

Abstract
Type 2 diabetes is characterized by poor glucose uptake in metabolic tissues and manifests when insulin secretion fails to cope with worsening insulin resistance. In addition to its effects on skeletal muscle, liver, and adipose tissue metabolism, it is evident that insulin resistance also affects pancreatic β-cells. To directly examine the alterations that occur in islet morphology as part of an adaptive mechanism to insulin resistance, we evaluated pancreas samples obtained during pancreatoduodenectomy from nondiabetic subjects who were insulin-resistant or insulin-sensitive. We also compared insulin sensitivity, insulin secretion, and incretin levels between the two groups. We report an increased islet size and an elevated number of β- and α-cells that resulted in an altered β-cell–to–α-cell area in the insulin- resistant group. Our data in this series of studies suggest that neogenesis from duct cells and transdifferentiation of α-cells are potential contributors to the β-cell compensatory response to insulin resistance in the absence of overt diabetes.
Introduction
Insulin resistance, along with β-cell inadequacy, represent the key features in the pathogenesis of type 2 diabetes, and that both are essential for the full manifestation of the disease is generally accepted (1).
A feature that has been recognized in rodents (2,3) and humans (4–6) is the ability of the pancreas to compensate for insulin resistance by an increase in β-cell mass and insulin secretion. Indeed, β-cell mass is dynamic and capable of adapting to physiological and pathological conditions to maintain normoglycemia (7–9). Studies in humans suggest that the number and mass of β-cells increase in response to obesity; however, the time of onset of the increase and the precise origin of such new β-cells are still unknown (7). It is also evident that a failure of this ability of the β-cells to compensate for insulin resistance leads to progressive hyperglycemia and glucose toxicity (10) and to overt diabetes (11). A challenge to identifying the pathways and investigating the mechanisms that underlie compensatory changes in islets is the lack of longitudinal access to human tissue samples of appropriate quality for analyses coupled with accurate metabolic and hormonal profiling.
We took advantage of the unique opportunity to collect pancreas samples from patients undergoing surgical removal of a tumor of the ampulla of Vater to explore the hypothesis that insulin resistance directly contributes to adaptive changes in β-cell mass and function. To this end, we measured insulin sensitivity, insulin secretion, and incretin levels in nondiabetic, nonobese subjects before and after pancreatoduodenectomy. We also evaluated markers of β-cell proliferation, apoptosis, hypertrophy, and islet neogenesis, as well as ductal cell markers. Our data indicate that alterations in insulin sensitivity are linked to markers of compensation in humans and suggest ductal cells and α-cell transdifferentation as sources for new β-cells.
Research Design and Methods
Selection and Description of Participants
The study recruited 18 patients (9 males and 9 females) scheduled to undergo pylorus-preserving pancreatoduodenectomy from the Hepato-Biliary Surgery Unit of the Department of Surgery (Agostino Gemelli University Hospital, Rome, Italy). The local ethics committee approved the study protocol, and all participants provided written informed consent, followed by a comprehensive medical evaluation.
Indication for surgery was tumor of the ampulla of Vater. None of the patients had a family history of diabetes, and all were classified as nondiabetic as determined by a 75-g oral glucose tolerance test and HbA1c according to the American Diabetes Association criteria (12). Only patients with normal cardiopulmonary and kidney functions, as determined by medical history, physical examination, electrocardiography, creatinine clearance, and urinalysis were included in the study. Altered serum lipase and amylase levels before surgery, as well as morphologic criteria for pancreatitis, were considered exclusion criteria. Potential patients who had severe obesity (BMI >40 kg/m2), uncontrolled hypertension, and/or hypercholesterolemia were excluded.
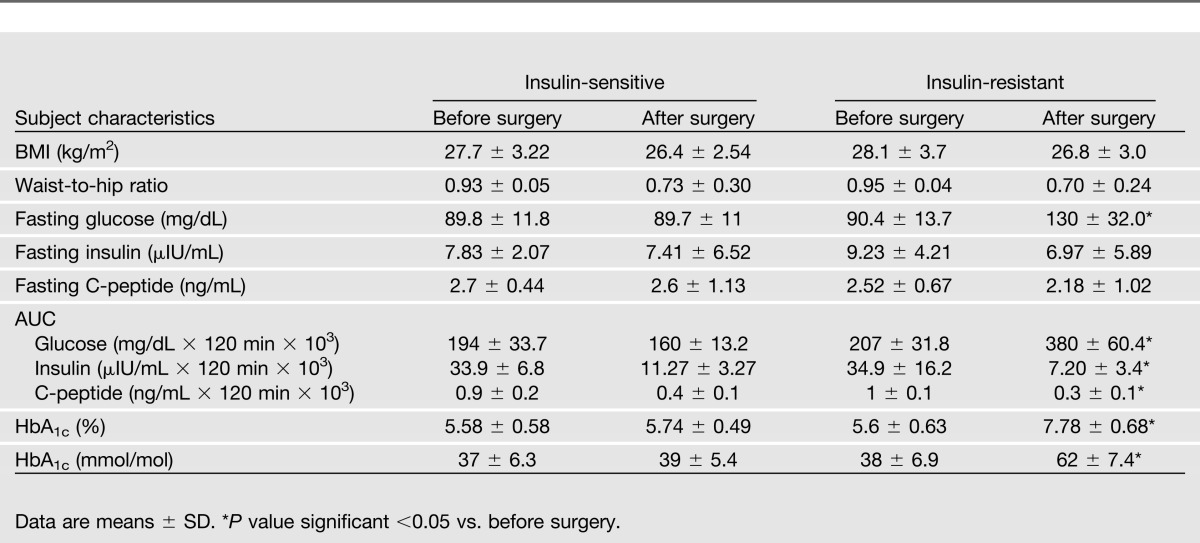
To assess differences in islet morphology in response to insulin-resistant versus insulin-sensitive states, patients were divided into insulin-resistant and insulin-sensitive groups according to their insulin sensitivity, as measured with the euglycemic hyperinsulinemic clamp procedure before surgery. As previously described (13), the cutoff for insulin sensitivity was the median value of glucose uptake in the overall cohort (4.9 mg ⋅ kg−1 ⋅ min−1); therefore, subjects whose glucose uptake exceeded the median value were classified as “more insulin sensitive” than subjects whose glucose uptake was less than the median; for ease of comprehension, the two groups were defined “insulin sensitive” or “insulin resistant.” Clinical and metabolic characteristics of the two groups are summarized in Table 2.
Table 2.
Clinical and metabolic characteristics of insulin-sensitive and insulin-resistant patients before and after surgery
Study Design and Experimental Procedures
Anthropometric parameters were determined according to standard procedures (14). BMI was calculated as weight in kilograms divided by height in meters squared (kg/m2). Blood samples were drawn from all patients for serum lipid assays (total cholesterol and HDL and LDL) in the morning after an overnight (8-h) fast. All procedures were performed with subjects supine throughout the experiments. Each subject underwent a hyperinsulinemic euglycemic clamp, a hyperglycemic clamp, and a mixed-meal test 1 week before the surgical procedure and after a variable period of recovery from the operation. A sufficient recovery period was judged on normalization of inflammatory parameters, such as C-reactive protein, erythrocyte sedimentation rate, stability of weight, and absence of symptoms of abnormal intestinal motility or exocrine pancreatic deficiency. During the clamp procedures, an intravenous catheter was inserted into each arm, one for infusions and the other for blood sampling.
Oral Glucose Tolerance Test
Normal glucose metabolism was confirmed by a standard 75-g oral glucose tolerance test measuring glycemia, insulin, and C-peptide at 0, 30, 60, 90, 120 min after the glucose load.
Hyperinsulinemic Euglycemic Clamp Procedure
The hyperinsulinemic euglycemic clamp test was performed after a 12-h overnight fast using insulin 40 mIU⋅m−2⋅min−1 of body surface according to DeFronzo et al. (15) A primed constant infusion of insulin was administered (Actrapid HM, 40 mIU⋅m−2⋅min−1; Novo Nordisk, Copenhagen, Denmark). The constant rate for the insulin infusion was reached within 10 min to achieve steady-state insulin levels. In the meantime, a variable infusion of 20% glucose was started with a separate infusion pump, and the rate was adjusted, on the basis of plasma glucose samples drawn every 5 min, to maintain the plasma glucose concentration at each participant’s fasting plasma glucose level. During the last 20 min of the clamp procedure, plasma samples from blood drawn at 5- to 10-min intervals were used to determine glucose and insulin concentrations. Whole-body peripheral glucose utilization was calculated during the last 30-min period of the steady-state insulin infusion and was measured as the mean glucose infusion rate (mg⋅kg−1⋅min−1).
Hyperglycemic Clamp Procedure
The plasma glucose was clamped at a stable level of 125 mg/dL above the fasting blood glucose concentration. The hyperglycemic clamp was started with a 200 mg/mL bolus dose of dextrose (150 mg/kg) administered into the antecubital vein. Blood was drawn from a cannulated dorsal hand vein on the opposite arm. Venous plasma glucose was analyzed every 5 min with a glucose analyzer, and the infusion of 20% glucose was adjusted to achieve a stable glucose level of 125 mg/dL above the fasting value. Serum samples for insulin and C-peptide were drawn at 0, 2.5, 5, 7.5, 10, 15, 30, 60, 90, 120, 130, 140, and 150 min.
The first-phase insulin release, reflecting the early insulin peak secreted from the pancreatic β-cell in response to glucose stimulation, was calculated as the area under the curve (AUC) during the first 10 min of the clamp by using the trapezium rule. The second-phase insulin release, reflecting β-cell function under sustained elevated glucose levels, was calculated as the AUC from 10 to 120 min. Subsequently a 5-g arginine bolus was administered to measure maximum C-peptide secretory capacity at a steady-state blood glucose concentration of 250 mg/dL. Combined hyperglycemia- and arginine-stimulated β-cell secretory capacity was calculated as the insulin AUC during the 30 min after the arginine bolus (Fig. 1A).
Figure 1.

Decreased insulin secretion but unaltered insulin sensitivity after pancreatoduodenectomy. A: Schematic of the hyperglycemic clamp experiment. l-arginine bolus (5 g) was injected at 120 min. B: Insulin secretion during hyperglycemic clamp. C: Changes in the AUC of first-phase, second-phase, and phase of insulin secretion after l-arginine stimulus (Arginine) detected during hyperglycemic clamp. A significant reduction was found in all phases of insulin secretion. D: Hyperinsulinemic euglycemic clamp performed before and after surgery. E: Mixed-meal test performed before and after surgery. Changes in the AUC of GLP-1, GIP, and glucagon. *P < 0.05, **P ≤ 0.001.
Mixed-Meal Test
Patients were instructed to consume a meal of 830 kcal (107 kcal from protein, 353 kcal from fat, and 360 kcal from carbohydrates) within 15 min. Blood samples were drawn twice in the fasting state and at 30-min intervals over the following 240 min (sample time 0, 30, 60, 90, 120, 150, 180, 210, and 240 min) for the measurement of plasma glucose, insulin, C-peptide, glucagon, and glucagon-like peptide 1 (GLP-1) or glucose-dependent insulinotropic polypeptide (GIP) concentrations. Blood samples for glucagon, total GLP-1, or intact GIP were sampled in tubes containing EDTA and a dipeptidyl peptidase-4 inhibitor (Millipore, Billerica, MA); after centrifugation (1,000 rpm for 10 min at 4°C), samples were stored at −80°C until analyses. Insulin levels were determined using a commercial radioimmunoassay kit (Medical System, Immulite DPC, Los Angeles, CA). Plasma glucose concentrations were determined by the glucose oxidase technique, using a glucose analyzer (Beckman Instruments, Palo Alto, CA). Plasma C-peptide was measured by AutoDELPHIA automatic fluoroimmunoassay (Wallac, Turku, Finland), with a detection limit of 17 pmol/L. Immunoreactive glucagon was measured in ethanol-extracted plasma by radioimmunoassay using antibody code no. 4305, which is directed against the C-terminus of glucagon and reacts specifically with pancreatic glucagon (16). Total GLP-1 concentrations were measured using antiserum no. 89390, reacting equally with intact GLP-1 (7-36) amide and its primary N-terminally truncated metabolite GLP-1 (9-36) amide. Intact GIP was measured using antiserum no. 98171, reacting with the N-terminus of GIP, but not with the metabolite, GIP 3-42 (17).
Surgical Procedures
Pancreatoduodenectomy was performed according to the pylorus-preserving technique (18,19). Briefly, the pancreatic head, the entire duodenum, common bile duct, and gallbladder were removed en bloc, leaving a functioning pylorus intact at the gastric outlet. All adjacent lymph nodes were carefully removed. The continuity of the gastrointestinal tract was restored by an end-to-side invaginated pancreatojejunostomy. Further downstream, an end-to-side hepaticojejunostomy and side-to side gastroenterostomy or an end-to-side pylorus-jejunostomy was made. The removed volume of pancreas during the surgery was constant (∼50%), as previously reported by Schrader et al. (20). A pancreas sample was collected during the surgery from the downstream edge of the surgical cut.
Immunohistochemical Analysis of Pancreas Samples
Pancreatic Tissue Processing
Pancreas samples were fixed in formaldehyde and embedded in paraffin for subsequent analysis. Five-micrometer sections were stained with hematoxylin and eosin or by immunohistochemistry for islet hormones using a cocktail of antibodies to insulin, glucagon, or somatostatin (21). In addition, sections were immunostained for insulin, Ki67, or DAPI (nucleus) to assess proliferation, TUNEL for apoptosis, and for duct marker using anti-CK19 antibodies, and GLP-1 to identify incretin immunoreactivity. The hematoxylin and eosin slides were examined in all cases by two pathologists to exclude those with pancreatitis, autolysis, and tumor infiltration.
Primary antibodies included insulin (guinea pig antibody, 1:200; Abcam), glucagon (mouse mono, 1:500; Sigma-Aldrich), somatostatin (rabbit poly, 1:500; Abcam), or Ki67 (mouse mono antibody, 1:50; BD Biosciences), GLP-1 (rabbit antibody, 1:1000; J.F. Habener, MD, Massachusetts General Hospital, Boston, MA), and CK19 (rabbit poly, 1:100; Abcam). For TUNEL we used an Apoptag Fluorescein in situ apoptosis detection kit (Roche) and PDX1 (rabbit poly, 1:200; Cell Signaling).
Secondary antibodies were donkey anti–GP-594, donkey anti–mouse-AMCA, donkey anti–rabbit-488, donkey anti–mouse-AMCA, and biotinylated donkey anti-rabbit (all from Jackson ImmunoResearch, West Grove, PA), and peroxidase labeled polymer (Dako).
Morphometric Analysis
Analysis of β-, α-, and δ-cell area was done as described previously (22). Each section was analyzed separately by measuring total insulin-, glucagon-, or somatostatin-positive areas, as well as the total pancreas section area, using Image Pro Plus 4.5.1 software (Media Cybernetics, Silver Springs, MD). The β-, α-, or δ-cell areas were expressed as a percentage of the total pancreas section area. The islet size was calculated as the sum of the individual β-, α-, and δ-cell areas divided by the number of islets counted in each pancreas section. Islet density was quantified by measuring the total area of the pancreas using Image Pro Plus and then counting the number of islets contained within that pancreas area, the results being expressed as islets per mm2. Islet size distribution was determined using the insulin-stained sections of pancreas counterstained with DAPI. At least 100 islets per section were examined and classified according to the number of insulin-positive cells (i.e., 1–8 cells, 9–19 cells, 20–49 cells, and 50 or more β-cells) and the data expressed as a percentage of islets. The ratio of the β-cell to α-cell area was evaluated for each section, dividing the individual percentage of β-cell area by the α-cell area. To evaluate β-cell size and nuclear area, five randomly selected islets per case were immunostained for insulin or DAPI and imaged at original magnification ×400 (×40 objective). The insulin-positive area for each islet was measured, and the number of nuclei present in the insulin-stained area (μm2) was manually counted to calculate the individual β-cell cross-sectional area (μm2). The number of β-cells was manually counted for each section and expressed as the ratio of β-cells per total pancreas section. To measure the β-cell nuclear area, insulin-stained sections of pancreas counterstained with DAPI were used; five randomly selected islets per case were photographed at original magnification ×400. Then, five representative β-cell nuclei were identified in each islet. Selection criteria included clear presence of the nucleus within a β-cell, the ability to clearly visualize nuclear boundaries, circular shape (similar dimensions in all directions), and the appearance to the observer that the nucleus had been sectioned through its maximum diameter. Once the identified, nucleus was encircled, and the nuclear area (μm2) was measured using Image Pro Plus software. The number of insulin and glucagon double-positive cells was manually counted in sections costained for insulin or glucagon. A mean ± SE of 1,172 ± 269 endocrine cells was evaluated per subject, and the resulting data were expressed as percentage of endocrine cells. The double-positive cells were confirmed in randomly selected islets by confocal microscopy. All data were expressed as the mean ± SE for each group.
Quantification of Scattered Islet and Exocrine Duct Cells Positive for Insulin
As previously described (23), clusters of less than eight endocrine cells were considered as new islets (neogenesis). On sections stained specifically for insulin, clusters with less than eight insulin-positive cells were manually counted and considered as scattered islets and then expressed as the ratio of the number of scattered islets per total pancreas area. Sections costained for pancreatic ductal marker CK19 or insulin were imaged by confocal microscopy, and the number of CK19- and insulin-positive cells was manually counted. A mean ± SE of 1,107 ± 475 duct cells were evaluated for each section. The resulting data were expressed as a percentage of duct cells positive for insulin in each pancreas. All data were expressed as the mean ± SE for each group.
Proliferation and Apoptosis
To determine replication in β-cells, the number of β-cells costaining with Ki67 was counted and expressed as the percentage of the total number of β-cells (at least 2,000 β-cells were counted in each case). For the evaluation of apoptosis, the number of β-cells costaining with TUNEL was counted and expressed as percentage of the total number of β-cells (at least 2,000 β-cells for each case). The entire analysis was performed by a single observer in a blinded fashion.
Statistics
All data are expressed as mean ± SE, unless indicated otherwise. Because samples were normally distributed, differences in means were tested by the two-tailed Student t test. The relationship between variables was derived with linear regression analysis using SPSS 9 software (SPSS, Chicago, IL). A P value of <0.05 was considered statistically significant.
Study Approval
This study was approved by the ethical committee of the Catholic University of the Sacred Heart, Rome, Italy. All study subjects provided written informed consent before screening and participating in the study.
Results
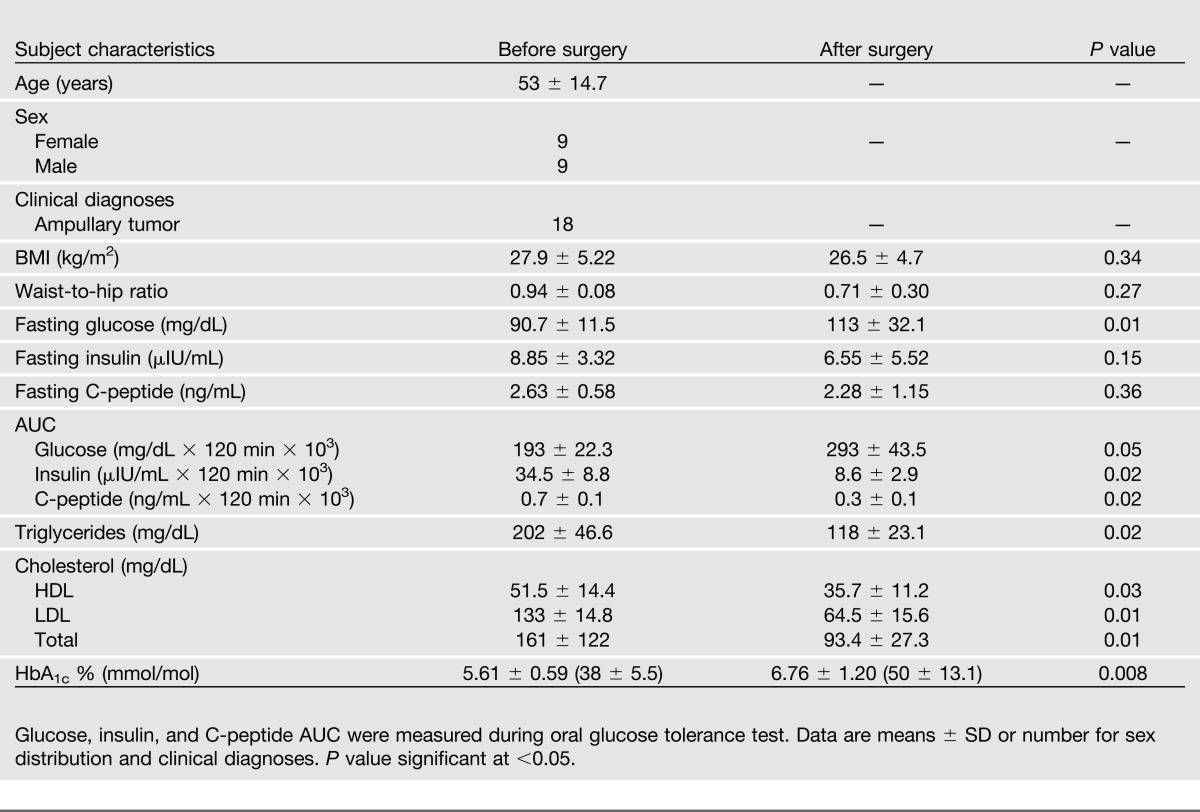
The current study included 18 patients (9 female and 9 male; mean age, 53 ± 15 years) undergoing pylorus-preserving pancreatoduodenectomy for a tumor of the ampulla of Vater. Clinical and metabolic characteristics of study subjects are provided in Table 1.
Table 1.
Clinical and metabolic characteristics of patients before and after surgery
Hemipancreatectomy Induced a Marked Reduction in Insulin Secretion, Without Affecting Insulin Sensitivity, and Resulted in the Onset of Diabetes Only in Insulin-Resistant Subjects
Subjects were evaluated 1 week before surgery and at 40 ± 7 days (range 34–48) after surgery. To evaluate the insulin secretory capacity, we performed hyperglycemic clamps over 2 h, followed by an acute stimulation with l-arginine (5 g). As expected, insulin secretion was significantly reduced after surgery (P < 0.001, Fig. 1B). The response to arginine (121 to 150 min after glucose infusion) revealed an even higher (76%) reduction of insulin secretion (Fig. 1C). Conversely, insulin sensitivity, as assessed by the hyperinsulinemic euglycemic clamp (15), did not change significantly after surgery (Fig. 1D).
Evaluation of glucose homeostasis by standard oral glucose tolerance tests (75 g) in the overall cohort revealed worsening of glucose tolerance after surgery (Table 1).
To further characterize changes in glucose tolerance after removal of ∼50% of the pancreas and to assess differences in islet morphology, patients were divided into insulin-resistant and insulin-sensitive groups (Table 2). Despite the removal of the head of the pancreas, which includes ∼50% of the β-cell mass, patients identified as insulin-sensitive before surgery preserved their glucose tolerance, whereas seven of nine (77.7%) insulin-resistant patients developed diabetes, as confirmed by a 75-g oral glucose tolerance test and an HbA1c higher than 7% (53 mmol/mol; Table 2).
GLP-1 secretion in response to a mixed meal significantly increased (AUC GLP-1 before vs. after surgery: 10.4 ± 3.2 nmol ⋅ L−1 ⋅ min vs. 15.6 ± 4.3 nmol ⋅ L−1 ⋅ min, P = 0.01; Fig. 1E), whereas GIP response was significantly reduced (AUC GIP before vs. after surgery: 19.1 ± 8.1 nmol ⋅ L−1 ⋅ min vs. 7.7 ± 2.8 nmol ⋅ L−1 ⋅ min, P < 0.001; Fig. 1E) after surgery.
Although 50% pancreatectomy led to a decrease in insulin secretion in insulin-sensitive and insulin-resistant patients (Fig. 2A), the latter exhibited a greater attenuation in all of the phases of insulin secretion (insulin-sensitive 924 ± 900 µIU ⋅ mL−1 ⋅ min vs. insulin-resistant 6,952 ± 1,951 µIU ⋅ mL−1 ⋅ min; ΔAUC insulin secretion, P = 0.04; Fig. 2C). Furthermore, the increase in glucagon secretion during the mixed-meal test after surgery was higher in insulin-resistant patients (insulin-sensitive 112 ± 101 nmol ⋅ L−1 ⋅ min vs. insulin-resistant 1,812 ± 326.3 nmol ⋅ L−1 ⋅ min; ΔAUC glucagon secretion, P = 0.02; Fig. 2B and D).
Figure 2.

After pancreatoduodenectomy, insulin-resistant subjects exhibited a reduction in insulin secretion and an increase in glucagon secretion. A: AUC of insulin secretion evaluated during the hyperglycemic clamp before and after surgery in insulin-sensitive and insulin-resistant subjects. Insulin secretion was reduced in both groups and for all of the insulin-secretion phases. B: AUC of glucagon secretion evaluated during the mixed-meal test before and after surgery in insulin-sensitive and insulin-resistant subjects. C: Absolute reductions from baseline (before surgery) in insulin secretion AUCs during the hyperglycemic clamp. D: Absolute increases from baseline (before surgery) in glucagon secretion AUCs during the mixed-meal test. *P < 0.05.
Insulin-Resistant Individuals Exhibit Increased Islet Size
To evaluate changes in islet morphology, we performed immunohistochemical analyses of sections of pancreas removed during surgery. Compared with insulin-sensitive individuals, insulin-resistant subjects exhibited an increased percentage of insulin area (insulin-sensitive 0.58 ± 0.17% vs. insulin-resistant 1.10 ± 0.23%, P = 0.05), percentage of glucagon area (insulin-sensitive 0.04 ± 0.01% vs. insulin-resistant 0.23 ± 0.06%, P < 0.01) and percentage of somatostatin area (insulin-sensitive 0.01 ± 0.00% vs. insulin resistant 0.03 ± 0.01%, P = 0.01; Fig. 3A). Overall, these differences resulted in an increased mean islet size in insulin-resistant subjects compared with the insulin-sensitive ones (mean islet size: insulin sensitive 2,456 ± 332 µm2 vs. insulin resistant 5,156 ± 944 µm2, P < 0.001; Fig. 3B). Further, we observed a strong inverse correlation between islet size and glucose uptake in the entire cohort (r = −0.74, P < 0.01; Fig. 3C), which suggests that changes in islet morphology are influenced by insulin sensitivity.
Figure 3.

Increased islet size, β-cell nuclear size, and glucagon area in insulin-resistant subjects. A: Insulin, glucagon, and somatostatin areas evaluated as fraction of total pancreatic section area. B: Mean islet size in insulin-sensitive and insulin-resistant subjects. C: Glucose uptake vs. islet size. Correlation between insulin sensitivity index and islet size is shown in all of the subjects. D: Mean β-cell area in insulin-sensitive and insulin-resistant subjects. E: Mean β-cell nuclear area in insulin-sensitive and insulin-resistant subjects. F: Islet density in insulin-sensitive and insulin-resistant subjects. *P ≤ 0.05, **P ≤ 0.001.
Increased Islet Size Is Likely Caused by β-Cell Hyperplasia
To identify potential mechanism(s) underlying the increased islet size in insulin resistance, we examined β-cell replication, apoptosis, and cell size. Among a total of 37,845 β-cells counted in the entire cohort, replication was undetectable, as determined by Ki67 immunostaining. β-Cell apoptosis was also infrequent among the 39,600 cells examined in the entire cohort. Although positive cells were undetectable in the insulin-sensitive group, we detected 24 positive apoptotic cells in three insulin-resistant patients (mean percentage of β-cell apoptosis among the insulin-resistant group, 0.1%); however, the differences between groups were not significant (P = 0.20). By measuring the mean individual β-cell cross-sectional area in the two groups, we also ruled out a possible contribution of cell hypertrophy to the increase in islet size (insulin-sensitive 127 ± 9 µm2 vs. insulin-resistant 129 ± 17 µm2, P = 0.79; Fig. 3D). Insulin-resistant patients showed an increased number of β-cells per mm2 of pancreas area (insulin-sensitive 22.7 ± 2.7 β-cells/mm2 vs. insulin-resistant 80.9 ± 15.8 β-cells/mm2, P < 0.01). These data suggest that the increase in islet size is due to increased number of cells (i.e., hyperplasia) rather than altered β-cell volume. Further, β-cell nuclear size was increased in the insulin-resistant group (insulin-sensitive 34.5 ± 1.1 µm2 vs. insulin resistant 42.4 ± 6.2 µm2, P = 0.03; Fig. 3E), suggesting that secretory β-cells are relatively young (24,25).
Insulin Resistance Is Associated With Increased Islet Neogenesis
On the basis of the results discussed above and the finding of increased islet density (insulin-sensitive 3.5 ± 0.5 islets/mm2 vs. insulin resistant 7.5 ± 1.4 islets/mm2, P < 0.01; Fig. 3F), we hypothesized that neogenesis, rather than proliferation, contributes to β-cell hyperplasia in insulin-resistant patients. To explore whether the pancreas shows evidence of neogenesis, we quantified the number of scattered islets with less than eight nuclei (23) and observed an increase of such islets in insulin-resistant subjects (insulin-sensitive 1.80 ± 0.18 nuclei/mm2 vs. insulin resistant 4.65 ± 1.16 nuclei/mm2, P = 0.04; Fig. 4A). Although these scattered islets were distributed within the exocrine tissue, we cannot ascertain whether these cells arise directly from acinar cells without further detailed investigation.
Figure 4.

Increased neogenesis in insulin-resistant patients. A: Clusters of islets (with less than eight nuclei) were counted and expressed per mm2 of pancreas section. The number of scattered islets was increased in insulin-resistant subjects. B: Percentage of duct cells marked by CK19 positive for insulin. C: Frequency distribution of β-cells per islet in sections from insulin-resistant and insulin-sensitive subjects. There was a marked shift toward small islets and very large islets in insulin-resistant compared with insulin-sensitive subjects. D: Insulin immunoreactivity in duct cells marked by CK19. Confocal microscopy analysis of CK19 (green) and insulin (red) showed duct cells positive for insulin in insulin-resistant (upper panel) and insulin-sensitive (lower panel) subjects. Scale bars = 100 µm. *P < 0.05, **P < 0.01.
Further, because previous studies have reported potential formation of new islets from duct cells, we evaluated the number of cells that were double-positive for the duct marker CK19 and insulin. The mean percentage of CK19/insulin double-positive cells was increased in insulin-resistant subjects (insulin-sensitive 0.28 ± 0.12% vs. insulin-resistant 1.47 ± 0.26%, P < 0.001; Fig. 4B and D). As shown in Fig. 4C, insulin-resistant subjects displayed a greater number of small clusters and islets with more than 50 cells.
Insulin Resistance and Alterations in α-Cells
The fractional α-cell area was greater in the insulin-resistant compared with the insulin-sensitive group and was inversely correlated with glucose uptake (r = −0.65, P = 0.03; Fig. 5A). The mean ratio of β-cell to α-cell areas was lower in the insulin-resistant subjects (insulin-sensitive 0.13 ± 0.01 vs. insulin-resistant 0.08 ± 0.01, P = 0.05; Fig. 5B), suggesting a relative increase in the α-cell area.
Figure 5.

Insulin resistance is associated with a decreased β-cell–to–α-cell ratio. A: Correlation between the percentage of glucagon area and insulin sensitivity. B: The ratio of β-cell to α-cell areas was significantly lower in the insulin-resistant subjects, suggesting an unbalanced proportion between β-cell and α-cell areas. C: Percentage of insulin and glucagon double-positive cells in the sections of insulin-sensitive and insulin-resistant subjects. D: Confocal microscopy analysis of insulin (red) and glucagon (green) immunostaining. ×25 objective, scale bar = 100 µm (1–6); ×3 amplification. Scale bar = 20 µm (insets). *P ≤ 0.05. GLU, glucagon; INS, insulin.
Because previous reports have suggested transdifferentiation of α-cells as a mechanism that contributes to alterations in β-cell mass, we immunostained pancreas sections to identify insulin and glucagon coexpressing cells. Interestingly, we detected an increased number of double-positive cells in insulin-resistant subjects compared with insulin-sensitive subjects (mean percentage of double-positive cells: insulin sensitive 4.51 ± 1.07% vs. insulin resistant 10.86 ± 2.17%, P = 0.02; Fig. 5C and D) (Supplementary Figs. 1 and 2). We also examined PDX1 immunoreactivity in the insulin and glucagon double-positive cells and detected PDX1-positive cells in insulin-sensitive and insulin-resistant subjects (Supplementary Fig. 3).
Further immunohistochemical analyses using a specific anti-GLP-1 antibody, which is highly selective for processed amidated GLP-1 directed to the COOH-terminal, revealed that glucagon colocalizes with GLP-1 in both groups (Fig. 6A). Interestingly, the α-cell area correlated with GLP-1 (r = 0.63, P = 0.04; Fig. 6B) but not GIP secretion (r = 0.08, P = 0.79). The AUC of GLP-1 secretion during the mixed-meal test also correlated with glucose uptake in the entire cohort (r = −0.57, P < 0.01). Whether these correlations indicate a link between circulating GLP-1 and α-cell biology requires further study.
Figure 6.

GLP-1 costains with glucagon, and its plasma values correlate with islet glucagon area and (inversely) with whole-body insulin sensitivity. Immunostaining is shown for glucagon (red), GLP-1 (green), and DAPI (blue). A: Representative picture of an islet shows an overlap of glucagon and GLP-1 immunoreactivity. Scale bar = 100 µm. B: Correlation between percentage of glucagon area and GLP-1 secretion evaluated during the mixed-meal test and expressed as AUC. C: Correlation between glucose uptake and GLP-1 secretion evaluated during the mixed-meal test and expressed as AUC and insulin sensitivity.
The relative increase in α-cell area could lead to an increase in β-cells by transdifferentiation and also to an increase in intraislet GLP-1 production. Furthermore, it is possible that this change in the relative proportion of α-cells could be a first step toward hyperglucagonemia, a hallmark of type 2 diabetes.
Discussion
In the current study, we evaluated pancreas samples obtained from nondiabetic subjects to investigate the effects of altered insulin sensitivity on islet morphology. The major finding of our study is that insulin-resistant subjects exhibited increased islet size, which was strongly inversely correlated with insulin sensitivity (r = −0.74, P < 0.001). This suggests that insulin resistance directly impacts islet biology in nondiabetic humans by inducing an increase in β-cell area to compensate for the increased insulin demand. These findings are consistent with reports in humans and mouse models (26,27) in which defects in insulin-signaling pathways in β-cells have been suggested to be responsible for a decrease in mass and reduced secretory function. Indeed, impaired β-cell responsiveness to insulin has been shown in insulin-resistant patients (28). Consistent with previous reports in insulin-resistant obese patients (7), who exhibit an increase in islet size due to increased cell number, β-cells from insulin-resistant individuals in our study also exhibited an increased number. In addition, the β-cells showed an increased nuclear area, suggesting that cells were relatively young and with increased secretory capacity (29).
Several studies (7,30,31) have investigated proliferation of β-cells in humans mostly in pancreata from autopsy samples. Although some variability is evident, most reports agree that the rate of β-cell proliferation is extremely low in the adult human pancreas. Consistently, we noted virtually undetectable β-cell proliferation by Ki67 immunostaining in pancreas sections from the entire group. We also failed to detect β-cell apoptosis, in contrast to the increase seen in patients with type 2 diabetes (5). Our data showing the lack of alterations between groups in Ki67 and TUNEL staining suggest that neither proliferation nor apoptosis contributes significantly to the β-cell adaptive response to insulin resistance in this cohort of patients.
Recent reports have suggested that plasticity of the adult β-cell mass is linked to neogenesis during different periods of life (early postnatal life, pregnancy, and aging) as well as in obesity, impaired glucose tolerance, and in individuals with newly diagnosed diabetes (32,33). Other reports suggest that cells lining the ducts or acinar cells may serve as a source of new β-cells (34). Indeed, our findings of a higher number of islet clusters and the presence of insulin-positive duct cells suggest that these pathways underlie the alteration in islet size associated with insulin resistance.
Because inappropriate glucagon secretion is a feature of patients with diabetes (4,35) and previous reports indicate a role for insulin signaling in the regulation of α cell function (36), we explored the link between α-cells and insulin sensitivity. Analyses of islet morphology revealed that the ratio between β- and α-cell area was lower in insulin-resistant subjects due to a relatively greater increase in α-cell area. The relative low α-cell number in the insulin-sensitive group is a feature of the significantly higher insulin sensitivity, whereas the increased number of α-cells in insulin-resistant subjects occurs as compensation for insulin resistance. Similar changes are also evident in diabetic patients (37) and insulin-resistant primates (38). Furthermore, we found a strong inverse correlation between α-cell area and insulin sensitivity (r = −0.65, P = 0.003). These observations raise several questions. For example, do alterations in α-cell biology precede changes in β-cell mass? Could transdifferentiation of α-cells contribute to an increase in β-cells? Does the imbalance between β- and α-cells result from dedifferentiation (39)? Despite the cross-sectional nature of our study, it is worth noting that our data are timely and highly relevant to islet biology because an ideal longitudinal study on human pancreata is extremely difficult to undertake due to ethical limitations.
A second link between insulin sensitivity and α-cell mass was evident from the correlation with GLP-1 secretion. As previously reported by our group (19), patients who underwent pancreatoduodenectomy and a mixed-meal test exhibited a significant increase in GLP-1 secretion in contrast to a significant decrease in GIP. Although the latter is likely due to removal of the duodenum, a major site of production of GIP (40), the mechanism contributing to greater GLP-1 secretion is not fully understood and suggests hypersecretion by existing intestinal L-cells and/or other potential sources of the incretin hormone.
Our observations on GLP-1 gain significance in light of previous reports (41) suggesting α-cells are a potential source of the incretin hormone, which, in turn, can exert a local paracrine effect on islet function, as previously suggested by Marchetti et al. (41). Indeed, the presence of GLP-1 in the islet has been suggested to have multiple effects, including differentiation of progenitor cells into β-cells in the pancreatic duct epithelium (42,43) and direct stimulation of β-cell proliferation and inhibition of apoptosis (44,45). In addition, pancreas extracts from glucagon receptor knockout mice (46) exhibit an increase in GLP-1 that is associated with an up to 10-fold increase in circulating GLP-1 amide, the active form of the incretin hormone. The lack of change in GLP-1 in intestinal extracts suggests that the pancreas is one of the sources contributing to circulating GLP-1.
In our study, none of the insulin-sensitive patients developed diabetes after surgery, whereas 77.7% of insulin-resistant patients became diabetic. The latter displayed a greater reduction in all phases of insulin secretion and a higher increase in glucagon secretion in response to a mixed-meal test after surgery. It is tempting to speculate that these alterations are secondary to insulin resistance in α-cells and, as a consequence, to an inability of ambient insulin to adequately suppress glucagon secretion (34,47).
In conclusion, our findings suggest that neogenesis from duct cells and/or transdifferentiation from α-cells are likely explanations for the alterations in β-cell mass observed in insulin-resistant subjects. Our study provides an example of a unique approach in the investigation of islet morphology in nondiabetic patients. A strength of this approach is the comprehensive evaluation of metabolic parameters in conjunction with analyses of pancreatic tissue from living donors, which allows comparison of in vivo and ex vivo studies that would otherwise not be possible unless sequential biopsies throughout life are performed. These findings provide a platform to plan studies to directly identify the source of new β-cells and determine the molecular mechanisms responsible for the dynamic changes that impact β-cell mass over the time course of progression of type 2 diabetes, with the long-term goal of enhancing islet compensation to insulin resistance.
Supplementary Material
Article Information
Acknowledgments. The authors thank S. Bonner-Weir, PhD, (Joslin Diabetes Center) for advice and assistance with histological examination of the samples; J.F. Habener, MD, (Massachusetts General Hospital) for the gift of the anti-GLP-1 antibody; C. Cahill and S. Sioletic, MD, PhD, for expert technical assistance; and A. Molven, PhD, A. El Ouaamari, PhD, and C. Conte, MD, for constructive comments.
Funding. This study was supported by grants to A.G. from Università Cattolica del Sacro Cuore (Fondi Ateneo Linea D.3.2 Sindrome Metabolica), from the Italian Ministry of Education, University and Research (PRIN 2010JS3PMZ_011) and from Fondazione Don Gnocchi. T.M. is the recipient of the Albert Reynolds Travel Fellowship by the European Association for the Study of Diabetes and the Fellowship Prize by the Società Italiana di Diabetologia. G.P.S. is the recipient of a fellowship from Laboratori Guidotti. Some of the reagents used in the study were supported by National Institutes of Health grant R01-DK-67536 to R.N.K.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. T.M. generated the data and wrote the manuscript. G.M. and A.P. contributed to discussion and reviewed and edited the manuscript. G.P.S., G.C., and J.H. researched data. J.J.H. generated data. A.G. generated the protocol and reviewed and edited the manuscript. R.N.K. reviewed and edited the manuscript and supervised the work at the Joslin Diabetes Center. T.M., A.G., and R.N.K. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analyses.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-1013/-/DC1.
See accompanying article, p. 832.
References
- 1.Nolan CJ, Damm P, Prentki M. Type 2 diabetes across generations: from pathophysiology to prevention and management. Lancet 2011;378:169–181 [DOI] [PubMed] [Google Scholar]
- 2.Pick A, Clark J, Kubstrup C, et al. Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes 1998;47:358–364 [DOI] [PubMed] [Google Scholar]
- 3.Brüning JC, Winnay J, Bonner-Weir S, Taylor SI, Accili D, Kahn CR. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell 1997;88:561–572 [DOI] [PubMed] [Google Scholar]
- 4.Yoon KH, Ko SH, Cho JH, et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab 2003;88:2300–2308 [DOI] [PubMed] [Google Scholar]
- 5.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003;52:102–110 [DOI] [PubMed] [Google Scholar]
- 6.Polonsky KS, Given BD, Hirsch L, et al. Quantitative study of insulin secretion and clearance in normal and obese subjects. J Clin Invest 1988;81:435–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care 2013;36:111–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butler AE, Cao-Minh L, Galasso R, et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010;53:2167–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patti ME, McMahon G, Mun EC, et al. Severe hypoglycaemia post-gastric bypass requiring partial pancreatectomy: evidence for inappropriate insulin secretion and pancreatic islet hyperplasia. Diabetologia 2005;48:2236–2240 [DOI] [PubMed] [Google Scholar]
- 10.Giaccari A, Sorice G, Muscogiuri G. Glucose toxicity: the leading actor in the pathogenesis and clinical history of type 2 diabetes–mechanisms and potentials for treatment. Nutr Metab Cardiovasc Dis 2009;19:365–377 [DOI] [PubMed] [Google Scholar]
- 11.Weir GC, Laybutt DR, Kaneto H, Bonner-Weir S, Sharma A. Beta-cell adaptation and decompensation during the progression of diabetes. Diabetes 2001;50(Suppl. 1):S154–S159 [DOI] [PubMed] [Google Scholar]
- 12.American Diabetes Association Diagnosis and classification of diabetes mellitus. Diabetes Care 2013;36(Suppl. 1):S67–S74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muscogiuri G, Sorice GP, Prioletta A, et al. 25-Hydroxyvitamin D concentration correlates with insulin-sensitivity and BMI in obesity. Obesity (Silver Spring) 2010;18:1906–1910 [DOI] [PubMed] [Google Scholar]
- 14.Lohman TG, Roche AF, Martorelli R. Anthropometric Standardization Reference Manual. Champaign, IL, Human Kinetics Books, 1988, p. 177 [Google Scholar]
- 15.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 1979;237:E214–E223 [DOI] [PubMed] [Google Scholar]
- 16.Meier JJ, Gallwitz B, Siepmann N, et al. Gastric inhibitory polypeptide (GIP) dose-dependently stimulates glucagon secretion in healthy human subjects at euglycaemia. Diabetologia 2003;46:798–801 [DOI] [PubMed] [Google Scholar]
- 17.Deacon CF, Nauck MA, Meier J, Hücking K, Holst JJ. Degradation of endogenous and exogenous gastric inhibitory polypeptide in healthy and in type 2 diabetic subjects as revealed using a new assay for the intact peptide. J Clin Endocrinol Metab 2000;85:3575–3581 [DOI] [PubMed] [Google Scholar]
- 18.Traverso LW, Longmire WP., Jr Preservation of the pylorus in pancreaticoduodenectomy a follow-up evaluation. Ann Surg 1980;192:306–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muscogiuri G, Mezza T, Prioletta A, et al. Removal of duodenum elicits GLP-1 secretion. Diabetes Care 2013;36:1641–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schrader H, Menge BA, Breuer TG, et al. Impaired glucose-induced glucagon suppression after partial pancreatectomy. J Clin Endocrinol Metab 2009;94:2857–2863 [DOI] [PubMed] [Google Scholar]
- 21.Bonner-Weir S, Deery D, Leahy JL, Weir GC. Compensatory growth of pancreatic beta-cells in adult rats after short-term glucose infusion. Diabetes 1989;38:49–53 [DOI] [PubMed] [Google Scholar]
- 22.Kulkarni RN, Holzenberger M, Shih DQ, et al. beta-cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta-cell mass. Nat Genet 2002;31:111–115 [DOI] [PubMed] [Google Scholar]
- 23.Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes 1999;48:2270–2276 [DOI] [PubMed] [Google Scholar]
- 24.Norman JT, Bohman RE, Fischmann G, et al. Patterns of mRNA expression during early cell growth differ in kidney epithelial cells destined to undergo compensatory hypertrophy versus regenerative hyperplasia. Proc Natl Acad Sci U S A 1988;85:6768–6772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Studer H, Derwahl M. Mechanisms of nonneoplastic endocrine hyperplasia—a changing concept: a review focused on the thyroid gland. Endocr Rev 1995;16:411–426 [DOI] [PubMed] [Google Scholar]
- 26.Kulkarni RN, Brüning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 1999;96:329–339 [DOI] [PubMed] [Google Scholar]
- 27.Assmann A, Hinault C, Kulkarni RN. Growth factor control of pancreatic islet regeneration and function. Pediatr Diabetes 2009;10:14–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halperin F, Lopez X, Manning R, Kahn CR, Kulkarni RN, Goldfine AB. Insulin augmentation of glucose-stimulated insulin secretion is impaired in insulin-resistant humans. Diabetes 2012;61:301–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meier JJ, Butler AE, Galasso R, Butler PC. Hyperinsulinemic hypoglycemia after gastric bypass surgery is not accompanied by islet hyperplasia or increased beta-cell turnover. Diabetes Care 2006;29:1554–1559 [DOI] [PubMed] [Google Scholar]
- 30.Caballero F, Siniakowicz K, Hollister-Lock J, et al. Birth and death of human β-cells in pancreas from cadaver donors, autopsies, surgical specimens, and islets transplanted into mice. Cell Transplant 2 January 2013. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Folli F, Okada T, Perego C, et al. Altered insulin receptor signalling and β-cell cycle dynamics in type 2 diabetes mellitus. PLoS ONE 2011;6:e28050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gregg BE, Moore PC, Demozay D, et al. Formation of a human β-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab 2012;97:3197–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoneda S, Uno S, Iwahashi H, et al. Predominance of β-cell neogenesis rather than replication in humans with an impaired glucose tolerance and newly diagnosed diabetes. J Clin Endocrinol Metab 2013;98:2053–2061 [DOI] [PubMed] [Google Scholar]
- 34.Juhl K, Bonner-Weir S, Sharma A. Regenerating pancreatic beta-cells: plasticity of adult pancreatic cells and the feasibility of in-vivo neogenesis. Curr Opin Organ Transplant 2010;15:79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Unger RH, Orci L. Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci U S A 2010;107:16009–16012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawamori D, Kurpad AJ, Hu J, et al. Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metab 2009;9:350–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henquin JC, Rahier J. Pancreatic alpha cell mass in European subjects with type 2 diabetes. Diabetologia 2011;54:1720–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guardado-Mendoza R, Jimenez-Ceja L, Majluf-Cruz A, et al. Impact of obesity severity and duration on pancreatic β- and α-cell dynamics in normoglycemic non-human primates. Int J Obes (Lond) 2013;37:1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012;150:1223–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holst JJ. On the physiology of GIP and GLP-1. Horm Metab Res 2004;36:747–754 [DOI] [PubMed] [Google Scholar]
- 41.Marchetti P, Lupi R, Bugliani M, et al. A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia 2012;55:3262–3272 [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Cao X, Li LX, Brubaker PL, Edlund H, Drucker DJ. beta-Cell Pdx1 expression is essential for the glucoregulatory, proliferative, and cytoprotective actions of glucagon-like peptide-1. Diabetes 2005;54:482–491 [DOI] [PubMed] [Google Scholar]
- 43.Zhu X, Zhou A, Dey A, et al. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc Natl Acad Sci U S A 2002;99:10293–10298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farilla L, Bulotta A, Hirshberg B, et al. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 2003;144:5149–5158 [DOI] [PubMed] [Google Scholar]
- 45.Buteau J, El-Assaad W, Rhodes CJ, Rosenberg L, Joly E, Prentki M. Glucagon-like peptide-1 prevents beta cell glucolipotoxicity. Diabetologia 2004;47:806–815 [DOI] [PubMed] [Google Scholar]
- 46.Gelling RW, Du XQ, Dichmann DS, et al. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci U S A 2003;100:1438–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akiyama M, Liew CW, Lu S, et al. X-box binding protein 1 is essential for insulin regulation of pancreatic α-cell function. Diabetes 2013;62:2439–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.