

Highlights
-
•
Coronavirus entry, proteolytic processing and RNA synthesis enzymes are attractive targets for antivirals.
-
•
Cell-based screens have been used to identify and evaluate antivirals against SARS-CoV and MERS-CoV.
-
•
New approaches are available to characterize pan-coronavirus inhibitors against CoV proteases.
-
•
Antivirals against SARS-CoV may have efficacy against MERS-CoV and other emerging coronaviruses.
Keywords: SARS-CoV, MERS-CoV, Entry inhibitors, Replicase inhibitors, Antivirals, pGlo
Abstract
To combat the public health threat from emerging coronaviruses (CoV), the development of antiviral therapies with either virus-specific or pan-coronaviral activities is necessary. An important step in antiviral drug development is the screening of potential inhibitors in cell-based systems. The recent emergence of Middle East respiratory syndrome coronavirus (MERS-CoV) necessitates adapting methods that have been used to identify antivirals against severe acute respiratory syndrome coronavirus (SARS-CoV) and developing new approaches to more efficiently screen antiviral drugs. In this article we review cell-based assays using infectious virus (BSL-3) and surrogate assays (BSL-2) that can be implemented to accelerate antiviral development against MERS-CoV and future emergent coronaviruses. This paper forms part of a series of invited articles in Antiviral Research on “From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses.”
1. Introduction
The emergence of zoonotic coronaviruses with human to human transmission potential has highlighted the need for effective antivirals against coronavirus replication (introduced in Hilgenfeld and Peiris, 2013). Since the identification of Middle East respiratory syndrome coronavirus (MERS-CoV), there have been 150 confirmed cases and 64 deaths; a fatality/case ratio of over 40% (World Health Organization, 2013). Currently it is unclear if there are additional asymptomatic cases of infection with MERS-CoV. MERS is reminiscent of the outbreak of severe acute respiratory syndrome (SARS) in the Guangdong province in China in 2002–2003 (reviewed in Cheng et al., 2013).
The causative agent of SARS, severe acute respiratory syndrome coronavirus (SARS-CoV) likely emerged from a bat reservoir and was transmitted through an intermediate reservoir (civet cats), before obtaining the spike mutations to efficiently infect humans and cause severe respiratory disease (reviewed Li, 2013). Recently, a SARS-like bat coronavirus was isolated that can directly infect human cells (Ge et al., 2013), suggesting that emerging coronaviruses may be transmitted directly from bats to humans. MERS-CoV is capable of infecting bat and human cells directly; as both the bat and human forms of the receptor, dipeptidyl peptidase IV, support viral entry (Muller et al., 2012, Raj et al., 2013). Additionally, a fragment of the RNA-dependent RNA polymerase gene that is genetically identical to MERS-CoV was detected in one Egyptian Tomb bat (Memish et al., 2013) and dromedary camels were found to have neutralizing antibodies against MERS-CoV, perhaps linking an endemic source of the virus and an intermediate host to human MERS-CoV infections (Perera et al., 2013, Reusken et al., 2013).
Since the outbreak of SARS-CoV, there have been extensive efforts on antiviral drug development, but no FDA approved antiviral drugs or vaccines exist against any human coronaviruses (reviewed in Barnard and Kumaki, 2011). However, much has been learned from studies evaluating inhibitors directed against SARS-CoV that can be applied to new emergent coronaviruses. Understanding the experimental methods and results of studies performed to screen for SARS-CoV antivirals can guide the current work to develop drugs against MERS-CoV or to ultimately identify pan-coronavirus inhibitors. Cell-based assays are of great importance when developing effective antivirals, as these assays can identify possible antivirals from candidates initially selected from in vitro screens. The focus of this review will be to outline potential drug targets in the coronavirus life cycle, describe cell-based assays used to test antivirals against SARS-CoV, highlight novel techniques used to evaluate potential antivirals against MERS-CoV and discuss the challenges facing anti-coronaviral drug development.
2. Druggable targets of coronaviruses
The coronavirus genome encodes many druggable targets, and these targets are highlighted in their role in the replication cycle life cycle (Fig. 1 ). Human dipeptidyl peptidase IV (DDP4, CD26) has been discovered as the receptor for MERS-CoV (Raj et al., 2013), the receptor-binding domain (RBD) of the spike protein has been identified and structurally characterized (Chen et al., 2013, Du et al., 2013, Mou et al., 2013) and the crystal structure of the complex between DPP4 and the RBD has been determined (Lu et al., 2013, Wang et al., 2013 and reviewed in Li, 2013). The interactions between viral glycoproteins and receptors have been targeted in other viruses, including SARS-CoV. Coronaviruses can enter cells through receptor mediated endocytosis or by membrane fusion with the plasma membrane. Endocytosis of the receptor–virus complex can occur, and upon acidification of the endosome, the host protease cathepsin L is activated and can cleave the viral spike protein to initiate viral fusion. The coronaviral spike can also be activated by extracellular proteases (trypsin) or proteases present on the cell surface (type II transmembrane serine protease or TMPRSS2), and this cleavage allows coronaviruses to enter cells in an cathepsin-independent manner (Glowacka et al., 2011, Matsuyama et al., 2010, Shulla et al., 2011 and reviewed in Simmons et al., 2013). Upon viral entry and fusion of the viral and host cell membranes, the positive sense RNA genome, which is 5′ methyl-capped and poly-adenylated, is translated in the cytoplasm. This translation yields two large polyproteins, pp1a and pp1b, which are then cleaved into 16 non-structural proteins by the papain-like protease, encoded within nsp3, and the 3C-like protease, encoded by nsp5. The proteases are drug targets, as the proteolysis of the non-structural proteins is required for replication of the virus. Further, the papain-like protease of SARS-CoV and other coronaviruses has been shown to antagonize host innate immune responses, so inhibiting the papain-like protease will stop viral replication and may prevent antagonism of host innate immune responses (Barretto et al., 2005, Chen et al., 2007, Devaraj et al., 2007, Frieman et al., 2009, Sun et al., 2012). Successful inhibitors have been generated against both SARS-CoV PLpro and 3CLpro.
Fig. 1.
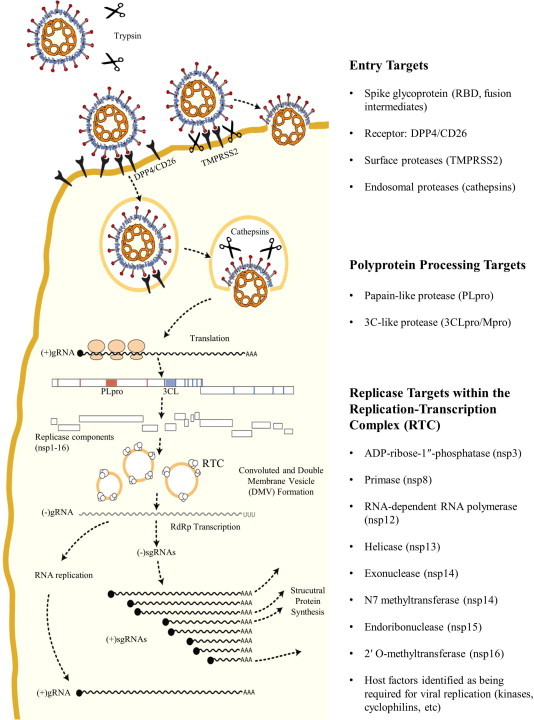
Coronavirus entry and RNA replication targets for antiviral drug development. Targets for viral entry include the viral spike-host receptor interaction, and host proteases that cleave the viral spike to mediate fusion. Viral replicase polyprotein processing can be targeted by inhibiting the papain-like or 3C-like proteases. The enzymatic activities of the replication-transcription complexes (RTCs) on convoluted membranes and double-membrane vesicles are also attractive targets for inhibitors.
To generate more genome copies and subgenomic mRNAs for synthesis of structural genes, the viral genome must be replicated by a series of enzymes that comprise the membrane-associated replication and transcription complex (RTC). The ADP-ribose-1″-phosphatase (nsp3), primase (nsp8), RNA-dependent RNA polymerase (RdRp, nsp12), helicase (nsp13), exonuclease and N7 methyltransferase (nsp14), endoribonuclease (nsp15), and 2′ O-methyltransferase (nsp16) are all proteins that have enzymatic activity that can be targeted by antivirals. In fact, inhibitors have been identified that can block the activity of SARS-CoV RdRp, helicase, and 2′ O-methyltransferase. After replication of the genome and generation of subgenomic mRNAs (sgmRNAs), structural and accessary proteins are translated from these sgmRNAs, assembly of the virion occurs at the endoplasmic reticulum–Golgi intermediate compartment (ERGIC), and the virion egresses through the exosomal pathway. Assembly and egress mechanisms have been targeted for inhibition in other viruses, but this strategy has not been explored for the development of coronavirus antivirals.
3. Cell-based screens for SARS-CoV antivirals
3.1. SARS-CoV entry inhibitor screens
Viral glycoprotein binding with its cognate receptor and the spike protein mediating viral envelope fusion with cellular membranes are necessary for infection. These steps in infection have been successfully targeted in other viruses, with two FDA approved antivirals targeting HIV-1 entry in clinical use (reviewed in Henrich and Kuritzkes, 2013). The antiviral Maraviroc is a small-molecule CCR5 antagonist that inhibits the HIV-1 glycoprotein from binding to its receptor CCR5. Using a different mechanism, the antiviral enfuvirtide inhibits viral fusion by interrupting the interaction between heptad repeat regions within the HIV-1 glycoprotein gp41. Partially based on the success of this strategy, both small-molecule and peptide inhibitors have been identified that target the entry of SARS-CoV. Here, we will review and describe the studies that used cell-based screening methods to identify these inhibitors.
The ability to evaluate of SARS-CoV entry specific inhibitors advanced with the demonstration that pseudotyped virion particles incorporating the S protein from SARS-CoV were competent for entry (Fukushi et al., 2005, Giroglou et al., 2004, Hofmann et al., 2004, Moore et al., 2004). In contrast to the previous cell-based screens based on virally induced CPE, pseudotyped lentiviral virions delivering a genome expressing GFP, luciferase, or other reporters allow for quantitative measurement and evaluation of coronaviral entry inhibitors. These assays can be used in BSL-2 laboratories and do not involve work with infectious CoVs. Using these techniques, the first demonstration of SARS-CoV entry inhibition was achieved using pieces of the SARS-CoV S or peptides designed to interact with the S, with the goal of both approaches to block viral membrane fusion intermediates (Yuan et al., 2004, Zhu et al., 2004). These studies demonstrated that small peptides that can bind to the heptad repeat regions of the SARS-CoV S (parallel in mechanism to FDA approved HIV entry inhibitor Enfuvirtide) could inhibit viral entry/fusion.
In addition to peptide studies based on the amino acid sequence of the SARS-CoV S, compound screens were performed against SARS-CoV entry. Using a commercial screen of diverse compounds (50,240 compound screen from ChemBridge) a small-molecule inhibitor was found to inhibit SARS-CoV entry based first on virally induced CPE reduction and then prevention of pseudotyped virion entry (Kao et al., 2004). Shortly after the demonstration that small-molecule compounds could target SARS-CoV entry, Yi et al. demonstrated that natural compound screens could also be used to inhibit SARS-CoV entry (Yi et al., 2004). They showed inhibition by small-molecules in a pseudotyped virion screen and identified two candidates that inhibited entry in the micromolar range. Utilizing a luciferase expressing lentiviral vector pseudotyped with S protein, molecules were screened based on luciferase expression levels of transduced cells. Two molecules, TGG and leuteolin (screened from a Chinese herbal library) were shown to have antiviral activity in the pseudotype assay and confirmed with live virus using an MTT colorimetric assay for cytotoxicity. These initial studies have evolved into three classes of inhibitor studies; small peptides that block viral fusion by interacting with SARS-CoV S, inhibition of cellular proteases that cleave the viral glycoprotein to mediate entry, and small chemical compounds that interact with the SARS-CoV S and inhibit entry.
The peptide studies have advanced to identify necessary peptides with very high affinity interaction and potent inhibition (Liu et al., 2009, Ni et al., 2005, Struck et al., 2012, Ujike et al., 2008, Zheng et al., 2005). Zheng et al. developed 20-mer peptides corresponding to small fragments of the SARS-CoV S protein and identified three synthetic peptides with inhibitory properties. These peptides also had additive inhibitory effects when pre-incubated in combination with cells prior to infection. To elucidate the mechanism of inhibition, the authors used molecular modeling and speculated that these 20-mer peptides interacted with faces of SARS-CoV S that are required for interaction between the monomers of S to form functional, trimeric S. Inhibition at these interfaces has not been previously observed, and suggested a novel targeting strategy for prevention of S mediated viral entry.
In addition to peptides designed to bind to the S protein, Han et al. took the opposite approach and designed peptides that corresponded to residues critical for entry mediated by ACE2 (Han et al., 2006). Using a pseudotyped MuLV and HeLa cells expressing wild-type or alanine point mutants within ACE2, they determined that many of the charged residues within alpha helices 1 and 2 (residues 22–57) have inhibitory effects on viral entry. Importantly, their studies determined that these effects could only be seen with a 20 min adsorption, and that the commonly used 60 min adsorption time led to a high background and could not properly identify inhibitors in the pseudotype assay. Based on these studies, the authors created peptides corresponding to the amino acids from residues 22–57 and determined that these peptides had specific inhibitory activities against SARS-CoV pseudotype entry, but not entry of VSV-G psuedotypes. Overall, peptides corresponding to SARS-CoV S or ACE2 are effective at limiting entry of pseudotyped and wild-type SARS-CoV; however, none of these inhibitors have been tested further in animal models.
The inhibition of cellular factors required for SARS-CoV entry are attractive targets, as they could be less susceptible to viral escape due to mutations within the viral glycoprotein. Catalytic mutants of ACE2 had no effect on viral entry, suggesting that the enzymatic activity of coronaviral receptors is not required for entry (Moore et al., 2004). The SARS-CoV S protein requires cleavage either extracellularly by trypsin or TMPRRS2 or within the endosomal compartment by cathepsin L (Fig. 1). Simmons et al. determined that cathepsin L mediates SARS-CoV S protein cleavage and that it can be pharmacologically targeted to prevent SARS-CoV S cleavage and entry (Simmons et al., 2005). In this study, a unique virus–virus membrane fusion assay was utilized. This assay involves two psuedotyped virions, one decorated with SARS-CoV S and the Avian sarcoma leukosis virus (ASLV) envelope protein, and one containing ACE2 and a luciferase expression construct. These particles can be induced to undergo fusion in vitro, and particles able to enter cells containing the ASLV receptor and express luciferase must have undergone viral membrane fusion upon mixing of the two pseudoparticle populations. If cathepsin L is responsible for S cleavage within endosomes, then inhibition of purified cathepsin L in vitro will prevent viral–viral membrane fusion and no pseudoparticle expressed luciferase will be detected. The authors used this assay to demonstrate cathepsin L activation of SARS-CoV S and also that other endosomal cathepsins were not responsible for mediating SARS-CoV entry. Cathepsin L has been the continued target of antiviral development not only for SARS-CoV but other enveloped viruses that have a cleavage requirement within their glycoproteins, such as Ebola virus (Shah et al., 2010).
Endosomal proteases are not the only proteases that are able to activate SARS-CoV S. TMPRRS2 can activate S at the host cell plasma membrane, allowing SARS-CoV to enter without utilizing cathepsin L (Fig. 1). Cell–cell fusion assays have been used to identify and characterize plasma membrane surface factors, and can be used to investigate cell surface fusion inhibitors. Cells expressing SARS-CoV S and a T7 polymerase are plated with cells expressing ACE2 and a luciferase or GFP reporter plasmid driven by a T7-dependent promoter. Upon receptor interaction and fusion, the cytoplasm from both cell types mixes, allowing for the transcription and translation of the luciferase or GFP (Shulla et al., 2011, Simmons et al., 2005). Pseudotyped virions were used by Kawase et al. in their work identifying commercially available protease inhibitors targeting TMPRRS2 (Kawase et al., 2012). Inhibiting only serine proteases, which includes TMPRRS2, partially inhibited the entry of pseudotyped particles; however, also inhibiting cysteine proteases (which includes cathepsins) fully inhibited the entry of both pseudoparticles and authentic SARS-CoV. Targeting these host cell factors has been shown to be an effective way to prevent viral fusion and entry in cell culture.
Finally, small-molecule inhibitors have been screened and developed to target SARS-CoV entry. Recently, Adedeji et al. used a SARS-CoV S pseudotyped HIV-Luc virus to screen a 3000 compound library (Adedeji et al., 2013). Forty-four hits were taken from this library and analyzed for specificity against SARS-CoV entry as compared to inhibition of a VSV-G enveloped HIV-Luc pseudovirus. Out of the initial 44 compounds, only 3 were found to be specific for inhibition of SARS-CoV. These counter screens highlight the importance of identifying specific compounds before undergoing further analysis of positive hits in initial screens. Further in vitro analysis was performed and the three compounds were identified to separately inhibit cathepsin L activity, inhibit interaction between S and ACE2, and inhibit a fusion step downstream of S-ACE2 recognition. The feasibility of chemical compound inhibition of virus entry is an established strategy to develop efficacious antivirals, especially for HIV; however, small compounds remain a relatively unexplored strategy with respect to coronaviruses. Overall, targeting viral entry of coronaviruses (especially SARS-CoV) has been demonstrated using specific cell-based assays outlined here. These strategies can be utilized for the evaluation of peptides or chemical inhibitors of S protein-receptor interaction, S-mediated fusion, or host cell factors necessary for mediating MERS-CoV and other emerging coronavirus entry.
3.2. SARS-CoV protease and replicase inhibitor screens
After entry and fusion, the SARS-CoV genome is translated into two large polyproteins from ORF1a and ORF1b. For replication to continue, these polyproteins must be cleaved by viral proteases, either 1 or 2 papain-like proteases (designated PLpro or PLP1/PLP2) and the 3C-like protease (3CLpro or Mpro for main protease). This step in the replicative process has been the target of the most extensive work done to identify inhibitors for SARS-CoV replication (reviewed in Barnard and Kumaki, 2011). SARS-CoV PLpro is responsible for three cleavages while SARS-CoV 3CLpro cleaves the polyprotein at 11 sites (Fig. 1). Because the use of proteases is conserved across coronaviruses, both PLpro and 3CLpro are considered attractive targets for antiviral drug development. Much of the antiviral work performed with the coronaviral proteases has been done using in vitro, cell free conditions, and this work is crucial in laying the foundation for continued antiviral development (reviewed by Mesecar, Hilgenfeld, and their colleagues in this series). In this section, we will continue to focus on highlighting important cell based assays and techniques to efficiently identify and validate antiviral candidates against coronavirus replication.
Some of the first SARS-CoV PLpro inhibitors discovered were first screened against purified protein in vitro and then further characterized in infection and cell lysate assays (Chou et al., 2008, Ratia et al., 2008). To determine if the inhibitors characterized were specific for SARS-CoV PLpro, Ratia et al. utilized a cell lysate experiment that combined purified SARS-CoV PLpro with cell lysates, a tagged ubiquitin-vinyl sulfone, and the inhibitor. In this experiment, cell lysates were incubated with the tagged ubiquitin-vinyl sulfone, and any deubiquitinating enzyme will be covalently linked via the vinyl sulfone linkage to the tagged ubiquitin. These lysates can then be analyzed by western blot to determine if deubiquitinating enzyme binding to the ubiquitin vinyl-sulfone is affected in the presence of inhibitors. In the presence of the specific SARS-CoV inhibitor, only SARS-CoV PLpro binding to the tagged ubiquitin vinyl sulfone was shown to be decreased. While not strictly a cell-based method, this is a useful technique for determining the specificity of inhibitors of papain-like protease activity.
Frieman et al. took an entirely cell-based approach in their screening, identification, and mechanistic evaluation of a small compound library against SARS-CoV PLpro (Frieman et al., 2011). The expression and activity of SARS-CoV PLpro within Sacchromyces cerevisiae led to a slow growth phenotype, and the basis for the screen was that inhibition of PLpro activity by small compounds could reverse this slow growth phenotype. Growth was measured by optical density, and from a 2000 compound library provided by the NIH, the authors generated candidate compounds that were then analyzed for their effect on PLpro expression or toxicity within S. cerevisiae. Based on these results, the authors further screened the compounds in VeroE6 and MA104 cells for activity against SARS-CoV replication. These compounds were also tested against three different activities of SARS-CoV papain-like protease. First, the drugs were tested for their ability to inhibit cleavage of a synthetic PLpro polyprotein substrate, nsp2–3, by SARS-CoV PLpro. This assay uses a tagged nsp2–3 and the cleavage product, which is dependent of PLpro catalytic activity, and can be visualized by western blot. The second assay uses the deubiquitinating activity of PLpro as a readout for PLpro activity and inhibition. Cells are transfected with PLpro and a tagged ubiquitin and then treated with inhibitors. PLpro will cleave the ubiquitin molecules, resulting in a decreased smear of ubiquitinated proteins and an increased smear if the inhibitors are blocking PLpro deubiquitinating activity. Finally, the authors assessed the ability of inhibitors to block the interferon antagonism properties of PLpro using an IFNβ luciferase assay. Cells transfected with either poly(I:C) or a constitutively active form of RIG-I, an IFNβ luciferase reporter, and PLpro, and then were treated with inhibitors. PLpro inhibits IFNβ promoter activation in response to stimulus, so if PLpro inhibitors block the ability of PLpro to inhibit IFNβ activation, an increase in IFNβ promoter activity will be seen. These cell based screening techniques are good tools to assess the mechanism of inhibitors and how they inhibit papain-like proteases within cells. However, these assays are not amenable to a higher-throughput analysis of compounds, and do not offer the specific quantitation of PLpro catalytic activity within cells, which is critical when attempting to optimize small compounds and build structure activity relationships.
SARS-CoV 3CLpro has also been the target of extensive efforts to generate specific antivirals. Modeling of SARS-CoV 3CLpro based on the structures of transmissible gastroenteritis virus (TGEV) and HCoV-229E 3CLpro presented the likelihood of structural conservation. Rhinoviral 3Cpro inhibitors bound efficiently to HCoV-229E 3CLpro, and were later shown to form complexes with SARS-CoV 3CLpro (Anand et al., 2003, Yang et al., 2003). Based on this biochemical characterization, and the enzymatic properties of proteases, much of the early work on 3CLpro inhibitors was performed in vitro in cell-free conditions. For example, Blanchard et al. used purified SARS-CoV 3CLpro and a FRET-based in vitro assay to screen a large compound library of compounds for 3CLpro inhibition (Blanchard et al., 2004). Multiple studies have used a live infection approach based on cell cytotoxicity, and then worked backwards from the hits to identify the mechanism of action of the identified inhibitors (Kao et al., 2004, Wu et al., 2004). Experiments like these are critical for the identification of lead compounds; however, cell-based assays designed to measure protease activity are needed to identify if these compounds could be efficacious in the context of viral infection.
To measure 3CLpro inhibitors in a cell-based assay, Lin et al. created a cis cleavage luciferase assay (Lin et al., 2004). This involved the creation of an expression construct that encoded for SARS-CoV 3CLpro in frame with an inserted 3CLpro cleavage site (SAVLQSGFRK) and a downstream luciferase construct. The rationale for this design was the knowledge that when luciferase is fused to larger proteins, its activity is substantially decreased. Upon the cis cleavage of the 3CLpro-luciferase polyprotein, the liberated luciferase should give an increased signal. The authors demonstrated that this luciferase construct was not very active when translationally fused with a mutated 3CLpro cleavage site. When the cleavage site was intact they witnessed a 5–25-fold increase in luciferase activity. This assay was the first demonstration of a quantitative readout for 3CLpro activity when the protease is transiently expressed within cells.
Replicons have been developed for SARS-CoV and have been used as a platform for screening anti-coronaviral compounds (Almazan et al., 2006, Ge et al., 2007, Ge et al., 2008). These replicons contain all of ORF1a/b from SARS-CoV in addition to the N gene (required for replication) and a reporter cassette. The N gene and reporter are both transcribed off of separate sub-genomic messenger RNAs, similar to the replication of the virus (Fig. 1), meaning that the reporter can only be synthesized when the replicon is replicating correctly. These SARS-CoV replicons can be either transfected or incorporated into stable cell lines for antiviral screening. While these replicons are useful, this approach does not address the specificity of compounds screened. To determine mechanisms of action, the candidate compounds must be screened against replicase proteins in another system.
Antivirals have also been developed against the SARS-CoV helicase (Adedeji et al., 2012, Tanner et al., 2005, Yu et al., 2012), polymerase (Ahn et al., 2011, te Velthuis et al., 2010), and 2′-O-methyltransferase (Ke et al., 2012). A previous study by van Hemert et al. established a protocol to isolate and monitor the activity SARS-CoV replication complexes in vitro (van Hemert et al., 2008). The study performed by te Velthuis et al. used this protocol and showed that zinc-ionophores inhibited the synthesis of RNA by SARS-CoV RdRp. This method allows for the study of cell-produced replication complexes in vitro.
4. Applications and development of cell-based screens for MERS-CoV antiviral screening
Since the outbreak of MERS-CoV, there has been adaptation of methods used to screen for SARS-CoV inhibitors to MERS-CoV and the development of novel methods to identify potential antiviral inhibitors against MERS-CoV (Table 1 ). The entry of some coronaviruses, including SARS-CoV, is known to be mediated by TMPRSS2 (Fig. 1). To determine if TMPRSS2 inhibition is a viable strategy to prevent MERS-CoV entry, Shirato et al. targeted TMPRSS2 activity with camostat and assessed the ability of MERS-CoV to cause syncytia and to enter cells using qRT-PCR for viral RNA. They demonstrated that inhibition of TMPRSS2 activity could inhibit MERS-CoV syncytia formation and that camostat, when combined with an inhibitor of cathepsin L (Fig. 1) potently inhibited MERS-CoV entry into cells (Shirato et al., 2013). These data represent the first chemical inhibition of MERS-CoV entry using infectious MERS-CoV by targeting host proteases. Targeting both the MERS-CoV S and receptor (CD26/DPP4) have been shown to be effective approaches, as Gao et al. designed viral fusion peptide inhibitors against the heptad repeat region HR2 of the MERS-CoV S and Ohnuma et al. created monoclonal antibodies against CD26 that were able to block MERS-CoV entry (Gao et al., 2013, Ohnuma et al., 2013).
Table 1.
Cell-based assays used for antiviral drug development against MERS-CoV.
| Cell-based Assay | For evaluation of: | References |
|---|---|---|
| Infectious virus (BSL-3) | ||
| Cytopathic effect | TMPRSS2 MERS-CoV S cleavage | Shirato et al. (2013) |
| Cyclophilins required for MERS-CoV replication | de Wilde et al. (2013) | |
| Immunofluorescence | Blocking MERS-CoV S and CD26 interaction | Ohnuma et al. (2013) |
| Viral titer | Host kinases up-regulated by MERS-CoV | Josset et al. (2013) |
| qRT-PCR | TMPRSS2 MERS-CoV S cleavage | Shirato et al. (2013) |
| Reporter virus | Synthetic RFP expressing MERS-CoV for antiviral screening | Scobey et al. (2013) |
| Surrogate assays (BSL-2) | ||
| Spike pseudotyped virus | Peptide blockage of HR2 | Gao et al. (2013) |
| Neutralization with patient serum | Gierer et al. (2013) | |
| Neutralization and entry inhibition by small molecules | Zhao et al. (2013) | |
| Protease cleavage bioassay | Small molecules targeting PLpro and 3CLpro | Kilianski et al. (2013) |
To understand how MERS-CoV infection influences and interacts with host cells, Josset et al. infected a lung epithelial cell line, Calu3, with MERS-CoV and SARS-CoV and analyzed the host transcriptome (Josset et al., 2013). This approach identified host factors to which inhibitory compounds exist, that could be exploited as antiviral therapeutics. To validate these putative inhibitors, the authors infected cells with MERS-CoV and showed that the host kinase inhibitor SB203580 had inhibitory effects on both SARS-CoV and MERS-CoV replication. This approach is useful for identifying host cell factors beneficial for replication, determining if they are druggable targets, and identifying existing inhibitors to these targets.
Based on the knowledge that cyclophilins play an important role in coronaviral infections (Pfefferle et al., 2011), de Wilde et al. used cyclosporine A to determine if cytotoxicity as a result of MERS-CoV infection is a possible readout for antiviral screening (de Wilde et al., 2013). Cyclosporin A was able to prevent MERS-CoV cytopathic effect in both Vero and Huh7 cells, verifying CPE as a read out of MERS-CoV infection, and a possible way to screen antiviral compounds against MERS-CoV. To aid in the study of MERS-CoV biology and antiviral screening, reverse genetics systems for MERS-CoV have been developed by two groups (Almazan et al., 2013, Scobey et al., 2013). The use of reverse genetics will allows for the creation of viruses expressing reporters as readouts of viral replication, and can be used to screen antiviral compounds in a more quantitative fashion. This was demonstrated by Scobey et al., who engineered a MERS-CoV encoding an RFP gene that is expressed during viral replication.
Showing inhibitory effects using the infectious virus is the critical step in developing antivirals, but screening large compound libraries or determining inhibitor mechanisms using infectious virus is not practical. Many approaches rely on using infectious MERS-CoV as a way to determine if compounds had inhibitory effects. As MERS-CoV is highly pathogenic and is transmittable from human to human, all assays must be performed in a BSL-3 laboratory. So far, two approaches have been developed for assaying antiviral compounds at either the entry or viral protease stage of the MERS-CoV lifecycle without using infectious MERS-CoV. The first approach involves the familiar pseudotype assay that has been used to evaluate SARS-CoV entry inhibitors. Gierer et al. used VSV-luciferase pseudotyped with the MERS-CoV S to determine that MERS-CoV does not utilize any other coronaviral receptors, that TMPRSS2 and endosomal cathepsins facilitate viral entry, and that the S protein can be neutralized with MERS-CoV infected patient serum (Gierer et al., 2013). Zhao et al. also used pseudotyped virions, but for their experiments they utilized an HIV-luciferase virus pseudotyped with MERS-CoV S (Zhao et al., 2013). They used this system to also show that the S could be neutralized by antibodies and that a small-molecule inhibitor of HIV entry had inhibitory properties on the transduction of their pseudovirions. Gao et al. used pseudotyped virions to determine that HR targeting peptides inhibit viral entry (Gao et al., 2013).
A novel, cell-based approach to assay for coronaviral protease activity was developed by Kilianski et al. and used to determine if previously identified SARS-CoV protease inhibitors had activity against MERS-CoV proteases (Kilianski et al., 2013). These experiments utilized a circularly permuted luciferase construct with an inserted cleavage site corresponding to either the papain-like or 3C-like protease recognition sites. Expression vectors for both MERS-CoV PLpro and 3CLpro were able to cleave their respective biosensors when transfected together. An endpoint dual-luciferase assay with renilla luciferase as a transfection control and a live cell assay using a cell permeable luciferase substrate both showed luciferase activation when the MERS-CoV proteases were present. These assays were used to test previously identified SARS-CoV PLpro and 3CLpro inhibitors. The PLpro inhibitor had no effect on the activity of MERS-CoV PLpro, while the 3CLpro inhibitor CE-5 did show inhibitory activity against MERS-CoV 3CLpro. This was the first demonstration of an efficacious antiviral compound specific to a MERS-CoV enzyme, and will lead to SARS-CoV specific compounds being further tested. The applications of this assay are not limited to MERS-CoV, as the viral papain-like protease from SARS-CoV also activated the luciferase construct. This cell-based biosensor assay is especially useful as it can be used at BSL-2 level to study the effects of compound inhibitors on coronaviral proteases in the context of a host cell.
5. Current state of coronaviral drug development
There are many challenges currently facing scientists who are developing coronaviral, and especially MERS-CoV, antiviral drugs. As highlighted above, there is a need for validated cell-based assays that can be used to accelerate antiviral compound discovery in the face of emerging coronavirus infections (Table 1). One issue with MERS-CoV antiviral design is that no suitable animal model exists. Rhesus macaques can be infected with MERS-CoV, exhibit symptoms, and respond to therapies (de Wit et al., 2013b, Falzarano et al., 2013, Munster et al., 2013); however, the symptoms are transient and do not completely reflect the severity of the disease in humans. Unlike SARS-CoV, de Wit et al. demonstrated that MERS-CoV does not establish a productive infection in Syrian hamsters (de Wit et al., 2013a). To date, there have been no published observations of MERS-CoV infection in small animal models, such as mice or ferrets. In addition, more work needs to be done to develop broad-spectrum inhibitors that would work against the common human coronaviral pathogens like HCoV-NL63, HCoV-229E, HCoV-OC43, or HCoV-HKU1. Development of inhibitors that also target these common viruses would allow for clinical trials on the effectiveness of antiviral drugs against endemic and possible emergent coronaviruses.
The goal of this review was to highlight not only the cell-based assays used for evaluation of coronavirus antivirals, but also to bring to light novel techniques being used by the field. Highlighting these methods and application of techniques will assist in generation of new assays and more rapid development of coronaviral inhibitors for future use. As the public health threat from MERS-CoV and future emergent coronaviruses continues to persist, adapting knowledge and methodology from SARS-CoV research while also developing novel methods to screen antivirals will be important for achieving the goal of creating broad-spectrum anti-coronaviral drugs that are approved for human use.
Acknowledgements
We thank Tom Gallagher, Anna Mielech, and Xufang Deng for their critical reading of the manuscript. Financial support for this work is provided by the National Institutes of Health (NIAID R01AI085089 to S.C.B.). A.K. was supported by NIH Training Grant in Experimental Immunology (NIH T32 AI512795).
References
- Adedeji A.O., Singh K., Calcaterra N.E., DeDiego M.L., Enjuanes L., Weiss S., Sarafianos S.G. Severe acute respiratory syndrome coronavirus replication inhibitor that interferes with the nucleic acid unwinding of the viral helicase. Antimicrobial Agents and Chemotherapy. 2012;56(9):4718–4728. doi: 10.1128/AAC.00957-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adedeji A.O., Severson W., Jonsson C., Singh K., Weiss S.R., Sarafianos S.G. Novel inhibitors of severe acute respiratory syndrome coronavirus entry that act by three distinct mechanisms. Journal of Virology. 2013;87(14):8017–8028. doi: 10.1128/JVI.00998-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn D.G., Lee W., Choi J.K., Kim S.J., Plant E.P., Almazan F., Oh J.W. Interference of ribosomal frameshifting by antisense peptide nucleic acids suppresses SARS coronavirus replication. Antiviral Research. 2011;91(1):1–10. doi: 10.1016/j.antiviral.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almazan F., Dediego M.L., Galan C., Escors D., Alvarez E., Ortego J., Enjuanes L. Construction of a severe acute respiratory syndrome coronavirus infectious cDNA clone and a replicon to study coronavirus RNA synthesis. Journal of Virology. 2006;80(21):10900–10906. doi: 10.1128/JVI.00385-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almazan F., DeDiego M.L., Sola I., Zuniga S., Nieto-Torres J.L., Marquez-Jurado S., Enjuanes L. Engineering a replication-competent, propagation-defective Middle East respiratory syndrome coronavirus as a vaccine candidate. mBio. 2013;4(5):e00650–13. doi: 10.1128/mBio.00650-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science (New York, N.Y.) 2003;300(5626):1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Barnard D.L., Kumaki Y. Recent developments in anti-severe acute respiratory syndrome coronavirus chemotherapy. Future Virology. 2011;6(5):615–631. doi: 10.2217/fvl.11.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A.D., Baker S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. Journal of Virology. 2005;79(24):15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard J.E., Elowe N.H., Huitema C., Fortin P.D., Cechetto J.D., Eltis L.D., Brown E.D. High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chemistry & Biology. 2004;11(10):1445–1453. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Wang Y., Ratia K., Mesecar A.D., Wilkinson K.D., Baker S.C. Proteolytic processing and deubiquitinating activity of papain-like proteases of human coronavirus NL63. Journal of Virology. 2007;81(11):6007–6018. doi: 10.1128/JVI.02747-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Rajashankar K.R., Yang Y., Agnihothram S.S., Liu C., Lin Y.L., Li F. Crystal structure of the receptor-binding domain from newly emerged middle east respiratory syndrome coronavirus. Journal of Virology. 2013;87(19):10777–10783. doi: 10.1128/JVI.01756-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng V.C., Chan J.F., To K.K., Yuen K.Y. Clinical management and infection control of SARS: lessons learned. Antiviral Research. 2013;100(2):407–419. doi: 10.1016/j.antiviral.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C.Y., Chien C.H., Han Y.S., Prebanda M.T., Hsieh H.P., Turk B., Chen X. Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochemical Pharmacology. 2008;75(8):1601–1609. doi: 10.1016/j.bcp.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wilde A.H., Ray V.S., Oudshoorn D., Bestebroer T.M., van Nieuwkoop S., Limpens R.W., van den Hoogen B.G. Human coronavirus-EMC replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-alpha treatment. The Journal of General Virology. 2013 doi: 10.1099/vir.0.052910-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E., Prescott J., Baseler L., Bushmaker T., Thomas T., Lackemeyer M.G., Munster V.J. The Middle East respiratory syndrome coronavirus (MERS-CoV) does not replicate in syrian hamsters. PloS One. 2013;8(7):e69127. doi: 10.1371/journal.pone.0069127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E., Rasmussen A.L., Falzarano D., Bushmaker T., Feldmann F., Brining D.L., Munster V.J. Middle East respiratory syndrome coronavirus (MERS-CoV) causes transient lower respiratory tract infection in rhesus macaques. Proceedings of the National Academy of Sciences of the United States of America. 2013 doi: 10.1073/pnas.1310744110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj S.G., Wang N., Chen Z., Chen Z., Tseng M., Barretto N., Li K. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. The Journal of Biological Chemistry. 2007;282(44):32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L., Zhao G., Kou Z., Ma C., Sun S., Poon V.K., Jiang S. Identification of receptor-binding domain in S protein of the novel human coronavirus MERS-CoV as an essential target for vaccine development. Journal of Virology. 2013 doi: 10.1128/JVI.01048-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzarano D., de Wit E., Rasmussen A.L., Feldmann F., Okumura A., Scott D.P., Feldmann H. Treatment with interferon-alpha2b and ribavirin improves outcome in MERS-CoV-infected rhesus macaques. Nature Medicine. 2013 doi: 10.1038/nm.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieman M., Ratia K., Johnston R.E., Mesecar A.D., Baric R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. Journal of Virology. 2009;83(13):6689–6705. doi: 10.1128/JVI.02220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieman M., Basu D., Matthews K., Taylor J., Jones G., Pickles R., Engel D.A. Yeast based small molecule screen for inhibitors of SARS-CoV. PloS One. 2011;6(12):e28479. doi: 10.1371/journal.pone.0028479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushi S., Mizutani T., Saijo M., Matsuyama S., Miyajima N., Taguchi F., Morikawa S. Vesicular stomatitis virus pseudotyped with severe acute respiratory syndrome coronavirus spike protein. The Journal of General Virology. 2005;86(Pt 8):2269–2274. doi: 10.1099/vir.0.80955-0. [DOI] [PubMed] [Google Scholar]
- Gao J., Lu G., Qi J., Li Y., Wu Y., Deng Y., Gao G.F. Structure of the fusion core and inhibition of fusion by a heptad-repeat peptide derived from the S protein of MERS-CoV. Journal of Virology. 2013 doi: 10.1128/JVI.02433-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge F., Luo Y., Liew P.X., Hung E. Derivation of a novel SARS-coronavirus replicon cell line and its application for anti-SARS drug screening. Virology. 2007;360(1):150–158. doi: 10.1016/j.virol.2006.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge F., Xiong S., Lin F.S., Zhang Z.P., Zhang X.E. High-throughput assay using a GFP-expressing replicon for SARS-CoV drug discovery. Antiviral Research. 2008;80(2):107–113. doi: 10.1016/j.antiviral.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X.Y., Li J.L., Yang X.L., Chmura A.A., Zhu G., Epstein J.H., Shi Z.L. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013 doi: 10.1038/nature12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierer S., Bertram S., Kaup F., Wrensch F., Heurich A., Kramer-Kuhl A., Pohlmann S. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. Journal of Virology. 2013;87(10):5502–5511. doi: 10.1128/JVI.00128-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giroglou T., Cinatl J., Jr., Rabenau H., Drosten C., Schwalbe H., Doerr H.W., von Laer D. Retroviral vectors pseudotyped with severe acute respiratory syndrome coronavirus S protein. Journal of Virology. 2004;78(17):9007–9015. doi: 10.1128/JVI.78.17.9007-9015.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowacka I., Bertram S., Muller M.A., Allen P., Soilleux E., Pfefferle S., Pohlmann S. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. Journal of Virology. 2011;85(9):4122–4134. doi: 10.1128/JVI.02232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D.P., Penn-Nicholson A., Cho M.W. Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology. 2006;350(1):15–25. doi: 10.1016/j.virol.2006.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrich T.J., Kuritzkes D.R. HIV-1 entry inhibitors: recent development and clinical use. Current Opinion in Virology. 2013;3(1):51–57. doi: 10.1016/j.coviro.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgenfeld R., Peiris M. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antiviral Research. 2013;100(1):286–295. doi: 10.1016/j.antiviral.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H., Hattermann K., Marzi A., Gramberg T., Geier M., Krumbiegel M., Pohlmann S. S protein of severe acute respiratory syndrome-associated coronavirus mediates entry into hepatoma cell lines and is targeted by neutralizing antibodies in infected patients. Journal of Virology. 2004;78(12):6134–6142. doi: 10.1128/JVI.78.12.6134-6142.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josset L., Menachery V.D., Gralinski L.E., Agnihothram S., Sova P., Carter V.S., Katze M.G. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. mBio. 2013;4(3) doi: 10.1128/mBio.00165-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao R.Y., Tsui W.H., Lee T.S., Tanner J.A., Watt R.M., Huang J.D., Yuen K.Y. Identification of novel small-molecule inhibitors of severe acute respiratory syndrome-associated coronavirus by chemical genetics. Chemistry & Biology. 2004;11(9):1293–1299. doi: 10.1016/j.chembiol.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawase M., Shirato K., van der Hoek L., Taguchi F., Matsuyama S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. Journal of Virology. 2012;86(12):6537–6545. doi: 10.1128/JVI.00094-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke M., Chen Y., Wu A., Sun Y., Su C., Wu H., Guo D. Short peptides derived from the interaction domain of SARS coronavirus nonstructural protein nsp10 can suppress the 2′-O-methyltransferase activity of nsp10/nsp16 complex. Virus Research. 2012;167(2):322–328. doi: 10.1016/j.virusres.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilianski A., Mielech A., Deng X., Baker S.C. Assessing activity and inhibition of MERS-CoV papain-like and 3C-like proteases using luciferase-based biosensors. Journal of Virology. 2013 doi: 10.1128/JVI.02105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Receptor recognition and cross-species infections of SARS coronavirus. Antiviral Research. 2013;100(1):246–254. doi: 10.1016/j.antiviral.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.W., Tsai C.H., Tsai F.J., Chen P.J., Lai C.C., Wan L., Lin K.H. Characterization of trans- and cis-cleavage activity of the SARS coronavirus 3CLpro protease: basis for the in vitro screening of anti-SARS drugs. FEBS Letters. 2004;574(1–3):131–137. doi: 10.1016/j.febslet.2004.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu I.J., Kao C.L., Hsieh S.C., Wey M.T., Kan L.S., Wang W.K. Identification of a minimal peptide derived from heptad repeat (HR) 2 of spike protein of SARS-CoV and combination of HR1-derived peptides as fusion inhibitors. Antiviral Research. 2009;81(1):82–87. doi: 10.1016/j.antiviral.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G., Hu Y., Wang Q., Qi J., Gao F., Li Y., Gao G.F. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature. 2013 doi: 10.1038/nature12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S., Nagata N., Shirato K., Kawase M., Takeda M., Taguchi F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. Journal of Virology. 2010;84(24):12658–12664. doi: 10.1128/JVI.01542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memish Z.A., Mishra N., Olival K.J., Fagbo S.F., Kapoor V., Epstein J.H. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerging Infectious Diseases. 2013 doi: 10.3201/eid1911.131172. 0.3201/eid1911.131172 [Internet] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M.J., Dorfman T., Li W., Wong S.K., Li Y., Kuhn J.H., Choe H. Retroviruses pseudotyped with the severe acute respiratory syndrome coronavirus spike protein efficiently infect cells expressing angiotensin-converting enzyme 2. Journal of Virology. 2004;78(19):10628–10635. doi: 10.1128/JVI.78.19.10628-10635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou H., Raj V.S., van Kuppeveld F.J., Rottier P.J., Haagmans B.L., Bosch B.J. The receptor binding domain of the new MERS coronavirus maps to a 231-residue region in the spike protein that efficiently elicits neutralizing antibodies. Journal of Virology. 2013 doi: 10.1128/JVI.01277-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M.A., Raj V.S., Muth D., Meyer B., Kallies S., Smits S.L., Drosten C. Human coronavirus EMC does not require the SARS-coronavirus receptor and maintains broad replicative capability in mammalian cell lines. mBio. 2012;3(6) doi: 10.1128/mBio.00515-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munster V.J., de Wit E., Feldmann H. Pneumonia from human coronavirus in a macaque model. The New England Journal of Medicine. 2013;368(16):1560–1562. doi: 10.1056/NEJMc1215691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni L., Zhu J., Zhang J., Yan M., Gao G.F., Tien P. Design of recombinant protein-based SARS-CoV entry inhibitors targeting the heptad-repeat regions of the spike protein S2 domain. Biochemical and Biophysical Research Communications. 2005;330(1):39–45. doi: 10.1016/j.bbrc.2005.02.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnuma K., Haagmans B.L., Hatano R., Raj V.S., Mou H., Iwata S., Morimoto C. Inhibition of middle east respiratory syndrome coronavirus (MERS-CoV) infection by anti-CD26 monoclonal antibody. Journal of Virology. 2013 doi: 10.1128/JVI.02448-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera R., Wang P., Gomaa M., El-Shesheny R., Kandeil A., Bagato O., Kayali G. Seroepidemiology for MERS coronavirus using microneutralisation and pseudoparticle virus neutralisation assays reveal a high prevalence of antibody in dromedary camels in Egypt, June 2013. Euro Surveillance: Bulletin Europeen Sur Les Maladies Transmissibles = European Communicable Disease Bulletin. 2013;18(36):20574. doi: 10.2807/1560-7917.es2013.18.36.20574. [DOI] [PubMed] [Google Scholar]
- Pfefferle S., Schopf J., Kogl M., Friedel C.C., Muller M.A., Carbajo-Lozoya J., von Brunn A. The SARS-coronavirus-host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathogens. 2011;7(10):e1002331. doi: 10.1371/journal.ppat.1002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj V.S., Mou H., Smits S.L., Dekkers D.H., Muller M.A., Dijkman R., Haagmans B.L. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495(7440):251–254. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Mesecar A.D. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(42):16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusken C.B., Haagmans B.L., Muller M.A., Gutierrez C., Godeke G.J., Meyer B., Koopmans M.P. Middle east respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. The Lancet Infectious Diseases. 2013 doi: 10.1016/S1473-3099(13)70164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scobey T., Yount B.L., Sims A.C., Donaldson E.F., Agnihothram S.S., Menachery V.D., Baric R.S. Reverse genetics with a full-length infectious cDNA of the middle east respiratory syndrome coronavirus. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(40):16157–16162. doi: 10.1073/pnas.1311542110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P.P., Wang T., Kaletsky R.L., Myers M.C., Purvis J.E., Jing H., Diamond S.L. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and Ebola pseudotype virus infection into human embryonic kidney 293T cells. Molecular Pharmacology. 2010;78(2):319–324. doi: 10.1124/mol.110.064261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirato K., Kawase M., Matsuyama S. Middle East respiratory syndrome coronavirus (MERS-CoV) infection mediated by the transmembrane serine protease TMPRSS2. Journal of Virology. 2013 doi: 10.1128/JVI.01890-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulla A., Heald-Sargent T., Subramanya G., Zhao J., Perlman S., Gallagher T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. Journal of Virology. 2011;85(2):873–882. doi: 10.1128/JVI.02062-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Gosalia D.N., Rennekamp A.J., Reeves J.D., Diamond S.L., Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(33):11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Zmora P., Gierer S., Heurich A., Pohlmann S. Proteolytic activation of the SARS-coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antiviral Research. 2013 doi: 10.1016/j.antiviral.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struck A.W., Axmann M., Pfefferle S., Drosten C., Meyer B. A hexapeptide of the receptor-binding domain of SARS corona virus spike protein blocks viral entry into host cells via the human receptor ACE2. Antiviral Research. 2012;94(3):288–296. doi: 10.1016/j.antiviral.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Xing Y., Chen X., Zheng Y., Yang Y., Nichols D.B., Chen Z. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PloS One. 2012;7(2):e30802. doi: 10.1371/journal.pone.0030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner J.A., Zheng B.J., Zhou J., Watt R.M., Jiang J.Q., Wong K.L., Huang J.D. The adamantane-derived bananins are potent inhibitors of the helicase activities and replication of SARS coronavirus. Chemistry & Biology. 2005;12(3):303–311. doi: 10.1016/j.chembiol.2005.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Velthuis A.J., van den Worm S.H., Sims A.C., Baric R.S., Snijder E.J., van Hemert M.J. Zn(2+) inhibits coronavirus and arterivirus RNA polymerase activity in vitro and zinc ionophores block the replication of these viruses in cell culture. PLoS Pathogens. 2010;6(11):e1001176. doi: 10.1371/journal.ppat.1001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujike M., Nishikawa H., Otaka A., Yamamoto N., Yamamoto N., Matsuoka M., Taguchi F. Heptad repeat-derived peptides block protease-mediated direct entry from the cell surface of severe acute respiratory syndrome coronavirus but not entry via the endosomal pathway. Journal of Virology. 2008;82(1):588–592. doi: 10.1128/JVI.01697-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hemert M.J., van den Worm S.H., Knoops K., Mommaas A.M., Gorbalenya A.E., Snijder E.J. SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathogens. 2008;4(5):e1000054. doi: 10.1371/journal.ppat.1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N., Shi X., Jiang L., Zhang S., Wang D., Tong P., Wang X. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Research. 2013;23(8):986–993. doi: 10.1038/cr.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (WHO), (2013). Middle East respiratory syndrome coronavirus (MERS-CoV) – update (Nov 4, 2013). Global Alert and Response (GAR). Retrieved November, 4, 2013, from 〈http://www.who.int/csr/don/2013_11_04/en/index.html〉.
- Wu C.Y., Jan J.T., Ma S.H., Kuo C.J., Juan H.F., Cheng Y.S., Wong C.H. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(27):10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(23):13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi L., Li Z., Yuan K., Qu X., Chen J., Wang G., Xu X. Small molecules blocking the entry of severe acute respiratory syndrome coronavirus into host cells. Journal of Virology. 2004;78(20):11334–11339. doi: 10.1128/JVI.78.20.11334-11339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M.S., Lee J., Lee J.M., Kim Y., Chin Y.W., Jee J.G., Jeong Y.J. Identification of myricetin and scutellarein as novel chemical inhibitors of the SARS coronavirus helicase, nsP13. Bioorganic & Medicinal Chemistry Letters. 2012;22(12):4049–4054. doi: 10.1016/j.bmcl.2012.04.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan K., Yi L., Chen J., Qu X., Qing T., Rao X., Deng H. Suppression of SARS-CoV entry by peptides corresponding to heptad regions on spike glycoprotein. Biochemical and Biophysical Research Communications. 2004;319(3):746–752. doi: 10.1016/j.bbrc.2004.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G., Du L., Ma C., Li Y., Li L., Poon V., Zhou Y. A safe and convenient pseudovirus-based inhibition assay to detect neutralizing antibodies and screen for viral entry inhibitors against the novel human coronavirus MERS-CoV. Virology Journal. 2013;10(1):266. doi: 10.1186/1743-422X-10-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B.J., Guan Y., Hez M.L., Sun H., Du L., Zheng Y., Huang J.D. Synthetic peptides outside the spike protein heptad repeat regions as potent inhibitors of SARS-associated coronavirus. Antiviral Therapy. 2005;10(3):393–403. [PubMed] [Google Scholar]
- Zhu J., Xiao G., Xu Y., Yuan F., Zheng C., Liu Y., Gao G.F. Following the rule: formation of the 6-helix bundle of the fusion core from severe acute respiratory syndrome coronavirus spike protein and identification of potent peptide inhibitors. Biochemical and Biophysical Research Communications. 2004;319(1):283–288. doi: 10.1016/j.bbrc.2004.04.141. [DOI] [PMC free article] [PubMed] [Google Scholar]