

Abstract
OBJECTIVE
To determine the occurrence of extremely low HDL cholesterol (HDL-C) among participants in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Lipid Trial and to examine the relationship of this finding with treatment with fenofibrate and thiazolidinedione (TZD).
RESEARCH DESIGN AND METHODS
The ACCORD Lipid Trial was a randomized, double-blind, placebo-controlled study conducted in patients with type 2 diabetes at 77 clinical centers across the U.S. and Canada in a 5,518-patient subset of the larger 10,251 ACCORD Glycemia Trial. Patients were enrolled from 11 January 2001 to 29 October 2005 and followed until the end of study visits between 1 March and 30 June 2009. Follow-up in the ACCORD Lipid Trial was 4–8 years (mean 4.7 years). Patients were treated with blinded fenofibrate or placebo on a background of simvastatin therapy. The main outcome measures for these descriptive, post hoc analyses was the occurrence of extremely low HDL-C (defined as <25 mg/dL [0.647 mmol/L]) during the trial.
RESULTS
Among ACCORD Lipid Trial participants, the occurrence of extremely low HDL-C ever during study follow-up was 106% higher among those randomized to fenofibrate (10.1% fenofibrate vs. 4.9% placebo, P < 0.001). The occurrence of low HDL-C was associated with concurrent treatment with fenofibrate and TZD (7.0% for both vs. 2.2% for neither at 48 months postrandomization).
CONCLUSIONS
Idiosyncratic and marked reduction in HDL-C can occur in some patients treated with both fenofibrate and TZD. Practitioners should recognize this important potential idiosyncratic reaction and take appropriate corrective action.
Introduction
The Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial was a National Heart, Lung, and Blood Institute–funded study that evaluated the effect of intensive control of blood glucose, blood pressure, and lipoproteins on cardiovascular risk in patients with type 2 diabetes (1). Embedded within the overall ACCORD trial was a lipid treatment trial in which study participants were randomized to receive either fenofibrate or placebo administered with background LDL cholesterol (LDL-C)–lowering therapy with simvastatin. During the trial, the new onset of a marked reduction in HDL cholesterol (HDL-C) levels among some lipid trial participants was noted. Individual cases have been reported in the literature of paradoxical substantial reductions in HDL-C of up to 50% in patients receiving fenofibrate, thiazolidinedione (TZD), or both (2–5). With use of data collected in the ACCORD Lipid Trial, we examined the relationship between concomitant administration of fenofibrate and TZD, primarily rosiglitazone, and the occurrence of low HDL-C.
Research Design and Methods
The rationale, design, and primary results of the ACCORD Lipid Trial have been previously reported (1,6–8). Overall in ACCORD, 10,251 participants were randomly assigned to receive either intensive glycemic control (targeting an HbA1c value <6.0% [42 mmol/mol]) or standard therapy (targeting an HbA1c of 7.0–7.9% [53–63 mmol/mol]) (1). With use of a double 2 × 2 factorial design, 4,733 of the ACCORD participants were also enrolled in the ACCORD Blood Pressure Trial, and 5,518 were enrolled in the ACCORD Lipid Trial. The hypothesis tested in the lipid trial was whether combination treatment with fenofibrate (to both raise HDL-C and lower triglyceride levels) and a statin (to reduce LDL-C) would reduce cardiovascular disease (CVD) event rates in high-risk people with type 2 diabetes compared with treatment with only a statin (1).
After institutional review board approval, participants were recruited from 77 clinical centers across the U.S. and Canada. All lipid trial participants were treated with simvastatin 20–40 mg/day and then randomly assigned to receive fenofibrate or matching placebo in a double-blind design. Clinic personnel were blinded to lipid measurements throughout the trial. Randomizations occurred from 11 January 2001 to 29 October 2005 using permuted blocks to maintain allocation concealment. End-of-study visits were scheduled between 1 March and 30 June 2009. Follow-up in the lipid trial was 4–8 years (mean 4.7 years). To be eligible for ACCORD, participants had documented type 2 diabetes and an HbA1c ≥7.5% and were either age 40–79 years with evidence of clinical CVD or age 55–79 years with evidence of subclinical CVD or at least two additional CVD risk factors. Participants were eligible for the embedded ACCORD Lipid Trial if they met the following additional entry criteria based on lipid measurements obtained within the previous year: 1) LDL-C between 60 and 180 mg/dL, inclusive; 2) HDL-C <55 mg/dL for women and blacks or <50 mg/dL for all other groups; and 3) triglycerides <750 mg/dL if not on a lipid medication or <400 mg/dL if on a lipid medication. Among the ACCORD Lipid Trial exclusion criteria were the use of a medication known to interact with statins or fibrate; history of pancreatitis, myositis/myopathy, or gallbladder disease; and refusal to discontinue any current lipid-altering treatment.
Open-labeled simvastatin therapy began at the randomization visit, and the blinded fenofibrate/placebo medication was initiated 1 month later. The initial dose of simvastatin complied with current national lipid guidelines at the time the study began and was modified over time in response to changing guidelines (7). The maximum daily dose of simvastatin used was 40 mg (7). At the beginning of the trial, the initial dose of fenofibrate was 160 mg/day. Because of an observed rise in serum creatinine levels in some participants receiving fenofibrate, the dose of fenofibrate was reduced to 48 mg/day in individuals with an estimated glomerular filtration rate ≤50 mL/min/m2 (9). Whereas rosiglitazone was the predominant TZD used in ACCORD, the analyses presented here also represent limited use of pioglitazone (e.g., only 23 participants were receiving the combination of pioglitazone and fenofibrate at postrandomization month 24, and only 72 were receiving the combination at month 48).
In the lipid trial, a blinded fasting plasma lipid profile was measured at the ACCORD central laboratory at baseline; 4, 8, 12 months postrandomization and annually thereafter; and study end. During follow-up, some participants obtained unblinded lipid measurements from their private health-care providers and informed ACCORD clinic staff of low HDL-C values, which sometimes occurred after the initiation of a TZD for the glycemia trial. After review of unblinded analyses by the ACCORD data and safety monitoring board (DSMB), an alert system was put in place for clinic personnel to be notified by the ACCORD central laboratory if a participant had an extremely low HDL-C level (defined as consistently <20 mg/dL [517 mmol/L]) during ACCORD study follow-up.
The prespecified primary outcome for all ACCORD trials was the first postrandomization occurrence of a major cardiovascular event, specifically nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes. All-cause mortality was a prespecified secondary outcome. Deaths, myocardial infarctions, and strokes were adjudicated by a central committee whose members were unaware of treatment group assignment (1,10).
All analyses were conducted at the coordinating center with the use of S-Plus version 8.0 (Insightful) or SAS version 9.3 (SAS Institute) software. The postrandomization occurrence and prevalence (at selected visits) of low HDL-C was examined with simple counts and percentages of participants by ACCORD trial, treatment group, and (within the lipid trial) TZD use. An HDL-C cutoff of <25 mg/dL (0.647 mmol/L) was chosen before the initiation of analyses. All analyses were repeated with a cutoff of <20 mg/dL (0.517 mmol/L) to assess the sensitivity of the results. In an exploration of how lipid trial participants who ever had a low HDL-C level during follow-up differed from all other lipid trial participants, baseline characteristics were compared with χ2 and two-sample t tests. Two multivariate generalized linear regression models were used to examine the relationship between medication use (fenofibrate and TZDs) and 24- and 48-month postrandomization HDL-C levels, with the interaction term “lipid treatment assignment × TZD use at time of blood draw” as the primary independent variable of interest and controlling for lipid trial treatment assignment, current TZD use, and other covariates identified in the baseline comparison analysis as follows: sex, age in years, race (white/nonwhite), and baseline values of HDL-C, LDL-C, triglycerides, and HbA1c. The analyses presented are for descriptive and exploratory purposes and are hypothesis generating only. No adjustments were made for multiple testing. Nominal P values are reported throughout as simple guides to possible associations.
Results
Restricting lipid measurements to those obtained at the annual visits (common to all ACCORD participants), 627 of all 10,251 randomized ACCORD participants (6.1%) had an HDL-C reported by the central laboratory of <25 mg/dL (0.647 mmol/L) at an annual follow-up visit (Table 1). There was no difference in the postrandomization occurrence of low HDL-C between the glycemia treatment groups (6.1% intensive vs. 6.2% standard, P = 0.83) or between the blood pressure treatment groups (4.8% intensive vs. 4.2% standard, P = 0.28). However, a greater proportion of lipid trial participants had a low HDL-C recorded at a follow-up annual visit compared with blood pressure trial participants (7.5% lipid vs. 4.5% blood pressure, P < 0.001). Among lipid trial participants, the occurrence of low HDL-C was 106% higher among those randomized to fenofibrate (10.1% fenofibrate vs. 4.9% placebo, P < 0.001). Because ACCORD Lipid Trial participants had additional measurements at 4 and 8 months, the occurrence of any follow-up visit low HDL-C was re-examined; 561 of the 5,518 lipid trial participants had an HLD-C <25 mg/dL at some point during follow-up (364 [13.2%] fenofibrate group, 197 [7.2%] placebo group).
Table 1.
ACCORD participants who ever had a postrandomization HDL-C <25 mg/dL (0.647 mmol/L) at an annual follow-up visit by trial and treatment group assignment
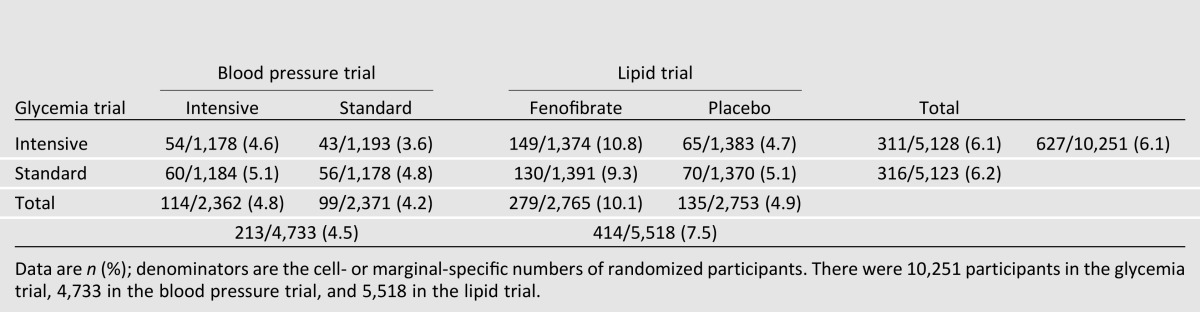
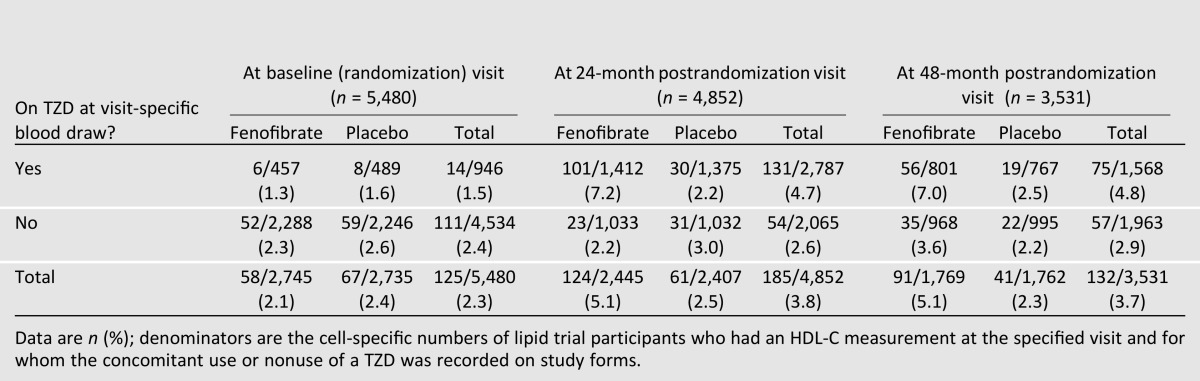
Given the prior reports of iatrogenic lowering of HDL-C with concomitant fenofibrate and TZD treatment (2,4,5,11–13), we examined the cross-sectional prevalence of low HDL-C among lipid trial participants at baseline (before initiation of fenofibrate treatment) and at postrandomization months 24 and 48, stratified by fenofibrate treatment assignment and TZD use at each time point (Table 2). At baseline, there was no difference in the prevalence of low HDL-C between the assigned lipid treatment groups (2.1% for those subsequently assigned to fenofibrate vs. 2.4% assigned to placebo) or between those receiving (1.5%) or not receiving a TZD (2.4%). However, at both months 24 and 48, the prevalence of a low HDL-C was generally twice as great for participants randomized to fenofibrate versus placebo (5.1 vs. 2.5% and 5.1 vs. 2.3% at months 24 and 48 postrandomization, respectively), but these higher proportions were a result of the increases in low HDL-C in the groups receiving fenofibrate and TZD (e.g., 7.2% for both medications vs. 2.2% for only fenofibrate at month 24, 7.0% for both medications vs. 3.6% for only fenofibrate at month 48). Indeed, the prevalence of HDL-C <25 mg/dL did not increase significantly in the groups not receiving a TZD (Table 2). To assess the sensitivity of the results, all these analyses were repeated with a cutoff of <20 mg/dL, and the trends were found to be the same.
Table 2.
ACCORD Lipid Trial participants who had an HDL-C <25 mg/dL (0.647 mmol/L) at baseline and 24- and 48-month postrandomization visits by treatment group and TZD use at visit-specific blood draws
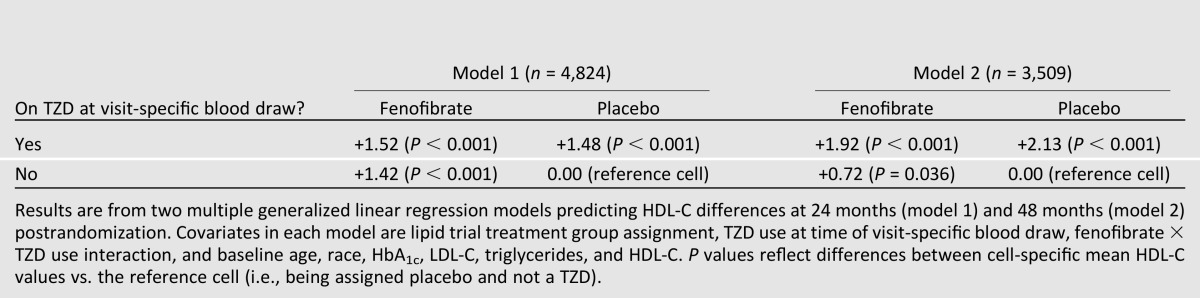
Because low HDL-C levels were associated with fenofibrate and/or TZD use, the overall effects of these medications on HDL-C were examined in two covariate-adjusted linear regression analyses predicting HDL-C levels at months 24 and 48, with the interaction term “lipid treatment assignment X TZD use at time of blood draw” as the primary independent variable of interest (Table 3). Examination of the regression coefficients for month 24 (model 1) indicated that 1) in the absence of a TZD, fenofibrate was associated with a 1.42 mg/dL (0.037 mmol/L) higher HDL-C; 2) in the absence of fenofibrate, TZD use was associated with a 1.48 mg/dL (0.038 mmol/L) higher HDL-C; and 3) the combination fenofibrate/TZD use was associated with a 1.53 mg/dL (0.040 mmol/L) higher HDL-C compared with receiving neither medication. At the 48-month visit, the same general trends existed, although there was a suggestion that TZD use alone was associated with a greater increase (2.13 mg/dL [0.055 mmol/L]) in HDL-C than fenofibrate alone (0.72 mg/dL [0.019 mmol/L]).
Table 3.
Covariate adjusted mean differences in postrandomization HDL-C values among lipid trial treatment and TZD groups relative to being on neither medication
Results of average HDL-C levels at various time points can be misleading when HDL-C values or changes in HDL-C values are widely distributed in a population. To explore this, we compared the distribution of HDL-C values at the 24- and 48-month postrandomization visits among participants receiving placebo/no TZD versus those receiving fenofibrate alone, TZD alone, or both (Fig. 1). At both time points and compared with those receiving placebo/no TZD, there was a shift toward higher HDL-C values in study participants treated with fenofibrate, TZD, or both. However, a greater proportion of participants receiving fenofibrate/TZD had lower HDL-C levels at both follow-up visits than all other participants. For example, at month 24 (as shown in the left-side tails of Fig. 1 and in Table 2), 7.1% of the participants receiving the combination had an HDL-C value ≤25 mg/dL compared with 2.4% of all other participants. In addition to these observations regarding the actual HDL-C values at these follow-up visits, these patterns remained when the absolute changes in HDL-C from baseline to 24 or 48 months were examined in the four medication groups (Supplementary Fig. 1).
Figure 1.

Cumulative percentage of participants at the 24-month (A) and 48-month (B) postrandomization follow-up visits with an HDL-C equal to or less than the value specified on the x-axis by lipid treatment group assignment and use of a TZD at the time of visit-specific blood draw. Eighty-eight percent and 64% of all randomized ACCORD Lipid Trial participants were available for this analysis at month 24 and 48, respectively. Mn, mean group HDL-C value at visit (in milligrams per deciliter). To convert HDL-C values to millimoles per liter, multiply by 0.02586.
To gain further insight into the potential relevance of the observed changes in postrandomization HDL-C levels, we examined in the four medication groups the prevalence of a 30% relative reduction in HDL-C from baseline to the 24- and 48-month follow-up visits as well as the coincident prevalence of HDL-C <25 mg/dL and a 30% reduction (Supplementary Table 1). In both analyses, it was clear that the combination of fenofibrate and TZD was associated with about fivefold increases in the prevalence of these outcomes.
To determine whether iatrogenic reductions in HDL-C might be associated with an increased risk of CVD events, postrandomization analyses examined the effect of fenofibrate and low HDL-C values on all-cause mortality. Consistent with the known inverse relationship between HDL-C and CVD risk, overall with both lipid treatment groups combined, all-cause mortality was 35% higher in participants who ever had a postrandomization HDL-C <25 mg/dL than in those who did not (10.0 vs. 7.4 deaths per 100 participants, respectively). This 35% increase in mortality was true regardless of whether the participants were assigned to the fenofibrate group (9.6 vs. 7.0%) or the placebo group (10.7 vs. 7.8%).
Conclusions
Treatment of patients with diabetes and other chronic diseases often involves complex medical regimens. We tend to evaluate pharmacological interventions by concentrating on the biological effects of a single agent on the disease process and often fail to recognize drug–drug interactions. Fenofibrate would be expected to ameliorate diabetic dyslipidemia by lowering triglycerides and raising HDL-C. Similarly, modest increases in HDL-C have been reported with TZD use (14,15). In this descriptive post hoc analysis of the ACCORD Lipid Trial population as a whole, fenofibrate exhibited the expected effect with an increase in mean HDL-C compared with baseline values. TZD use was also associated with a modest overall increase in HDL-C, as was the combination of the two agents (Table 3). Supplementing the observations of these average responses, however, we also observed a dramatically divergent HDL-C response in the tails of the distribution to combined fenofibrate and TZD treatment. As presented in Fig. 1, a greater proportion of participants receiving the combination treatment at 24 months (18.2%) had an HDL-C of up to 50 mg/dL (1.293 mmol/L) compared with only 13.8% of all other participants, but at the other tail of the distribution, a greater proportion of the same group of participants (7.1%) also had an HDL-C ≤25 mg/dL compared with only 4% of all other participants.
Aside from concomitant TZD treatment, low HDL-C at baseline, a history of coronary heart disease, and the baseline prevalence of lower alcohol intake were associated with low HDL-C levels during ACCORD follow-up (data not shown). Because the approach used to assess the prevalence of low HDL-C in this study favored selection of participants whose baseline HDL-C was low and, therefore, more likely to fall to <25 mg/dL, it is difficult to attribute these factors as contributing to iatrogenic reduction in HDL-C or as predictors of a paradoxical response to fenofibrate. A prior study of 43 patients with fenofibrate-related reduction in HDL-C found only low baseline HDL-C as a predictor of fenofibrate response (16); however, this analysis has a similar bias to that of the present study. Thus, it appears that a paradoxical reduction in HDL-C in a subset of patients treated with concomitant TZD and fenofibrate is truly an idiosyncratic reaction that is difficult to predict.
For simple analytic purposes, we defined extremely low HDL-C as a level <25 mg/dL. Although this analysis clearly conveys the prevalence of extremely low HDL-C among study participants, it is biased toward selection of participants whose HDL-C was lower at baseline and, therefore, more likely to fall below the 25 mg/dL threshold at the follow-up visits. Given the similar baseline HDL-C levels of study participants, it is important to note that the occurrence of excess cases of extremely low HDL-C was seen in study participants randomized to fenofibrate either alone or, in particular, in combination with TZD. In all, the proportion of lipid trial participants who ever had an extremely low HDL-C during ACCORD was more than two times greater among those randomized to fenofibrate than among those randomized to placebo (10.1 vs. 4.9%) (Table 1). Further analysis indicates that the majority of these cases occurred among those concurrently treated with fenofibrate and a TZD (Table 2). These data suggest that iatrogenic lowering of HDL-C could occur with a frequency of up to 5% or 1 in 20 patients with type 2 diabetes treated with combined fenofibrate and TZD therapy. This prevalence was supported by alternative analyses that determined the number of ACCORD participants experiencing a significant (30%) decrease in HDL-C from baseline and those experiencing a coincident 30% decrease in HDL-C and a follow-up HDL-C of <25 mg/dL (Supplementary Table 1). These findings are particularly significant in that the number of cases reported here are greater than all previous case reports combined and allows a determination of the prevalence of this idiosyncratic reaction. Additionally, because this was a randomized controlled trial of fenofibrate therapy, study participants were randomly assigned to fenofibrate treatment regardless of baseline HDL-C levels, thereby reducing the potential for selection bias.
Although the present analyses cannot exclude the possibility that paradoxical lowering of HDL-C also occurred in some patients treated with either fenofibrate or TZD alone, there was no increase in overall prevalence of low HDL-C in groups not receiving TZD compared with baseline (Table 2), suggesting that the risk of such HDL-C lowering was greatest in those receiving both agents. This finding is concordant with the findings of a prior retrospective pharmacoepidemiologic survey of diabetic patients (17). On the other hand, although most case reports of HDL-C lowering involved combined treatment with fibrate and TZDs (5,11–13,16,18), this effect has also been observed with either TZD or fibrate treatment alone (5,16). Although rosiglitazone was the predominant TZD used in the ACCORD Lipid Trial, paradoxical HDL-C lowering has also been reported with other TZDs, including pioglitazone (5). (As noted previously, pioglitazone use in ACCORD was too low to allow us to analyze the effects of each TZD separately.) Similarly, decreased HDL-C has also been reported with other fibrates (ciprofibrate and bezafibrate) with the sole exception being gemfibrozil (5). These case reports have shown that HDL-C returns to baseline levels after discontinuing either fibrate or TZD (11,18). Of note, HDL-C levels increased to >25 mg/dL in 73.1% of these ACCORD study participants after the site investigator received a laboratory alert containing instructions to discontinue TZD and/or fenofibrate.
Although apolipoprotein A-I (apoA1) levels were not assessed in ACCORD, other case reports have noted concomitant reduction in apoAI with reduced HDL-C, indicating decreased HDL-C particle numbers in addition to reduced cholesterol content of HDL-C (12). These findings highlight the biological diversity in response to medical interventions and raise the question of how drug–drug interactions may play a role in clinical responses.
The idiosyncratic occurrence of paradoxical lowering of HDL-C with fibrate and/or TZD suggests that some individuals may be predisposed to this effect, possibly because of polymorphisms of one or more genes related to HDL-C metabolism. Fibrates increase apoAI production in the human through activation of a peroxisome proliferator–activated receptor (PPAR) response element in the apoAI promoter (19). In contrast, apoAI promoters, which lack a functional PPAR response element either because of species variation or experimental manipulation, are negatively regulated by fibrates (20–22). Accordingly, mutation in the apoAI gene or, alternatively, the PPARα nuclear receptor itself may underlie genetic susceptibility to paradoxical HDL-C lowering with fibrate treatment. Alternatively, paradoxical lowering of HDL-C could also result from altered expression of genes related to HDL-C catabolism, for example, the scavenger receptor B1 (SRB1), lecithin:cholesterol acyltransferase (LCAT), or cholesteryl ester transfer protein (CETP) (23). Relevant to this issue is a study that reported a paradoxical, but reproducible decrease of HDL-C in a patient after ciprofibrate treatment that was exclusively a result of an increased catabolism (24).
Although gene polymorphisms involving the apoAI/C3/A4/A5 gene cluster, PPARα, apoE, and lipoprotein lipase have been associated with altered lipoprotein response to fenofibrate (25–27), no gene polymorphism has been specifically linked to paradoxical lowering of HDL-C with fibrate. Of note, although the present findings might implicate the combination of PPARα/γ ligands in paradoxical lowering of HDL-C, this effect has not been described among dual PPARα/γ agonists currently in development (28,29).
Epidemiological studies have long shown an increased risk of cardiac events associated with lower HDL-C. In an observational cohort study of 30,067 patients with type 2 diabetes, Nichols et al. (30) used a categorical analysis centered on baseline HDL-C to show that a >6.5 mg/dL decrease in HDL-C was associated with an 11% increase in CVD risk. Given the vigilance of the ACCORD investigators, the observed fall in HDL-C in select patients was noted early in the study and then carefully analyzed by the data and safety monitoring board at each of its meetings. In addition, the investigators were notified of participants with extremely low HDL-C values and advised to discontinue either TZD or fenofibrate/placebo. When examined in crude, postrandomization post hoc analyses, we observed that all-cause mortality was higher in the group of patients who ever had an HDL-C <25 mg/dL compared with those who did not, which is not entirely unexpected given the higher prevalence of coronary disease and lower baseline HDL-C in this group. However, this finding was true regardless of whether these participants were treated with fenofibrate or placebo. Thus, we were unable to detect an adverse effect of fenofibrate-related reduction in HDL-C. It is important to note that the study was not designed or powered to evaluate the impact of this unexpected response to treatment on mortality. Nevertheless, these observations should alert practitioners to the potential for a paradoxical reduction in HDL-C with fenofibrate treatment, especially when used concomitantly with a TZD. Although the mechanism of iatrogenic reduction in HDL-C with combined fenofibrate and TZD treatment is unknown and there is as yet no definitive proof of lack of harm, if the magnitude of the reduction in HDL-C is significant, it may be appropriate to discontinue either fenofibrate or TZD and monitor HDL-C levels to confirm return to baseline levels.
Supplementary Material
Article Information
Funding. The ACCORD Trial was supported by grants (N01-HC-95178, N01-HC-95179, N01-HC-95180, N01-HC-95181, N01-HC-95182, N01-HC-95183, N01-HC-95184, IAA-Y1-HC-9035, and IAA-Y1-HC-1010) from the National Heart, Lung, and Blood Institute; by other components of the National Institutes of Health, including the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute on Aging, and the National Eye Institute; by the Centers for Disease Control and Prevention; and by General Clinical Research Centers.
Views expressed in this report are those of the authors and do not reflect official policy or position of the U.S. Navy, Department of Defense, Department of Veterans Affairs, and the U.S. Government.
Duality of Interest. All authors have completed and submitted to R.P.B. (in February/March 2013) the International Committee of Medical Journal Editors Form for Disclosure of Potential Conflicts of Interest. P.E.L. owns common stock from Pfizer, Novartis, AstraZeneca, and Roche. R.P.B. is a member of a data safety monitoring board for Eli Lilly. P.J.O. received (or may receive) funding from the National Institutes of Health, and he and his institution may receive monies, royalties, and stocks from Diabetes Decision Support for a patent. L.A.L. received consultant fees from AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Merck, Novartis, Novo Nordisk, Sanofi, Servier, Roche, Amgen, Takeda, and GlaxoSmithKline; received (or may receive) payments for grant work for AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Eli Lilly, Merck, Novartis, Novo Nordisk, Sanofi, GlaxoSmithKline, Roche, and Amgen; received payments for lectures for AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Eli Lilly, Merck, Novo Nordisk, Sanofi, and Novartis; and received payment for the development of educational presentations for AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Eli Lilly, Merck, and Novartis. D.W. received consulting fees from Sanofi; received (or will receive) payments for grant work for MannKind, Bristol-Myers Squibb, Keryx, Eisai, InteKrin Therapeutics, Orexigen Therapeutics, Boehringer Ingelheim, Sanofi, Amylin, Novo Nordisk, NephroGenex, Genzyme/Isis, Amarin, Roche, Weight Watchers International, Eli Lilly, and Reata Pharmaceuticals; received payment for lectures for Amarin, GlaxoSmithKline, Bristol-Myers Squibb, AstraZeneca, Takeda, Sanofi, Eli Lilly, Novo Nordisk, Santarus, and Vivus; received payment for developing educational materials for the American Association of Clinical Endocrinologists; and owns stock in Bristol-Myers Squibb and Sanofi. R.W.F. received grant funding from Sanofi, Amylin, and Merck. F.I.-B. received payments for consulting for Novo Nordisk; has grants with the National Institutes of Health and Novo Nordisk; and has stock with Thermalin. H.N.G. received consultant fees from Merck, Pfizer, AstraZeneca, Bristol-Myers Squibb, Novartis, Boehringer Ingelheim, Sanofi, Regeneron, Amgen, and Roche-Genetech and received (or will have received) grant funding from Merck, Genzyme Sanofi, and Regeneron Sanofi. M.B.E. received consultant fees from Abbott and lecture payments from Merck Schering-Plough and Abbott-Solvay. No other potential conflicts of interest relevant to this article were reported.
The following companies provided study medications, equipment, or supplies: Abbott Laboratories, Amylin Pharmaceuticals, AstraZeneca Pharmaceuticals, Bayer HealthCare, Closer Healthcare, GlaxoSmithKline, King Pharmaceuticals, Merck, Novartis Pharmaceuticals, Novo Nordisk, Omron Healthcare, Sanofi U.S., Schering-Plough, and Takeda Pharmaceuticals. No company had review rights on this article.
Author Contributions. P.E.L. wrote the manuscript. L.C.L. researched the data and performed the statistical analysis. R.P.B. researched the data and wrote, reviewed, and edited the manuscript. P.J.O., L.A.L., and F.I.-B. contributed to the discussion and reviewed and edited the manuscript. D.W., R.W.F., J.R.C., D.L.S., V.P., and H.N.G. reviewed and edited the manuscript. M.B.E. wrote, reviewed, and edited the manuscript. R.P.B. is guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Clinical trial reg. no. NCT00000620, clinicaltrials.gov.
This article contains Supplementary Data online at http://care.diabetesjournals.org/lookup/suppl/doi:10.2337/dc13-0790/-/DC1.
References
- 1.Buse JB, Bigger JT, Byington RP, et al. ACCORD Study Group Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial: design and methods. Am J Cardiol 2007;99:21i–33i [DOI] [PubMed] [Google Scholar]
- 2.Sarker A, Semple RK, Dinneen SF, O’Rahilly S, Martin SC. Severe hypo-alpha-lipoproteinemia during treatment with rosiglitazone. Diabetes Care 2004;27:2577–2580 [DOI] [PubMed] [Google Scholar]
- 3.Savage RL, Kiuru A. Thiazolidinediones and lowered HDL cholesterol. Diabetes Care 2005;28:2329–2330 [DOI] [PubMed] [Google Scholar]
- 4.Normén L, Frohlich J, Montaner J, Harris M, Elliott T, Bondy G. Combination therapy with fenofibrate and rosiglitazone paradoxically lowers serum HDL cholesterol. Diabetes Care 2004;27:2241–2242 [DOI] [PubMed] [Google Scholar]
- 5.Mymin D, Dembinski T, Friesen MH. Iatrogenic severe depression of high-density lipoprotein cholesterol. J Clin Pharmacol 2009;49:865–871 [DOI] [PubMed] [Google Scholar]
- 6.Goff DC, Jr., Gerstein HC, Ginsberg HN, et al. ACCORD Study Group Prevention of cardiovascular disease in persons with type 2 diabetes mellitus: current knowledge and rationale for the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007;99:4i–20i [DOI] [PubMed] [Google Scholar]
- 7.Ginsberg HN, Bonds DE, Lovato LC, et al. ACCORD Study Group Evolution of the lipid trial protocol of the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007;99:56i–67i [DOI] [PubMed] [Google Scholar]
- 8.Cushman WC, Grimm RH, Jr., Cutler JA, et al. ACCORD Study Group Rationale and design for the blood pressure intervention of the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007;99:44i–55i [DOI] [PubMed] [Google Scholar]
- 9.Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D, Modification of Diet in Renal Disease Study Group A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Ann Intern Med 1999;130:461–470 [DOI] [PubMed] [Google Scholar]
- 10.Gerstein HC, Miller ME, Byington RP, et al. Action to Control Cardiovascular Risk in Diabetes Study Group Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008;358:2545–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venero CV, Thompson PD, Fernandez AB. Reduced high-density lipoprotein cholesterol in patients receiving rosiglitazone and fenofibrate. Am J Med 2008;121:e3–e4 [DOI] [PubMed] [Google Scholar]
- 12.Shetty C, Balasubramani M, Capps N, Milles J, Ramachandran S. Paradoxical HDL-C reduction during rosiglitazone and fibrate treatment. Diabet Med 2007;24:94–97 [DOI] [PubMed] [Google Scholar]
- 13.Schwing W, Hustak L, Taylor HC. Paradoxical severe decrease in high-density lipoprotein cholesterol due to rosiglitazone-fenofibrate interaction. Endocr Pract 2010;16:382–388 [DOI] [PubMed] [Google Scholar]
- 14.Lebovitz HE. Differentiating members of the thiazolidinedione class: a focus on safety. Diabetes Metab Res Rev 2002;18(Suppl. 2):S23–S29 [DOI] [PubMed] [Google Scholar]
- 15.Goldberg RB, Kendall DM, Deeg MA, et al. GLAI Study Investigators A comparison of lipid and glycemic effects of pioglitazone and rosiglitazone in patients with type 2 diabetes and dyslipidemia. Diabetes Care 2005;28:1547–1554 [DOI] [PubMed] [Google Scholar]
- 16.Magee G, Sharpe PC. Paradoxical decreases in high-density lipoprotein cholesterol with fenofibrate: a quite common phenomenon. J Clin Pathol 2009;62:250–253 [DOI] [PubMed] [Google Scholar]
- 17.Keidar S, Guttmann H, Stam T, Fishman I, Shapira C. High incidence of reduced plasma HDL cholesterol in diabetic patients treated with rosiglitazone and fibrate. Pharmacoepidemiol Drug Saf 2007;16:1192–1194 [DOI] [PubMed] [Google Scholar]
- 18.Gutschi LM, Malcolm JC, Favreau CM, Ooi TC. Paradoxically decreased HDL-cholesterol levels associated with rosiglitazone therapy. Ann Pharmacother 2006;40:1672–1676 [DOI] [PubMed] [Google Scholar]
- 19.Shah A, Rader DJ, Millar JS. The effect of PPAR-alpha agonism on apolipoprotein metabolism in humans. Atherosclerosis 2010;210:35–40 [DOI] [PubMed] [Google Scholar]
- 20.Staels B, Auwerx J. Regulation of apo A-I gene expression by fibrates. Atherosclerosis 1998;137(Suppl.):S19–S23 [DOI] [PubMed] [Google Scholar]
- 21.Vu-Dac N, Chopin-Delannoy S, Gervois P, et al. The nuclear receptors peroxisome proliferator-activated receptor alpha and Rev-erbalpha mediate the species-specific regulation of apolipoprotein A-I expression by fibrates. J Biol Chem 1998;273:25713–25720 [DOI] [PubMed] [Google Scholar]
- 22.Vu-Dac N, Schoonjans K, Laine B, Fruchart JC, Auwerx J, Staels B. Negative regulation of the human apolipoprotein A-I promoter by fibrates can be attenuated by the interaction of the peroxisome proliferator-activated receptor with its response element. J Biol Chem 1994;269:31012–31018 [PubMed] [Google Scholar]
- 23.Krause BR, Auerbach BJ. Reverse cholesterol transport and future pharmacological approaches to the treatment of atherosclerosis. Curr Opin Investig Drugs 2001;2:375–381 [PubMed] [Google Scholar]
- 24.Beghin L, Capps N, Duhal N, Davies J, Staels B, Luc G. Metabolism of apolipoproteins AI and AII in a patient with paradoxical reduction in high-density lipoprotein due to ciprofibrate. Ann Clin Biochem 1999;36:523–525 [DOI] [PubMed] [Google Scholar]
- 25.Foucher C, Rattier S, Flavell DM, et al. DAIS Investigators Response to micronized fenofibrate treatment is associated with the peroxisome-proliferator-activated receptors alpha G/C intron7 polymorphism in subjects with type 2 diabetes. Pharmacogenetics 2004;14:823–829 [DOI] [PubMed] [Google Scholar]
- 26.Brisson D, Ledoux K, Bossé Y, et al. Effect of apolipoprotein E, peroxisome proliferator-activated receptor alpha and lipoprotein lipase gene mutations on the ability of fenofibrate to improve lipid profiles and reach clinical guideline targets among hypertriglyceridemic patients. Pharmacogenetics 2002;12:313–320 [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Ordovas JM, Gao G, et al. Pharmacogenetic association of the APOA1/C3/A4/A5 gene cluster and lipid responses to fenofibrate: the genetics of lipid-lowering drugs and diet network study. Pharmacogenet Genomics 2009;19:161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henry RR, Lincoff AM, Mudaliar S, Rabbia M, Chognot C, Herz M. Effect of the dual peroxisome proliferator-activated receptor-alpha/gamma agonist aleglitazar on risk of cardiovascular disease in patients with type 2 diabetes (SYNCHRONY): a phase II, randomised, dose-ranging study. Lancet 2009;374:126–135 [DOI] [PubMed] [Google Scholar]
- 29.Cavender MA, Lincoff AM. Therapeutic potential of aleglitazar, a new dual PPAR-α/γ agonist: implications for cardiovascular disease in patients with diabetes mellitus. Am J Cardiovasc Drugs 2010;10:209–216 [DOI] [PubMed] [Google Scholar]
- 30.Nichols GA, Vupputuri S, Rosales AG. Change in high-density lipoprotein cholesterol and risk of subsequent hospitalization for coronary artery disease or stroke among patients with type 2 diabetes mellitus. Am J Cardiol 2011;108:1124–1128 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.