

Abstract
Heterozygosity–fitness correlations (HFCs) are often used to link individual genetic variation to differences in fitness. However, most studies examining HFCs find weak or no correlations. Here, we derive broad theoretical predictions about how many loci are needed to adequately measure genomic heterozygosity assuming different levels of identity disequilibrium (ID), a proxy for inbreeding. We then evaluate the expected ability to detect HFCs using an empirical data set of 200 microsatellites and 412 single nucleotide polymorphisms (SNPs) genotyped in two populations of bighorn sheep (Ovis canadensis), with different demographic histories. In both populations, heterozygosity was significantly correlated across marker types, although the strength of the correlation was weaker in a native population compared with one founded via translocation and later supplemented with additional individuals. Despite being bi-allelic, SNPs had similar correlations to genome-wide heterozygosity as microsatellites in both populations. For both marker types, this association became stronger and less variable as more markers were considered. Both populations had significant levels of ID; however, estimates were an order of magnitude lower in the native population. As with heterozygosity, SNPs performed similarly to microsatellites, and precision and accuracy of the estimates of ID increased as more loci were considered. Although dependent on the demographic history of the population considered, these results illustrate that genome-wide heterozygosity, and therefore HFCs, are best measured by a large number of markers, a feat now more realistically accomplished with SNPs than microsatellites.
Keywords: SNP, microsatellite, standardized multilocus heterozygosity, identity disequilibrium, bighorn sheep, Ovis canadensis
Introduction
Individual heterozygosity can be easily measured using genetic markers and is often used as proxy for inbreeding (Hansson and Westerberg, 2002; Balloux et al., 2004). Many studies have examined the relationship between individual genetic diversity and fitness using heterozygosity–fitness correlations (HFCs). HFCs appeal to wildlife and conservation biologists who cannot easily reconstruct pedigrees and directly measure inbreeding in natural populations, especially for endangered species (Balloux et al., 2004; Grueber et al., 2008; Chapman et al., 2009). Meta-analyses of HFCs, however, have revealed that effect sizes are often weak (Coltman and Slate, 2003; Chapman et al., 2009; Szulkin et al., 2010). The modest numbers of genetic markers typically employed may provide inaccurate estimates of genome-wide heterozygosity. It is also unclear whether different types of markers provide similar information content.
Microsatellites have been the most commonly used markers to investigate HFCs. They are relatively abundant in the genome and are highly polymorphic. However, their mutational mechanism is not well understood, and the high mutation rate likely leads to elevated levels of homoplasy, which can underestimate true heterozygosity (Hansson and Westerberg, 2002). In addition, the process of isolating and characterizing novel loci often selects for the most polymorphic markers, resulting in ascertainment bias and an upwardly skewed estimate of genome-wide diversity (Brandstrom and Ellegren, 2008).
Despite their growing use in molecular ecology and evolutionary biology, single nucleotide polymorphisms (SNPs) have been less widely used in HFC studies, perhaps because they are almost exclusively bi-allelic. However, they have some advantages over microsatellites: they are more abundant in the genome, have a well-understood mutational mechanism with low levels of homoplasy and are amenable to high throughput genotyping (Morin et al., 2004). Several authors contend that SNPs may be more suitable than microsatellites for HFCs. Tsitrone et al. (2001) used extensive simulation studies to examine the effect of different mutational patterns (corresponding to SNPs and microsatellites) and demographic history on the expected correlation between heterozygosity and fitness. Their results point to a complex interplay between these two factors. The high mutation rate of microsatellites should make them more suitable to detect HFCs that result from recent inbreeding due to crosses between relatives or small population size. The lower mutation rates typical of SNPs may make them better than microsatellites to detect HFCs resulting from ancient inbreeding, such as when two subpopulations accumulate genetic differentiation during a long period of isolation and then come back into contact (Tsitrone et al., 2001). Chakraborty (1981) and DeWoody and DeWoody (2005) argued that correlations between heterozygosity at a set of loci and genomic heterozygosity would be high only when the set of marker loci represents a high fraction of all polymorphisms in the genome. However, they modeled populations with no inbreeding and no correlations among loci, a condition under which HFC does not occur unless the marker loci themselves are coding for fitness traits, which is not the case for most recently published HFC studies. It remains unclear whether many markers with low genetic diversity (SNPs) or fewer markers with higher diversity (microsatellites) are more suitable to explore general-effect HFCs. This question becomes important as new technologies allow for the development of larger genome-wide marker sets of both SNPs and microsatellites (Baird et al., 2008; Davey et al., 2011).
Studies of HFCs commonly use 10–30 loci (Chapman et al., 2009). However, as the demographic history of a population will heavily influence correlations in marker heterozygosity within individuals (Ljungqvist et al., 2010; Szulkin et al., 2010), such modest numbers of markers may sometimes be insufficient (Balloux et al., 2004; Väli et al., 2008; Ljungqvist et al., 2010; Forstmeier et al., 2012). For example, Väli et al. (2008) looked at the correlation between heterozygosity at 10–17 microsatellites and allelic diversity in 10 introns across eight populations of carnivores. They found a positive correlation between average heterozygosity and allelic diversity among populations but not between individual heterozygosity at SNPs and microsatellites. In general, HFCs are not expected within populations without measurable identity disequilibrium (ID), a correlation in identity by descent among markers (Slate et al., 2004; David et al., 2007; Szulkin et al., 2010). ID arises from departures from random mating (for example, inbreeding) or demographic events (for example, a population bottleneck or admixture) that cause the heterozygosity of loci to become associated (Bierne et al., 2000; Szulkin et al., 2010). In the absence of ID, HFCs will be detected only if one or more markers are directly associated with a gene influencing fitness, so-called local or direct effects (Hansson and Westerberg, 2002). These direct effect correlations are difficult to detect because they depend on the specific marker set used in a study.
Here, we examine the contrasting effects of the number of markers considered and marker type on the ability to detect general-effect HFCs. We first use existing models of HFC to derive broad theoretical predictions about how many loci are needed to adequately measure genomic heterozygosity assuming different levels of inbreeding and marker genetic diversity. We then use large sets of both microsatellites and SNPs genotyped in two populations of bighorn sheep (Ovis canadensis) to approach this question empirically. Our two study populations have very different demographic histories: one was founded in the 1920's with 12 individuals and experienced a prolonged bottleneck post founding, then recent admixture following a ‘genetic rescue' where 15 individuals were intentionally introduced into the population. The other is a native population with no genetic evidence of a comparable bottleneck. These contrasting histories should affect the magnitude of ID and hence our ability to detect HFCs. In the population subject to ‘genetic rescue', ID is expected to be higher, arising both from historical inbreeding and admixture following the introductions. In the native population, ID will likely track demography, arising if heterozygosity decreases because of inbreeding. Therefore, power to detect HFCs should be greater in the bottlenecked population than the native one. To test these hypotheses, we first sought to measure the strength of correlations between estimates of heterozygosity from microsatellites and SNPs within individuals. We then examined how many markers are needed to accurately reflect genome-wide heterozygosity and ID in these two populations.
Theory
General-effect HFCs arise as the product of two correlations: the correlation between fitness (W) and inbreeding (f), and the correlation between f and heterozygosity (h) (Slate et al., 2004; Szulkin et al., 2010), such that:
For the purposes of this paper, we do not consider the correlation between W and f. Rather, we focus on the power of different marker sets to detect the correlation between h and f.
Two sources of sampling variance may affect HFCs. One is the sampling of individuals: if a small sample is taken in a population that contains a small proportion of inbred individuals, the proportion of inbred individuals in the sample is subjected to a large variance that directly affects HFC estimates. The estimated HFC will be stronger or weaker than true the HFC in the population simply because the proportion of inbred individuals in the sample happens to be higher or lower than their frequency in the population. This source of error can be large but the only way to reduce it is to sample more individuals. The second source of variance arises from the fact that heterozygosity measured at a set of marker loci is not perfectly correlated with genomic heterozygosity and/or with individual inbreeding level. This error depends on the characteristics of the marker loci (number and genetic diversity). We will mainly concentrate on this second type of error, assuming that all efforts have been made to reduce the first source of error.
The problem is now to estimate how well inbreeding is measured by (or correlated to) heterozygosity in a sample of markers. We consider standardized heterozygosity (sensu Coltman et al. (1999) denoted H** for consistency with notations in Szulkin et al. (2010)) at a set of loci L, of size A, where hi is the observed heterozygosity at locus i and upper bar denotes expectations
 |
based on Szulkin et al. (2010), the expected correlation between H** and inbreeding level (f) is
 |
where g2 is the covariance of heterozygosity between markers standardized by their average heterozygosity (David et al., 2007) and
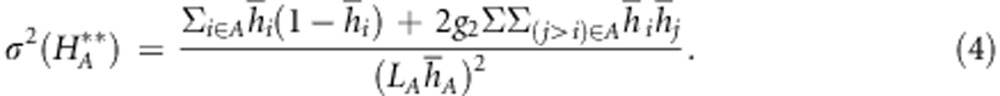 |
Assuming that all loci in the set LA have the same average heterozygosity hA (for simplicity), this gives
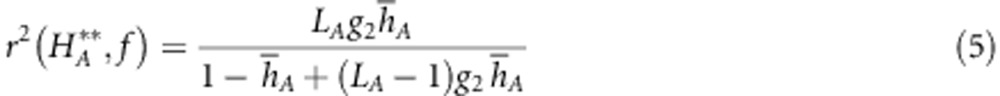 |
from this it can be seen that the correlation approaches unity as the number of markers increases, and depends mostly on the product of the number of loci and their average heterozygosity: 100 loci with h=0.1 are equivalent to 20 loci with h=0.5. The rate at which the correlation approaches 1 increases with the ID (represented by g2). When g2 is null, the correlation is necessarily zero because inbreeding does not vary in the population. In such cases, trying to estimate genome-wide heterozygosity from a small set of markers is pointless because all the variance comes from sampling error.
Often one does not have an independent measure of inbreeding (for example, pedigrees), and therefore the above formula cannot be checked directly. Instead, what can be performed (and will be performed below using real data) is (i) to check the consistency between two different subsets of marker loci (for example, SNPs and microsatellites) and (ii) to check how fast estimates of heterozygosity based on increasing numbers of loci converge to the most precise estimate available (which uses all loci). Theoretical predictions can be obtained for (i) and (ii). The first is simply the correlation between heterozygosities at two non-overlapping sets of loci; it can be simply computed based on the assumption (underlying the general-effect model) that this correlation emerges only as a result of the common dependency of heterozygosities in both sets of markers on the extent of inbreeding. Therefore,
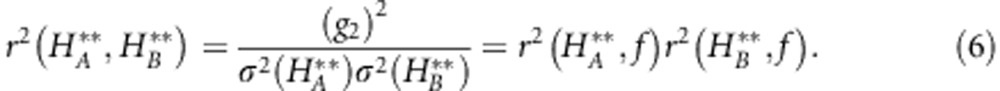 |
The quantity relevant to point (ii) is the correlation between heterozygosity at L loci and heterozygosity at a subset (S) of these loci, which contain a fraction pS of the total number of loci. For simplicity, we model a situation in which there are no missing data and all individuals are typed at the same set of loci. If individuals are typed at sets of loci that substantially differ in their average heterozygosity correlations will be reduced; however, this is unlikely to be the case unless substantial amounts of data are missing. As correlations are insensitive to scaling by a constant, we can work here with raw heterozygosities H (not standardized heterozygosity H**). Using raw heterozygosity, total heterozygosity H is the sum of heterozygosity at the S loci (HS) and at the remaining loci (HR). Thus,
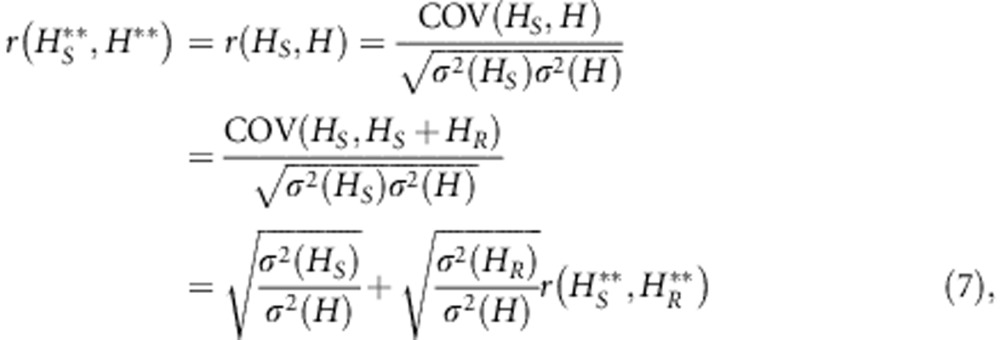 |
where σ2(HS) is the numerator of Equation 4 (σ2(HR) and σ2(H) can be computed similarly, making the summations over the appropriate sets of loci). Assuming that all loci have the same heterozygosity h one obtains, after some algebra
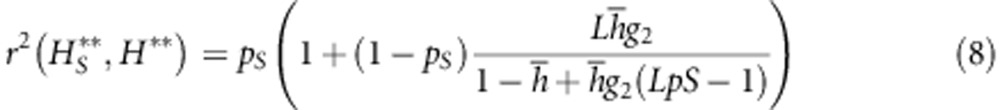 |
in this formula, one can distinguish two terms: the first is simply the proportion of loci included in the subset (pS) and reflects the fact that subset S will always capture a proportion of the variance in total heterozygosity at all loci because they are part of the total. The ability of the S subset to inform about the other loci (hence about the genome in general) is reflected by the second term, which relies on the existence of ID (g2): through this disequilibrium, the loci in S inform about the state of other loci and hence capture a more than proportional share of total variance in heterozygosity.
Materials and methods
Study populations
We examined patterns of heterozygosity in bighorn sheep at the National Bison Range (Montana, USA; NBR) and at Ram Mountain (Alberta, Canada; RM). In both populations, long-term studies follow individuals throughout their lives. The National Bison Range population was founded in 1922 via translocation of 12 individuals from Banff National Park (Alberta, Canada). Individual monitoring started in 1979, with genetic sampling beginning in 1988. Beginning in 1985, NBR experienced a ‘genetic rescue' via intentional translocation of 15 individuals from neighboring populations to prevent local extinction after years of isolation and inbreeding (Hogg et al., 2006; Miller et al., 2012). Prior to the introduction, census size and growth rate had been steadily declining (average census size of 48 sheep between 1922 and 1985). Following the supplementation, there has been an increase both in census size (142 sheep in late 2012) and genetic diversity (Hogg et al., 2006; Miller et al., 2012).
In contrast, Ram Mountain is a native population in which individual-based monitoring began in 1972 with genetic sampling starting in 1988 (Jorgenson et al., 1997; Coltman et al., 2002). Between 1988 and 2010, census size fluctuated between 38 and 210 sheep, declining recently because of low recruitment (Jorgenson et al., 1997) and cougar (Puma concolor) predation (Festa-Bianchet et al., 2006).
Marker genotyping and selection
SNP genotypes used in this study were generated by typing 27 individuals from NBR and 50 from RM on the OvineSNP50 BeadChip (Miller et al., 2011), yielding 853 variable loci. For this study, we excluded loci that were genotyped in <90% of individuals in both populations (N=38), had <5% minor allele frequency (N=392) and did not conform to Hardy–Weinberg expectations following a Bonferroni correction (N=2). We also excluded any loci mapped to the X chromosome (N=9). This resulted in a final data set of 412 SNPs (Supplementary Table 1). We included loci polymorphic in one population but monomorphic in the other. Note that because of their discovery via cross-species application of the OvineSNP50 BeadChip, the SNPs used in this study are widely distributed in the genome, and are mostly intergenic as few are expected to be in or near genes based on annotation of the domestic sheep genome (Miller et al., 2011).
Microsatellite loci used in analyses were a subset (N=200; Supplementary Table 2) of those used to construct a bighorn sheep linkage map (Poissant et al., 2010). Primer information and PCR conditions for the markers can be found in Poissant et al. (2009, 2010) and references therein. All loci conformed to Hardy–Weinberg expectations following a Bonferroni correction. Loci were retained only if they were genotyped in both populations and had <25% missing genotypes in the samples from either population. As with the SNP set, loci on the X chromosome were excluded.
In total, 26 individuals from NBR and 48 from RM were genotyped at both sets of markers and included in subsequent analyses. The individuals from NBR were born between 1981 and 2004 and include descendants of the original founders of the population (N=4), transplanted individuals (N=2) and their progeny (N=20).
Statistical analyses
We calculated individual standardized multilocus heterozygosity (stMLH) following Coltman et al. (1999). The relationship between individual stMLH from each marker set was assessed using reduced major axis regression with 1000 jackknife iterations, as implemented in RMA version 1.21 (Bohonak and van der Linde, 2004). Reduced major axis regression was chosen to account for the uncertainty associated with stMLH measures used as both dependent and independent variables. For resampling tests, 100 random subsets of markers were sampled without replacement from the full data sets using the ‘sample' function in R version 2.13.0 (R Development Core Team, 2005). For microsatellites, subsets of 5, 10, 20, 30, 50, 75, 100, 125, 150 and 175 loci were extracted, whereas for SNPs the subsets consisted of 20, 50, 75, 100, 150, 200, 250, 300, 350 and 400 loci. We then calculated stMLH for each subset using a custom Perl script. The coefficients of determination (r2) were compared between the stMLH calculated for each subset and a total stMLH calculated from the concatenation of the SNP and microsatellite data sets (all 612 loci). In addition, we calculated r2 between each subset and total stMLH for all loci of their respective marker type. We then compared these results to the theoretical predictions described in the previous section.
Estimates of ID and expected power to detect HFCs
To measure ID, we used the program RMES (David et al., 2007) to calculate the g2 statistic. Significant covariance among marker genotypes can be attributed to inbreeding, admixture or a bottleneck (David et al., 2007; Szulkin et al., 2010). Assessment of significant levels of ID (g2 >0) utilized 1000 resampling iterations. Calculations were performed for both populations on the full microsatellite set, the full SNP set, the concatenated marker set and all marker subsets used in the stMLH resampling calculations. We also examined the effect of the number of individuals sampled on the accuracy of g2 estimates. For this analysis, we bootstrapped both the full microsatellite and full SNP data sets in each population, generating 100 replicates containing different numbers of individuals: 5, 10, 20, 30, 50 or 100 individuals.
To explore the power of different marker sets to detect HFCs, we calculated the expected correlation between f and stMLH using Equation 3 based on empirical estimation of variance in heterozygosity as well as its theoretical value based only on the number and average heterozygosity of the markers (Equation 5). Equation 5 has the advantage that it can be applied to assess the power of a study before actually performing it, as it requires only approximated parameter values. As with our estimates of g2, we calculated r2(f, h) for the full SNP, microsatellite and concatenated data sets, as well as all subsets. When the estimate of g2 was negative we set r2(f, h) to 0.
Results
Summary statistics of markers
In NBR, the average (±s.d.) minor allele frequency for SNP loci was 0.197 (±0.160), and average observed heterozygosity (Ho) was 0.279 (±0.202). In RM, average minor allele frequency was 0.212 (±0.151) and Ho 0.292 (±0.178). For microsatellite loci, Ho was 0.643 (±0.161), and number of alleles per locus ranged from 2 to 9 (average 4.39±1.43) in NBR, whereas in RM Ho was 0.610 (±0.157) and the number of alleles per locus ranged from 2 to 10 (average 4.21±1.48).
Estimates of ID and expected power to detect HFCs
All estimates of g2 based on total marker sets were greater than zero for both populations (P<0.001; Table 1). However, g2 was much stronger in NBR than RM. Across both populations, the full SNP set produced higher estimates of g2 than the full microsatellite set, and the combined data sets were intermediate. Our subsampling analyses showed that average values of g2 on par with the genome-wide estimates of that same marker type were obtained even when few markers were considered (Figure 1a–d). However, there was considerable variation around these estimates. For example, in RM the s.d. estimates were larger than the average values of g2 when fewer than 75 microsatellites or 150 SNPs were examined (Figure 1b and d).
Table 1. Estimate of identity disequilibrium (g 2) and expected r 2 between inbreeding (f) and stMLH (H A **) for the different full marker sets in each population of sheep.
| s.d. of stMLH | g2 | s.d. of g2 | Expected r2 (HA**, f) | |
|---|---|---|---|---|
| NBR | ||||
| Microsatellites | 0.22245 | 0.05554 | 0.02773 | 0.95695 |
| SNPs | 0.23662 | 0.06481 | 0.03081 | 0.91378 |
| Combined | 0.24891 | 0.06074 | 0.02920 | 0.96095 |
| RM | ||||
| Microsatellites | 0.07086 | 0.00198 | 0.00412 | 0.38321 |
| SNPs | 0.09843 | 0.00456 | 0.00519 | 0.43703 |
| Combined | 0.07093 | 0.00309 | 0.00254 | 0.54937 |
Abbreviations: SNP, single nucleotide polymorphism; stMLH, standardized multilocus heterozygosity.
Figure 1.

Box plots showing the average level of identity disequilibrium (g2) for the different marker subsets. Each subset was generated by sampling markers from the total data set 100 times. Plots A and C correspond to NBR at microsatellites (a) and SNPs (c), whereas plots B and D correspond to RM at microsatellites (b) and SNPs (d).
Bootstrapping the full data sets similarly showed that a stable average value of g2 can be estimated with small sample sizes; however, larger sample sizes increase the precision (Supplementary Table 3). One might have expected the effects of increasing sample size to be more apparent in NBR, where the data set contains a few highly inbred individuals and the chances of sampling them would therefore lead to large s.d. around the estimates of g2. However, when scaled as a percentage of the average estimate, the effect of sampling is greater for RM than NBR. Even when 100 individuals were assumed, s.d. values representing >14% of the average g2 estimate in NBR and >30% in RM were seen.
Expected r2 between f and stMLH for the various full marker sets (Table 1) were stronger in NBR than RM. In both populations, the strongest r2 was seen from the combined marker data set. The expected r2 increased and the variation around estimates decreased as the number of markers increased (Figure 2a–d). There was an apparent upward bias of the r2 between f and stMLH when measured by the full marker sets compared with the subsets. This bias is likely an artifact given that there is only one estimate based on the full marker sets (rather than 100 permutations) and that the correlation is based on a g2 statistic that still has error associated with it (Table 1).
Figure 2.

Box plots showing the average expected r2 between inbreeding and heterozygosity for the different marker subsets. Plots A and C show correlation in NBR at microsatellites (a) and SNPs (c), whereas plots B and D show correlations in RM at microsatellites (b) and SNPs (d). Solid lines show predicted correlations based on Equation 5.
Correlations between marker types and among subsets
Individual stMLH was significantly positively correlated between marker types in both populations (Figure 3). However, the correlation was much stronger in NBR (NBR r=0.954, t24 =15.656, P<<0.001; RM r=0.360, t46 =2.620, P=0.011). Based on Equation 6, the expected correlations were r=1.15 and r=0.44 for NBR and RM, respectively. Slight differences between the predicted and observed values (as well as the r>1) likely arise because the combined g2 value is measured with error (Table 1). To ensure that the larger sample size in RM was not the main driver of the difference in correlation between RM and NBR we jackknifed our data from RM resampling 100 sets of 26 individuals, this yielded an average correlation of 0.355±0.121 (s.d.).
Figure 3.

Correlation between individual heterozygosity at SNPs and microsatellites in RM and NBR. Reduced major axis regression lines are shown: y=0.7199x+0.2802 for RM (solid line); y =0.9401x+0.0631 for NBR (dashed line).
In our resampling analyses, r2 values were stronger in NBR than RM regardless of the number or type of markers examined (Figures 4a–d). In both the SNP and microsatellite data sets, the correlation with the total measure of stMLH strengthened with increasing number of markers (Figures 4a and c). In NBR, correlations between microsatellite subsets and total stMLH were higher than those for an equal number of SNPs (Figure 4c). However, an asymptote of strong correlation (r2>0.9) was reached with as few as 75 microsatellites or 200 SNPs. These differences between marker types disappeared when marker subsets were compared with stMLH of only that same marker type and scaled as a proportion of the total number of either SNPs or microsatellites considered (Figure 4d). For RM, when equal numbers of markers were compared, microsatellites produced a marginally higher average correlation to total stMLH than SNPs (Figure 4a), although these differences were not significant. The two marker types gave near-identical correlations when marker subsets were compared with stMLH values from only the same marker type (Figure 4b). Average correlations to marker-specific estimates of stMLH were stronger than when subsets were compared with total stMLH (average increase of 0.12 for SNPs and 0.20 for microsatellites). For both populations, all empirical correlations exceeded the null expectations and closely paralleled predicted correlations (Figure 4).
Figure 4.

Average r2 between marker subset stMLH and genome-wide stMLH. Each subset was generated by sampling markers from the total data set 100 times; error bars show s.d. Plots A and C show correlations for SNPs (open squares) and microsatellites (filled triangles) when all 612 loci are considered in RM and NBR, respectively. Plots B and D show correlations when subsets are compared with stMLH exclusively from the same marker type in RM (b) and NBR (d). Note that the x axis is now scaled as a proportion of the total number of either SNPs or microsatellites. Predicted correlations based on Equation 8 are shown for SNPs (solid lines) and microsatellites (dotted lines), dashed lines show predicted correlations among subsets in the absence of identity disequilibrium.
Discussion
Influence of population history
As expected, across all analyses the strength of association between marker heterozygosities as well as the expected ability to detect HFCs was highly dependent on the demographic history of the population (Ljungqvist et al., 2010; Szulkin et al., 2010). Bighorn sheep tend to be philopatric and have a highly polygynous mating system, in which a few dominant males sire the majority of offspring (Hogg and Forbes, 1997; Coltman et al., 2002). Thus, even in a native population such as RM a certain level of ID is to be expected. In addition, RM is relatively isolated and rarely receives immigrants (Rioux-Paquette et al., 2010), furthering the likelihood of non-random association of alleles because of inbreeding. Disequilibrium is even more likely in NBR given its population history. Descendants of NBR founders are expected to have low overall genetic diversity after years of inbreeding, translocated individuals from neighboring herds will have relatively higher levels of diversity and their progeny are expected to have the highest heterozygosity as a result of the admixture between the founder and translocated individuals (Hogg et al., 2006; Miller et al., 2012).
Theory predicts that population history as well as mating system (that is, partial inbreeding or selfing), as summarized through the g2 parameter, determines how well heterozygosity at a set of markers reflects heterozygosity at other loci and, by extension, genomic heterozygosity and inbreeding (Szulkin et al., 2010, our Equations 3, 5 and 6). Theoretical predictions correctly match the observed correlations between heterozygosity at SNPs and heterozygosity at microsatellites in our data. Although significant correlations were seen in both populations, it was much tighter in NBR (Figure 3). In contrast, Väli et al. (2008) found no significant correlation between individual heterozygosity at SNPs and microsatellites at the level of the individual in four populations of wolves (Canis lupus) and one of coyotes (C. latrans). However, both of these species have high dispersal rates and large effective population sizes (Pilot et al., 2006), which may not allow for such correlations to develop. In addition, Väli et al. (2008) used only 10–17 microsatellites and 25–51 SNPs in 10 introns, which our results suggest may not have had the power to detect an association in a population with low g2 (for example, 0.001–0.005). The contrasting effect of demographic history is equally apparent when trying to estimate genome-wide heterozygosity from a subset of markers (Figure 4).
Interaction between the number of markers and marker type on correlations in stMLH
In NBR, microsatellites were more highly correlated to total stMLH than SNPs when equal numbers of markers were compared: 20 microsatellites predicted inbreeding as well as 75 SNPs—as expected given their higher average heterozygosity (0.643 compared with 0.279) and our theoretical equations (Equation 8). However, even a small number of either type of marker was highly correlated with inbreeding level and with total heterozygosity at all markers because of the high g2. The situation was slightly different in RM. Here, SNPs and microsatellites gave essentially the same correlations to our total measure of stMLH when equal numbers of markers were compared. However, given the low g2, a much larger number of loci (microsatellites or SNPs) is needed to adequately measure inbreeding in RM.
In short, microsatellites are more informative than SNPs because they have higher genetic diversity per locus; however, to find the most efficient strategy, one must consider that it is now becoming technically easier to develop and type a large number of SNPs than an equivalent number of microsatellites (Baird et al., 2008; Davey et al., 2011; Guichoux et al., 2011). These results agree with previous theoretical and empirical studies that suggested that highly heterozygous multi-allelic markers will have higher correlation between MLH and genome-wide heterozygosity than bi-allelic ones (Slate et al., 2004; Ljungqvist et al., 2010; Online Appendix 2 in Szulkin et al., 2010; Forstmeier et al., 2012) except in special cases (Tsitrone et al., 2001).
One factor that could seem to limit the robustness of our conclusions is that correlations to total heterozygosity could be biased because the data set contains a larger number of SNPs than microsatellites (412 versus 200 loci). We do not feel that this is a problem for several reasons. First, individual heterozygosity between the two marker types was correlated (Figure 3) and should therefore show the same patterns no matter the ratio of loci examined. Moreover, if the relative proportion of markers biased our estimates, one would expect correlations to the measure of total stMLH to be constrained by the marker's abundance in the total data set—for example, r2<0.33 for microsatellites. Figure 4 shows that this is not the case: both sets of loci quickly rose to essentially perfect correlations in NBR and increased to levels well above their relative proportions in RM. Finally, in general, SNPs are more abundant in a genome than microsatellites, and although the ratio is not 2:1, our data set reflects this difference in abundance. Together, these points suggest that there should not be any substantial bias based on the relative composition of the markers.
ID and expected correlations between f and stMLH
On average, modest numbers of markers seemed to accurately estimate the levels of ID; however, variability was high when only a few markers were considered. Several recent studies have estimated ID for both wild and captive populations (Küpper et al., 2010; Borrell et al., 2011; Grueber et al., 2011; Olano-Marin et al., 2011; Jourdan-Pineau et al., 2012; Wetzel et al., 2012). In all but one case (Olano-Marin et al., 2011), these estimates were nonsignificant, even in the highly endangered takahe (Porphyrio hochstetteri) that had experienced a bottleneck reducing the population to 17 individuals (Grueber et al., 2011). However, all of the studies showing nonsignificant results used between 7 and 24 microsatellite loci (average 18.2), which we have shown can give an inaccurate picture of diversity, depending on the specific loci examined and the demographic history of the population. In contrast, Olano-Marin et al. (2011) used 80 microsatellites to study a wild population of blue tits (Cyanistes caeruleus).
Time to move towards SNPs for use in HFCs?
Our results suggest that SNPs are more suited for HFCs than previously thought. First, significant correlations between individual stMLH at SNPs and microsatellites indicate that there is no loss of information when using a bi-allelic rather than a multi-allelic marker to estimate heterozygosity. Second, SNPs may be more suited for examining the various hypotheses that underlie HFCs, such as direct effects and local effects (Hansson and Westerberg, 2002), given that they are more abundant in the genome than in microsatellites and can be more readily genotyped at ultra-high density. However, to perform the same job as microsatellites, SNPs need to be more numerous as they are on average less heterozygous. The exact number of markers needed to obtain high correlations depended heavily on the demographic history of the population. For populations such as NBR that have experienced a severe bottleneck or admixture fewer markers will be needed to obtain significant g2 estimates and detect HFCs. In this situation, it will be more beneficial for researchers to type additional individuals, getting an accurate estimate of the variance in inbreeding and fitness, rather than typing more markers in fewer individuals. For populations with no history of a bottleneck or severe inbreeding, such as RM, significantly more markers will be needed to accurately estimate genome-wide heterozygosity. It is then questionable whether lots of effort should be invested into typing the required number of markers, whatever their type, given that the signal (HFC and inbreeding) is necessarily very weak in such situations. Our equations can be directly used to assess the required number of loci needed to achieve a given accuracy in the measure of inbreeding (or genomic heterozygosity), provided a value of g2 is available (or can be estimated from preliminary data with fewer loci). For example, in RM using the combined g2 estimate from all markers (the most precise value available), Equation 5 can be used to predict that no less than 1853 microsatellites or 7053 SNPs would be needed for stMLH to be highly correlated (r2=0.9) to inbreeding.
Although SNPs are still moderately expensive to develop for wild species, new methods allow rapid discovery of large marker panels (Baird et al., 2008; Davey et al., 2011) at diminishing costs. Once discovered, new technologies, such as array-based genotyping assays (Shen et al., 2005) and genotype-by-sequencing approaches (Baird et al., 2008), will allow for SNP data sets to be rapidly genotyped in many individuals. Comparable methods for scaling up the genotyping of microsatellites are not currently available.
Although we were unable to directly compare SNPs and microsatellites in terms of their ability to detect HFCs for specific traits because of the small number of individuals genotyped at the sets of SNPs and microsatellites used here, we now have an indication of the number of markers that would be needed for future efforts. More broadly, our results highlight that accurate calculations of stMLH, assessment of ID, and thereby detection of HFCs will likely require a large number of markers, be they SNPs or microsatellites. However, the exact number is highly dependent on the demographic history (or mating system) of the population being examined, the key parameter being the ID (which can be estimated with g2). Efforts should be directed towards precisely estimating this parameter in natural populations. To this end, assuming that the number of loci available in population genetic studies will continue to increase, the main limitation will become the sample size in terms of numbers of individuals.
Data archiving
Tables of individual heterozygosity and homozygoisty for both populations and marker types have been deposited in Dryad, doi: 10.5061/dryad.6vk48.
Acknowledgments
Funding for lab work conducted in this study came from Alberta Conservation Association Grant in Biodiversity (ACA), the Natural Sciences and Engineering Research Council of Canada (NSERC), and Alberta Innovates Technology Futures (AI). JMM and RMM's graduate research was funded by AI graduate scholarships, the University of Alberta, and JMM was an NSERC Vanier scholar. JP was supported by NSERC and Marie Curie postdoctoral fellowships. The NBR field study was supported by grants from The Charles Engelhard Foundation, Eppley Foundation for Research and National Geographic Society. The RM field study was supported by NSERC, Alberta Fish and Wildlife, and ACA. We thank the U.S. Fish and Wildlife Service for ongoing cooperation and assistance at the NBR study site and the numerous field assistants at both RM and NBR for their help throughout the years. We would like to thank an anonymous editor and two reviewer for helpful comments on the manuscript.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Heredity website (http://www.nature.com/hdy)
Supplementary Material
References
- Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3:e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balloux F, Amos W, Coulson T. Does heterozygosity estimate inbreeding in real populations. Mol Ecol. 2004;13:3021–3031. doi: 10.1111/j.1365-294X.2004.02318.x. [DOI] [PubMed] [Google Scholar]
- Bierne N, Tsitrone A, David P. An inbreeding model of associative overdominance during a population bottleneck. Genetics. 2000;155:1981–1990. doi: 10.1093/genetics/155.4.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohonak AJ, van der Linde K.2004. RMA: Software for Reduced Major Axis regression, Java version. Website http://www.kimvdlinde.com/professional/rma.html .
- Borrell YJ, Carleos CE, Sánchez JA, Vázquez E, Gallego V, Asturiano JF, et al. Heterozygosity–fitness correlations in the gilthead sea bream Sparus aurata using microsatellite loci from unknown and gene-rich genomic locations. J Fish Biol. 2011;79:1111–1129. doi: 10.1111/j.1095-8649.2011.03099.x. [DOI] [PubMed] [Google Scholar]
- Brandstrom M, Ellegren H. Genome-wide analysis of microsatellite polymorphism in chicken circumventing the ascertainment bias. Genome Res. 2008;18:881–887. doi: 10.1101/gr.075242.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty R. The distribution of the number of heterozygous loci in an individual in natural-populations. Genetics. 1981;98:461–466. doi: 10.1093/genetics/98.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Nakagawa S, Coltman DW, Slate J, Sheldon BC. A quantitative review of heterozygosity–fitness correlations in animal populations. Mol Ecol. 2009;18:2746–2765. doi: 10.1111/j.1365-294X.2009.04247.x. [DOI] [PubMed] [Google Scholar]
- Coltman D, Pilkington J, Smith J, Pemberton J. Parasite-mediated selection against inbred Soay sheep in a free-living, island population. Evolution. 1999;53:1259–1267. doi: 10.1111/j.1558-5646.1999.tb04538.x. [DOI] [PubMed] [Google Scholar]
- Coltman D, Slate J. Microsatellite measures of inbreeding: a meta-analysis. Evolution. 2003;57:971–983. doi: 10.1111/j.0014-3820.2003.tb00309.x. [DOI] [PubMed] [Google Scholar]
- Coltman DW, Festa-Bianchet M, Jorgenson JT, Strobeck C. Age-dependent sexual selection in bighorn rams. Proc Biol Sci. 2002;269:165–172. doi: 10.1098/rspb.2001.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet. 2011;12:499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- David P, Pujol B, Viard F, Castella V, Goudet J. Reliable selfing rate estimates from imperfect population genetic data. Mol Ecol. 2007;16:2474–2487. doi: 10.1111/j.1365-294X.2007.03330.x. [DOI] [PubMed] [Google Scholar]
- DeWoody Y, DeWoody J. On the estimation of genome-wide heterozygosity using molecular markers. J Heredity. 2005;96:85–88. doi: 10.1093/jhered/esi017. [DOI] [PubMed] [Google Scholar]
- Festa-Bianchet M, Coulson T, Gaillard JM, Hogg JT, Pelletier F. Stochastic predation events and population persistence in bighorn sheep. Proc Biol Sci. 2006;273:1537–1543. doi: 10.1098/rspb.2006.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstmeier W, Schielzeth H, Mueller JC, Ellegren H, Kempenaers B. Heterozygosity–fitness correlations in zebra finches: microsatellite markers can be better than their reputation. Mol Ecol. 2012;21:3237–3249. doi: 10.1111/j.1365-294X.2012.05593.x. [DOI] [PubMed] [Google Scholar]
- Grueber C, Waters J, Jamieson I. The imprecision of heterozygosity-fitness correlations hinders the detection of inbreeding and inbreeding depression in a threatened species. Mol Ecol. 2011;20:67–79. doi: 10.1111/j.1365-294X.2010.04930.x. [DOI] [PubMed] [Google Scholar]
- Grueber CE, Wallis GP, Jamieson IG. Heterozygosity–fitness correlations and their relevance to studies on inbreeding depression in threatened species. Mol Ecol. 2008;17:3978–3984. doi: 10.1111/j.1365-294x.2008.03910.x. [DOI] [PubMed] [Google Scholar]
- Guichoux E, Lagache L, Wagner S, Chaumeil P, Léger P, Lepais O, et al. Current trends in microsatellite genotyping. Mol Ecol Res. 2011;11:591–611. doi: 10.1111/j.1755-0998.2011.03014.x. [DOI] [PubMed] [Google Scholar]
- Hansson B, Westerberg L. On the correlation between heterozygosity and fitness in natural populations. Mol Ecol. 2002;11:2467–2474. doi: 10.1046/j.1365-294x.2002.01644.x. [DOI] [PubMed] [Google Scholar]
- Hogg JT, Forbes SH. Mating in bighorn sheep: frequent male reproduction via a high-risk “unconventional” tactic. Behav Ecol Sociobiol. 1997;41:33–48. [Google Scholar]
- Hogg JT, Forbes SH, Steele BM, Luikart G. Genetic rescue of an insular population of large mammals. Proc Biol Sci. 2006;273:1491–1499. doi: 10.1098/rspb.2006.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgenson JT, Festa-Bianchet M, Gaillard J-M, Wishart WD. Effects of age, sex, disease, and density on survival of bighorn sheep. Ecology. 1997;78:1019–1032. [Google Scholar]
- Jourdan-Pineau H, David P, Crochet P-A. Phenotypic plasticity allows the Mediterranean parsley frog Pelodytes punctatus to exploit two temporal niches under continuous gene flow. Mol Ecol. 2012;21:876–886. doi: 10.1111/j.1365-294X.2011.05420.x. [DOI] [PubMed] [Google Scholar]
- Küpper C, Kosztolányi A, Augustin J, Dawson DA, Burke T, Székely T, et al. Heterozygosity-fitness correlations of conserved microsatellite markers in Kentish plovers Charadrius alexandrinus. Mol Ecol. 2010;19:5172–5185. doi: 10.1111/j.1365-294X.2010.04867.x. [DOI] [PubMed] [Google Scholar]
- Ljungqvist M, ÅKesson M, Hansson B. Do microsatellites reflect genome-wide genetic diversity in natural populations? A comment on Väli et al. (2008) Mol Ecol. 2010;19:851–855. doi: 10.1111/j.1365-294X.2010.04522.x. [DOI] [PubMed] [Google Scholar]
- Miller JM, Poissant J, Hogg JT, Coltman DW. Genomic consequences of genetic rescue in an insular population of bighorn sheep (Ovis canadensis) Mol Ecol. 2012;21:1583–1596. doi: 10.1111/j.1365-294X.2011.05427.x. [DOI] [PubMed] [Google Scholar]
- Miller JM, Poissant J, Kijas J, Coltman DW, Consortium TISG A genome-wide set of SNPs detects population substructure and long range linkage disequilibrium in wild sheep. Mol Ecol Res. 2011;11:314–322. doi: 10.1111/j.1755-0998.2010.02918.x. [DOI] [PubMed] [Google Scholar]
- Morin PA, Luikart G, Wayne RK, Grp SNPW SNPs in ecology, evolution and conservation. Trends Ecol Evol. 2004;19:208–216. [Google Scholar]
- Olano-Marin J, Mueller JC, Kempenaers B. Correlations between heterozygosity and reproductive success in the blue tit (Cyanistes caeruleus): an analysis of inbreeding and single locus effects. Evolution. 2011;65:3175–3194. doi: 10.1111/j.1558-5646.2011.01369.x. [DOI] [PubMed] [Google Scholar]
- Pilot M, Jedrzejewski W, Branicki W, Sidorovich VE, Jedrzejewska B, Stachura K, et al. Ecological factors influence population genetic structure of European grey wolves. Mol Ecol. 2006;15:4533–4553. doi: 10.1111/j.1365-294X.2006.03110.x. [DOI] [PubMed] [Google Scholar]
- Poissant J, Hogg JT, Davis CS, Miller JM, Maddox JF, Coltman DW, et al. Genetic linkage map of a wild genome: genomic structure, recombination and sexual dimorphism in bighorn sheep. BMC Genomics. 2010;11:524. doi: 10.1186/1471-2164-11-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poissant J, Shafer ABA, Davis CS, Mainguy J, Hogg JT, Côté SD, et al. Genome-wide cross-amplification of domestic sheep microsatellites in bighorn sheep and mountain goats. Mol Ecol Res. 2009;9:1121–1126. doi: 10.1111/j.1755-0998.2009.02575.x. [DOI] [PubMed] [Google Scholar]
- R Development Core Team . R: A Language and Environment for Statistical Computing, Reference Index Version 2.13.0. R Foundation for Statistical Computing: Vienna, Austria; 2005. [Google Scholar]
- Rioux-Paquette E, Festa-Bianchet M, Coltman D. No inbreeding avoidance in an isolated population of bighorn sheep. Animal Behav. 2010;80:865–871. [Google Scholar]
- Shen R, Fan J-B, Campbell D, Chang W, Chen J, Doucet D, et al. High-throughput SNP genotyping on universal bead arrays. Mutat Res. 2005;573:70–82. doi: 10.1016/j.mrfmmm.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Slate J, David P, Dodds KG, Veenvliet BA, Glass BC, Broad TE, et al. Understanding the relationship between the inbreeding coefficient and multilocus heterozygosity: theoretical expectations and empirical data. Heredity. 2004;93:255–265. doi: 10.1038/sj.hdy.6800485. [DOI] [PubMed] [Google Scholar]
- Szulkin M, Bierne N, David P. Heterozygosity-fitness correlations: Aatime for reapprasial. Evolution. 2010;64:1202–1217. doi: 10.1111/j.1558-5646.2010.00966.x. [DOI] [PubMed] [Google Scholar]
- Tsitrone A, Rousset F, David P. Heterosis, marker mutational processes and population inbreeding history. Genetics. 2001;159:1845–1859. doi: 10.1093/genetics/159.4.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väli Ü, Einarsson A, Waits L, Ellegren H. To what extent do microsatellite markers reflect genome-wide genetic diversity in natural populations. Mol Ecol. 2008;17:3808–3817. doi: 10.1111/j.1365-294X.2008.03876.x. [DOI] [PubMed] [Google Scholar]
- Wetzel DP, Stewart IRK, Westneat DF. Heterozygosity predicts clutch and egg size but not plasticity in a house sparrow population with no. Mol Ecol. 2012;21:406–420. doi: 10.1111/j.1365-294X.2011.05380.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.