13, respectively, equatorial with respect to the tetrahydropyran ring. Thus, H-13 exhibited similar magnitude NOE interactions with both diastereotopic hydrogens H2-14, suggesting a chair conformation for the tetrahydropyran ring.
Waltherione D (3) was isolated as an off-white powder. The molecular formula, C27H30NO9, was deduced from the HRESIMS ([M + H]+ at m/z 512.1930 (calcd 512.1921). Waltherione D is the 3-O-demethyl-β-glycosylated, 10-hydroxylated analogue of alkaloid 2. The 13C and 1H NMR data of 3 were found to be more similar to 2 than to 1 (Table 1). There was, however, a loss in 3 of a signal of a methoxy group attached to C-3 in both 1 and 2. Instead, a glucose residue was attached to the oxygen of C-3. The presence of glucose was evident from the ESIMS, which showed an in-source fragment ion at m/z 350 ([M+H-162]+), and can be explained by the loss of the glucosyl moiety. This was confirmed by acid hydrolysis of alkaloid 3 and analysis of the sugar fraction by TLC and polarimetry. Co-elution on TLC of the aqueous extract from the acid hydrolysis with an authentic D-glucose sample proved that the sugar residue is glucose. The positive optical activity of this aqueous extract proved that the glucosyl group has the D-configuration. The position of the glucosyl moiety was established from the HMBC spectra of 3 showing a correlation between the anomeric proton H-1″ and C-3 (Figure 2). An NOE between H-1″ and the methyl protons attached to C-2 was also observed from the ROESY spectrum (Figure 3B). The glucose residue was in an O-β-glycosidic linkage as evident from the coupling constant of H-1″ to H- 2″ (J = 7 Hz), indicating that H-1″ is in the axial position. In addition, ROESY correlations were observed from H-1″ to both of H-3″ and H-5″, consistent with an O-β-glucosyl residue (Figure 3B). Relevant HMBC correlations are shown in Figure 2.
Figure 2.
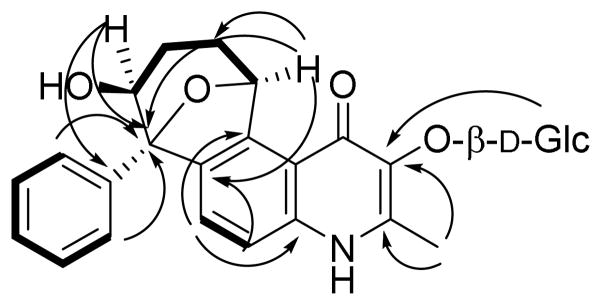
Key COSY (solid lines) and HMBC (arrows) correlations in alkaloid 3.
Figure 3.
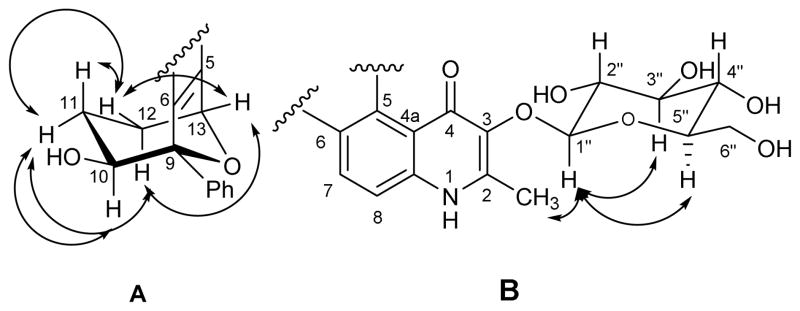
Key NOESY correlations in alkaloid 3.
Position C-10 in alkaloid 3 is oxygenated as in 1. However, 3 has the same six-membered fused ether ring encompassing C-9 to C-13 as in 2. This was determined from the HMBC correlation between H-13 and C-9 (Figure 2). The relative configuration of 3 was deduced from the 1H NMR and ROESY data. The six-membered tetrahydropyran ring of C-9 to C-13 appears to adopt a chair conformation to which the aromatic ring of the quinolone system is fused diaxially (Figure 3A) as in 2. Also as in 2, both the phenyl and hydrogen substituents to C-9 and C-13, respectively, are equatorial with respect to the tetrahydropyran ring. H-13 was hence observed as a doublet with J = 2 Hz proving its equatorial orientation. H-10 on the other hand, was seen to be in the axial position due to its double doublet signal with J = 9.5 Hz and J = 6 Hz corresponding to its coupling with H-11ax and with H-11eq, respectively. A cross peak in the ROESY spectrum showed an NOE between axial H-10 and H-2′/H-6′ of the equatorial phenyl ring suggesting that H-10 and the aromatic ring are gauche to one another (Figure 3A). Additional data from the ROESY spectra confirmed the proposed chair conformation, namely, H-10 (δH 4.27) showed a correlation with H-11eq (δH 1.88) but not with H-11ax (δH 0.74); H-11ax exhibited a correlation with H-12eq (δH 1.90) but not with H- 12ax (δH 2.10); and H-13 showed two correlations with H-12eq and H-12ax of equal intensity (Figure 3A). It was not possible to relate the relative configuration of the sugar moiety to the bicyclic ether, and thus the relative configurations depicted in the structural diagram of 3 for the corresponding groups are independent of one another.
Two of the alkaloids, waltheriones A (1) and C (2), showed significant cytoprotection4 against HIV-1 in infected CEM-TART cells in vitro (Table 2). A second assay measuring the production of HIV capsid protein P2415 in infected T cells confirmed the anti-HIV activity of 1 and 2 (Figures 5 and 6) showing > 50% inhibition at 1.7 and 0.95 μM, respectively. Each of these anti-HIV assays also provides secondary outcome measures of cytotoxicity. While no significant toxicity was observed with alkaloids 1-3 in the 48 h P24 assay, narrower therapeutic indices were observed in the 96 h cytoprotection assay (Table 2). To quantify these theoretical therapeutic indices, the MTT cytotoxicity assay (determined at approximately 72 h) was performed. The EC50/LC50 ratio for 1 was approximately 2; for compound 2 it was a more promising 13-fold.
Table 2.
Cytoprotection and Toxicity Assay Data of Alkaloids 1-3.
| alkaloid | cytoprotection EC50 (μM)a [Standard Deviation] | cytotoxicity LC50 (μM)b [Standard Deviation] |
|---|---|---|
| 1 | 56.2 [12.3] | 102 [2.25] |
| 2 | 0.84 [0.8] | 11 [1.36] |
| 3 | Not effectivea | Not toxicc |
| AZT | 1.30 [0.12] | 89.5 [10.8] |
Protection of CEM-TART cells from killing by HIV infection at 96 h (EC50)
Survival was measured by MTT metabolism by viable cells (LC50).
Not effective or toxic at the highest concentration tested (100 μM).
Figure 5.

Dose dependent inhibition of P24 production by AZT and waltheriones in CEMTART cells at 48 hours. P24 production was measured by indirect immunoflourescent staining of HIV P24.
Alkaloids 1 and 2 showed in vitro anti-HIV activity, with 2 showing less toxicity than 1. Alkaloid 3 did not show any significant activity. It would seem that the methoxy group at C-3, the site of glycosylation in 3, as well as the bicyclic ring system fused to a 4-quinolone ring are important determinants of the anti-HIV activity of the waltheriones. The shift in the ether bridgehead of the bicyclic ring system from C-10 to C-9 seems to marginally enhance anti-HIV activity and decrease toxicity as seen from the greater efficacy of 2 compared with 1. The methoxy group at C-2′ does not seem to be important for anti-HIV activity since 2, which does not have this functional group, exhibited activity in both cytoprotection and P24 inhibition assays. The waltheriones represent a structurally distinct class from both the HIV integrase-inhibitory and the HIV reverse transcriptase inhibitory classes of 4-quinolones. Further work on these alkaloids may improve our understanding of the structure-activity relationship of anti-HIV activity of the waltheriones.
EXPERIMENTAL SECTION
General Experimental Procedures
Specific rotations were recorded on a Perkin- Elmer (Downers Grove, IL, USA) 343 digital polarimeter in MeOH at 22 °C. UV spectra were measured on a Hewlett-Packard 8452A diode array spectrophotometer (Agilent, Santa Clara, CA, USA). IR spectra were recorded using a JASCO (Easton, MD, USA) FT/IR-400 spectrometer. NMR spectra were recorded on a Varian (Palo Alto, CA, USA) INOVA at 500 MHz for 1H and 125 MHz for 13C using vendor-supplied pulse sequences. Residual solvent (CD3OD) signals were used as reference (δH 3.31; δC 49.2). Accurate mass measurements were performed by HRESIMS on a Micromass Q-tof Micro (Waters, MA, USA) using the positive-ion mode, and a FTMS (LTQ-FT, ThermoFisher, Waltham, MA, USA). HPLC was performed on an Agilent 1200 series equipped with PDA detector (Agilent Technologies, Santa Clara, CA, USA). A Luna 5 μm C18 column 250 × 10 mm (Phenomenex, Torrance, CA, USA) was used for the isolation of the three alkaloids. Supelco Diaion HP20SS was purchased from Sigma-Aldrich (St. Louis, MO, USA). TLC was conducted using Kieselgel 60 F254 (Merck, Whitehouse Station, NJ, USA).
Plant Material
Stems and twigs of Melochia odorata were collected near Pauluaku Village, Buin District, Autonomous Regions of Bougainville, Papua New Guinea, in January 2004, as previously described16 as part of an ICBG agreement. Voucher specimens (U20246- 180) have been deposited at the University of PNG Herbarium, Port Moresby, and the PNG Forest Research Institute, Lae, PNG, and the plant was identified by Dr. Osia G. Gideon and Pius Piskaut of the University of PNG Herbarium.
Extraction and Isolation
Air-dried stems and twigs (30.0 g) of Melochia odorata were ground and extracted with 500 mL of MeOH (2 x 24 h).The crude methanol extract (2.8 g) was dissolved in MeOH and mixed with 8.4 g of Diaion HP20SS and dried. The resin was loaded into a column (8.5 cm x 2.0 cm ID) and fractionated using 40 mL each of the following solvents: 100% water, 75% H2O/25% 2-propanol, 50% H2O/50% 2-propanol, 25% H2O/75% 2-propanol, and 100% MeOH to yield five fractions, designated FW, F1, F2, F3, and F4.17 The fractions were collected and solvents evaporated using a rotary evaporator.
Bioassay guided fractionation showed cytoprotective anti-HIV activity for F1 (0.205 g) and F2 (0.196 g). Fraction 2 was found to be the more potent protector of T cells against HIV-mediated lysis at 10 μg/mL (p <0.005), while not displaying cytotoxicity to human T cells below 50 μg/mL. F2 was fractionated by semi-preparative HPLC using a reversed-phase C18 column employing the following method at 3.5 mL/min flow rate: 30% methanol/70% water from 0 to 5 min, and linear gradient from 30% to 100% methanol from 5 to 30 min. Compound 3 (2.4 mg) eluted at 14.7 min, compound 1 (1.4 mg) at 20.8 min and compound 2 (2.4 mg) at 25.3 min.
Additional material for the acid hydrolysis and determination of the absolute configuration of the monosaccharide in alkaloid 3 was obtained from F1. F1 (0.205 g) was separated by Sephadex LH-20 (MeOH) chromatography, affording four fractions: F1-1 to F1- 4. F1-2 (77 mg) was purified by semi-preparative HPLC using the same method described above for F2 to yield 3.5 mg of 3.
Waltherione C (2)
Off-white solid; [α]D 22 -17.0 (c 0.15, MeOH); UV (MeOH) λmax nm (log ε) 212 (3.08), 246 (3.14), 334 (2.53), 348 (2.52) nm; IR (NaCl disk) νmax 3392, 2924, 2360, 1634, 1560, 1508, 1022 cm−1; 1H NMR (CD3OD, 500 MHz) and 13C NMR (CD3OD, 125 MHz) see Table 1;HRESIMS m/z 348.1600 [M + H]+ (calcd for C22H22NO3 348.15942; Δ+2.0 ppm).
Waltherione D (3)
Off-white solid; [α]D22 -30.8 (c 0.12, MeOH); UV (MeOH) λmax nm (log ε) 212 (3.68), 238 (3.47), 282 (2.91), 330 (2.78), 344 (2.72) nm; IR (NaCl disk) νmax 3400, 2928, 1631, 1604, 1563, 1513, 1272 cm−1; 1H NMR (CD3OD, 500 MHz) and 13C NMR (CD3OD, 125 MHz) see Table 1; ESIMS positive mode m/z 512 [M + H]+, 350 [M + H- 162]+; HRESIMS m/z 512.1930 [M + H]+ (calculated for C27H30NO9 512.1921; Δ+1.8 ppm).
Acid Hydrolysis of 3
A solution of alkaloid 3 (2.0 mg) with a mixture of concentrated HCl (0.5 mL), H2O (1.5 mL), and dioxane (3.0 mL) was refluxed on a boiling water bath for 2 h.18 After the completion of the reaction [monitored by following the disappearance of 3 on TLC (CHCl3-MeOH, 8:2) detected by UV], the reaction mixture was extracted with ethyl acetate (3 x 5 mL). The aqueous layer was neutralized with 5% NaHCO3, concentrated to dryness under reduced pressure, and purified by Sephadex LH-20 chromatography (MeOH) to give a sugar fraction. The sugar (0.7 mg) was confirmed as D-glucose by comparison with an authentic sample on TLC [CHCl3-MeOH-H2O (8:5:1), Rf 0.3, detected by spraying with anisaldehyde-MeOH-H2SO4 (0.5:0.5:9) spray reagent followed by heating] and measurement of its optical rotation ([α]20 D +43.5, c 0.023, H2O).
Cytoprotective HIV Assay
1A2 cells (a subclone of CEM-TART cells4 that are more sensitive to lysis upon HIV infection) were cultured in RPMI 1640/20% FBS and plated in 96 well plates.5 Controls included DMSO-treated cells without HIV, DMSO-treated cells infected with HIV, and cells infected with HIV and treated with azidothymidine (AZT) at final concentrations of 5 or 0.5 μg/mL.5 Treatment of control cells with DMSO or drugtreated cells with DMSO-drug (1% vol) was delayed 2 h after infection with HIV stock. This delay reduces the detection of entry-inhibitor drugs and promotes detection of drugs that protect T cells from HIV killing by intracellular mechanisms.19 Uninfected and HIV-infected cells were allowed to incubate for 96 h at 37 °C in 5% CO2, at which time cell viability was assessed with a standard MTT assay, initiated by the addition of 11 μL/well of 5 mg/mL MTT in PBS to each culture well. To solubilize the formazan precipitate after 3 to 4 h of further incubation, cells were pelleted, the medium aspirated, and 100 μL DMSO/0.1% SDS added.20 Plates were read on a Fisher Scientific Multiskan FC (Fisher Scientific, Waltham, MA, USA) plate reader at 570 nm absorbance. On each assay plate, each condition was performed in triplicate and p values were determined between groups to gauge the performance of the assay. For a compound or extract to be considered active in the screen it must rescue at least 50% as many infected cells as the more protective of the two AZT positive controls, and must provide statistically significant (p <0.05) better survival than untreated control HIV-infected cells.5 To calculate EC50 (effective concentration of a compound to achieve 50% of the protection afforded by 5 ]M AZT) and LC50 (concentration exhibiting 50% cell killing in HIV uninfected 1A2 cells), dose response experiments and standards of deviation,16 and standards of deviation data from multiple experiments (≥ 3) were combined and analyzed using ED50plus V1.0 (mhvargas@conacyt.mx).
P24 Inhibition Assay
Controls included cells without HIV, cells infected with HIV, and cells infected with HIV and treated with AZT at final concentrations of 5 or 0.5 μg/mL. Control and extract-treated cells were washed twice in PBS and incubated at 1:100 with murine myeloma IgG (m5284, Sigma-Aldrich) in PBS for 30 min at 4 °C. Cell were then washed in PBS, fixed in PBS/1% paraformaldehyde/0.05% Triton X100, washed in FACS buffer (PBS, 2.0% FBS, 0.05% Triton X100) and exposed to 1:50 dilution of AG3.0 mouse monoclonal21 anti-P24 antibody in FACS buffer for 30 min at 4°C.15
The antibody-treated cells were washed twice in FACS buffer and exposed to goat-anti- mouse FITC conjugated secondary antibody for 30 min at 4 °C, washed twice in FACS buffer and analyzed on a BD FACSCanto instrument (BD Bioscience San Jose, CA, USA) (Figure 3).
Cytotoxicity Assay
Cytotoxicity was determined in a 3-(4,5-dimethylthiazol-2-yl)- 2,5-diphenyltetrazolium bromide (MTT).5 Drugs were dissolved in DMSO at the required concentrations; the final concentration of DMSO was 0.5%. Drugs and 20,000 CEM-TART cells were seeded in 96 well plates in fresh RPMI medium supplemented with 20% FBS and antibiotic and allowed to incubate at 37 °C in a 5% CO2 atmosphere for 72 h, after which 11 μL of MTT (5 mg/mL in PBS) were added. Viable cells reduce MTT to a purple formazan product that was solubilized in DMSO/0.1% SDS and quantified on a plate reader as described above. Tests were performed in triplicate or quadruplicate.
Supplementary Material
Figure 4.

P24 production assay results +/− HIV and 1 - 3 in CEM-TART cells at 48 h. Abscissa: cell size as measured by forward scatter; ordinate: intracellular indirect immunofluorescent staining of HIV capsid protein P24 via FITC (see methods). Top row, left to right: Control, HIV infected, AZT treated (5 μg/mL). Bottom row, left to right: 3, 1, and 2 at 5 μg/mL.
Acknowledgments
The authors wish to gratefully acknowledge NIH support through the ICBG 5UO1TW006671-3. The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: CEM-TART from Drs. Herbert Chen, Terence Boyle, Michael Malim, Bryan Cullen, and H. Kim Lyerly; and monoclonal antibody to HIV- 1 p24 (AG3.0) from Dr. Jonathan Allan.
Footnotes
Conflict of Interest
All authors declare they have no conflict of interest concerning the work reported here.
Supporting Information Available 1H and 13C NMR spectra of compounds 2 and 3 are available.
References
- 1.Ahmed A, Daneshtalab M. J Pharm Pharmaceut Sci. 2012;15:52–72. doi: 10.18433/j3302n. [DOI] [PubMed] [Google Scholar]
- 2.Di Santo R, Costi R, Roux A, Artico M, Lavecchia A, Marinelli L, Novellino E, Palmisano L, Andreotti M, Amici R, Galluzzo CM, Nencioni L, Palamara AT, Pommier Y, Marchand C. J Med Chem. 2006;49:1939–1945. doi: 10.1021/jm0511583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng P, Gu Q, Liu W, Zou JF, Ou YY, Lou ZY, Zeng JG. Molecules. 2011;16:7649–7661. doi: 10.3390/molecules16097649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kiser R, Makovsky S, Terpening SJ, Laing N, Clanton DJ. J Virol Methods. 1996;58:99–109. doi: 10.1016/0166-0934(95)01998-7. [DOI] [PubMed] [Google Scholar]
- 5.Lu Z, Harper MK, Pond CD, Barrows LR, Ireland CM, Van Wagoner RM. J Nat Prod. 2012;75:1436–1440. doi: 10.1021/np300270p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoelzel SCSM, Vieira ER, Giacomelli SR, Dalcol II, Zanatta N, Morel AF. Phytochemistry. 2005;66:1163–1167. doi: 10.1016/j.phytochem.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 7.Gressler V, Stücker CZ, Diaz G, de OC, Dalcol, Burrow RA, Schmidt J, Wessjohann L, Morel AF. Phytochemistry. 2008;69:994–999. doi: 10.1016/j.phytochem.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 8.Dias GOC, Gressler V, Hoelzel SCSM, Silva UF, Dalcol II, Morel AF. Phytochemistry. 2007;68:668–672. doi: 10.1016/j.phytochem.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Emile A, Waikedre J, Herrenknecht C, Fourneau C, Gantier JC, Hnawia E, Cabalion P, Hocquemiller R, Fournet A. Phytother Res. 2007;21:398–400. doi: 10.1002/ptr.2078. [DOI] [PubMed] [Google Scholar]
- 10.Lima MMC, Lopez JA, David JM, Silva EP, Giulietti AM, de Queiroz LP, David JP. Planta Med. 2009;75:335–337. doi: 10.1055/s-0028-1112215. [DOI] [PubMed] [Google Scholar]
- 11.Buske A, Schmidt J, Hoffmann P. Phytochemistry. 2002;60:489–496. doi: 10.1016/s0031-9422(02)00117-6. [DOI] [PubMed] [Google Scholar]
- 12.Dias GOC, Porto C, Stüker CZ, Graessler V, Burrow RA, Dalcol II, da Silva UF, Morel AF. Planta Med. 2007;73:289–292. doi: 10.1055/s-2007-967107. [DOI] [PubMed] [Google Scholar]
- 13.Kapadia GJ, Paul BD, Silverton JV, Fales HM, Sokoloski EA. J Am Chem Soc. 1975;97:6814–6819. doi: 10.1021/ja00856a036. [DOI] [PubMed] [Google Scholar]
- 14.Warurai J, Sipana B, Koch M, Barrows LR, Maitanaho TK, Rai PP. J Ethnopharmacol. 2011;138:564–577. doi: 10.1016/j.jep.2011.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spector SA, Kennedy C, McCutchan JA, Bozzette SA, Straube RG, Connor JD, Richman DD. J Infect Dis. 1989;159:822–828. doi: 10.1093/infdis/159.5.822. [DOI] [PubMed] [Google Scholar]
- 16.Jadulco RC, Koch M, Van Wagoner RM, Pond C, Gideon OG, Matainaho T, Piskaut P, Barrows LR. Planta Med. 2011;77:1651–1654. doi: 10.1055/s-0030-1271027. [DOI] [PubMed] [Google Scholar]
- 17.Bugni TS, Harper MK, McCulloch MWB, Reppart J, Ireland CM. Molecules. 2008;13:1372–1383. doi: 10.3390/molecules13061372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan T, Wan C, Gonzáez-Sarrís A, Kandhi V, Cech NB, Seeram NP. J Nat Prod. 2011;74:2472–2476. doi: 10.1021/np200678n. [DOI] [PubMed] [Google Scholar]
- 19.Andjelic CD, Planelles V, Barrows LR. Mar Drugs. 2008;6:528–549. doi: 10.3390/md20080027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howett MK, Neely EB, Christensen ND, Wigdahl B, Krebs FC, Malamud D, Patrick SD, Pickel MJ, Welsh PA, Reed CA, Ward MG, Budgeon LR, Kreider JW. Antimicrob Agents Chemother. 1999;43:314–321. doi: 10.1128/aac.43.2.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanders-Beer BE, Eschricht M, Seifried J, Hirsch VM, Allan JS, Norley S. Virology. 2012;422:402–412. doi: 10.1016/j.virol.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.