

Abstract
Promoter melting by bacterial RNA polymerase is a key step in transcription initiation. We used Next Generation Sequencing (NGS) based approach to analyze in parallel promoter melting of all 4096 sequence variants of the 6 bp -10 promoter element. We used NGS read count for each sequence of a promoter library containing randomized -10 sequence as an observable to determine relative enrichment of -10 element sequence variants at different time points of promoter melting reaction. The analysis reinforced the dominating role of consensus bases at positions -11 and -7, demonstrated an enhanced preference for A at -11 among sequences exhibiting the fastest melting kinetics, and showed higher overall importance of the T at -7 compared to the A at -11 for efficient promoter melting. Sequences lacking the consensus bases at -7 or -11 could still melt fast if they contained compensatory base patterns at other positions. We observed a significant correlation between the duplex melting energy of -10 element and the kinetics of promoter melting that became more pronounced when the dominating base-specific interactions with RNAP were diminished. These observations indicate that promoter melting kinetics is determined by a combination of base-specific effects/interactions and sequence-dependent stability DNA duplex with the former playing a dominating role. Our data show that NGS can be a reliable, quantitative readout for highly parallel analysis of DNA template sequence dependence of activities of proteins that bind or operate on DNA template.
Transcription initiation in bacteria is a multistep process carried out by RNA polymerase (RNAP), a large multisubunit enzyme 1–5. The major steps of the initiation are promoter recognition by the enzyme, followed by a critical promoter-RNAP complex isomerization step that involves melting of the DNA duplex in the vicinity of the transcription start site. The resulting “open” complex, in the presence of NTP’s, can initiate synthesis of RNA 6. The sequence of the promoter DNA plays a critical role in transcription initiation in bacteria since RNAP is capable of forming an “open” complex with many promoters without the need of any additional factors or additional enzymatic activities. Therefore, the steps leading to and including open complex formation are orchestrated by RNAP-promoter interactions and are driven by the energy available from favorable RNAP-promoter contacts, which emphasizes the key role of the promoter DNA sequence 7–9. A typical E. coli promoter is defined by two conserved hexametric sequences (-35 and -10 elements) with consensus sequences of TTGACA and TATAAT, respectively 10, 11. The -35 and -10 promoter elements are separated by spacer DNA of conserved preferred length (17 bp) 11. Although the -35 and -10 elements are both believed to be recognized by RNAP in their double stranded form during initial RNAP-promoter complex formation, the -10 element was shown to play an essential role in subsequent promoter melting 6. The nontemplate strand of the -10 element is sequence-specifically bound by RNAP 12–16, which could drive promoter melting in the “open” complex. The upstream boundary of the transcription bubble in the open complex is located around positions -11/-12. Thus, a majority of the -10 element is single stranded in the open complex 6. Sequence determinants for the recognition of the -10 elements in the single-stranded form were studied in detail demonstrating essential roles for -11A and -7T for high affinity binding to RNAP 17 (consistent with their highest level of sequence conservation among bacterial promoters 11) and the cooperativity between RNAP interactions with individual -10 element bases18. Specific function for A at position -11 in promoter melting nucleation was suggested by studies with base analogues, which demonstrated that this base could be recognized by RNAP only when flipped out of the DNA base stack17. Base flipping of -11A in the initial steps of promoter melting is consistent with: fluorescence data demonstrating a tight association of -11A with RNAP19, the observed correlation between stability of the base-pair at the position -11 and the promoter melting rate20, the evidence for an intermediate involving base unstacking and tyrosine-adenine stacking 21, and identification of a RNAP residue that could promote -11A flipping 22. Recent X-ray crystallographic analysis of the complex between the fragment of RNAP and ss -10 element DNA revealed a specific protein pocket for the recognition of -11A in the unstacked conformation, which further reinforces -11A flipping as an important aspect of a promoter melting reaction23. A specific role for -7T is less clear. The location of this base downstream from -11, together with the observation that promoter melting expands unidirectionally from the site of nucleation24, suggests a possible function as a check point to assure downstream expansion of the transcription bubble 17. Structural analysis revealed that similar to -11A, -7T is bound to RNAP in a flipped out conformation 23.
Understanding the exact roles of the DNA sequence of the -10 promoter element is complicated by the fact that this region, during the open complex formation, in addition to interacting with RNAP, undergoes transition from ds to ss DNA. A change in DNA sequence could thus affect base-specific interactions with RNAP or the stability of the DNA duplex. To enhance the insights into the role of the -10 element sequence in promoter melting, we developed a Next Generation Sequencing 25 (NGS) based experimental approach that allows parallel simultaneous investigation of promoter melting kinetics of all 4096 sequence variants of the -10 sequence. Our data reinforce important roles of -11A and -7T in the melting reaction. The A at position -11 is important for fast promoter melting while -7T is more important overall for melting compared to -11A. Melting kinetics appear to be determined by a combination of base-specific effects/interactions and a sequence-dependent stability of DNA duplex with the former playing a dominating role. Additionally, our results illustrate excellent performance of NGS as a quantitative readout in biophysical experiments. Its use allows highly parallel investigations of biophysical and biochemical functions of a large number of nucleic acid sequence variants.
Materials and Methods
Materials
Oligonucleotides were obtained from IDT (Coralville, IA) or Keck Oligonucleotide Facility at Yale University. E. coli core RNAP with His-Tag on the C-terminal of the β’ subunit was expressed in BL21(DE3) cells using the pSV10 expression vector (a gift from Dr. Irina Artsimovitch, The Ohio Sate University) and purified as described in 26. Wt σ70 was expressed and purified as described previously 27. Holoenzyme was reconstituted by incubating 2 μM core with 5 μM σ70 for 15min at 37°C. Sybr Green was purchased from Life Technologies (Grand Island, NY). KlenTaq PCR mix was from Sigma (Saint Louis, MO). All other reagents were of the highest purity commercially available.
Oligonucleotides
The following oligonucleotides were used in this work:
O1 ATC TAT CAC CGC AAG GGA TAA ATA TCT AAC ACC GTG CGT GTT GAC TAT TTT ACC TCT GGC GGT NNN NNN GGT TGC ATG TAG TAA GG
where NNNNNN denotes a 6 bp random sequence that was incorporated in place of -10 promoter element.
O2 TCA GTT GCC GCT TTC TTT CTT GCT GAC TGC TTA ATC GCT TCT AGG GAT ATA GGT AAT TCC ATA CCA CCT CCT TAC TAC ATG CAA CC
O3 CCCTACACGACGCTCTTCCGATCTxxxxxxACCTCTGGCGGT
where xxxxxx denotes a 6 bp “barcode” sequence that allowed pooling all samples for NGS analysis.
O4 AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC T
O5 CAA GCA GAA GAC GGC ATA CGA GAT TTC AGT TGC CGC TTT CTT TCT
O6 ATC TAT CAC CGC AAG GGA TAA ATA TCT AAC ACC GTG CGT GTT GAC TAT TTT ACC TCT GGC GGT GAT AAT GGT TGC A1G
where 1 denotes fluorescein-dT.
O7 GCA TAA TCT TTC AGG GTT ATG CGT TGT TCC ATA CAA CCT CCT TAG TAC ATG CAA CC2 TTA TCA CCG C
where 2 denotes base analogue incorporated into -7 position of the template strand.
O8 GCA TAA TCT TTC AGG GTT ATG CGT TGT TCC ATA CAA CCT CCT TAG TAC ATG CAA CCA TTA TC2 CCG C
where 2 denotes base analogue incorporated into the -13 position of the template strand. Bases and base analogues that were incorporated into O7 and O8 oligonucleotides were: dT, dG, 5-nitroindole, nebularine and 2-amino-dA. Phosphoramidates for incorporating base analogues were from Glen Research (Sterling, VA).
Preparation of promoter DNA library
A promoter library containing all 4096 sequence variants of the -10 element was prepared from O1 and O2 synthetic oligonucleotides roughly corresponding to halves of the top and bottom strands of the promoter with 17 bp complementary overlapping sequence at the 3’ ends. Oligonucleotides were mixed at 2 μM concentration and were annealed by heating to 95°C for 2 minutes and slowly cooling down to room temperature. Annealed partial duplex (0.25 μM) was extended by a 2-cycle PCR extension with Klentaq enzyme. Extended duplex DNA was purified with Wizard SV DNA purification kit (Promega).
RNAP-dependent melting of promoter DNA library
Heparin-resistant complexes formed at different time points after addition of RNAP to the promoter DNA library were isolated by native polyacrylamide gel electrophoresis. The experiment was performed at room temperature in 20 mM Tris-HCl (pH 8.0) buffer containing 150 mM NaCl, 10 mM MgCl2, 0.1 mg/ml BSA and 5% glycerol. For each time point the reaction was initiated by mixing 2.5 μl of 100 nM promoter DNA with 2.5 μl of 150 nM holoenzyme. Reactions were terminated at 6, 20, 90, 270 and 810 sec by adding 2 μl of a heparin-Ficoll mixture. Final concentration of heparin was 0.2 mg/ml. 7 μl of the reaction mixtures were loaded on 7.5% native polyacrylamide gel running at constant voltage (90 V) in TBE buffer. A sample of promoter DNA without the holoenzyme but otherwise treated the same way was also loaded. The gel was stained with Sybr Green and imaged on Molecular Imager FX (Bio-Rad). The bands corresponding to heparin-resistant complex and the DNA only sample were excised from the gel. DNA was eluted from the gel by incubating gel pieces in 100 mM Tris-HCl (pH 8.0), 0.5 M NaCl and 5 mM EDTA at 50°C for 2 hrs. Eluted DNA was filtered, precipitated with ethanol and used for preparing Illumina Next Generation Sequencing (NGS) compatible DNA templates.
NGS sequencing and data processing
Custom, Illumina NGS sequencing compatible, DNA templates were prepared from promoter DNA samples eluted from the gel by a two step low-cycle PCR procedure. The first step involved amplification of promoter DNA with O5 and O3 primers, which added a 6 bp barcode sequence (to allow multiplexing many DNA sequences in a single NGS experiment 28), and a downstream Illumina sequencing compatible end. DNA product of this PCR reaction was used as a template in a second PCR reaction with O5 and O4 primers that added an upstream Illumina sequencing compatible end. The final DNA products were purified with Wizard SV DNA purification kit (Promega) and their concentration was determined using Qubit R ds DNA BR Assay Kit (Invitrogen). Illumina 50 bp single end NGS sequencing was performed at the DNA Core facility at the University of Missouri in Columbia. Raw sequencing data were trimmed to 6 bp corresponding to the -10 element and were filtered to remove sequences with quality factor <20 at any position. All pre-processing was performed using Galaxy tools (http://galaxyproject.org/). The read count for each of 4096 variants of the 6 bp sequence was obtained using a custom script written for high performance parallel computing hardware to facilitate tabulating read counts for a large number of sequences of interest.
Calculation of sequence dependent DNA duplex stability
The free energy of DNA duplex melting for -10 element sequence variants was calculated using UNAFold software on DINAMelt server (http://mfold.rna.albany.edu/) 29, 30. Calculations were performed for 25°C and 100 mM NaCl.
Open complex formation kinetics with promoter constructs containing base substitutions at -7 and -13 of the template strand
Promoter DNA duplexes containing base substitutions at -7 or -13 of the template strand were prepared from appropriate O6/O7 (or O6/O8) oligonucleotides using Klenow extension and purified by ion exchange chromatography on a 1-ml Resource Q column (Amersham Biosciences) as described previously 20. Kinetics of heparin-resistant complex formation was determined by the electrophoretic mobility shift assay as described in 20. Final concentrations of DNA and the holoenzyme in these experiments were 15 nM and 100 nM, respectively. 5% TBE Ready-Gel precast gels (Biorad) were used in these experiments. The gels were imaged on Molecular Imager FX (Bio-Rad). Total fluorescence of free DNA bands was determined using scanner software, and the rate constants were obtained by fitting the data to a double-exponential decay kinetic model. Amplitude-weighted average of the rate constants for the two exponentials was used as a measure of the kinetics of open complex formation.
Results
The sequence of the promoter library that we used for NGS-based analysis of promoter melting kinetics is shown in Fig. 1A. The library was based on the λPR promoter. The 6 bp -10 element was replaced with a random sequence resulting in a promoter library containing 4096 possible sequence variants. The promoter DNA library was mixed with RNAP, and at various time points (0 to 810 sec) heparin was added to stop the promoter melting reaction. Heparin rapidly dissociates RNAP from DNA in a duplex form but open RNAP-promoter complexes (where promoter DNA has been melted) are relatively resistant to the heparin challenge. At each time point, RNAP-promoter complexes that survived the heparin challenge (and thus contained promoter sequences that underwent melting by RNAP) were isolated by native polyacrylamide electrophoresis (Fig. 1B&C). The bands corresponding to the RNAP-DNA complex were cut out of the gel, the DNA was purified and was used as a template for preparing Illumina NGS sequencing compatible DNA samples. NGS sequencing data for each time point were analyzed to determine the read count for each of the 4096 -10 element sequence variants. Read counts for each sequence variant were normalized by dividing them with a sum of the reads for all sequence variants.
Figure 1.

Design of promoter melting experiments employing promoter library containing all sequence variants of -10 promoter element. (A) DNA sequence of promoter library. (B) Native polyacrylamide gel electrophoresis analysis of the kinetics of open complex formation employing promoter DNA library. The bands marked “complex” were cut out of the gel and DNA eluted from the gel slices was subjected to NGS analysis. (C) Kinetics of the decrease in free DNA as a result of open complex formation.
We first investigated reproducibility and accuracy of NGS as a readout of relative amounts of promoter sequence variants under the experimental protocol illustrated in Fig. 1. Fig. 2A-C summarizes results of NGS analysis of 3 independent repeats of the zero time DNA sample (DNA that wasn’t bound to RNAP but was otherwise processed the same way as the DNA from each time point of promoter melting reaction). Outstanding correlation between read counts for each sequence between the 3 repeats of the experiment was observed. The coefficient of variation (CV) (average of CV’s for all 4096 sequences) for the read count was 5.5 +/− 4.3 %. The average read count for all 4096 sequences was 1373 +/− 1260 indicating a wide distribution of read counts between various -10 element sequence variants. This wide distribution of read counts reflects a cumulative effect of differences in oligonucleotide sequence synthesis efficiency, sequencing bias and possible differences in elution from polyacrylamide gel between different sequence variants. The distribution of read counts had no significant effect on the analysis since read counts for each sequence were highly reproducible (Fig. 2) and the ratio of read counts for each sequence at a given time point to zero time (no RNAP) sample was used for further analysis. Therefore, we concluded that NGS analysis provides highly reproducible quantitative readout for the experimental design depicted in Fig. 1.
Figure 2.

(A-C) Reproducibility of NGS analysis of DNA samples eluted from the experiment depicted in Fig. 1B. Correlation between normalized read counts for each -10 element sequence variant between the pairs of samples (free DNA band) from three independent experiments is plotted. (D) Relative enrichment of each of 4096 sequence variants of -10 element at 6 s time point (ratio of normalized read count for each sequence at 6 s time point to its normalized read count for no RNAP (0 s time point) sample). The point corresponding to the sequence with the highest relative enrichment value (TATAAT) is marked with a red circle. (E) Correlation between relative enrichment values at 6 s and 20 s time points. (F) Correlation between relative enrichment values at 6 s and 90 s time points.
In order to follow melting kinetics of the -10 element sequence variants, we calculated the relative enrichment value (a ratio of normalized read count at a given time point to the read count at time 0) for each sequence at each time point of the promoter melting reaction. Fig. 2D illustrates the results of such calculation for the 6 sec time point. For the majority of sequences, the value of this ratio was ≤1 indicating that these sequences were depleted in the DNA isolated from the open complex. In principle, the sequences that were not melted should not be present at all in the DNA isolated from the open complex. However, free DNA always travels through the area of the gel from which eventually open complex band is cut out (Fig. 1B) leaving a smear that provides enough material for PCR amplification and detection by NGS analysis. When the distribution of relative enrichment values was plotted (not shown), the peak (most frequently observed relative enrichment value) was 0.83 (depicted by the red line in Fig. 2A). We thus interpreted the relative enrichment values equal or lower to 0.83 as representing sequences that were either were melted at a slow rate or not melted at all. The fact that a great majority of the sequences belonged to this category at 6 sec time point was expected since RNAP should be able to melt only a limited number of sequences at such a short time. A smaller subset of sequences was specifically enriched in DNA isolated from the open complex exhibiting the read ratio significantly higher than 0.83.
These sequences were preferentially melted by RNAP at 6 sec reaction time point. The time course of promoter library melting reaction (Fig. 1C) appeared biphasic suggesting existence of at least two classes of sequences differing significantly in their rates of promoter melting. This is better illustrated in Fig. 2E&F where correlation between relative enrichment for all sequences between early time points (6 sec and 20 sec, Fig. 2E) and time point further along the reaction progress (90 sec, Fig. 2F) is compared. The sequence with the highest value of the relative enrichment at 6 sec (TATAAT, Fig. 2D) corresponds to the consensus sequence of the -10 promoter element 11. Thus, the consensus -10 element indeed represents the combination of bases within the -10 element that can be melted by RNAP most rapidly. Fig. 3A shows the relative enrichment (after 6 seconds) of consensus -10 sequence and its three mutants at most conserved positions of -10 element compared to the analogous data for wt λPR -10 element sequence. The observed pattern of relative enrichment changes due to mutations is consistent with what could be expected from the previously reported effects of such mutations on the binding affinity of RNA to model fork DNA constructs17. Very similar effects of mutations on relative enrichment values were observed in the context of the consensus and the wt λPR -10 element sequence (Fig. 3A; with relative enrichment values for the wt λPR -10 element lower compared to the corresponding consensus sequences) further illustrating excellent quality of NGS data.
Figure 3.
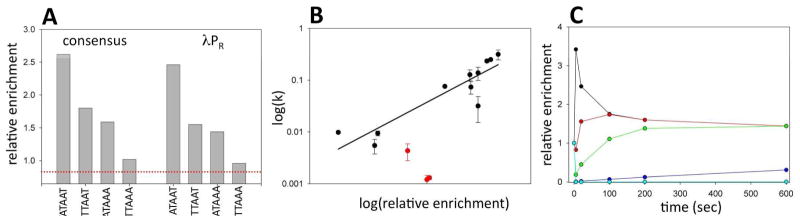
(A) Relative enrichment factors for indicated mutants of consensus -10 element (TATAAT) and λPR wt -10 element sequence (GATAAT). (B) Correlation between open complex formation rates determined by standard methodology for promoters containing 14 sequence variants of the -10 element (data from 20) and enrichment factors at 6 s time point for the same sequences. Data for mutants at -11 are in red. (C) Calculated time course of enrichment factor changes for a mixture of 5 DNA templates with the following rates constants of open complex formation: 0.5 s−1 (black), 0.05 s−1 (red), 0.01 s−1 (green), 0.004 s−1 (blue), and 0.000005 s−1 (magenta).
To further validate NGS approach, Fig. 3B shows a comparison of previously published data20 on melting kinetics for 14 sequence variants of λPR promoter measured using standard approaches with the NGS-based analysis. There is a good correlation between the two approaches (R=0.91) but the data on mutants at position -11 significantly deviates from this correlation (red symbols in Fig. 3B; with these data included R=0.71). We don’t know the explanation for this; further investigation will be required to explain this phenomenon. We used relative enrichment values at early time point (6 sec) to construct the plot in Fig. 3B. In principle, time dependent changes in relative enrichment values for each sequence in the library could be used to calculate promoter melting rate constants. Rate constants will provide a more precise description of melting kinetics for each sequence compared to the enrichment values at a single time point. However, extracting the rate constants from NGS data is not straightforward. The challenge of extracting rate constants from NGS data is that NGS read numbers for each sequence reflect fraction of each sequence rather then the amount of the sequence in the sample. Thus, the time dependence of the relative enrichment values for each sequence cannot be fitted to obtain the rate constant because the shape of these curves is affected by the kinetics of all other sequences in the samples. To illustrate this, in Fig. 3C we plotted calculated relative enrichment values for a hypothetical mixture of 5 sequences with a range of melting rate constants. The distortion of the curves is most easily visible for the fastest melting sequence. The enrichment value for this sequence reaches the maximum value at the shortest time and then progressively decreases with time. This is because all of this sequence melts rapidly and its relative ratio will decrease with time due to the slower build up with time of slow melting sequences. Thus, in order to obtain rate constants from NGS data, simultaneous fitting of all data for the rate constants for all sequences needs to be done. We are developing the analytical procedures for extracting rate constants from NGS data and they will described elsewhere.
In order to determine common properties of -10 element sequence that promote efficient melting of the promoter by RNAP we sorted the sequences into three groups according to the range of promoter melting kinetics observed in the experiment. The first group corresponded to sequences that appeared to melt fast (i.e. were already significantly enriched at the shortest time point (6 sec)). We chose to use a 1.5-fold enrichment as a cut off value because it is roughly a half point between no enrichment and maximum enrichment observed (Fig. 2D). There were 362 sequences enriched >1.5 times at the 6 s time point and the plot of their average relative enrichment as a function of time (Fig. 4A) confirms their rapid melting kinetics. The second group corresponds to sequences that require longer times to melt and thus are not significantly enriched at the shortest reaction time but become enriched at longer reaction times (i.e. the sequences with enrichment factor <1.5 at 6 sec and > 1.5 at 270 sec). Their average relative enrichment when plotted as a function of time (Fig. 4A) shows a slow buildup of open complex over time. The third group corresponded to sequences that are either not melted or melted very slowly (i.e. the sequences with enrichment factor <1.5 at 6 sec and <1.5 at 270 sec. Their average enrichment decreases and remains low for the duration of the experiment (Fig. 4A). The relative enrichment value for this set of sequences was ~ 0.8, consistent with the value for slow or non-melting sequences (0.83) identified by the analysis of distribution of relative enrichment values described previously.
Figure 4.

(A) Time course of average relative enrichment values changes for “fast” (relative enrichment > 1.5 at 6 sec), “slow” (relative enrichment < 1.5 at 6 sec but > 1.5 at 270 sec) and “no melting” (relative enrichment < 1.5 at 6 sec and < 1.5 at 270 sec) sequences. (D) Sequence logo for “fast” sequences. (E) Sequence logo for “slow” sequences. (F) Sequence logo for “no melting” sequences.
A strong preference for A at -11 and an even stronger preference for T at the -7 position is an obvious property of “fast” melting sequences (Fig. 4B). Essentially identical base preferences are observed when the data at the 20 sec time point were examined (not shown). The A at -11 and T at -7 are the most evolutionary conserved positions within the -10 element 11. Previously published data demonstrated their important role in bacterial promoter function and data shown in Fig. 4B are consistent with such notion. The flipping of the -11 A out of the DNA duplex was proposed to nucleate promoter melting by RNAP 17, 19–23, 31. One could thus expect that this base should be especially important for fast melting of the promoter. However, examination of Fig. 4B would suggests that -7 T is more important for melting compared to -11A. To investigate this issue in more detail, we analyzed base preferences for sequences selected using progressively more stringent cut off values for the relative enrichment at 6 sec time point (Fig. 5). The higher the cut off point for the relative enrichment, the faster the melting kinetics of the sequences selected with such cut off point would likely be. While there is a stronger preference for T at -7 compared to A at -11 with less stringent cut off values (Fig. 5B and Fig. 5A), this changes to stronger preference for A at -11 than T at -7 when the stringency of cut off values for relative enrichment increases (Fig. 5 B-D). Also, a clear preference for the consensus T at position -12 emerges with the increase in stringency of relative enrichment cut off value (Fig. 5 C&D). The simplest interpretation of these observations is that -11A is more important for fast melting whereas -7T plays more general role in melting. Such conclusion is consistent with sequence features of “slow” melting sequences (Fig. 4C). A great majority of these sequences have a T at position -7, but relatively few required A at position -11, suggesting that while -11A is important for very fast melting, it is not absolutely essential for melting as long as there is T at -7. Sequences that did not melt or melted very slowly exhibited clear preference for non-consensus bases at the most conserved positions of the -10 element (-12, -11, and -7; Fig. 4D).
Figure 5.
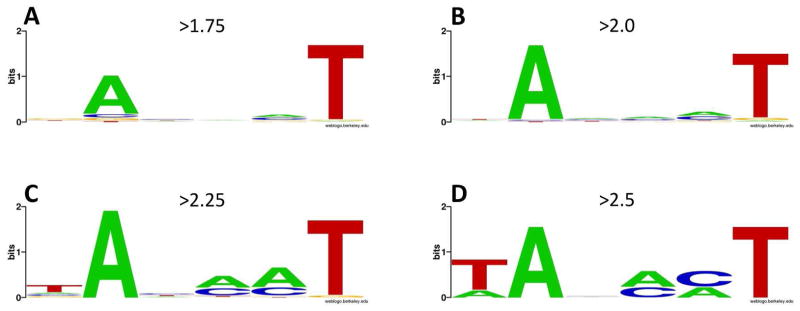
Sequence logos for -10 element variants selected at the 6 sec time point using progressively more stringent relative enrichment cut off values: (A) 1.75; (B) 2.0; (C) 2.25; (D) 2.5.
Figs. 6&7 illustrate how the presence of non-consensus bases at positions -7 and -11 affects sequence preferences at the remaining positions of the -10 element in “fast” melting sequences. When the -7 position was not T, a strong preference for T at position -8 and A at position-12 is observed. The stringency of the requirement for -8 T and -12 A is dependent on the nature of the base at position -7. With G at position -7, all “fast” melting sequences have T at -8 and A at -12 (Fig. 6). With A or C at -7, a majority of “fast” melting sequences have T at -8 and A at -12, but some instead had a mandatory A at -11 and also A’s at positions -9 and -8 (Fig. 6). T at -8 and A at -12 recreate the pattern of bases that characterizes “fast” melting sequences (-7 T and -11A) but shifted 1 bp upstream thus shortening the length of promoter spacer linking -10 and -35 promoter element to 16 bp (from 17 bp, which is the most preferred length for a bacterial promoter 11). It appears, that the nonconsensus, shorter length of the spacer DNA is better tolerated than the nonconsensus base at position -7 for efficient melting, further emphasizing the important role of -7T for promoter melting. Analogous analysis for the -11 position (Fig. 7) demonstrated significantly different relationships. In this case, when -11 is not the consensus A, the requirement for “fast” melting for the majority of sequences is to simply have T at position -7 (Fig. 7). A smaller set of sequences has the -8T, -12A sequence pattern that was predominant in the case of non-consensus bases at position -7. Again, these data together with data in Fig. 6 are consistent with -7T playing overall more important role for promoter melting compared to -11A.
Figure 6.

Sequence logos for sequences with relative enrichment at 6 sec > 1.5 and the indicated base at position -7. Sequences from which the logos were derived are shown at the bottom of the figure grouped into those that had T at -8 and A at -12 and those that did not show this base pattern.
Figure 7.

Sequence logos for sequences with relative enrichment at 6 sec > 1.5 and an indicated base at position -11. Sequences from which the logos were derived are shown at the bottom of the figure grouped into those that had T at -8 and A at -12 and those that did not show this base pattern.
A majority of the bases (from position -11 to position -7) in the -10 element undergo transition from DNA duplex to ss DNA upon open complex formation. Energetic cost of melting the DNA duplex depends on the DNA sequence and could thus contribute to the sequence dependence of promoter melting kinetics. Previous studies demonstrated that local stability of the base-pair at position -11 correlated with the kinetics of open complex formation 20. DNA sequence-dependent contributions to promoter melting kinetics due to base-specific interactions with RNAP and due to the modulation of DNA duplex stability are difficult to sort out. However, the availability of promoter melting kinetics data for all sequence variants of the -10 element provides an excellent opportunity to explore this issue. Fig. 8A shows a correlation between relative enrichment for all sequences (at 6 sec time point) and their corresponding calculated stability of DNA duplex. A weak but significant correlation (R=0.42) is observed, suggesting a significant but not dominating contribution of sequence-dependent energetic cost of promoter -10 element melting to the kinetics of open complex formation. This correlation is worse for sequences that melt “fast” (relative enrichment at 6 sec > 1.5 fold) (Fig. 8B) compared to those with slower melting kinetics (relative enrichment at 6 sec < 1.5 fold) (Fig. 8C). We interpret this as a further indication that base-specific interactions with RNAP play a dominant role compared to the sequence-dependent stability of the DNA duplex in the -10 element in open complex formation. Such conclusion is consistent with further analysis illustrated in Fig. 8D. We plotted there a correlation between relative enrichment (at 6 sec time point) for all sequences with A at -11 and with -7 and -8 not being the T. By sorting sequences this way, we eliminated from the data one of the two strongest base-specific contributions to the open complex formation kinetics. A much higher correlation between relative enrichment at the 6 sec time point and DNA duplex stability was observed for this group of sequences, consistent with the notion that when dominating base-specific effects are reduced, contribution of DNA duplex melting energy to the overall kinetics of promoter melting becomes more pronounced. When correlations between the relative enrichment and free energy of DNA duplex stability at the 270 s time point were examined, a very similar pattern to the one observed at 6 s time point (Fig. 8) was obtained (data not shown). However overall, the correlation at 270 s between relative enrichment values and DNA duplex stability was significantly worse (the corresponding R values for data analogous to those in Fig. 8A, B, C & D were 0.29, 0.04, 0.28 and 0.37, respectively), consistent with the notion that the energetic cost of melting of DNA duplex can have a more significant effect on the kinetics of promoter melting rather then on the overall extent of melting reaction.
Figure 8.

(A) Correlation between relative enrichment at 6 sec time point and free energy of DNA duplex melting for all 4096 -10 element sequence variants. (B) Correlation between relative enrichment at 6 sec time point and free energy of DNA duplex melting for sequence with relative enrichment > 1.5. (C) Correlation between relative enrichment at 6 sec time point and free energy of DNA duplex melting for sequence with relative enrichment < 1.5. (D) Correlation between relative enrichment at 6 sec time point and free energy of DNA duplex melting for sequences with A at -11 and A or G or C at positions -8 and -7.
Base-specific interactions of RNAP with -11A and -7T most likely occur when these bases are flipped out of the DNA double helix. The observed correlation between the -10 element DNA duplex stability and promoter melting kinetics may thus have two components. The first would be a more general effect reflecting the impact of the energetic cost of melting of the DNA duplex on the rate of open complex formation. The second could be a more specific effect where the local duplex stability in the vicinity of the bases at positions -11 and -7 would affect base flipping and thus affect promoter melting through modulation of RNAP interactions with these bases. We have previously demonstrated that subtle local perturbation of the stability of the base-pair at position -11 correlated well with the changes in the rate of open complex formation 20. Therefore, we performed experiments to investigate if perturbations to the local stability of the base-pair at position -7 would produce similar outcomes. A series of base analogues were incorporated at the template strand -7 position and the rates of open complex formation were measured using a native gel electrophoresis based assay (Fig. 9A). Excellent correlation between promoter melting rate constants and the changes in free energy of duplex stability introduced by the base analogues was observed (Fig. 9B). The same base analogues when incorporated into position -13 (that does not undergo melting during open complex formation) had a very small effect on promoter melting kinetics (Fig. 9B).
Figure 9.

The effect of destabilizing the base-pair at position -7 on the kinetics of open complex formation. (A) Examples of the gel images for reactions with the wt promoter and the promoter containing T-G mismatch at position -7. (B) Correlation between the rates of open complex formation and free energy of DNA duplex destabilization due to base analogue substitutions at positions -7 and -13 on the template strand.
Discussion
There were two major goals of this work. The first was to test and verify the utility of Next Generation Sequencing as readout in biophysical experiments where nucleic acid sequence dependence of the process of interest could be studied for a large number of sequence variants in parallel. Our data confirm that NGS analysis can be used as a robust, quantitative and highly reproducible readout in biophysical experiments providing a detailed picture of nucleic-acid sequence dependence of the process of interest as long as an appropriate nucleic acid selection screen could be designed. We used native gel-electrophoresis based isolation of heparin resistant open complexes to select for promoter sequences that underwent rapid promoter melting. While resistance to heparin challenge is a widely accepted property of open complexes, unstable heparin-sensitive open complexes could escape our analysis. Native gel electrophoresis, which we used for isolating heparin-resistant complexes, could in principle produce further bias towards slow dissociating complexes. While possible, such bias is not very likely since protein-DNA complexes have been shown to be stabilized during electrophoresis due to gel sequestration effect32–34. Faster separation techniques for protein-DNA complexes of interest could be used to enhance analytical capabilities of NGS-based approach for labile complexes. For example, in the case of studies of open complexes with bacterial RNAP, rapid capture of open complex by filtration on nitrocellulose membrane could be employed. Also, in addition to the analysis of RNAP-DNA complex band on the native gel, the free DNA band from the same lane could be analyzed. Analysis of free DNA should produce data complementary to RNAP-DNA complex and would be free from potential bias due to the dissociation of the RNAP-DNA complexes during gel electrophoresis.
We used NGS-based approach to analyze time-dependence of a specific protein-DNA complex formation. The time resolution of the experiment was determined by manual mixing that we used. However, faster processes could be analyzed if fast mixing and quenching techniques are employed. The approach is not limited to the studies of time-dependence of the process of interest. The protocols to analyze DNA sequence dependence of other activities (such as, for example, binding affinity or binding cooperativity) for a variety of proteins that bind or operate on DNA could be envisioned.
The robustness of the NGS quantitative readout is in large part a result of the way by which the read count is used as a signal. Read count signal is internally calibrated by normalizing it to the total read count for all sequences present in the sample that makes it relatively insensitive to the signal variability due to the losses of the material, pipetting errors, etc. The detailed data depicting sequence dependence of the process of interest can be interrogated computationally to uncover the mechanistic aspects of sequence-dependence. While we were able to follow simultaneously melting kinetics of 4096 sequence variants of promoter DNA, it will be possible to analyze simultaneously a much higher number of sequences. The total number of sequence reads for a given experiment will define the number of sequences that could be analyzed simultaneously. In experiments described in this work we pooled (using a barcoding approach 28) 30 samples for one NGS sequencing run resulting in ~5 million reads per experiment. Assuming that at least 10 reads for each sequence would be needed for quantitation of relative amount of each sequence in the sample, simultaneous analysis of ~ 500,000 sequences should be feasible with the NGS analysis design employed in this work. This estimation is validated by our recent analysis, where we used the same NGS analysis design to analyze simultaneously promoter escape properties of 262,144 sequence variants of the promoter (Heyduk, E. and Heyduk, T., unpublished). Higher number of sequences could be analyzed if smaller number of barcoded samples is pooled for NGS analysis, resulting in a higher number of reads per experiment.
The second goal of this work was to perform an exhaustive analysis of -10 element sequence dependence of promoter melting kinetics. Our results show a dominating role of -11A and -7T for the melting of promoter DNA by RNAP. This is in line with the high sequence conservation of these two positions 11 and with the essential role of these two positions in binding by RNAP of the -10 element in a single-stranded form 17. Several studies have suggested a specific role for -11A in nucleating promoter melting through flipping this base out of DNA helix early in the promoter melting reaction 17, 19–23, 31. Our results showing enhanced preference for A at -11 among sequences exhibiting the fastest melting kinetics (Fig. 4) are in agreement with the role for this base in early steps of promoter melting. Thus, the finding by NGS analysis of all sequence variants of -10 element that -11A position is important for fast melting is quite logical and expected. The stronger general role of -7T compared to -11A for promoter melting was interesting and less expected. Recent X-ray structure of RNAP in a complex with ss DNA containing a -10 promoter element demonstrated that both -11A and -7T bind to the pockets of RNAP in a flipped out conformation 23. Thus, it is likely that nucleation of promoter melting doesn’t have to be localized at the -11 base-pair but might be more delocalized within the -10 element, with -7T flipping out of DNA base-stack as an alternate route to initiation of promoter melting. The excellent correlation observed between promoter melting kinetics and local perturbation of -7 base-pair stability (Fig. 9) is consistent with such possible function of -7T. It is still an open question if nucleation of promoter melting through base flipping is an active process where RNAP catalyzes base flipping or if it is a process where RNAP rather more passively uses its binding pockets specific for flipped out bases to trap spontaneously formed DNA bubbles within the -10 promoter element. The latter less ordered mechanism would benefit from a possible duplication in DNA bubble trapping activity derived from two alternative (and possible cooperative) contacts stabilizing the trapped DNA bubble. The cooperativity between RNAP interactions with -10 element residues has been recently reported18. Stabilization of the downstream spread of the DNA bubble initiated around position -11 is another possible function for -7T that would also be consistent with our data.
Alignment of bacterial promoters showed that the region around the -10 promoter element is a hot-spot for DNA duplex instability 35. Our data show that thermodynamic stability of the -10 element DNA duplex plays a role in the kinetics of open complex formation most likely directly (through the impact of the energetic cost of melting the -10 element on promoter melting kinetics) and indirectly (by modulating base flipping at positions -11 and –7, which in turn would affect base-specific interactions of these bases with RNAP). However, the base-specific interactions involving positions -11 and -7 are a much more dominating factor compared to the duplex stability for rapid formation of the open complex. The relative role of energetic cost of melting the -10 promoter element will be the strongest for promoters with the weakest base-specific interactions with RNAP.
Acknowledgments
This study was partially supported by grant from NIH (GM50514).
We thank Dr. David Gohara (Saint Louis University) for the script to obtain read counts from NGS sequencing files for a large number of sequences of interest and the members of the lab for the helpful comments regarding the manuscript.
Abbreviations
- NGS
Next Generation Sequencing
- RNAP
RNA polymerase
Bibliography
- 1.Burgess RR, Anthony L. How sigma docks to RNA polymerase and what sigma does. Curr Opin Microbiol. 2001;4:126–131. doi: 10.1016/s1369-5274(00)00177-6. [DOI] [PubMed] [Google Scholar]
- 2.Murakami KS, Darst SA. Bacterial RNA polymerases: the wholo story. Curr Opin Struct Biol. 2003;13:31–39. doi: 10.1016/s0959-440x(02)00005-2. [DOI] [PubMed] [Google Scholar]
- 3.Borukhov S, Nudler E. RNA polymerase holoenzyme: structure, function and biological implications. Curr Opin Microbiol. 2003;6:93–100. doi: 10.1016/s1369-5274(03)00036-5. [DOI] [PubMed] [Google Scholar]
- 4.Record MT, Jr, RWS, Craig ML, McQuade KL, Schlax PJ. In: Escherichia coli and Salmonella: cellular and molecular biology. 2. Neidhardt FC, Curtis R III, Ingraham JL, Lin ECC, Low KR, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE, editors. ASM Press; Washington, DC: 1996. pp. 792–820. [Google Scholar]
- 5.Busby S, Kolb A, Buc H. Where it all begins: An Overview of Promoter Recognition and Open Complex Formation. Royal Society of Chemsitry; 2009. [Google Scholar]
- 6.Saecker RM, Record MT, Jr, Dehaseth PL. Mechanism of bacterial transcription initiation: RNA polymerase - promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J Mol Biol. 2011;412:754–771. doi: 10.1016/j.jmb.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Haseth PL, Helmann JD. Open complex formation by Escherichia coli RNA polymerase: the mechanism of polymerase-induced strand separation of double helical DNA. Mol Microbiol. 1995;16:817–824. doi: 10.1111/j.1365-2958.1995.tb02309.x. [DOI] [PubMed] [Google Scholar]
- 8.Helmann JD, de Haseth PL. Protein-nucleic acid interactions during open complex formation investigated by systematic alteration of the protein and DNA binding partners. Biochemistry. 1999;38:5959–5967. doi: 10.1021/bi990206g. [DOI] [PubMed] [Google Scholar]
- 9.Helmann JD, Chamberlin MJ. Structure and function of bacterial sigma factors. Annu Rev Biochem. 1988;57:839–872. doi: 10.1146/annurev.bi.57.070188.004203. [DOI] [PubMed] [Google Scholar]
- 10.Pribnow D. Nucleotide sequence of an RNA polymerase binding site at an early T7 promoter. Proc Natl Acad Sci U S A. 1975;72:784–788. doi: 10.1073/pnas.72.3.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shultzaberger RK, Chen Z, Lewis KA, Schneider TD. Anatomy of Escherichia coli sigma70 promoters. Nucleic Acids Res. 2007;35:771–788. doi: 10.1093/nar/gkl956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savinkova LK, Baranova LV, Knorre VL, Salganik RI. Binding of RNA-polymerase from Escherichia coli with oligodeoxyribonucleotides homologous to transcribed and non-transcribed DNA stands in the "-10"-promoter region of bacterial genes. Mol Biol (Mosk) 1988;22:807–812. [PubMed] [Google Scholar]
- 13.Marr MT, Roberts JW. Promoter recognition as measured by binding of polymerase to nontemplate strand oligonucleotide. Science. 1997;276:1258–1260. doi: 10.1126/science.276.5316.1258. [DOI] [PubMed] [Google Scholar]
- 14.Roberts CW, Roberts JW. Base-specific recognition of the nontemplate strand of promoter DNA by E. coli RNA polymerase. Cell. 1996;86:495–501. doi: 10.1016/s0092-8674(00)80122-1. [DOI] [PubMed] [Google Scholar]
- 15.Callaci S, Heyduk T. Conformation and DNA binding properties of a single-stranded DNA binding region of sigma 70 subunit from Escherichia coli RNA polymerase are modulated by an interaction with the core enzyme. Biochemistry. 1998;37:3312–3320. doi: 10.1021/bi972041m. [DOI] [PubMed] [Google Scholar]
- 16.Ring BZ, Yarnell WS, Roberts JW. Function of E. coli RNA polymerase sigma factor sigma 70 in promoter-proximal pausing. Cell. 1996;86:485–493. doi: 10.1016/s0092-8674(00)80121-x. [DOI] [PubMed] [Google Scholar]
- 17.Matlock DL, Heyduk T. Sequence determinants for the recognition of the fork junction DNA containing the -10 region of promoter DNA by E. coli RNA polymerase. Biochemistry. 2000;39:12274–12283. doi: 10.1021/bi001433h. [DOI] [PubMed] [Google Scholar]
- 18.Mekler V, Severinov K. Cooperativity and interaction energy threshold effects in recognition of the -10 promoter element by bacterial RNA polymerase. Nucleic Acids Res. 2013;41:7276–7285. doi: 10.1093/nar/gkt541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsujikawa L, Strainic MG, Watrob H, Barkley MD, DeHaseth PL. RNA polymerase alters the mobility of an A-residue crucial to polymerase-induced melting of promoter DNA. Biochemistry. 2002;41:15334–15341. doi: 10.1021/bi026539m. [DOI] [PubMed] [Google Scholar]
- 20.Heyduk E, Kuznedelov K, Severinov K, Heyduk T. A consensus adenine at position -11 of the nontemplate strand of bacterial promoter is important for nucleation of promoter melting. J Biol Chem. 2006;281:12362–12369. doi: 10.1074/jbc.M601364200. [DOI] [PubMed] [Google Scholar]
- 21.Schroeder LA, Gries TJ, Saecker RM, Record MT, Jr, Harris ME, DeHaseth PL. Evidence for a tyrosine-adenine stacking interaction and for a short-lived open intermediate subsequent to initial binding of Escherichia coli RNA polymerase to promoter DNA. J Mol Biol. 2009;385:339–349. doi: 10.1016/j.jmb.2008.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schroeder LA, Karpen ME, de Haseth PL. Threonine 429 of Escherichia coli sigma 70 is a key participant in promoter DNA melting by RNA polymerase. J Mol Biol. 2008;376:153–165. doi: 10.1016/j.jmb.2007.11.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feklistov A, Darst SA. Structural basis for promoter-10 element recognition by the bacterial RNA polymerase sigma subunit. Cell. 2011;147:1257–1269. doi: 10.1016/j.cell.2011.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen YF, Helmann JD. DNA-melting at the Bacillus subtilis flagellin promoter nucleates near -10 and expands unidirectionally. J Mol Biol. 1997;267:47–59. doi: 10.1006/jmbi.1996.0853. [DOI] [PubMed] [Google Scholar]
- 25.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 26.Artsimovitch I, Svetlov V, Murakami KS, Landick R. Co-overexpression of Escherichia coli RNA polymerase subunits allows isolation and analysis of mutant enzymes lacking lineage-specific sequence insertions. J Biol Chem. 2003;278:12344–12355. doi: 10.1074/jbc.M211214200. [DOI] [PubMed] [Google Scholar]
- 27.Callaci S, Heyduk E, Heyduk T. Conformational changes of Escherichia coli RNA polymerase sigma70 factor induced by binding to the core enzyme. J Biol Chem. 1998;273:32995–33001. doi: 10.1074/jbc.273.49.32995. [DOI] [PubMed] [Google Scholar]
- 28.Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010;2010 doi: 10.1101/pdb.prot5448. pdb prot5448. [DOI] [PubMed] [Google Scholar]
- 29.Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol. 2008;453:3–31. doi: 10.1007/978-1-60327-429-6_1. [DOI] [PubMed] [Google Scholar]
- 30.Markham NR, Zuker M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 2005;33:W577–581. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schroeder LA, Choi AJ, DeHaseth PL. The -11A of promoter DNA and two conserved amino acids in the melting region of sigma70 both directly affect the rate limiting step in formation of the stable RNA polymerase-promoter complex, but they do not necessarily interact. Nucleic Acids Res. 2007;35:4141–4153. doi: 10.1093/nar/gkm431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fried MG, Liu G. Molecular sequestration stabilizes CAP-DNA complexes during polyacrylamide gel electrophoresis. Nucleic Acids Res. 1994;22:5054–5059. doi: 10.1093/nar/22.23.5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fried MG, Bromberg JL. Factors that affect the stability of protein-DNA complexes during gel electrophoresis. Electrophoresis. 1997;18:6–11. doi: 10.1002/elps.1150180103. [DOI] [PubMed] [Google Scholar]
- 34.Vossen KM, Fried MG. Sequestration stabilizes lac repressor-DNA complexes during gel electrophoresis. Anal Biochem. 1997;245:85–92. doi: 10.1006/abio.1996.9944. [DOI] [PubMed] [Google Scholar]
- 35.Margalit H, Shapiro BA, Nussinov R, Owens J, Jernigan RL. Helix stability in prokaryotic promoter regions. Biochemistry. 1988;27:5179–5188. doi: 10.1021/bi00414a035. [DOI] [PubMed] [Google Scholar]