

Abstract
Using an integrative approach in which genetic variation, gene expression, and clinical phenotypes are assessed in relevant tissues may help functionally characterize the contribution of genetics to disease susceptibility. We sought to identify genetic variation influencing skeletal muscle gene expression (expression quantitative trait loci [eQTLs]) as well as expression associated with measures of insulin sensitivity. We investigated associations of 3,799,401 genetic variants in expression of >7,000 genes from three cohorts (n = 104). We identified 287 genes with cis-acting eQTLs (false discovery rate [FDR] <5%; P < 1.96 × 10−5) and 49 expression–insulin sensitivity phenotype associations (i.e., fasting insulin, homeostasis model assessment–insulin resistance, and BMI) (FDR <5%; P = 1.34 × 10−4). One of these associations, fasting insulin/phosphofructokinase (PFKM), overlaps with an eQTL. Furthermore, the expression of PFKM, a rate-limiting enzyme in glycolysis, was nominally associated with glucose uptake in skeletal muscle (P = 0.026; n = 42) and overexpressed (Bonferroni-corrected P = 0.03) in skeletal muscle of patients with T2D (n = 102) compared with normoglycemic controls (n = 87). The PFKM eQTL (rs4547172; P = 7.69 × 10−6) was nominally associated with glucose uptake, glucose oxidation rate, intramuscular triglyceride content, and metabolic flexibility (P = 0.016–0.048; n = 178). We explored eQTL results using published data from genome-wide association studies (DIAGRAM and MAGIC), and a proxy for the PFKM eQTL (rs11168327; r2 = 0.75) was nominally associated with T2D (DIAGRAM P = 2.7 × 10−3). Taken together, our analysis highlights PFKM as a potential regulator of skeletal muscle insulin sensitivity.
Introduction
Although genome-wide association studies (GWASs) have identified thousands of single nucleotide polymorphisms (SNPs) associated with traits and diseases, the molecular mechanisms underlying these associations remain largely unknown. Changes in gene expression can affect phenotypic variation; as a consequence, understanding genetic regulation of gene expression through the identification of expression quantitative trait loci (eQTLs) could elucidate the mechanisms underlying genotype-phenotype associations. While cis-eQTL analyses have been carried out in a number of human tissues, their discovery in human skeletal muscle has been limited. A large study of skeletal muscle eQTLs (n = 225) was carried out in healthy Pima Indians to assess the causes and prevalence of bimodal gene expression and suggests that bimodality may be an underlying factor in disease development (1). In a more recent study, cis-eQTL effects were compared between blood and four nonblood tissues (including liver, subcutaneous, and visceral adipose tissue and skeletal muscle), which provided new insights into the mechanisms by which genetic variants mediate tissue-dependent gene expression (2).
Insulin resistance (IR) is a physiological condition where insulin-mediated glucose disposal in skeletal muscle is inhibited (3), and it plays a key role in the pathogenesis of type 2 diabetes (T2D) (4). The molecular mechanisms underlying IR are largely unknown, but mitochondrial dysfunction coupled with metabolic inflexibility has been implicated as a key contributor (reviewed by Szendroedi et al. [5]). One proposed hypothesis for the etiology of IR is a redistribution of lipid stores from adipose to nonadipose tissues (e.g., skeletal muscle, liver, and insulin-producing pancreatic β-cells), resulting in accumulation of intracellular fatty acids. Accumulation of intracellular fatty acids in turn inhibits insulin-stimulated glucose transport by stimulating phosphorylation of serine sites on insulin receptor substrate 1 (6). The aim of this study was to identify skeletal muscle eQTLs, as well as expression associated with measures of insulin sensitivity, to elucidate genetic contributions to skeletal muscle expression and insulin sensitivity.
Research Design and Methods
Cohort Descriptions
Malmö Exercise Intervention
The Malmö Exercise Intervention (MEI) cohort consists of 50 male subjects from southern Sweden, all of whom have European ancestry. Of these 50 subjects, 25 have and 25 do not have a first-degree relative with T2D (7). All participants had normal glucose tolerance and Vo2max of 32.0 ± 5.0 mL/kg/min. All participants also completed a 6-month aerobic training period, aiming at 3 group training sessions per week (∼60 min training/session, supervised by members of the research group). Information from the baseline screening visit before the intervention was used for the work presented in this article.
Malmö Men
The Malmö men (MM) cohort is a subset of 203 nonobese Swedish men from the Malmö Prospective Project (MPP) who were asked to participate in a training intervention (8,9). The MPP was initiated in 1974 as an intervention project to prevent T2D in men born between 1926 and 1935. Upon inclusion in the MPP, all participants in MM had normal glucose tolerance; at the baseline screening for the MM intervention, however, some of them had developed impaired glucose tolerance or T2D. Information from the baseline screening visit before the intervention was used for the work presented in this article.
Multiple Tissue Human Expression Resource
The Multiple Tissue Human Expression Resource (MuTHER) study consists of 856 women of European descent (336 monozygotic and 520 dizygotic twins), recruited from the UK Adult Twin Registry (TwinsUK) (10). MuTHER participants (n = 39) had both muscle tissue expression profiles and genome-wide genotypes available. The age at inclusion ranged from 40 to 87 years, with a median age of 62 years. Metabolic phenotypes were measured at the same time the biopsies were collected. Because of the twin structure of the data, the minimum effective sample size for the MuTHER skeletal muscle samples was calculated and used in the analysis. Since there were 3 monozygotic twins sharing 100% of their genetic material, 11 dizygotic twins sharing 50% of their genetic material, and 11 singletons, we calculated the minimum effective sample size to be 3 + (11 + 5.5) + 11 = 30.5.
Phenotype Selection
Fasting insulin and homeostasis model assessment–insulin resistance (HOMA-IR) were selected as measurements of peripheral insulin sensitivity that were consistently measured in all three studies and where large-scale GWAS meta-analyses have been performed. These measures will capture not only skeletal muscle insulin sensitivity but also hepatic insulin sensitivity. Because of the strong association between BMI and T2D we also included BMI as a third phenotype. From the MuTHER cohort, two nonfasted and/or diabetic individuals were excluded from phenotype association analyses carried out on insulin and HOMA-IR; 12 individuals were similarly excluded from the MM cohort.
cis-eQTL Analysis
A cis-eQTL analysis was performed within each cohort on the 7,006 genes common to all three studies. The analysis was restricted to nondiabetic individuals, leaving for analysis 26, 39, and 39 individuals from the MEI, MM, and MuTHER cohorts, respectively. We investigated associations between expression levels and all SNPs within 1Mb up- or downstream from the transcription start site (TSS) of each of these genes. The Malmö studies (MEI and MM) were analyzed using a linear model adjusting for age as implemented in the R Matrix eQTL package (11). In MuTHER, the analysis was performed using a linear mixed effect model implemented in R by the lmer function in the lme4 package. The model was adjusted for age and experimental batch (fixed effects), as well as family relationship (twin pairing) and zygosity (random effects).
A meta-analysis of the cis-eQTL results from each study was carried out using METAL (12). Because gene expression levels were measured using different platforms in each study, the effect estimates of the cis-eQTLs are not comparable across studies. Thus, the “samplesize” scheme in METAL, which uses P values and direction of effect, weighted by sample size, was used for the meta-analysis. Heterogeneity was assessed using the I2 statistic. We used a false discovery rate (FDR) <5% of the threshold for the cis-eQTL meta-analysis, computed using the QVALUE package in R (13). To calculate the number of independent cis-eQTLs, we looked for the SNP that was most strongly associated with each gene (top SNP per gene) among all significant cis-eQTLs.
Gene Expression–Phenotype Association
Associations between gene expression levels and the selected phenotypes were investigated separately within each study. All participants who were diabetic were excluded from the analysis, and values within each study were inverse normal transformed (Blom) before analysis. A secondary analysis adjusting for BMI also was performed to test for associations independent of the effect of overall obesity. The same models used in the cis-eQTL analyses were used to test for these associations.
To be able to perform fixed and random effect meta-analyses on expression-phenotype associations, gene expression levels were inverse normal transformed before analysis. Meta-analyses were performed using METAL for all phenotypes, and a combined FDR <5% of the threshold was applied. We then looked for significant cis-eQTLs, where the gene was significantly associated with a phenotype to investigate evidence for genetic control of phenotypic variation through gene expression. Further information on quality control of genotyping, imputation, and gene expression can be found in the Supplemental Material.
Results
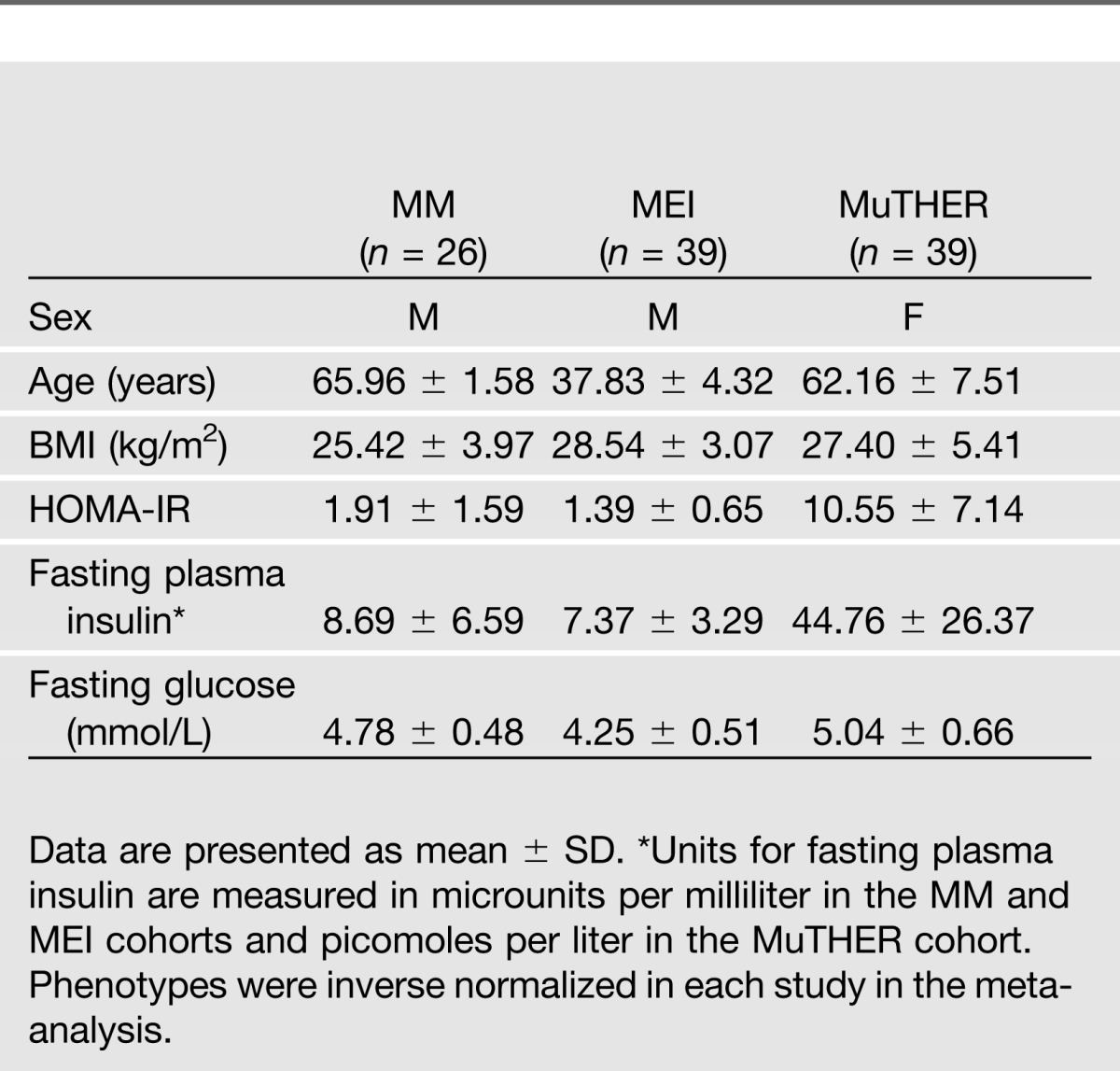
We combined genome-wide SNP, gene expression, and phenotype data from three independent cohorts of European origin: the MM study (n = 26) (8), the MEI study (n = 39) (7), and the MuTHER consortium (n = 39) (14). The methods used here are outlined in Supplementary Fig. 1, and the sample size and characteristics for each study are shown in Table 1. For the gene expression analysis we used a gene-centric approach by selecting in each study only probes that mapped (using National Center for Biotechnology Information build 37) to genes common to all three cohorts. This limited our analysis to the overlap of genes present on all the different arrays used (7,006 genes) that were analyzed for association with 3,799,401 genetic variants. Furthermore, we used only uniquely mapping probes with no mismatches and without common SNPs (minor allele frequency >5%) to limit false-positive findings in the analysis.
Table 1.
Characteristics of the 104 individuals (effective n = 95.5) separated by study cohort
eQTLs (n = 287) Identified in Human Skeletal Muscle
First, we investigated the associations between genetic variation (SNPs) and gene expression within an arbitrary ±1 Mb window around every gene. The summary statistics for skeletal muscle eQTLs from the three cohorts (effective n = 95.5; see Research Design and Methods for this calculation) were meta-analyzed. We identify 287 genes with at least one significant eQTL (top eQTL SNP per gene; FDR <5%; P < 1.96 × 10−5) (Supplementary Table 1). The most significant eQTL was observed for μ-crystallin (CRYM), a NADPH-regulated thyroid hormone–binding protein previously reported to be regulated by blood glucose concentrations (15) (Fig. 1A). Among the other significant eQTLs were signal transducer and activator of transcription 3 (STAT3), endoplasmic reticulum aminopeptidase 2 (ERAP2), and muscle phosphofructokinase (PFKM) (Fig. 1B and C).
Figure 1.

LocusZoom plots of selected eQTL regional association results for the three genes CRYM (A), ERAP2 (B), and PFKM (C).
To describe the distribution of eQTL P values in relation to gene proximity, we plotted the distribution of distances between the SNP with the lowest P value and the TSS for each gene for the combined analysis (Fig. 2). We found an enrichment of low P values closer to TSSs (approximately ±250 kb), showing that cis-eQTLs are more likely to be found within this distance (P = 9.71 × 10−6, binomial test). Moreover, within a 250-kb distance, there is a higher likelihood that it is the nearest gene that is influenced in cis by a SNP (P = 2.93 × 10−6, binomial test).
Figure 2.

Distance distribution from each gene’s most strongly associated SNP to its TSS.
To assess the potential functional significance of the eQTLs, genes were annotated for the top eQTL SNP per gene and compared with all SNPs tested in the dataset. As shown in Fig. 3, most of the eQTL SNPs are located in intronic regions (62.4%); interestingly, we observed an enrichment for noncoding regions upstream/downstream and in untranslated regions but a depletion for intergenic and coding regions (test of equal proportions; Bonferroni corrected P = 0.0041). To quantify further the potential regulatory significance of the noncoding eQTL SNPs, RegulomeDB, a database derived from the ENCODE project, was used to annotate the variants (16). By using scores of functional regional significance, we found that eQTL SNPs were enriched in the score category “likely to affect transcription factor binding and linked to expression of a gene target” (test of equal proportions; Bonferroni corrected P = 0.013). A gene set enrichment analysis of the eQTL genes (FDR <1%) performed in DAVID (17) showed nominal enrichment in gene ontologies related to posttranscriptional regulation of gene expression (P = 0.009, Fisher exact test).
Figure 3.

Functional annotation of eQTL SNPs. The bars indicate the percentages of eQTL SNPs per gene functional unit and their enrichment or depletion relative to all SNPs tested in this study. The SNPs were annotated with snpEff. Downstream and upstream regions are defined as being within a 5-kb distance from a gene.
Further support for the identified muscle eQTLs was obtained by probing databases containing published eQTLs. In particular, the Phenotype-Genotype Integrator (https://www.ncbi.nlm.nih.gov/gap/PheGenI), Pritchard’s laboratory eQTL browser (http://eqtl.uchicago.edu/cgi-bin/gbrowse/eqtl/), and Genevar (18) were searched and a skeletal muscle eQTL study performed in Pima Indians (1) was reviewed. We found that 19% of our eQTLs were detected in at least one of these resources (Supplementary Table 1). The observed discrepancy might be explained in part by the use of different genotyping and expression platforms, the known high degree of tissue specificity of eQTLs (2), and the fact that only one of those databases contains eQTL data from skeletal muscle.
Associations of Significant eQTL SNPs in GWAS Data From MAGIC and DIAGRAM
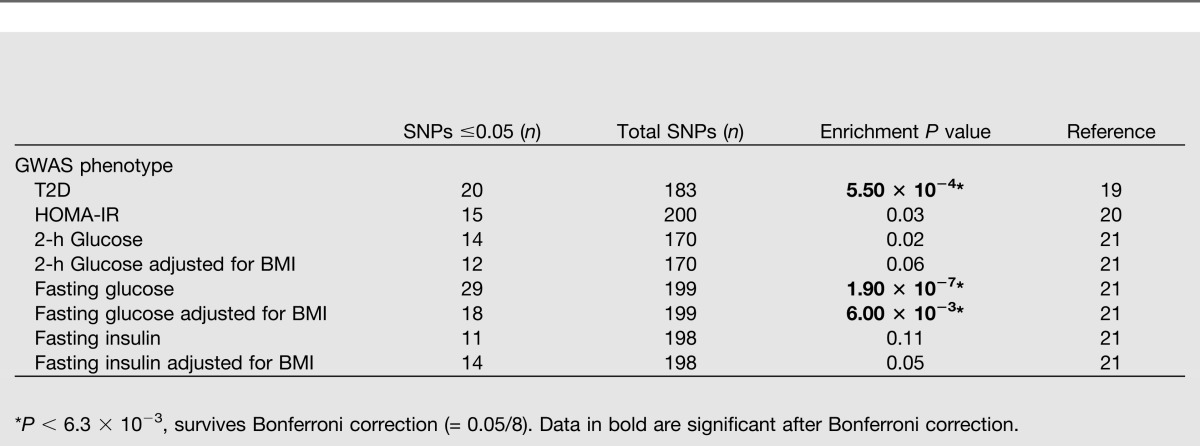
To explore the results of the eQTL analysis further we looked up our 287 significant (FDR <5%; P < 1.96 × 10−5) eQTLs in GWAS data from the DIAGRAM consortium for T2D (19) and the MAGIC (Meta-Analyses of Glucose- and Insulin-related Traits Consortium) for HOMA-IR (20), 2-h glucose, 2-h glucose adjusted for BMI, fasting insulin, fasting insulin adjusted for BMI, fasting glucose (FG), and FG adjusted for BMI (21). To investigate whether eQTLs are overrepresented in associations found in the published DIAGRAM and MAGIC GWAS results, we calculated the enrichment of nominally significant (P ≤ 0.05) GWAS associations among our significant eQTLs using a binomial test (Table 2). The eQTL SNPs were enriched (uncorrectedP ≤ 0.05) in GWAS signals for six of the eight investigated phenotypes, that is, T2D, HOMA-IR, fasting insulin adjusted for BMI, FG, FG adjusted for BMI, and 2-h glucose. Three phenotypes persisted after Bonferroni correction. One of the eQTL SNPs (rs1019503, significantly associated with the expression level of ERAP2; P = 7.17 × 10−10) is also significantly associated with 2-h glucose (P = 8.87 × 10−9) and 2-h glucose adjusted for BMI (P = 5.10 × 10−9) in the MAGIC consortium dataset. Another eQTL (rs4547172, associated with PFKM; P = 7.69 × 10−6) has a proxy (rs11168327; r2 = 0.75), which was nominally associated with T2D in DIAGRAM (P = 2.70 × 10−3).
Table 2.
Binomial test for enrichment (exact match) of GWAS signals for T2D, HOMA-IR, 2-h glucose, 2-h glucose adjusted for BMI, fasting glucose, fasting glucose adjusted for BMI, fasting insulin, and fasting insulin adjusted for BMI among significant (FDR <5%) cis-eQTL SNPs
Skeletal Muscle Expression–Phenotype Associations (n = 49) Identified
We meta-analyzed the association test statistics for gene expression (standardized units) with fasting plasma insulin, HOMA-IR, and BMI in each study. Significant associations (FDR <5%; P < 1.34 × 10−4) are presented in Supplementary Table 2. The expression of 18 and 11 genes were associated with fasting plasma insulin and HOMA-IR, respectively, and the expression levels of 8 of these genes were associated with both traits (Supplementary Table 2). The expression levels of 20 genes were associated with BMI, and none overlapped with the fasting plasma insulin– or HOMA-IR–associated genes. Association of expression levels with fasting insulin and HOMA-IR adjusted for BMI also were investigated to control for BMI (Supplementary Table 3) and putative confounding factors correlated with BMI (e.g., diet and physical activity level). With adjustment for BMI, the expression levels of eight and three genes were associated with fasting plasma insulin and HOMA-IR, respectively. Several of the genes for which expression levels were associated with measures of insulin sensitivity have previously been implicated in T2D and related traits, for example, calsequestrin 1 (CASQ1) (22), solute carrier family 30, member 10 (SLC30A10) (23), and growth arrest-specific 6 (GAS6) (24).
To identify genetic variations influencing clinical phenotypes through gene expression, we integrated significant results from the eQTL analysis (FDR <5%; P < 1.96 × 10−5) and gene expression-phenotype associations (FDR <5%; P < 1.34 × 10−4), highlighting PFKM as the only gene associated in both these analyses. Expression of PFKM (rs4547172 is eQTL lead SNP; P = 7.69 × 10−6; Fig. 1C) was associated with fasting plasma insulin levels (P = 1.34 × 10−4) in our cohorts, suggesting that genetic variation at this locus could be involved in the interplay between fasting plasma insulin levels and PFKM expression.
Extended Phenotype Association of PFKM in the MM Study
To extend the analysis of insulin sensitivity, we tested both the PFKM eQTL SNP (rs4547172) and PFKM expression for association with M-value (i.e., glucose uptake) using the euglycemic-hyperinsulinemic clamp technique (the gold standard for characterizing insulin sensitivity in vivo) (25) in the MM study. Both the eQTL SNP (rs4547172; β = 0.15 [0.03, 0.27] [square root {mg ∙ min−1 ∙ kg−1}]; P = 0.016; n = 178) and the transcription levels of PFKM (β = −0.000295 [−0.001, −0.000037] [square root {mg ∙ min−1 ∙ kg−1}]; P = 0.026; n = 42) were nominally associated with M-value.
Given that PFKM catalyzes a rate-limiting step in the glycolytic pathway, we investigated whether the eQTL SNP (rs4547172) was associated with measures of oxidative fuel partitioning, that is, metabolic flexibility measured as the δ respiratory quotient (RQ) (26). We examined the difference in RQ during the euglycemic-hyperinsulinemic clamp between the noninsulin-stimulated (basal) and the insulin-stimulated (clamp) states. The rs4547172 SNP was nominally associated with delta RQ (β = 0.011 [0.001, 0.02] [AU]; P = 0.030; n = 173). Also, the insulin-stimulated glucose oxidation rate in the insulin-stimulated state was nominally associated with the same SNP (β = 0.19 [0.025, 0.36] [mg ∙ body weight−1 ∙ min−1], per allele; P = 0.025; n = 178). We also investigated whether rs4547172 was associated with skeletal muscle energy stores, that is, intramuscular triglycerides and glycogen. The rs4547172 SNP was nominally associated with intramuscular triglycerides (β = 8.69 [0.26, 17.12] [AU]; P = 0.043; n = 167) but not with glycogen (P = 0.213). These results and descriptive data are summarized in Supplementary Tables 4 and 5.
Replication of the Expression-Phenotype Associations Using Publicly Available Data
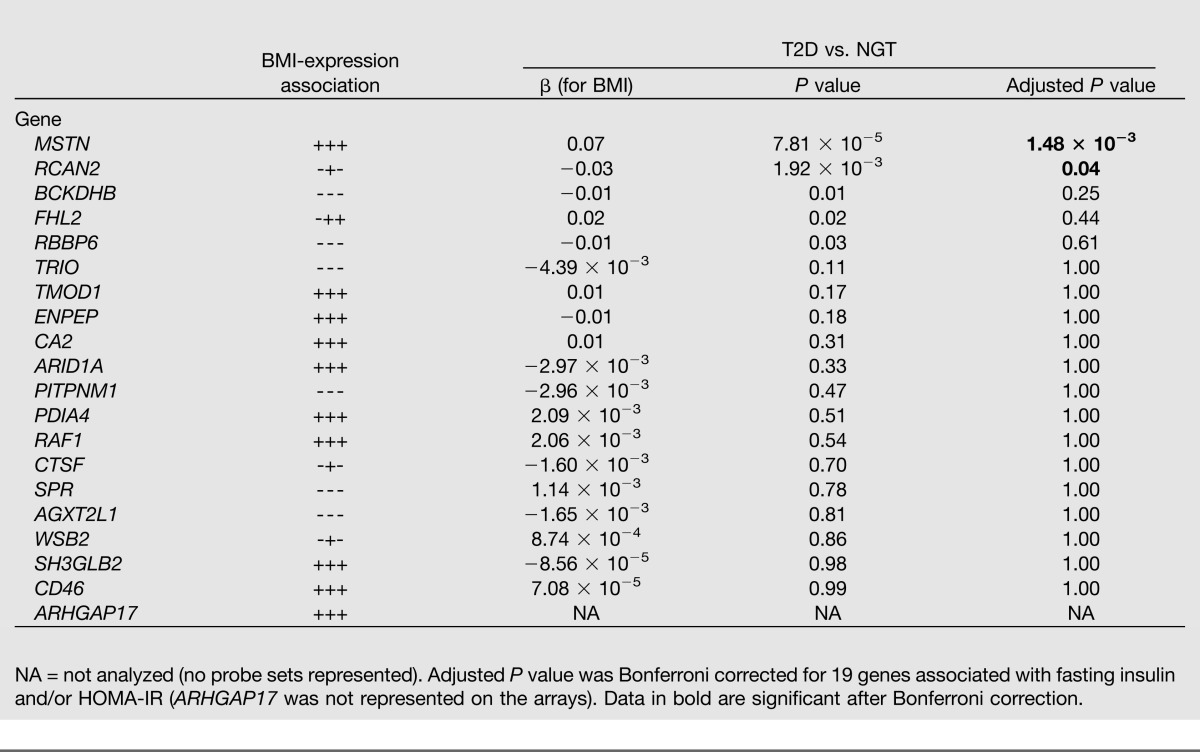
The 29 expression-phenotype associations (corresponding to 21 genes) (Supplementary Table 2) with fasting plasma insulin levels and/or HOMA-IR were investigated for differential expression between patients with T2D (n = 102) and normoglycemic-insulin sensitive controls (n = 87). This was done using publicly available microarray skeletal muscle expression data from three independent studies (27–29), which we retrieved from ArrayExpress (http://www.ebi.ac.uk/arrayexpress) and subsequently meta-analyzed. We found four genes (CASQ1, DBNDD1, DHRS7, and PFKM) that were associated with increased expression in muscle from patients with T2D versus normoglycemic-insulin sensitive controls after Bonferroni correction (Table 3). This supports our initial expression-phenotype associations (Supplementary Table 2). Furthermore, raw data on skeletal muscle expression from seven independent studies (30–36) with available BMI data (n = 185) were used to replicate our 20 expression-BMI associations (Supplementary Table 2). The expression of two genes—MSTN and RCAN2—was positively and negatively associated with BMI, respectively, after Bonferroni correction (Table 4).
Table 3.
Differential expression of 21 genes with association to at least one of the insulin sensitivity–related phenotypes (fasting insulin and/or HOMA-IR) in skeletal muscle of patients with T2D and normoglycemic/insulin-sensitive individuals (NGT)
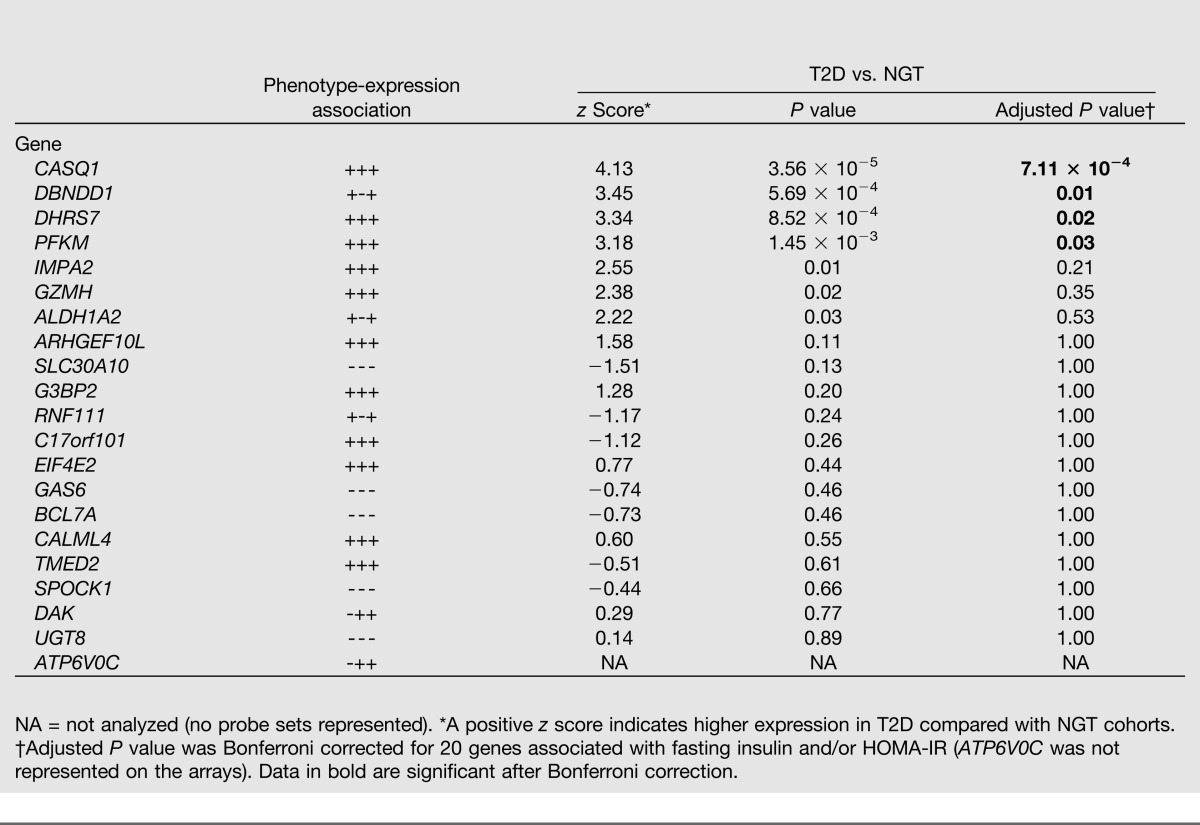
Table 4.
Replication of the skeletal muscle expression and BMI associations using publicly available data
Discussion
Here we have integrated genetic variation, skeletal muscle gene expression, and clinical phenotype data from 104 individuals to investigate the genetic contribution to gene expression in skeletal muscle and insulin sensitivity. We identified 287 muscle eQTLs and 49 associations between gene expression and measures related to insulin sensitivity.
We found a number of cis-eQTLs in our analysis equivalent to those found in previous studies (1,2). Our eQTL data suggest that significant associations between genetic variants and gene expression are more likely to be found within a 250-kb region of a gene. Previous studies have shown similar enrichment of significant eQTLs in this region (2,37,38). Proximal genes within this region are also more likely to be influenced by a polymorphism in cis than genes located farther away. Furthermore, when categorizing the known function of the genes with eQTLs, we found an enrichment of genes annotated as being involved in posttranslational regulation of gene expression. This suggests that the eQTL SNPs are not only affecting the expression of the nearest gene but also may regulate other genes and thereby form transregulatory cascades.
When interpreting GWAS data, it is often difficult to determine which genes/pathways are influenced by genetic variation. Using eQTL data as an intermediate phenotype is one possible way to address this problem. To this end, we investigated our significant muscle eQTL SNPs in GWAS data from MAGIC, thereby identifying an eQTL SNP for ERAP2 that is significantly associated with 2-h glucose both adjusted and unadjusted for BMI. ERAP2 is an endoplasmic reticulum aminopeptidase that functions as an antigen-trimming peptide and has been implicated as a regulator of blood pressure and angiogenesis (39). The rs1019503 SNP also influences the expression of ERAP2 in other tissues, such as human pancreatic islets (unpublished data), lymphoblastoid cell lines, primary fibroblasts, T-cells, and skin and adipose tissue (14,40–42). Our results suggest that differential expression of ERAP2 in skeletal muscle may be part of the molecular mechanism underlying this genome-wide significant association with 2-h glucose levels, but at this point we cannot exclude effects in other tissues. Nevertheless, the findings related to ERAP2 exemplify the efficacy of analyzing gene expression in disease-relevant tissues to disentangle the molecular mechanisms underlying GWAS results.
Several of the genes we found to be associated with measures of insulin sensitivity have previously been implicated in T2D and related traits; for example, CASQ1, a skeletal muscle protein expressed in the sarcoplasmic reticulum, is important for the regulation of calcium channel activity. We found CASQ1 expression to both be positively associated with fasting plasma insulin and have higher expression in individuals with T2D compared with normoglycemic individuals. SLC30A10 expression was negatively associated with both fasting insulin and HOMA-IR. SLC30A10 is a zinc transporter, and a SNP (rs4846567) proximal to this gene has previously been associated with waist-to-hip ratio (43) and measures of insulin sensitivity, for example, fasting plasma insulin, HOMA-IR, the Insulin Sensitivity Index, and the Matsuda index (23). This SNP (rs4846567) was not, however, identified as a skeletal muscle eQTL for SLC30A10 in this study. Three genes (GAS6, ALDH1A2, and CALML4) were found to be associated with fasting insulin with or without correcting for BMI. The relatively large influence of BMI on the associations between expression and measures of insulin sensitivity suggests that, for many genes, either BMI directly affects the expression of these genes and consequently affects skeletal muscle insulin sensitivity or that other BMI-correlated factors such as diet and physical activity level confound associations between expression levels and clinical phenotypes.
By cross-referencing the significant eQTL results with data from the gene expression–phenotype analysis, we found that SNP rs4547172 regulates the expression of PFKM, a gene whose expression also was positively associated with fasting plasma insulin. Since PFKM encodes for phosphofructokinase 1 (PFK1), the muscle isoform of phosphofructokinase, and is a key regulator of glycolysis (44), this gene is a strong candidate for skeletal muscle gene expression associated with glycemic traits. Mutations in PFKM have been shown to cause glycogen storage disease VII (Tarui disease), an autosomal-recessive metabolic disorder characterized clinically by exercise intolerance, muscle cramping, myopathy, and compensated hemolysis. To determine in more detail the role of PFKM in the regulation of insulin sensitivity, we extended these findings to additional phenotypes related to insulin sensitivity and skeletal muscle metabolism, for example, with data such as glucose uptake (M-value) from a euglycemic-hyperinsulinemic clamp setting (25). Both the A-allele (displaying increased expression of PFKM) and increased expression of PFKM were associated with reduced glucose uptake. Because PFK1 regulates a key step in glycolysis by fueling the mitochondria with carbohydrates, we examined the influence on metabolic flexibility and found that the A-allele was associated with reduced flexibility, possibly by promoting an elevation of the glucose oxidation rate in the fasted state. One of the first observations that instigated the concept of metabolic flexibility was the observation that administration of fat emulsions during an oral glucose tolerance test leads to increased glucose intolerance (45). Since then, metabolic flexibility has been defined as the ability to switch from high rates of fatty acid uptake and lipid oxidation to suppression of lipid metabolism with a parallel increase in glucose uptake, storage, and oxidation, for example, in response to feeding or high-intensity exercise (46). Reduced metabolic flexibility has been described in both patients with manifest T2D as well as prediabetic individuals (47).
The finding that increased expression of PFKM is associated with T2D and IR is somewhat paradoxical given that the encoded protein, PFK1, is rate-limiting to glycolysis. We can only speculate about the reasons for this increased expression, which we consider secondary to, rather than a cause of, IR. First, even in an insulin-resistant state glycolysis might be sensitive enough to the small amount of insulin required to maintain a normal flux; the median effective dose for stimulation of glycolysis/glucose oxidation is half that of what is required to stimulate glycogen synthesis (48). Therefore, under insulin-resistant conditions it is possible that glucose entering the cell can still be shunted through glycolysis to ensure sufficient production of energy in the citric acid cycle. Second, if the insulin-resistant state is associated with, or caused by, excess oxidation of free fatty acids, there will be a feedback inhibition of glycolysis, including PFK1, by accumulated citrate and acetyl-CoA. It is possible that the amount of PFKM transcript is increased in an attempt to overcome the allosteric inhibition of PFK1. It should be acknowledged, however, that this study was not designed to address these issues, which will require flux measurements using isotopes as well as measurement of enzyme activities. However, increased PFKM activity has been associated with increased deposition of muscle fat as measured by computed tomography (49), and increased glycolytic capacity in T2D has been suggested (49–54). These observations could possibly be attributed to fiber type composition with a shift toward increased glycolytic type IIx fibers in T2D (53). Our hypothesis that PFKM is involved in T2D pathogenesis is strengthened by the observation that PFKM is overexpressed in diabetic muscle compared with muscle from nondiabetic, normoglycemic individuals, and a proxy SNP (r2 = 0.75) for rs4547172 has a nominally significant association with T2D (none of the top eQTL SNPs for PFKM were represented in DIAGRAM).
In conclusion, the identified association of PFKM with skeletal muscle insulin sensitivity demonstrates how an integrative approach combining different levels of genomic data with clinical phenotype data from disease-relevant tissue may help to functionally characterize the genetic contribution to disease susceptibility.
Supplementary Material
Article Information
Acknowledgments. The authors thank the excellent technical assistance of Maria Sterner, Gabriella Gremsperger, Esa Laurila, and Tina Rönn at Lund University.
Funding. The MuTHER study was supported by a program grant from the Wellcome Trust (081917/Z/07/Z) and by core funding for the Wellcome Trust Centre for Human Genetics (090532). Additional support came from a Linnaeus grant from the Swedish Research Council to Lund University Diabetes Centre (Dnr 349-2006-237); a European Research Council (ERC) Advanced Researcher grant (GA 269045); the Wallenberg Foundation; the Påhlsson Foundation; the European Community’s Seventh Framework Programme (FP7/2007-2013); the ENGAGE project and grant agreement (HEALTH-F4-2007-2014139); the Swiss National Science Foundation and the NCCR Frontiers in Genetics; the Louis-Jeantet Foundation; and a U.S. National Institutes of Health–National Institute of Mental Health grant (GTEx project). C.M.L. is a Wellcome Trust Research Career Development Fellow (086596/Z/08/Z).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. S.K. and J.F. wrote the manuscript and performed analysis. C.L. and H.-F.Z. imputed data. Å.K.H., T.E., K.S.S., E.G., A.C.N., D.G., A.B., J.N., T.R., K.S., and K.-F.E. performed phenotyping/genotyping. J.B.R. imputed data and performed phenotyping/genotyping. I.P. analyzed data. T.D.S., E.T.D., P.D., M.I.M., and P.W.F. designed the study. J.R. designed the study and analyzed data. L.G. and C.M.L. designed the study, wrote the manuscript, and supervised the study. O.H. designed the study, wrote the manuscript, analyzed data, and supervised the study. O.H. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-1301/-/DC1.
References
- 1.Mason CC, Hanson RL, Ossowski V, et al. Bimodal distribution of RNA expression levels in human skeletal muscle tissue. BMC Genomics 2011;12:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fu J, Wolfs MG, Deelen P, et al. Unraveling the regulatory mechanisms underlying tissue-dependent genetic variation of gene expression. PLoS Genet 2012;8:e1002431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abel ED, Peroni O, Kim JK, et al. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 2001;409:729–733 [DOI] [PubMed] [Google Scholar]
- 4.Savage DB, Petersen KF, Shulman GI. Mechanisms of insulin resistance in humans and possible links with inflammation. Hypertension 2005;45:828–833 [DOI] [PubMed] [Google Scholar]
- 5.Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 2012;8:92–103 [DOI] [PubMed] [Google Scholar]
- 6.Dresner A, Laurent D, Marcucci M, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest 1999;103:253–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elgzyri T, Parikh H, Zhou Y, et al. First-degree relatives of type 2 diabetic patients have reduced expression of genes involved in fatty acid metabolism in skeletal muscle. J Clin Endocrinol Metab 2012;97:E1332–E1337 [DOI] [PubMed] [Google Scholar]
- 8.Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34:267–273 [DOI] [PubMed] [Google Scholar]
- 9.Tripathy D, Eriksson KF, Orho-Melander M, Fredriksson J, Ahlqvist G, Groop L. Parallel manifestation of insulin resistance and beta cell decompensation is compatible with a common defect in Type 2 diabetes. Diabetologia 2004;47:782–793 [DOI] [PubMed] [Google Scholar]
- 10.Moayyeri A, Hammond CJ, Hart DJ, Spector TD. The UK Adult Twin Registry (TwinsUK Resource). Twin Res Hum Genet 2013;16:144–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 2012;28:1353–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010;26:2190–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 2003;100:9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nica AC, Parts L, Glass D, et al. MuTHER Consortium The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet 2011;7:e1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al-Kafaji G, Malik AN. Hyperglycemia induces elevated expression of thyroid hormone binding protein in vivo in kidney and heart and in vitro in mesangial cells. Biochem Biophys Res Commun 2010;391:1585–1591 [DOI] [PubMed] [Google Scholar]
- 16.Boyle AP, Hong EL, Hariharan M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 2012;22:1790–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57 [DOI] [PubMed] [Google Scholar]
- 18.Yang TP, Beazley C, Montgomery SB, et al. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics 2010;26:2474–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris AP, Voight BF, Teslovich TM, et al. Wellcome Trust Case Control Consortium. Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) Investigators. Genetic Investigation of ANthropometric Traits (GIANT) Consortium. Asian Genetic Epidemiology Network–Type 2 Diabetes (AGEN-T2D) Consortium. South Asian Type 2 Diabetes (SAT2D) Consortium. DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 2012;44:981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dupuis J, Langenberg C, Prokopenko I, et al. DIAGRAM Consortium. GIANT Consortium. Global BPgen Consortium. Anders Hamsten on behalf of Procardis Consortium. MAGIC investigators New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 2010;42:105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott RA, Lagou V, Welch RP, et al. DIAbetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet 2012;44:991–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das SK, Chu W, Zhang Z, Hasstedt SJ, Elbein SC. Calsquestrin 1 (CASQ1) gene polymorphisms under chromosome 1q21 linkage peak are associated with type 2 diabetes in Northern European Caucasians. Diabetes 2004;53:3300–3306 [DOI] [PubMed] [Google Scholar]
- 23.Burgdorf KS, Gjesing AP, Grarup N, et al. Association studies of novel obesity-related gene variants with quantitative metabolic phenotypes in a population-based sample of 6,039 Danish individuals. Diabetologia 2012;55:105–113 [DOI] [PubMed] [Google Scholar]
- 24.Lee CH, Chu NF, Shieh YS, Hung YJ. The growth arrest-specific 6 (Gas6) gene polymorphism c.834+7G>A is associated with type 2 diabetes. Diabetes Res Clin Pract 2012;95:201–206 [DOI] [PubMed] [Google Scholar]
- 25.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 1979;237:E214–E223 [DOI] [PubMed] [Google Scholar]
- 26.Galgani JE, Moro C, Ravussin E. Metabolic flexibility and insulin resistance. Am J Physiol Endocrinol Metab 2008;295:E1009–E1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gallagher IJ, Scheele C, Keller P, et al. Integration of microRNA changes in vivo identifies novel molecular features of muscle insulin resistance in type 2 diabetes. Genome Med 2010;2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin W, Goldfine AB, Boes T, et al. Increased SRF transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. J Clin Invest 2011;121:918–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Tienen FH, Praet SF, de Feyter HM, et al. Physical activity is the key determinant of skeletal muscle mitochondrial function in type 2 diabetes. J Clin Endocrinol Metab 2012;97:3261–3269 [DOI] [PubMed] [Google Scholar]
- 30.Gordon PM, Liu D, Sartor MA, et al. Resistance exercise training influences skeletal muscle immune activation: a microarray analysis. J Appl Physiol (1985) 2012;112:443–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palsgaard J, Brøns C, Friedrichsen M, et al. Gene expression in skeletal muscle biopsies from people with type 2 diabetes and relatives: differential regulation of insulin signaling pathways. PLoS One 2009;4:e6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park JJ, Berggren JR, Hulver MW, Houmard JA, Hoffman EP. GRB14, GPD1, and GDF8 as potential network collaborators in weight loss-induced improvements in insulin action in human skeletal muscle. Physiol Genomics 2006;27:114–121 [DOI] [PubMed] [Google Scholar]
- 33.Pihlajamäki J, Lerin C, Itkonen P, et al. Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metab 2011;14:208–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skov V, Glintborg D, Knudsen S, et al. Reduced expression of nuclear-encoded genes involved in mitochondrial oxidative metabolism in skeletal muscle of insulin-resistant women with polycystic ovary syndrome. Diabetes 2007;56:2349–2355 [DOI] [PubMed] [Google Scholar]
- 35.Skov V, Glintborg D, Knudsen S, et al. Pioglitazone enhances mitochondrial biogenesis and ribosomal protein biosynthesis in skeletal muscle in polycystic ovary syndrome. PLoS ONE 2008;3:e2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stephens NA, Gallagher IJ, Rooyackers O, et al. Using transcriptomics to identify and validate novel biomarkers of human skeletal muscle cancer cachexia. Genome Med 2010;2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Innocenti F, Cooper GM, Stanaway IB, et al. Identification, replication, and functional fine-mapping of expression quantitative trait loci in primary human liver tissue. PLoS Genet 2011;7:e1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veyrieras JB, Kudaravalli S, Kim SY, et al. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet 2008;4:e1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cifaldi L, Romania P, Lorenzi S, Locatelli F, Fruci D. Role of endoplasmic reticulum aminopeptidases in health and disease: from infection to cancer. Int J Mol Sci 2012;13:8338–8352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dimas AS, Deutsch S, Stranger BE, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science 2009;325:1246–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grundberg E, Small KS, Hedman AK, et al. Multiple Tissue Human Expression Resource (MuTHER) Consortium Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet 2012;44:1084–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Montgomery SB, Dermitzakis ET. From expression QTLs to personalized transcriptomics. Nat Rev Genet 2011;12:277–282 [DOI] [PubMed] [Google Scholar]
- 43.Heid IM, Jackson AU, Randall JC, et al. MAGIC Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet 2010;42:949–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakajima H, Noguchi T, Yamasaki T, Kono N, Tanaka T, Tarui S. Cloning of human muscle phosphofructokinase cDNA. FEBS Lett 1987;223:113–116 [DOI] [PubMed] [Google Scholar]
- 45.Felber JP, Vannotti A. Effects of fat infusion on glucose tolerance and insulin plasma levels. Med Exp Int J Exp Med 1964;10:153–156 [DOI] [PubMed] [Google Scholar]
- 46.Kelley DE, Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes 2000;49:677–683 [DOI] [PubMed] [Google Scholar]
- 47.Russell RD, Kraemer RR, Nelson AG. Metabolic dysfunction in diabetic offspring: deviations in metabolic flexibility. Med Sci Sports Exerc 2013;45:8–15 [DOI] [PubMed] [Google Scholar]
- 48.Groop LC, Saloranta C, Shank M, Bonadonna RC, Ferrannini E, DeFronzo RA. The role of free fatty acid metabolism in the pathogenesis of insulin resistance in obesity and noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 1991;72:96–107 [DOI] [PubMed] [Google Scholar]
- 49.Simoneau JA, Colberg SR, Thaete FL, Kelley DE. Skeletal muscle glycolytic and oxidative enzyme capacities are determinants of insulin sensitivity and muscle composition in obese women. FASEB J 1995;9:273–278 [PubMed] [Google Scholar]
- 50.Bass A, Vondra K, Rath R, Vítek V, Havránek T. Metabolic changes in the quadriceps femoris muscle of obese people. Enzyme activity patterns of energy-supplying metabolism. Pflugers Arch 1975;359:325–334 [DOI] [PubMed] [Google Scholar]
- 51.Simoneau JA, Kelley DE. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. J Appl Physiol (1985) 1997;83:166–171 [DOI] [PubMed] [Google Scholar]
- 52.Lithell H, Lindgärde F, Hellsing K, et al. Body weight, skeletal muscle morphology, and enzyme activities in relation to fasting serum insulin concentration and glucose tolerance in 48-year-old men. Diabetes 1981;30:19–25 [DOI] [PubMed] [Google Scholar]
- 53.Oberbach A, Bossenz Y, Lehmann S, et al. Altered fiber distribution and fiber-specific glycolytic and oxidative enzyme activity in skeletal muscle of patients with type 2 diabetes. Diabetes Care 2006;29:895–900 [DOI] [PubMed] [Google Scholar]
- 54.Krotkiewski M, Bylund-Fallenius AC, Holm J, Björntorp P, Grimby G, Mandroukas K. Relationship between muscle morphology and metabolism in obese women: the effects of long-term physical training. Eur J Clin Invest 1983;13:5–12 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.