

Abstract
Epidermal growth factor receptor (EGFR) and HER2 are major prognosis biomarkers and drug targets overexpressed in various types of cancer cells. There is a pressing need to develop MRI contrast agents capable of enhancing the contrast between normal tissues and tumors with high relaxivity, capable of targeting tumors, and with high intratumoral distribution and minimal toxicity. In this review, we first discuss EGFR signaling and its role in tumor progression as a major drug target. We then report our progress in the development of protein contrast agents with significant improvement of both r1 and r2 relaxivities, pharmacokinetics, in vivo retention time, and in vivo dose efficiency. Finally, we report our effort in the development of EGFR-targeted protein contrast agents with the capability to cross the endothelial boundary and with good tissue distribution across the entire tumor mass. The noninvasive capability of MRI to visualize spatially and temporally the intratumoral distribution as well as quantify the levels of EGFR and HER2 would greatly improve our ability to track changes of the biomarkers during tumor progression, monitor treatment efficacy, aid in patient selection, and further develop novel targeted therapies for clinical application.
Keywords: MRI, Contrast agent, Molecular imaging, Protein engineering, Epidermal growth factor receptor/HER2
Introduction
Epidermal growth factor receptor (EGFR) and HER2 are major prognosis biomarkers overexpressed in various types of cancer cells. In numerous clinical studies, EGFR- and HER2-positive tumors are associated with a shorter, disease-free postclinical period and shorter overall survival for breast and ovarian cancer patients [1]. Both EGFR and HER2 are prognostic factors for multiple tumor types, including non-small-cell lung carcinoma and pancreatic cancer. Co-expression of EGFR and HER2 is found in 10–36 % of primary human breast carcinomas, and is generally associated with a poor prognosis compared with expression of a single receptor. Molecular imaging provides us with direct measurement of temporal and spatial biomarkers during disease progression and treatment, in addition to improved sensitivity and specificity [2–4]. Therefore, monitoring the spatial and temporal changes of several molecular biomarkers such as EGFR and HER2 sharing the same signaling pathway during cancer progression and treatment is the key for understanding the molecular basis of cancers. In addition, achieving noninvasive molecular imaging of EGFRs will have a broad impact on early and accurate diagnosis and the development of effective drugs with synergistic effects to treat these deadly diseases.
Magnetic resonance imaging (MRI) is one of the most powerful imaging techniques owing to its significant advantages of noninvasiveness and no tissue depth limitations [5]. MRI measures the water relaxation properties and interactions in an external magnetic field with high resolution [6]. Owing to its high resolution and sensitivity to soft tissues and especially the development of MRI contrast agents, clinical MRI has been applied as a major diagnostic and prognostic modality for various types of human diseases, including diseases of the brain, heart, liver, kidney, breast, ovary, and prostate [6–13].
MRI contrast agents are used to shorten the relaxation time (T1 and T2) of the protons in the tissue area on the basis of the mechanisms, and they can be divided into T1-weighted and T2-weighted contrast agents [14]. The most widely used T1-weighted contrast agents are based on gadolinium (Gd3+), since Gd3+ is a lanthanide metal ion with seven unpaired electrons, a large magnetic moment, and a long electron spin relaxation time [15]. Iron oxide based contrast agents are usually T2-weighted contrast agents [16]. Since free Gd3+ is highly toxic, with a median lethal dose of 0.2 mmol kg−1 in mice [17], it must be encapsulated by chelators. Current FDA-approved MRI contrast agents are based on small chelators. These clinical contrast agents have a relaxivity (the ability of MRI contrast agents to increase the relaxation rate of water protons) of approximately 5 mM−1 s−1. For example, gadolinium(III) diethylenetriaminepentaacetate (Gd-DTPA) has a r1 relaxivity of 3.8 M−1 s−1 at 20 MHz [7, 18]. To detect contrast changes due to the difference in proton relaxation times in organs clinically, a relaxation rate change of 0.5 s−1 is necessary [19]. Thus, a local contrast agent concentration of 100 μM is required [19]. In general, an injection dose of about 0.1–0.3 mmol kg−1 is needed to obtain high contrast in human and small-animal tissues.
The high concentration of contrast agent required for in vivo contrast is indicative of low-dose efficiency and results in increased risk of certain disorders. Nephrogenic systemic fibrosis, a disease found in patients with kidney disease, has been correlated with the use of gadolinium-based MRI contrast agents and is reported to be related to the release of free Gd3+ [20–23]. In addition, small-molecule contrast agents have a very short half-life and blood retention time. For example, Magnevist has a distribution half-life around 3 min and an elimination half-life of about 20 min in rats and 1.5 h in humans [8, 24, 25]. MRI of the liver, for example, is especially challenging owing to rapid secretion of contrast agents. The clinically approved liver contrast agent Eovist has an arterial phase of 20 s and a hepatocyte phase of 20 min in humans [26]. Thus, there is a very narrow time window for MRI data collection, which makes it difficult to obtain high-quality images. Repeated injections are necessary and risks of metal toxicity are raised. To date, clinical contrast agents have the capability to detect lesions larger than 2 cm with high confidence. However, no contrast agent to date has the capability to image molecular biomarkers. This is probably due to a lack of desired sensitivity associated with high relaxivity and desired pharmacokinetics and pharmacodynamics [27].
Native metalloproteins or engineered metal binding proteins have been used to develop MRI contrast agents [28]. Metalloproteins such as hemoglobin and the heme domain of cytochrome P450 BM3 containing paramagnetic metal ions (Cu2+, Fe2+, Fe3+, Mn2+, and Mn3+) with spin quantum numbers from 1/2 to 5/2 have been applied to probe brain activities [28, 29]. Paramagnetic transition metal ions can be caged in ferritin as T2-weighted MRI contrast agents [30, 31]. In addition, T1-weighted contrast agents have also been developed by noncovalent binding or covalent binding of small monomeric agents to proteins to improve the relaxivity by increasing the rotational correlation time τR [32–47]. As a major limiting factor for high relaxivity in clinical Gd3+-based MRI contrast agents is size, the resulting τR is less than 1 ns. Proteins with a size around 20 kDa have τR around 10 ns, which is optimized to achieve high relaxivity at clinical field strengths. A helix–loop–helix peptide motif of virus particles has been shown to have increased r1 relaxivity [48]. A fibrin-specific contrast agent with four Gd-DOTA molecules conjugated to a fibrin-targeting peptide has a relaxivity of 17.8 mM−1 s−1 on binding to its biomarker [49–51]. The development of MRI contrast agents with the capability of molecular imaging of EGFR/HER biomarkers is still in its infancy, despite the rapid development of molecular imaging contrast agents for the same biomarkers which has been achieved for positron emission tomography (PET)/single photon emission computed tomography (SPECT) and fluorescence imaging owing to their sensitivity being higher than that of MRI [52–55].
To achieve molecular imaging of disease biomarkers by MRI, several criteria should be considered. First, it is essential to improve the relaxivity, in order to have the sensitivity to detect surface biomarkers usually in the nanomolar to micromolar range. Most receptors or bio-markers have an expression level of less than 106 per cell. Assuming a cell has a volume of 1,000 μm3 and the bio-marker level is 106 per cell, the local concentration of this biomarker is 1 nM or less. In one calculation, approximately 5 × 106 Gd3+ ions per cell are required to obtain a clear contrast-enhanced image using currently approved contrast agents (e.g., Gd-DTPA) with a relaxivity of 5 mM−1 s−1. On the basis of the above reasoning, it is not possible to achieve molecular imaging using currently available MRI contrast agents [7, 56]. Mechanisms to enrich the local concentration of gadolinium contrast agents via rapid endocytosis, multivalent conjugations, liposomes with multiple copies of gadolinium contrast agents, and enzymatic amplification were explored to achieve molecular imaging [11, 12, 33, 57–60]. The target contrast agents should have good tissue penetration in addition to a fast circulation time for locating their targets. This is required for quantitative analysis of the receptor distributions and levels, as well as biomarker changes due to disease progression and drug treatment. Furthermore, the targeting moiety should have strong targeting specificity and affinity for the desired biomarkers. Surface receptors with high expression levels are preferred for targeting compared with intracellular bio-markers owing to their accessibility. Ideally, the epitope binding site on the biomarker should be different from the drug interaction site to avoid immunological interference with the drug treatment. Moreover, targeted contrast agents with multimodality for imaging, e.g., MRI with near-IR spectrophotometry, are preferred to ensure detection sensitivity and resolution for clinical studies.
In this review, we will first discuss EGFR signaling and its role in tumor progression and as a major drug target. Next we will report our progress in the development of protein contrast agents with significantly improved relaxivity. Finally, we will report our effort in the development of EGFR-targeted protein contrast agents.
EGFR structure and signaling
The EGFR family comprises four members: EGFR (also known as HER1), HER2 (also known as Neu), HER3, and HER4. They share similar structures, with an extracellular ligand binding domain, a transmembrane domain, and a functional intracellular tyrosine kinase domain (except for HER3). Different from the other three receptor family members, HER2 does not have a natural ligand and its extracellular domain is able to adopt an activated state to dimerize with EGFR or HER3 (EGFR/HER2, HER2/HER3). HER2 is the preferred dimerization partner in the EGFR family (Fig. 1a).
Fig. 1.

a Epidermal growth factor receptor (EGFR) family and its targeting molecules. All the members in EGFR family have an extracellular domain (ECD), a transmembrane domain, and an intracellular domain. HER2 is the one member that does not have a natural ligand for the ECD, and tends to form a dimer with other members. With some ligand binding in the ECD of either member in the former dimer, the inner kinase pathway may be activated, which will promote cell proliferation and angiogenesis. b Expression levels of EGFR and HER2 in various breast and ovarian cancer cells. c Current clinical diagnostic methods for patient selection. IHC immunohistochemistry
EGFR has many ligands, such as epidermal growth factor and transforming growth factor α. A ligand-induced conformational change occurs on dimerization of domain II, which in turn activates the tyrosine kinase sites located in the intracellular domain [61]. On the other hand, in the absence of direct ligand binding, HER2 interacts with other receptors of the same family [62]. The antibody drug trastuzumab (Herceptin) binds to the juxtamembrane region of HER2, which is likely to facilitate endocytosis by providing direct interaction between the steric barrier formed and the transmembrane regions [63]. Subsequent autophosphorylation of tyrosine residues leads to interaction of the activated receptor with SH2 or PTB adaptor proteins to promote downstream signaling, migration, differentiation, apoptosis, and cell motility. Activated receptors can be switched “off” through dephosphorylation, receptor ubiquitination, or removal of active receptors from the cell surface through endosomal sorting and lysosomal degradation.
Differential expression of EGFR family members in various cancers and cell lines
EGFR family proteins are also expressed on the cell membrane of normal tissue, with relatively low expression of EGFR except on normal skin epithelial cells [64]. The expression level of HER2 is about 103–104 per normal cell. On the other hand, EGFR is widely overexpressed in tumor epithelial cells, including those of breast cancers, ovarian cancers, pancreatic cancers, prostate cancers, and lung cancers [65, 66], likely owing to the amplification of EGFR genes in cancer cells involved in tumor metastasis and aggressiveness [66]. In about 30 % of breast cancers, HER2 is overexpressed. EGFR is overexpressed in many solid tumors to trigger the tyrosine kinase domain for the downstream signaling pathway [67, 68], such as promotion of cell proliferation and angiogenesis and inhibition of cell apoptosis [68]. HER2-positive breast cancer is correlated with a high metastasis and low survival rate. HER2 and EGFR are also major prognosis biomarkers overexpressed in various types of cancer cells [69] and tissue samples from cancer patients [70, 71]. In various carcinomas, such as glioma, bladder carcinomas, and lung cancers, EGFR proteins are over-expressed [72, 73]. Table 1 shows that up to 69 % of tumors have a high expression level of EGFR, especially in later stages, and HER2 is highly expressed in about 30 % of tumor cells. However, HER2 is also overexpressed in the early stages of these cancers, which could serve as a biomarker [74].
Table 1.
Epidermal growth factor receptor (EGFR) and HER2 expression rates in cancers
| Reference | Definition of EGFR overexpression | Tumors overexpressing EGFR (%) | Reference | Definition of HER2 overexpression | Tumors overexpressing HER2 (%) |
|---|---|---|---|---|---|
| Onn et al. [145] | 2 or 3 staining by IHC | 60 | Slamon et al. [151] | Twofold or more HER2 gene by Southern blotting | 30 |
| Selvaggi et al. [146] | 2+ or 3+ staining by IHC | 37 | Paik et al. [152] | Definite membrane staining by IHC in tumor cell | 29 |
| Ohtsuka et al. [147] | 2+ or 3+ by Western blotting | 40 | Owens et al. [153] | Twofold or more of HER2 gene by FISH | 23 |
| Dancer et al. [148] | Twofold or more EGFR gene by FISH | 65 | Owens et al. [153] | 2+ or higher result by IHC | 20 |
| Bloomston et al. [149] | 1+ or higher staining by IHC | 69 | Seshadri et al. [154] | Twofold or more HER2 gene by slot blotting | 21 |
| Thybusch-Bernhardt et al. [150] | Positive staining by IHC | 33 | Andrulis et al. [155] | Twofold or more HER2 gene by RT-PCR | 20 |
FISH fluorescent in situ hybridization, IHC immunohistochemistry, RT-PCR real-time polymerase chain reaction
Overexpression of EGFR and HER2 is associated with poor prognosis [75, 76]. HER2 and EGFR are overexpressed in various types of cancer cells [69]. HER2 is a negative prognostic factor [70, 71]. In numerous clinical studies, it has associated with shorter disease-free and overall survival for breast and ovarian cancers as well as increased risk of death. About 30 % of all breast cancer cases are associated with expression of HER2. High expression of HER2 closely correlates with a low survival rate [74, 77, 78]. The rate of HER2 overexpression was estimated to be 6–35 % in gastric cancer [79], 9–32 % in ovarian cancer [80], and up to 70 % in human pancreatic cancer [81]. EGFR also leads to increased cell proliferation and motility and decreased apoptosis [82]. EGFR is also a negative prognostic factor for multiple tumor types, including non-small-cell lung carcinoma and pancreatic cancer [83]. In addition, co-expression of EGFR and HER2 is found in 10–36 % of primary human breast carcinomas, and it is generally associated with a poor prognosis compared with the expression of a single receptor [57, 84–86].
There are more than 20 cell lines which have a high expression level of EGFR and HER2 (Fig. 1b) [87–89]. They mainly come from breast, ovarian, and pancreatic tumors. Among these cell lines, several pairs of positive and negative HER2 cell lines are widely used in research [90]. As shown in Fig. 1b, the expression is up to 106 per cell in cancer cells, especially in breast cancers.
Most of the cancer cells with EGFR/HER2 overexpression have tumorigenic properties and are suitable for xenografted cancer models in animals. Some of the cell lines, such as MCF-10DCIS, can generate tumors with biomarker level changes at different stages of cancers. The EGFR expression level will increase, but the HER2 expression level will decrease during tumor progression. Mouse HER2 is different from human HER2, and is also related to breast cancer in the mouse. Both NT5 and EMT-6 form mouse mammary cancer; HER2 is overexpressed in NT5.
Current EGFR clinical diagnosis and prognosis
Among all the members of the EGFR family, EGFR and HER2 are two well established biomarkers for clinical diagnosis, staging, and monitoring treatments, especially for breast cancer. In current techniques (Fig. 1c) to measure specific levels as protein, DNA, or RNA biomarkers, including immunohistochemistry (IHC), enzyme-linked immunosorbent assays, fluorescent in situ hybridization, and real-time polymerase chain reaction, the cancer tissue or cells have to be removed for biopsy. Today one in five HER2 clinical tests, including biopsy and immunostaining (IHC), provides inaccurate results [84]. This severely affects the selection of appropriate patients for personalized treatment using HER2/EGFR-targeted cancer therapies. This is likely due to sample errors as well as lack of accurate differentiation of tumor boundaries. Additionally, these diagnostic methods are invasive and not in real time. Thus, there is an urgent need to develop a noninvasive imaging method with the capability to monitor the expression level and location of EGFRs during disease progression and treatment as well as to understand the mechanism of disease development at the molecular level [91].
EGFR-targeted therapy and molecular imaging reagents
Different types of drugs have been developed to target various regions of EGFR/HER2. The targeted reagents can generally be divided into two types. One type of reagents targets the intracellular domain of EGFR, whereas another type targets the extracellular domain of EGFR [92, 93]. The intracellular targeting reagents are inhibitors of the tyrosine kinase domain. Small molecules such as lapatinib, which functions as a kinase domain inhibitor, can inhibit both EGFR and HER2.
Several targeted drugs such as monoclonal antibodies (trastuzumab) against the extracellular domain of EGFR and HER2 expressed at the cell surfaces of various cancers have been developed [94, 95]. Antibodies have a therapeutic function by several pathways. Because HER2 mediates cell signaling pathways such as the phosphati-dylinositol 3-kinase and mitogen-activated protein kinase pathways, antibody targeting of HER2 can cause antibody-dependent cell-mediated cytotoxicity, which can cause the cancer cells to be swallowed by macrophages [96]. At the same time, antibody targeting can inhibit proteolysis of the HER2 extracellular domain and inhibit HER2 DNA repair [97]. An HER2-specific antibody such as trastuzumab exclusively targets the extracellular domain of HER2, which can inhibit HER2 expression. The Fc domains of antibodies have been conjugated with drugs, toxic proteins, radioisotopes, and thermotherapy drugs. Differently from traditional cancer treatments such as radiation therapy and chemotherapy that affect all cells nonspecifically, protein drugs have the ability to target diseased cells with decreased toxicity [98].
Several major efforts have been devoted to achieve molecular imaging of EGFR signaling to provide information regarding biological processes at a cellular level before anatomical changes occur. Similarly to the approaches used to develop EGFR therapeutic agents, two strategies can be used for EGFR-targeted tumor imaging. The first class of imaging agents targets the extracellular domain of the receptor. Examples of these imaging agents include whole antibodies, antibody fragments, affibodies, and nanobodies. The EGFR ligands, which bind to the EGFR extracellular domain at high affinity, can also be used for this purpose. The second class of molecules includes tyrosine kinase inhibitors and analogs that bind reversibly or irreversibly to the intracellular kinase domain of the receptor.
Artemov et al. [57] have developed a multiple-step HER2-targeted MRI contrast agent using biotin-labeled HER2 antibody and avidin-conjugated Gd-DTPA complexes. Biotin-labeled HER2 antibody was injected into the animals 1 day before, in order for the antibody to bind to HER2 receptors in vivo. Avidin-conjugated Gd3+-DTPA was injected on the second day to search for and bind the biotin-labeled antibody [99, 100]. To facilitate in vivo targeting capabilities, minimal monovalent binding fragments, such as Fab (approximately 55 kDa) and single-chain Fv (scFv; approximately 28 kDa) with retained binding specificity, were used for molecular imaging studies [101]. EGFR-targeted single-chain antibody scFvEGFR was conjugated to nanoparticles bonded to ions to function as a T2-weighted MRI contrast agent.
Besides antibodies targeted to biomarkers, various flexible peptides have been screened or designed for specific biomarkers, such as gastrin releasing peptide for gastrin-releasing peptide receptor in prostate cancers [102]. High-affinity affibody molecules have been developed by phage display selections based on the 58 amino acids in the Z domain of staphylococcal protein. These affibody molecules provide a versatile moiety targeting HER2 [103, 104]. Owing to their small size (approximately 7 kDa), the high-affinity affibodies offer the unique advantage of deeper tissue penetration, which is an essential property for targeting cancer cell surface HER2. Biodistribution analyses with an SKOV-3 subcutaneous tumor demonstrated a relatively favorable tumor uptake by the affibody target [105]. HER2-targeting affibodies have been widely used in molecular imaging targeting HER2 by many imaging methods [64]. The affibodies, which target EGFR/HER2 overexpressed tumors, were directly linked to small molecular chelators with radioisotopes for PET or SPECT [105–107]. Imaging agents with 125I- and 18F-labeled HER2 affibodies have also been used in PET or SPECT of breast cancers [108, 109]. Recently, affibodies were conjugated to an iron oxide nanoparticle for MRI of breast cancer [110]. Despite some reported successes in molecular imaging targeting HER2 using affibodies, the major issue of bio-distribution remains, and few successful examples using affibodies in HER2-targeted MRI have been reported [99, 108].
Development of protein-based MRI contrast agents
The criteria for molecular imaging by MRI shows the importance of improving the relaxivity of contrast agents. On the basis of the distance between the water molecule and Gd3+, the relaxivity can be further divided into inner-sphere relaxivity, secondary-sphere relaxivity, and outer-sphere relaxivity. The overall relaxivity of contrast agents is the combination of inner-sphere, secondary-sphere, and outer-sphere relaxivity. Inner-sphere and secondary-sphere relaxivity can be characterized by the Solomon–Bloembergen–Morgan equation [14, 111–114]. The outer-sphere relaxivity of contrast agents can be characterized by a hard-sphere model [115]. In general, the inner-sphere relaxivity and the secondary-sphere relaxivity of contrast agents are influenced by the rotational correlation time (τR), the residence time of the bound water (τm), and number of water molecules interacting with Gd3+ (q) (for details, see the excellent reviews [18, 19, 116–118]).
τR is one of the key factors that determines the overall correlation time (τc; determined by the smallest value among τR, τm, and the longitudinal and transverse electron spin relaxation times of the metal ion) and further determines the relaxivity [19]. By increasing τR from 100 ps (for small chelators) to about 10 ns, one can dramatically increase the relaxivity. We rationalize that high relaxivity can be achieved by designing a Gd3+ binding site embedded into the protein frame such as domain 1 of CD2 with a correlation time around 10 ns. In addition, the gadolinium–protein vector rotated together allows us to eliminate the flexibility and uncontrolled local internal motion of the gadolinium–chelator complex associated with conjugation. In addition, this protein design approach enables us to increase the relaxivity by increasing the number of water molecules (q) without sacrificing the metal binding properties, which could cause undesired release of Gd3+ in vivo.
According to simulations by us [119] and others [120], the secondary-sphere and outer-sphere contribution to the relaxivity of protein contrast agents could be significant because of the large hydration surface of the protein. With a Gd3+–water distance of 5 Å, a rotational correlation time of 10 ns, a residence time of the bound water in the secondary sphere of 10 ns, and a secondary-sphere water number of 4, the secondary-sphere relaxivity of contrast agents can reach 3.3 and 8.8 mM−1 s−1 at 20 and 60 MHz, respectively [119].
Radically different from all other reported methods [46, 50, 56, 117, 118, 121, 122], the design of protein-based MRI contrast agents is based on our careful analysis of more than 500 structures of molecules which bind to Gd3+ and other lanthanide ions [123]. In solution, Gd3+ is usually associated with eight or nine oxygen atoms from water molecules [124]. A small chelator usually binds to Gd3+ with both oxygen and nitrogen ligands. For example, Gd-DTPA has five oxygen and three nitrogen ligands. Gd3+ always has one water ligand to allow robust water exchange. However, when binding to proteins, Gd3+ has a higher preference to bind to oxygen rather than nitrogen, with an average of 7.2 oxygen ligands for Ln3+ [118].
We chose domain 1 of CD2 as our scaffold to design protein-based MRI contrast agents. It is expressed on the surface of T cells and natural killer cells for cell adhesion and signal transduction. This protein has a rigid structure with high resistance to pH change and enzyme cleavage and is very tolerant to mutations [125–133]. On the basis of our knowledge of Gd3+ binding ligands and the properties of CD2, we designed a Gd3+ binding pocket formed by a group of carboxyl side chains (E15, D56, D58, D62, D64) from different β-sheets of CD2. We carefully designed one side of the Gd3+ binding pocket to open, allowing rapid Gd3+ water exchange (Fig. 2).
Fig. 2.
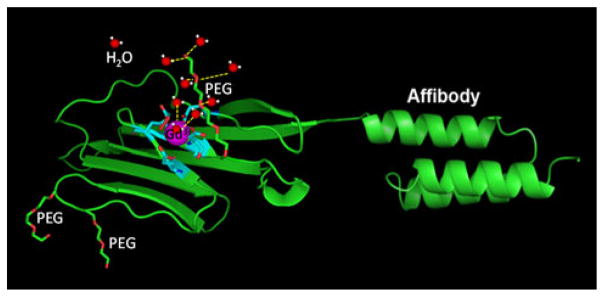
Modeled structure of the designed contrast agent ProCA1-CD2 shown with the Gd3+ (purple) binding site (cyan), and a cartoon structure of the secondary-sphere and outer-sphere water molecules (red and white) associated with the PEG chain (red and green). A multimodal HER2-targeted magnetic resonance imaging (MRI) probe was created by connecting a high-affinity HER2 affibody (ZHER2-342) at the C-terminal of a de novo designed protein contrast agent (ProCA1-CD2) with a designed Gd3+ binding site
We have shown the protein-based MRI contrast agent we developed (ProCA1-CD2) exhibits 14–20-fold improvement of both r1 and r2 relaxivities compared with those of the current clinically used contrast agents such as Gd-DTPA. To our knowledge, protein-based MRI contrast agents exhibit r1 and r2 relaxivities of 117 and 129 mM−1 s−1, respectively, per Gd3+ ion at 1.5 T [119]. Protein-based MRI contrast agents also have a greater than fourfold improvement at high field strength (r1 = 18.9 mM−1 s−1, r2 = 48.6 mM−1 s−1 for ProCA1-CD2 at 7 T). Further modification of the protein by PEGylation led to a 30–100 % increase of r1 and r2 relaxivity, supporting our notion that key factors such as the correlation time and the number of exchangeable water molecules from the first coordination shell and the secondary/outer sphere can be tuned by protein design and modification [119, 123, 134] (Fig. 2).
To prevent the competitive binding of physiological metal ions such as Ca2+, Mg2+, and Cu2+ to the contrast agent chelators, strong metal selectivity (kinetic metal stabilities) should be achieved when designing MRI contrast agents. The first generation of ProCA1-CD2 has a much higher metal selectivity (pGd/pCa > 9.84, pGd/pZn = 5.34, pGd/pMg > 10.06) than clinical MRI contrast agents (pGd/pCa = 11.70, pGd/pZn = 4.17, pGd/pMg = 4.25 for Gd-DTPA). Further assay shows that the potential chelators in serum, such as 50 mM phosphate, cannot remove Gd3+ from a Gd3+–metal complex [123].
We have further reported that a PEGylated protein-based MRI contrast agent, named ProCA1-CD2-m, has improved biocompatibility, such as improved solubility, improved dose efficiency, increased blood circulation time, and decreased immunogenicity with retention of metal binding and relaxation properties [134]. The blood retention time of ProCA1-CD2-m is more than 12 h compared with less than 30 min for Gd-DTPA in mice. Using a rabbit model, we further showed that ProCA1-CD2-m does not have significant immunogenicity without the use of adjuvants [134]. Further reduction in immunogenicity can be achieved by humanization of protein contrast agents. Figure 3a shows that using Pro-CA1-CD2-m with a 40-fold lower dose injection than for Gd-DTPA allows in vivo contrast enhancement of multiple organs owing to improved relaxivity and pharmacokinetics. No significant cellular toxicity and acute toxicity were detected. Therefore, we have shown that protein design technology (protein-based MRI contrast agents) (Fig. 2) enables us to significantly improve several key properties of an ideal MRI contrast agent, such as metal stability, relaxation, pharmacokinetics, and molecular recognition for molecular imaging individually or in parallel.
Fig. 3.

a Gradient echo transversal MR images collected prior to injection and at various time points post injection of 3.0 mM ProCA1-affi342-m which is PGEylated ProCA1-affi342 targeting to HER2 in HEPES saline via tail vein. The MRI signal on the positive tumor (SKOV-3, right) exhibits significant enhancement at 24 h post injection. Blocking results confirm the specific binding of ProCA1-affi342 to HER2-positive tumors. b Enhancement changes of MRI intensity in tumors and at 24 h post injection, highest enhancement was observed. c MR imaging of contrast agent ProCA1-affi1907 targeting to EGFR specifically. d Immunostaining show HER2 targeted ProCA1-affi342 has better tumor penetration than HER2 antibodies
Development of HER2/EGFR-targeted protein contrast agents
We have developed a novel multimodal molecular imaging probe to target the cancer marker HER2 using magnetic resonance based on our protein-based MRI contrast agent ProCA1 which exhibits significantly improved T1 relaxivity for MRI contrast enhancement and in vivo pharmacokinetics/pharmacodynamics compared with those of the commonly used Gd-DTPA. Figure 2 shows our designed HER2/EGFR-targeted protein contrast agent for molecular imaging. To meet the criteria for molecular imaging with good tissue and tumor penetration, we chose an affibody with a small protein domain (molecular mass of 16 kDa) instead of an antibody (molecular mass of 150 kDa). A high-affinity HER2 affibody capable of producing a molecular image of breast cancer by targeting the bio-marker HER2 [62, 104] was engineered into the C-terminal of the designed Gd3+-binding protein by a flexible linker. The small molecular size (16 kDa) provides good tissue penetration. To increase protein solubility and the blood circulation time and to reduce immunogenicity, the designed HER2-targeted protein contrast agent was PEGylated using PEG-40, a molecule with three branches of 12 PEG units (denoted as ProCA1-affi342-m) [135].
We tested whether our designed contrast agent would produce MRI contrast enhancement in nude mice xenograft models of two human cancer cell lines, SKOV3 and MDA-MB-231, with HER2 expression levels of 106 and 104, respectively (Fig. 1b). The HER2-targeted contrast agent was introduced via the tail vein at a concentration of approximately 5 mM (60–100-fold lower dose than for Gd-DTPA). Pre- and post-contrast magnetic resonance images were taken at different time points (Fig. 3). Figure 3a shows that the HER2-targeted contrast agent exhibits significantly greater enhancement at the SKOV3 tumor site, with high HER2 expression, than the MDA-MD-231 tumor, with lower expression of HER2 [136].
The in vivo HER2-targeting capability of the contrast agent we developed was further verified by a competition assay. HER2-specific MRI contrast enhancement was blocked by preinjection of HER2 affibody labeled with Cy5.5 at 12 h and 4 h before the injection of the protein contrast agent (Fig. 3a) [136].
After MRI, mouse organs were dissected for histological analysis. Using the antibody PAbPGCA1 against PEGylated ProCA1-CD2 with tissue slides made from the tissue samples of mice imaged, including tumors, the strongest staining was observed with liver and HER2-positive tumor tissue slides. Close examination of the staining patterns of the tumor slides revealed the contrast agent was evenly distributed throughout the entire tumor. In addition, a very high level of the targeted agent inside the cancer cells was observed, indicating cancer cell targeting and internalization (endocytosis) of the protein contrast agent. The results also suggested the contrast agent penetrated the tumor tissue, rather than being trapped in the tumor vasculature. This is an important property for HER2 targeting in the whole cancer mass (Fig. 3d). Co-immunostaining the slides made from HER2-positive tumor with the antibody PAb-PGCA1 and the antibody against CD31, an endothelial molecular marker, demonstrated that the protein contrast agent largely localized in tissue areas other than the tumor microvascular structure (CD31-positive areas). This staining pattern suggested the designed protein contrast agent had penetrated tissue rather than simply being trapped in the blood in the microvasculature of the tumor tissue. This staining pattern is in contrast with the distribution pattern of anti-HER2 antibody (Fig. 3d).
Summary and future perspectives
EGFR and HER2 are highly expressed as biomarkers in various cancers and play important roles in cancer progression and survival. They are also major drug targets. Several targeted drugs, such as monoclonal antibodies (trastuzumab) and small inhibitors (erlotinib), against HER2 and EGFR have been shown to be effective in patients overexpressing those biomarkers. Unfortunately, the clinical application of targeted therapy is largely limited by current methods for assessment of these cancer biomarkers using invasive methods such as biopsy. One in five HER2 clinical tests, including biopsy and IHC, provides incorrect results, severely affecting the selection of appropriate patients for personalized treatment using HER2/EGFR-targeted cancer therapies [84, 137].
To meet the urgent need to develop noninvasive and accurate methods for diagnosis and to monitor the levels and distribution of biomarkers and their changes on treatment with targeted drugs in cancer patients, we have developed a new class of protein-based MRI contrast agents by de novo design of Gd3+ binding sites in a stable host protein with significantly improved MRI relaxivity at field strengths used clinically [123]. We recently discovered that PEGylation of protein contrast agents leads to a further increase of r1 and r2 relaxivities and a 100-fold improvement in in vivo dose efficiency, in addition to a reduction in immunogenicity and an increase in solubility. Furthermore, we have successfully designed an HER2-targeted MRI contrast agent by adding a HER2-specific affibody moiety into the designed protein contrast agent. The newly designed HER2-targeted MRI contrast agent exhibits strong HER2-specific MRI enhancement in both tumor cells and xenograft mice models. Moreover, the agent we have developed exhibits disease-marker-dependent imaging enhancement and desirable penetration of tissue and the endothelial boundary, and has much better clearance and targeting than albumin or antibody cross-linked with Gd-DTPA.
Further development of an MRI method to allow non-invasive, semi-quantitative measurement of HER2 levels in breast cancers will significantly improve breast cancer diagnosis/prognosis [86]. MRI of cancer metastasis, especially early-stage metastasis, will be applicable to the detection of metastasis at various organ sites. In addition, the development of EGFR/HER2 imaging reagents will potentially provide insight into the mechanism of HER2-mediated cancer progression. For example, pre-invasive ductal carcinoma in situ represents 20–25 % of all newly diagnosed breast cancers with moderate EGFR and HER2 levels (approximately 104 per cell) and its natural history is largely unknown. Developing HER2- and EGFR-targeted contrast agents with improved sensitivity will provide insight into the clinical treatment of patients with ductal carcinoma in situ [138]. Since HER2 and EGFR are expressed in various types of cancers, including prostate, gastrointestinal and breast cancers, the contrast agents developed will aid in patient selection [139] and benefit HER-2 targeted diagnostics of cancer patients (Fig. 1) [140, 141]. Currently, there is no effective and noninvasive method to monitor the changes of HER2 response to the cancer therapies/treatment. Molecular imaging that monitors the changes in HER2 expression levels and patterns after drug treatments will significantly improve our capability to monitor treatment efficacy, especially early responses. It will also facilitate the design of new strategies for cancer treatments. Moreover, it is expected that the contrast agents developed and additional contrast agents targeting various molecular biomarkers will improve our capability in image-guided biopsy, local treatment, and drug delivery [2, 142–144].
Acknowledgments
We thank Katheryn Meenach, Anvi Patel, and Rose Auguste for careful editing of the manuscript, and Robert Long, Hua Yang, and Hans Grossniklaus for MRI and immunohistological experiments. This work was supported by research grants from the National Institutes of Health (EB007268 and GM62999) to J.J.Y.
Contributor Information
Jingjuan Qiao, Department of Chemistry, Georgia State University, 50 Decatur Street, 550 NSC, Atlanta, GA 30303, USA.
Shenghui Xue, Department of Chemistry, Georgia State University, 50 Decatur Street, 550 NSC, Atlanta, GA 30303, USA. Department of Biology, Georgia State University, Atlanta, GA 30303, USA.
Fan Pu, Department of Chemistry, Georgia State University, 50 Decatur Street, 550 NSC, Atlanta, GA 30303, USA.
Natalie White, Department of Chemistry, Georgia State University, 50 Decatur Street, 550 NSC, Atlanta, GA 30303, USA.
Jie Jiang, Department of Chemistry, Georgia State University, 50 Decatur Street, 550 NSC, Atlanta, GA 30303, USA.
Zhi-Ren Liu, Department of Biology, Georgia State University, Atlanta, GA 30303, USA. Center for Diagnostics and Therapeutics, Georgia State University, Atlanta, GA 30303, USA.
Jenny J. Yang, Email: jenny@gsu.edu, Department of Chemistry, Georgia State University, 50 Decatur Street, 550 NSC, Atlanta, GA 30303, USA. Center for Diagnostics and Therapeutics, Georgia State University, Atlanta, GA 30303, USA
References
- 1.Yewale C, Baradia D, Vhora I, Patil S, Misra A. Bio-materials. 2013;34(34):8690–8707. doi: 10.1016/j.biomaterials.2013.07.100. [DOI] [PubMed] [Google Scholar]
- 2.Kramer-Marek G, Longmire MR, Choyke PL, Kobayashi H. Curr Med Chem. 2012;19(28):4759–4766. doi: 10.2174/092986712803341584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang W, Ke S, Wu Q, Charnsangavej C, Gurfinkel M, Gelovani JG, Abbruzzese JL, Sevick-Muraca EM, Li C. Mol Imaging. 2004;3(4):343–351. doi: 10.1162/15353500200404148. [DOI] [PubMed] [Google Scholar]
- 4.Semmler W, Schwaiger M, editors. Handbook of Experimental Pharmacology. Springer; Berlin, Heidelberg: 2008. Molecular Imaging; pp. 167–224. [Google Scholar]
- 5.Johnston DL, Liu P, Lauffer RB, Newell JB, Wedeen VJ, Rosen BR, Brady TJ, Okada RD. J Nucl Med. 1987;28(5):871–877. [PubMed] [Google Scholar]
- 6.Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Chem Rev. 1999;99(9):2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- 7.Tweedle MF. Eur Radiol. 1997;7(Suppl 5):225–230. doi: 10.1007/pl00006897. [DOI] [PubMed] [Google Scholar]
- 8.Bousquet J-C, Saini S, Stark D, Hahn P, Nigam M, Wittenberg J, Ferrucci J. Radiology. 1988;166(3):693–698. doi: 10.1148/radiology.166.3.3340763. [DOI] [PubMed] [Google Scholar]
- 9.Vogl TJ, Kümmel S, Hammerstingl R, Schellenbeck M, Schumacher G, Balzer T, Schwarz W, Müller P, Bechstein WO, Mack MG. Radiology. 1996;200(1):59–67. doi: 10.1148/radiology.200.1.8657946. [DOI] [PubMed] [Google Scholar]
- 10.Kaiser W, Zeitler E. Radiology. 1989;170(3):681–686. doi: 10.1148/radiology.170.3.2916021. [DOI] [PubMed] [Google Scholar]
- 11.Flacke S, Fischer S, Scott MJ, Fuhrhop RJ, Allen JS, McLean M, Winter P, Sicard GA, Gaffney PJ, Wickline SA, Lanza GM. Circulation. 2001;104(11):1280–1285. doi: 10.1161/hc3601.094303. [DOI] [PubMed] [Google Scholar]
- 12.Terreno E, Castelli DD, Viale A, Aime S. Chem Rev. 2010;110(5):3019–3042. doi: 10.1021/cr100025t. [DOI] [PubMed] [Google Scholar]
- 13.Weinmann H-J, Ebert W, Misselwitz B, Schmitt-Willich H. Eur J Radiol. 2003;46(1):33–44. doi: 10.1016/s0720-048x(02)00332-7. [DOI] [PubMed] [Google Scholar]
- 14.Tóth E, Helm L, Merbach AE. Top Curr Chem. 2002;221:61–102. [Google Scholar]
- 15.Zhou G, Li Y, Liu Y, Ge C, Li W, Sun B, Li B, Gao Y, Chen C. J Nanosci Nanotechnol. 2010;10(12):8597–8602. doi: 10.1166/jnn.2010.2486. [DOI] [PubMed] [Google Scholar]
- 16.Arbab AS, Liu W, Frank JA. Expert Rev Med Devices. 2006;3(4):427–439. doi: 10.1586/17434440.3.4.427. [DOI] [PubMed] [Google Scholar]
- 17.Ranganathan RS, Raju N, Fan H, Zhang X, Tweedle MF, Desreux JF, Jacques V. Inorg Chem. 2002;41(25):6856–6866. doi: 10.1021/ic025695e. [DOI] [PubMed] [Google Scholar]
- 18.Villaraza AJ, Bumb A, Brechbiel MW. Chem Rev. 2010;110(5):2921–2959. doi: 10.1021/cr900232t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caravan P. Chem Soc Rev. 2006;35(6):512–523. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]
- 20.Gibby WA, Gibby KA. Invest Radiol. 2004;39(3):138–142. doi: 10.1097/01.rli.0000112789.57341.01. [DOI] [PubMed] [Google Scholar]
- 21.Prince MR, Zhang HL, Prowda JC, Grossman ME, Silvers DN. Radiographics. 2009;29(6):1565–1574. doi: 10.1148/rg.296095517. [DOI] [PubMed] [Google Scholar]
- 22.Grobner T. Nephrol Dial Transplant. 2006;21:1104–1108. doi: 10.1093/ndt/gfk062. [DOI] [PubMed] [Google Scholar]
- 23.Thomsen HS, Morcos SK, Dawson P. Clin Radiol. 2006;61(11):905–906. doi: 10.1016/j.crad.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Clarkson RB. Top Curr Chem. 2002;221:201–235. [Google Scholar]
- 25.McMurry TJ, Parmelee DJ, Sajiki H, Scott DM, Ouellet HS, Walovitch RC, Tyeklar Z, Dumas S, Bernard P, Nadler S, Midelfort K, Greenfield M, Troughton J, Lauffer RB. J Med Chem. 2002;45(16):3465–3474. doi: 10.1021/jm0102351. [DOI] [PubMed] [Google Scholar]
- 26.Ringe KI, Husarik DB, Sirlin CB, Merkle EM. AJR Am J Roentgenol. 2010;195(1):13–28. doi: 10.2214/AJR.10.4392. [DOI] [PubMed] [Google Scholar]
- 27.Wedeking P, Sotak CH, Telser J, Kumar K, Chang CA, Tweedle MF. Magn Reson Imaging. 1992;10(1):97–108. doi: 10.1016/0730-725x(92)90378-d. [DOI] [PubMed] [Google Scholar]
- 28.Matsumoto Y, Jasanoff A. FEBS Lett. 2013;587(8):1021–1029. doi: 10.1016/j.febslet.2013.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shapiro MG, Westmeyer GG, Romero PA, Szablowski JO, Kuster B, Shah A, Otey CR, Langer R, Arnold FH, Jasanoff A. Nat Biotechnol. 2010;28(3):264–270. doi: 10.1038/nbt.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sana B, Poh CL, Lim S. Chem Commun. 2012;48(6):862–864. doi: 10.1039/c1cc15189d. [DOI] [PubMed] [Google Scholar]
- 31.Sana B, Johnson E, Sheah K, Poh CL, Lim S. Biointerphases. 2010;5(3):FA48–FA52. doi: 10.1116/1.3483216. [DOI] [PubMed] [Google Scholar]
- 32.Bryant LH, Jr, Jordan EK, Bulte JW, Herynek V, Frank JA. Acad Radiol. 2002;9(Suppl 1):S29–S33. doi: 10.1016/s1076-6332(03)80390-2. [DOI] [PubMed] [Google Scholar]
- 33.Bryant LH, Jr, Brechbiel MW, Wu C, Bulte JW, Herynek V, Frank JA. J Magn Reson Imaging. 1999;9(2):348–352. doi: 10.1002/(sici)1522-2586(199902)9:2<348::aid-jmri30>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 34.Bryant LH, Jr, Hodges MW, Bryant RG. Inorg Chem. 1999;38(5):1002–1005. doi: 10.1021/ic981197n. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi H, Brechbiel MW. Curr Pharm Biotechnol. 2004;5(6):539–549. doi: 10.2174/1389201043376571. [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi H, Reijnders K, English S, Yordanov AT, Milenic DE, Sowers AL, Citrin D, Krishna MC, Waldmann TA, Mitchell JB, Brechbiel MW. Clin Cancer Res. 2004;10(22):7712–7720. doi: 10.1158/1078-0432.CCR-04-1175. [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi H, Kawamoto S, Saga T, Sato N, Hiraga A, Ishimori T, Konishi J, Togashi K, Brechbiel MW. Magn Reson Med. 2001;46(4):781–788. doi: 10.1002/mrm.1257. [DOI] [PubMed] [Google Scholar]
- 38.Kellar KE, Henrichs PM, Hollister R, Koenig SH, Eck J, Wei D. Magn Reson Med. 1997;38(5):712–716. doi: 10.1002/mrm.1910380506. [DOI] [PubMed] [Google Scholar]
- 39.Vander Elst L, Chapelle F, Laurent S, Muller RN. J Biol Inorg Chem. 2001;6(2):196–200. doi: 10.1007/s007750000195. [DOI] [PubMed] [Google Scholar]
- 40.Behra-Miellet J, Briand G, Kouach M, Gressier B, Cazin M, Cazin JC. Biomed Chromatogr. 1998;12(1):21–26. doi: 10.1002/(SICI)1099-0801(199801/02)12:1<21::AID-BMC714>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 41.Lauffer RB, Brady TJ. Magn Reson Imaging. 1985;3(1):11–16. doi: 10.1016/0730-725x(85)90004-9. [DOI] [PubMed] [Google Scholar]
- 42.Lauffer RB, Brady TJ, Brown RD, 3rd, Baglin C, Koenig SH. Magn Reson Med. 1986;3(4):541–548. doi: 10.1002/mrm.1910030407. [DOI] [PubMed] [Google Scholar]
- 43.Ogan MD. Invest Radiol. 1988;23(12):961. [PubMed] [Google Scholar]
- 44.Lauffer RB, Parmelee DJ, Dunham SU, Ouellet HS, Dolan RP, Witte S, McMurry TJ, Walovitch RC. Radiology. 1998;207(2):529–538. doi: 10.1148/radiology.207.2.9577506. [DOI] [PubMed] [Google Scholar]
- 45.Lauffer RB, Parmelee DJ, Ouellet HS, Dolan RP, Sajiki H, Scott DM, Bernard PJ, Buchanan EM, Ong KY, Tyeklar Z, Midelfort KS, McMurry TJ, Walovitch RC. Acad Radiol. 1996;3(Suppl 2):S356–S358. doi: 10.1016/s1076-6332(96)80583-6. [DOI] [PubMed] [Google Scholar]
- 46.Karfeld-Sulzer LS, Waters EA, Kohlmeir EK, Kissler H, Zhang X, Kaufman DB, Barron AE, Meade TJ. Magn Reson Med. 2011;65(1):220–228. doi: 10.1002/mrm.22587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liepold L, Anderson S, Willits D, Oltrogge L, Frank JA, Douglas T, Young M. Magn Reson Med. 2007;58(5):871–879. doi: 10.1002/mrm.21307. [DOI] [PubMed] [Google Scholar]
- 48.Caravan P, Greenwood JM, Welch JT, Franklin SJ. Chem Commun. 2003:2574–2575. doi: 10.1039/b307817e. [DOI] [PubMed] [Google Scholar]
- 49.Overoye-Chan K, Koerner S, Looby RJ, Kolodziej AF, Zech SG, Deng Q, Chasse JM, McMurry TJ, Caravan P. J Am Chem Soc. 2008;130(18):6025–6039. doi: 10.1021/ja800834y. [DOI] [PubMed] [Google Scholar]
- 50.Caravan P, Das B, Dumas S, Epstein FH, Helm PA, Jacques V, Koerner S, Kolodziej A, Shen L, Sun WC, Zhang Z. Angew Chem Int Ed. 2007;46(43):8171–8173. doi: 10.1002/anie.200700700. [DOI] [PubMed] [Google Scholar]
- 51.Uppal R, Medarova Z, Farrar CT, Dai G, Moore A, Caravan P. Invest Radiol. 2012;47(10):553–558. doi: 10.1097/RLI.0b013e31825dddfb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pomper MG. J Cell Biochem. 2002;87(Suppl 39):211–220. doi: 10.1002/jcb.10443. [DOI] [PubMed] [Google Scholar]
- 53.Jarrett BR, Correa C, Ma KL, Louie AY. PLoS ONE. 2010;5(10):13254. doi: 10.1371/journal.pone.0013254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tu C, Ma X, House A, Kauzlarich SM, Louie AY. ACS Med Chem Lett. 2011;2(4):285–288. doi: 10.1021/ml1002844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wadas TJ, Wong EH, Weisman GR, Anderson CJ. Chem Rev. 2010;110(5):2858–2902. doi: 10.1021/cr900325h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Leon-Rodriguez LM, Lubag AJ, Malloy CR, Martinez GV, Gillies RJ, Sherry AD. Acc Chem Res. 2009;42(7):948–957. doi: 10.1021/ar800237f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Artemov D, Mori N, Ravi R, Bhujwalla ZM. Cancer Res. 2003;63(11):2723–2727. [PubMed] [Google Scholar]
- 58.Zhu W, Okollie B, Bhujwalla ZM, Artemov D. Magn Reson Med. 2008;59(4):679–685. doi: 10.1002/mrm.21508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Louie AY, Huber MM, Ahrens ET, Rothbacher U, Moats R, Jacobs RE, Fraser SE, Meade TJ. Nat Biotechnol. 2000;18(3):321–325. doi: 10.1038/73780. [DOI] [PubMed] [Google Scholar]
- 60.Groth RD, Lindskog M, Thiagarajan TC, Li L, Tsien RW. Proc Natl Acad Sci USA. 2011;108(2):828–833. doi: 10.1073/pnas.1018022108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Feinmesser RL, Wicks SJ, Taverner CJ, Chantry A. J Biol Chem. 1999;274(23):16168–16173. doi: 10.1074/jbc.274.23.16168. [DOI] [PubMed] [Google Scholar]
- 62.Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, Jr, Leahy DJ. Nature. 2003;421(6924):756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 63.Burke P, Schooler K, Wiley HS. Mol Biol Cell. 2001;12(6):1897–1910. doi: 10.1091/mbc.12.6.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tolmachev V. Curr Pharm Des. 2008;14(28):2999–3019. doi: 10.2174/138161208786404290. [DOI] [PubMed] [Google Scholar]
- 65.Zhao X, Dai W, Zhu H, Zhang Y, Cao L, Ye Q, Lei P, Shen G. Cell Biol Int. 2006;30(8):653–658. doi: 10.1016/j.cellbi.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 66.Zhu H, Acquaviva J, Ramachandran P, Boskovitz A, Woolfenden S, Pfannl R, Bronson RT, Chen JW, Weissleder R, Housman DE, Charest A. Proc Natl Acad Sci USA. 2009;106(8):2712–2716. doi: 10.1073/pnas.0813314106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tang X, Shigematsu H, Bekele BN, Roth JA, Minna JD, Hong WK, Gazdar AF, Wistuba II. Cancer Res. 2005;65(17):7568–7572. doi: 10.1158/0008-5472.CAN-05-1705. [DOI] [PubMed] [Google Scholar]
- 68.Arteaga C. Semin Oncol. 2003;30(3 Suppl 7):3–14. [PubMed] [Google Scholar]
- 69.Ross JS, Linette GP, Stec J, Clark E, Ayers M, Leschly N, Symmans WF, Hortobagyi GN, Pusztai L. Expert Rev Mol Diagn. 2004;4(2):169–188. doi: 10.1586/14737159.4.2.169. [DOI] [PubMed] [Google Scholar]
- 70.Sternlicht MD. Breast Cancer Res. 2006;8(1):201. doi: 10.1186/bcr1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sternlicht MD, Sunnarborg SW, Kouros-Mehr H, Yu Y, Lee DC, Werb Z. Development. 2005;132(17):3923–3933. doi: 10.1242/dev.01966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mitsudomi T, Yatabe Y. FEBS J. 2010;277(2):301–308. doi: 10.1111/j.1742-4658.2009.07448.x. [DOI] [PubMed] [Google Scholar]
- 73.Okamoto I. FEBS J. 2010;277(2):309–315. doi: 10.1111/j.1742-4658.2009.07449.x. [DOI] [PubMed] [Google Scholar]
- 74.Menard S, Casalini P, Campiglio M, Pupa S, Agresti R, Tagliabue E. Ann Oncol. 2001;12(Suppl 1):S15–S19. doi: 10.1093/annonc/12.suppl_1.s15. [DOI] [PubMed] [Google Scholar]
- 75.Amin DN, Hida K, Bielenberg DR, Klagsbrun M. Cancer Res. 2006;66(4):2173–2180. doi: 10.1158/0008-5472.CAN-05-3387. [DOI] [PubMed] [Google Scholar]
- 76.Milanezi F, Carvalho S, Schmitt FC. Expert Rev Mol Diagn. 2008;8(4):417–434. doi: 10.1586/14737159.8.4.417. [DOI] [PubMed] [Google Scholar]
- 77.Cooke T, Reeves J, Lanigan A, Stanton P. Ann Oncol. 2001;12(Suppl 1):S23–S28. doi: 10.1093/annonc/12.suppl_1.s23. [DOI] [PubMed] [Google Scholar]
- 78.Tagliabue E, Agresti R, Ghirelli C, Morelli D, Menard S. Breast Cancer Res Treat. 2001;70(2):155–156. doi: 10.1023/a:1012955229031. [DOI] [PubMed] [Google Scholar]
- 79.Yonemura Y, Ninomiya I, Yamaguchi A, Fushida S, Kimura H, Ohoyama S, Miyazaki I, Endou Y, Tanaka M, Sasaki T. Cancer Res. 1991;51(3):1034–1038. [PubMed] [Google Scholar]
- 80.Hellstrom I, Goodman G, Pullman J, Yang Y, Hellstrom KE. Cancer Res. 2001;61(6):2420–2423. [PubMed] [Google Scholar]
- 81.Buchler P, Reber HA, Eibl G, Roth MA, Buchler MW, Friess H, Isacoff WH, Hines OJ. Int J Oncol. 2005;27(4):1125–1130. [PubMed] [Google Scholar]
- 82.Yarden Y. Oncology. 2001;61(Suppl 2):1–13. doi: 10.1159/000055396. [DOI] [PubMed] [Google Scholar]
- 83.Yamanaka Y, Friess H, Kobrin MS, Buchler M, Beger HG, Korc M. Anticancer Res. 1993;13(3):565–569. [PubMed] [Google Scholar]
- 84.Allison M. Nat Biotechnol. 2010;28(2):117–119. doi: 10.1038/nbt0210-117. [DOI] [PubMed] [Google Scholar]
- 85.Artemov D. J Cell Biochem. 2003;90(3):518–524. doi: 10.1002/jcb.10660. [DOI] [PubMed] [Google Scholar]
- 86.Artemov D, Mori N, Okollie B, Bhujwalla ZM. Magn Reson Med. 2003;49(3):403–408. doi: 10.1002/mrm.10406. [DOI] [PubMed] [Google Scholar]
- 87.Leonessa F, Green D, Licht T, Wright A, Wingate-Legette K, Lippman J, Gottesman M, Clarke R. Br J Cancer. 1996;73(2):154. doi: 10.1038/bjc.1996.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang D, Kuan C-T, Payne J, Kihara A, Murray A, Wang L-M, Alimandi M, Pierce JH, Pastan I, Lippman ME. Clin Cancer Res. 1998;4(4):993–1004. [PubMed] [Google Scholar]
- 89.Park J-G, Frucht H, LaRocca RV, Bliss DP, Kurita Y, Chen T-R, Henslee JG, Trepel JB, Jensen RT, Johnson BE. Cancer Res. 1990;50(9):2773–2780. [PubMed] [Google Scholar]
- 90.Wilken JA, Webster KT, Maihle NJ. J Ovarian Res. 2010;3:7. doi: 10.1186/1757-2215-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Winnard PT, Jr, Pathak AP, Dhara S, Cho SY, Raman V, Pomper MG. J Nucl Med. 2008;49(Suppl 2):96S–112S. doi: 10.2967/jnumed.107.045948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Diaz R, Nguewa PA, Parrondo R, Perez-Stable C, Manrique I, Redrado M, Catena R, Collantes M, Penuelas I, Diaz-Gonzalez JA, Calvo A. BMC Cancer. 2010;10:188. doi: 10.1186/1471-2407-10-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Eck MJ, Yun CH. Biochim Biophys Acta. 2010;1804(3):559–566. doi: 10.1016/j.bbapap.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Akita RW, Sliwkowski MX. Semin Oncol. 2003;30(3 Suppl 7):15–24. [PubMed] [Google Scholar]
- 95.Knuefermann C, Lu Y, Liu B, Jin W, Liang K, Wu L, Schmidt M, Mills GB, Mendelsohn J, Fan Z. Oncogene. 2003;22(21):3205–3212. doi: 10.1038/sj.onc.1206394. [DOI] [PubMed] [Google Scholar]
- 96.Baselga J, Norton L, Albanell J, Kim YM, Mendelsohn J. Cancer Res. 1998;58(13):2825–2831. [PubMed] [Google Scholar]
- 97.Dawood S, Broglio K, Buzdar AU, Hortobagyi GN, Giordano SH. J Clin Oncol. 2010;28(1):92–98. doi: 10.1200/JCO.2008.19.9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Leyland-Jones B, Gelmon K, Ayoub JP, Arnold A, Verma S, Dias R, Ghahramani P. J Clin Oncol. 2003;21(21):3965–3971. doi: 10.1200/JCO.2003.12.109. [DOI] [PubMed] [Google Scholar]
- 99.Gong H, Kovar J, Little G, Chen H, Olive DM. Neoplasia. 2010;12(2):139–149. doi: 10.1593/neo.91446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schier R, Bye J, Apell G, McCall A, Adams GP, Malmqvist M, Weiner LM, Marks JD. J Mol Biol. 1996;255(1):28–43. doi: 10.1006/jmbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 101.Yang L, Mao H, Wang YA, Cao Z, Peng X, Wang X, Duan H, Ni C, Yuan Q, Adams G, Smith MQ, Wood WC, Gao X, Nie S. Small. 2009;5(2):235–243. doi: 10.1002/smll.200800714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wei L, Li S, Yang J, Ye Y, Zou J, Wang L, Long R, Zurkiya O, Zhao T, Johnson J, Qiao J, Zhou W, Castiblanco A, Maor N, Chen Y, Mao H, Hu X, Yang JJ, Liu ZR. Mol Imaging Biol. 2010;13:416–423. doi: 10.1007/s11307-010-0342-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nygren PA. FEBS J. 2008;275(11):2668–2676. doi: 10.1111/j.1742-4658.2008.06438.x. [DOI] [PubMed] [Google Scholar]
- 104.Wikman M, Steffen AC, Gunneriusson E, Tolmachev V, Adams GP, Carlsson J, Stahl S. Protein Eng Des Sel. 2004;17(5):455–462. doi: 10.1093/protein/gzh053. [DOI] [PubMed] [Google Scholar]
- 105.Tolmachev V, Orlova A, Pehrson R, Galli J, Baastrup B, Andersson K, Sandstrom M, Rosik D, Carlsson J, Lundqvist H, Wennborg A, Nilsson FY. Cancer Res. 2007;67(6):2773–2782. doi: 10.1158/0008-5472.CAN-06-1630. [DOI] [PubMed] [Google Scholar]
- 106.Kramer-Marek G, Kiesewetter DO, Martiniova L, Jagoda E, Lee SB, Capala J. Eur J Nucl Med Mol Imaging. 2008;35(5):1008–1018. doi: 10.1007/s00259-007-0658-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Orlova A, Rosik D, Sandstrom M, Lundqvist H, Einarsson L, Tolmachev V. Q J Nucl Med Mol Imaging. 2007;51(4):314–323. [PubMed] [Google Scholar]
- 108.Orlova A, Magnusson M, Eriksson TL, Nilsson M, Larsson B, Hoiden-Guthenberg I, Widstrom C, Carlsson J, Tolmachev V, Stahl S, Nilsson FY. Cancer Res. 2006;66(8):4339–4348. doi: 10.1158/0008-5472.CAN-05-3521. [DOI] [PubMed] [Google Scholar]
- 109.Namavari M, Padilla De Jesus O, Cheng Z, De A, Kovacs E, Levi J, Zhang R, Hoerner JK, Grade H, Syud FA, Gambhir SS. Mol Imaging Biol. 2008;10(4):177–181. doi: 10.1007/s11307-008-0142-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen TJ, Cheng TH, Chen CY, Hsu SC, Cheng TL, Liu GC, Wang YM. J Biol Inorg Chem. 2008;14:253–260. doi: 10.1007/s00775-008-0445-9. [DOI] [PubMed] [Google Scholar]
- 111.Helm L. Future Med Chem. 2010;2(3):385–396. doi: 10.4155/fmc.09.174. [DOI] [PubMed] [Google Scholar]
- 112.Bloembergen N. J Chem Phys. 1957;27:572–573. [Google Scholar]
- 113.Bloembergen N, Morgan R. J Chem Phys. 1961;34:842. [Google Scholar]
- 114.Solomon I. Phys Rev. 1955;99:559–565. [Google Scholar]
- 115.Freed JH. J Chem Phys. 1978;68(9):4034–4037. [Google Scholar]
- 116.Aime S, Castelli DD, Crich SG, Gianolio E, Terreno E. Acc Chem Res. 2009;42(7):822–831. doi: 10.1021/ar800192p. [DOI] [PubMed] [Google Scholar]
- 117.Caravan P. Acc Chem Res. 2009;42(7):851–862. doi: 10.1021/ar800220p. [DOI] [PubMed] [Google Scholar]
- 118.Datta A, Raymond KN. Acc Chem Res. 2009;42(7):938–947. doi: 10.1021/ar800250h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xue S, Qiao J, Pu F, Cameron M, Yang JJ. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2013;5(2):163–179. doi: 10.1002/wnan.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Diakova G, Goddard Y, Korb JP, Bryant RG. J Magn Reson. 2011;208(2):195–203. doi: 10.1016/j.jmr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Strauch RC, Mastarone DJ, Sukerkar PA, Song Y, Ipsaro JJ, Meade TJ. J Am Chem Soc. 2011;133(41):16346–16349. doi: 10.1021/ja206134b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lubag AJ, De Leon-Rodriguez LM, Burgess SC, Sherry AD. Proc Natl Acad Sci USA. 2011;108(45):18400–18405. doi: 10.1073/pnas.1109649108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yang JJ, Yang J, Wei L, Zurkiya O, Yang W, Li S, Zou J, Zhou Y, Maniccia AL, Mao H, Zhao F, Malchow R, Zhao S, Johnson J, Hu X, Krogstad E, Liu ZR. J Am Chem Soc. 2008;130(29):9260–9267. doi: 10.1021/ja800736h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lu ZR, Parker DL, Goodrich KC, Wang X, Dalle JG, Buswell HR. Magn Reson Med. 2004;51(1):27–34. doi: 10.1002/mrm.10656. [DOI] [PubMed] [Google Scholar]
- 125.Yang JJ, Ye Y, Carroll A, Yang W, Lee HW. Curr Protein Pept Sci. 2001;2(1):1–17. doi: 10.2174/1389203013381251. [DOI] [PubMed] [Google Scholar]
- 126.Zhou Y, Xue S, Chen Y, Yang JJ. Methods Mol Biol. 2013;963:37–53. doi: 10.1007/978-1-62703-230-8_3. [DOI] [PubMed] [Google Scholar]
- 127.Zhou Y, Xue S, Yang JJ. Metallomics. 2013;5(1):29–42. doi: 10.1039/c2mt20009k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yanyi C, Shenghui X, Yubin Z, Jie YJ. Sci China Chem. 2010;53(1):52–60. doi: 10.1007/s11426-010-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yang W, Wilkins AL, Ye Y, Liu ZR, Li SY, Urbauer JL, Hellinga HW, Kearney A, van der Merwe PA, Yang JJ. J Am Chem Soc. 2005;127(7):2085–2093. doi: 10.1021/ja0431307. [DOI] [PubMed] [Google Scholar]
- 130.Yang W, Jones LM, Isley L, Ye Y, Lee HW, Wilkins A, Liu ZR, Hellinga HW, Malchow R, Ghazi M, Yang JJ. J Am Chem Soc. 2003;125(20):6165–6171. doi: 10.1021/ja034724x. [DOI] [PubMed] [Google Scholar]
- 131.Yang W, Wilkins AL, Li S, Ye Y, Yang JJ. Biochemistry. 2005;44(23):8267–8273. doi: 10.1021/bi050463n. [DOI] [PubMed] [Google Scholar]
- 132.Wilkins AL, Ye Y, Yang W, Lee HW, Liu ZR, Yang JJ. Protein Eng. 2002;15(7):571–574. doi: 10.1093/protein/15.7.571. [DOI] [PubMed] [Google Scholar]
- 133.Wang X, Kirberger M, Qiu F, Chen G, Yang JJ. Proteins. 2009;75(4):787–798. doi: 10.1002/prot.22285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li S, Jiang J, Zou J, Qiao J, Xue S, Wei L, Long R, Wang L, Castiblanco A, White N, Ngo J, Mao H, Liu ZR, Yang JJ. J Inorg Biochem. 2012;107(1):111–118. doi: 10.1016/j.jinorgbio.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nord K, Gunneriusson E, Ringdahl J, Stahl S, Uhlen M, Nygren PA. Nat Biotechnol. 1997;15(8):772–777. doi: 10.1038/nbt0897-772. [DOI] [PubMed] [Google Scholar]
- 136.Qiao J, Li S, Wei L, Jiang J, Long R, Mao H, Wei L, Wang L, Yang H, Grossniklaus HE, Liu ZR, Yang JJ. PLoS ONE. 2011;6(3):e18103. doi: 10.1371/journal.pone.0018103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Morse DL, Gillies RJ. Biochem Pharmacol. 2010;80(5):731–738. doi: 10.1016/j.bcp.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, Fujisawa T, Feng Z, Roth JA, Herz J, Minna JD, Gazdar AF. J Natl Cancer Inst. 2005;97(5):339–346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 139.Toi M, Sperinde J, Huang W, Saji S, Winslow J, Jin X, Tan Y, Ohno S, Nakamura S, Iwata H, Masuda N, Aogi K, Morita S, Petropoulos C, Bates M. BMC Cancer. 2010;10:56. doi: 10.1186/1471-2407-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Aime S, Botta M, Garino E, Crich SG, Giovenzana G, Pagliarin R, Palmisano G, Sisti M. Chemistry. 2000;6(14):2609–2617. doi: 10.1002/1521-3765(20000717)6:14<2609::aid-chem2609>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 141.Mankoff DA, Eary JF. Clin Cancer Res. 2008;14(22):7159–7160. doi: 10.1158/1078-0432.CCR-08-2233. [DOI] [PubMed] [Google Scholar]
- 142.Srinivas M, Aarntzen EH, Bulte JW, Oyen WJ, Heerschap A, de Vries IJ, Figdor CG. Adv Drug Deliv Rev. 2010;62(11):1080–1093. doi: 10.1016/j.addr.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 143.Lanza GM, Yu X, Winter PM, Abendschein DR, Karukstis KK, Scott MJ, Chinen LK, Fuhrhop RW, Scherrer DE, Wickline SA. Circulation. 2002;106(22):2842–2847. doi: 10.1161/01.cir.0000044020.27990.32. [DOI] [PubMed] [Google Scholar]
- 144.Pan D, Caruthers SD, Hu G, Senpan A, Scott MJ, Gaffney PJ, Wickline SA, Lanza GM. J Am Chem Soc. 2008;130(29):9186–9187. doi: 10.1021/ja801482d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Onn A, Correa AM, Gilcrease M, Isobe T, Massarelli E, Bucana CD, O’Reilly MS, Hong WK, Fidler IJ, Putnam JB, Herbst RS. Clin Cancer Res. 2004;10(1 Pt 1):136–143. doi: 10.1158/1078-0432.ccr-0373-3. [DOI] [PubMed] [Google Scholar]
- 146.Selvaggi G, Novello S, Torri V, Leonardo E, De Giuli P, Borasio P, Mossetti C, Ardissone F, Lausi P, Scagliotti GV. Ann Oncol. 2004;15(1):28–32. doi: 10.1093/annonc/mdh011. [DOI] [PubMed] [Google Scholar]
- 147.Ohtsuka K, Ohnishi H, Furuyashiki G, Nogami H, Koshiishi Y, Ooide A, Matsushima S, Watanabe T, Goya T. J Thorac Oncol. 2006;1(8):787–795. [PubMed] [Google Scholar]
- 148.Dancer J, Takei H, Ro JY, Lowery-Nordberg M. Oncol Rep. 2007;18(1):151–155. [PubMed] [Google Scholar]
- 149.Bloomston M, Bhardwaj A, Ellison EC, Frankel WL. Dig Surg. 2006;23(1–2):74–79. doi: 10.1159/000093497. [DOI] [PubMed] [Google Scholar]
- 150.Thybusch-Bernhardt A, Beckmann S, Juhl H. Int J Surg Investig. 2001;2(5):393–400. [PubMed] [Google Scholar]
- 151.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Science. 1987;235(4785):177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 152.Paik S, Bryant J, Tan-Chiu E, Yothers G, Park C, Wickerham DL, Wolmark N. J Natl Cancer Inst. 2000;92(24):1991–1998. doi: 10.1093/jnci/92.24.1991. [DOI] [PubMed] [Google Scholar]
- 153.Owens MA, Horten BC, Da Silva MM. Clin Breast Cancer. 2004;5(1):63–69. doi: 10.3816/cbc.2004.n.011. [DOI] [PubMed] [Google Scholar]
- 154.Seshadri R, Firgaira FA, Horsfall DJ, McCaul K, Setlur V, Kitchen P. J Clin Oncol. 1993;11(10):1936–1942. doi: 10.1200/JCO.1993.11.10.1936. [DOI] [PubMed] [Google Scholar]
- 155.Andrulis IL, Bull SB, Blackstein ME, Sutherland D, Mak C, Sidlofsky S, Pritzker KP, Hartwick RW, Hanna W, Lickley L, Wilkinson R, Qizilbash A, Ambus U, Lipa M, Weizel H, Katz A, Baida M, Mariz S, Stoik G, Dacamara P, Strongitharm D, Geddie W, McCready D. J Clin Oncol. 1998;16(4):1340–1349. doi: 10.1200/JCO.1998.16.4.1340. [DOI] [PubMed] [Google Scholar]