

Abstract
Oxidative stress is purported to be involved in the pathogenesis of obesity-associated insulin resistance. We evaluated whether alterations in levels of circulating uric acid (UA), a systemic antioxidant, affects the following: 1) systemic (plasma and saliva) nonenzymatic antioxidant capacity (NEAC); 2) markers of systemic (urinary 8-iso-prostaglandin-F2α) and muscle (carbonylated protein content) oxidative stress; and 3) whole-body insulin sensitivity (percentage increase in glucose uptake during a hyperinsulinemic-euglycemic clamp procedure). Thirty-one obese subjects (BMI 37.1 ± 0.7 kg/m2) with either high serum UA (HUA; 7.1 ± 0.4 mg/dL; n = 15) or normal serum UA (NUA; 4.5 ± 0.2 mg/dL; n = 16) levels were studied; 13 subjects with HUA levels were studied again after reduction of serum UA levels to 0 by infusing a recombinant urate oxidase. HUA subjects had 20–90% greater NEAC, but lower insulin sensitivity (40%) and levels of markers of oxidative stress (30%) than subjects in the NUA group (all P < 0.05). Acute UA reduction caused a 45–95% decrease in NEAC and a 25–40% increase in levels of systemic and muscle markers of oxidative stress (all P < 0.05), but did not affect insulin sensitivity (from 168 ± 25% to 156 ± 17%, P = NS). These results demonstrate that circulating UA is a major antioxidant and might help protect against free-radical oxidative damage. However, oxidative stress is not a major determinant of insulin action in vivo.
Introduction
Obesity is associated with an increase in oxidative stress, which is defined as an increased load of free radicals composed of reactive oxygen and nitrogen species generated during cellular metabolism (1). These free radicals are chemically reactive molecules that can damage cell proteins, membranes, and DNA. The increase in oxidative stress is purported to be involved in the pathogenesis of insulin resistance and type 2 diabetes associated with obesity (2–4). Data from studies conducted in cell culture systems have shown that products of oxidative stress impair insulin-mediated translocation of GLUT4 in myotubes and adipocytes (2,3), and suppress gene transcription of insulin in β-cells (5) and adiponectin in adipocytes (4).
Uric acid (UA) is a powerful scavenger of free radicals and provides ∼60% of free-radical scavenging capacity in plasma (6). Although the antioxidant effect of UA suggests that it might have therapeutic effects, high serum UA concentration is associated with obesity and insulin resistance (7,8), and hyperuricemia has even been proposed as a component of the metabolic syndrome (9). However, it is possible that this increase in circulating levels of UA represents an adaptive response to protect against the detrimental effects of excessive free radicals and oxidative stress (6).
The purpose of the current study was to test the hypothesis that increased levels of plasma UA is an adaptive response to insulin resistance, because of its therapeutic antioxidant effects. Therefore, a decrease in serum UA level will decrease nonenzymatic antioxidant capacity (NEAC), increase the levels of markers of oxidative stress, and decrease insulin sensitivity in obese people. Accordingly, we evaluated insulin sensitivity by using the hyperinsulinemic clamp procedure, antioxidant capacity in plasma and saliva by using the total radical-trapping antioxidant potential (TRAP) and ferric-reducing antioxidant potential (FRAP) assays, and oxidative stress by measuring urinary products of membrane phospholipids peroxidation and skeletal muscle protein carbonylation in obese men and women who have normal and high serum UA concentrations and in the subjects with high serum UA levels before and after acute pharmacological reduction in serum UA levels.
Research Design and Methods
Subjects
A total of 31 obese adults (22 women, 9 men; age 47 ± 2 years; BMI 37.1 ± 0.7 kg/m2), 16 of whom had normal (<5 mg/dL; n = 16) and high (>6 mg/dL; n = 15) serum UA concentrations, participated in this study. Subjects who were receiving treatment with medications that could affect our outcome measures, or had a history of xanthinuria, had glucose-6-phosphate dehydrogenase deficiency, were pregnant or lactating, or had diabetes or other significant diseases, were excluded from the study. Subjects gave written informed consent before participating in this study, which was approved by the Institutional Review Board of Washington University School of Medicine.
Body Composition
Body fat mass and fat-free mass were determined by using dual-energy X-ray absorptiometry. Intra-abdominal visceral adipose tissue volume was quantified by magnetic resonance imaging.
Metabolic Studies
Subjects were admitted to the Clinical Research Unit the evening before the metabolic study and were given a standard meal at 1900 h. Subjects then fasted, except for water, until completion of the study the next day. Urine was collected for the 12 h overnight period to measure urinary isoprostane levels. At 0600 h, catheters were inserted into an antecubital vein of one arm for isotope infusion, and a second intravenous catheter was inserted into a hand vein, which was heated to obtain arterialized blood samples (10). At 0700 h, a primed (22.5 µmol/kg) constant (0.25 µmol ⋅ min−1 ⋅ kg−1) infusion of [6,6 2H2]glucose was started and continued for 7 h. After 3.5 h of tracer infusion (basal period), insulin was infused at 50 mU ⋅ m−2 ⋅ min−1 for 3.5 h, initiated by a two-step priming dose of insulin for 10 min (200 mU ⋅ m−2 ⋅ min−1 for 5 min, followed by 100 mU ⋅ m−2 ⋅ min−1 for 5 min). Euglycemia (∼5.6 mmol/L) was maintained by a variable rate infusion of 20% dextrose enriched to ∼2.5% with [6,6 2H2]glucose. Blood and saliva samples were obtained before beginning the isotope infusion to determine NEAC and background substrate tracer-to-tracee ratios. Blood samples were obtained every 10 min during the last 30 min of the basal period, and insulin infusion was performed to determine glucose kinetics and plasma insulin concentrations. Muscle samples were obtained from the quadriceps femoris muscle 60 min after starting the glucose tracer infusion, as described previously (11).
Thirteen subjects in the high-UA group were readmitted to the Clinical Research Unit 2–21 days after completing the first metabolic study. At 1800 h, rasburicase (Elitek; Sanofi-Synthelabo, Inc.) (0.19 mg/kg fat-free mass) was infused over 30 min. Rasburicase is a recombinant urate oxidase that reduces UA by ∼90% within 4 h after infusion (12). At 1900 h, a standard meal was served, and the subjects completed the same metabolic study that had been performed previously, but muscle biopsy samples were obtained in only seven subjects.
Sample Analyses
Serum UA concentrations were determined by using a uricase-colorimetric method (Roche Diagnostic Corporation, Indianapolis, IN). Plasma insulin concentration was measured by using a chemiluminescent immunoassay (Immulite 1000; Diagnostic Products Corporation, Los Angeles, CA). Plasma glucose tracer-to-tracee ratio was determined by using gas chromatography–mass spectrometry (13). Antioxidant capacity in plasma and saliva was determined by using the TRAP assay (based on the ability of plasma/saliva to prevent the decay of R-phycoerythrin induced by peroxyl radicals) and the FRAP assay (based on the ability of plasma/saliva to reduce ferric-tripyridyltriazine to its ferrous colored form) (14).
Urinary 8-iso-prostaglandin F2α (8-iso-PGF2α), a product of nonenzymatic peroxidation of arachidonic acid in membrane phospholipids, was measured by using mass spectrometry (15). The ratio of total carbonylated proteins to the loading control protein Ran in skeletal muscle provides an assessment of nonreversible modification of amino acid side chains by lipid peroxidation end products (16) and is a marker of intramuscular oxidative stress. Lysates were prepared and treated with hydrazide-activated biotin (Pierce, Rockford, IL) to label reactive carbonyl residues. Samples were separated by SDS-PAGE (Invitrogen, Carlsbad, CA), electroblotted to polyvinylidene fluoride (Millipore, Billerica, MA), and membranes developed with either IRDye-labeled streptavidin (carbonylated proteins; LI-COR, Lincoln, NE) or with anti-Ran antibody (Santa Cruz Biotechnology, Santa Cruz, CA) by using the LI-COR Odyssey detection system (16).
Calculations
The homeostasis model assessment of insulin resistance (HOMA-IR) and substrate kinetics were calculated as previously described (11). Whole-body (primarily skeletal muscle) insulin sensitivity was assessed as the increase in glucose Rd from plasma during insulin infusion.
Statistical Analyses
The differences in markers of oxidative stress, NEAC, and insulin-stimulated glucose Rd between groups at baseline were evaluated using a Student t test for unpaired samples, and differences between subjects with high serum UA concentrations before and after rasburicase treatment were evaluated using a Student t test for paired samples. When variables were not normally distributed and could not be normalized by using standard mathematical transformations, data sets were ranked for statistical analyses. Results are presented as the mean ± SEM. A P value ≤0.05 was considered statistically significant. Based on our own reproducibility data of insulin sensitivity, assessed by using the hyperinsulinemic-euglycemic clamp procedure in obese people (17), we estimated that 13 subjects would be needed to detect a 20% change in insulin sensitivity, with a power of 0.9 and an α value of 0.05. This effect on insulin action is a minimal clinically relevant target, and is less than the usual improvement observed after moderate weight loss (17).
Results
Metabolic Variables
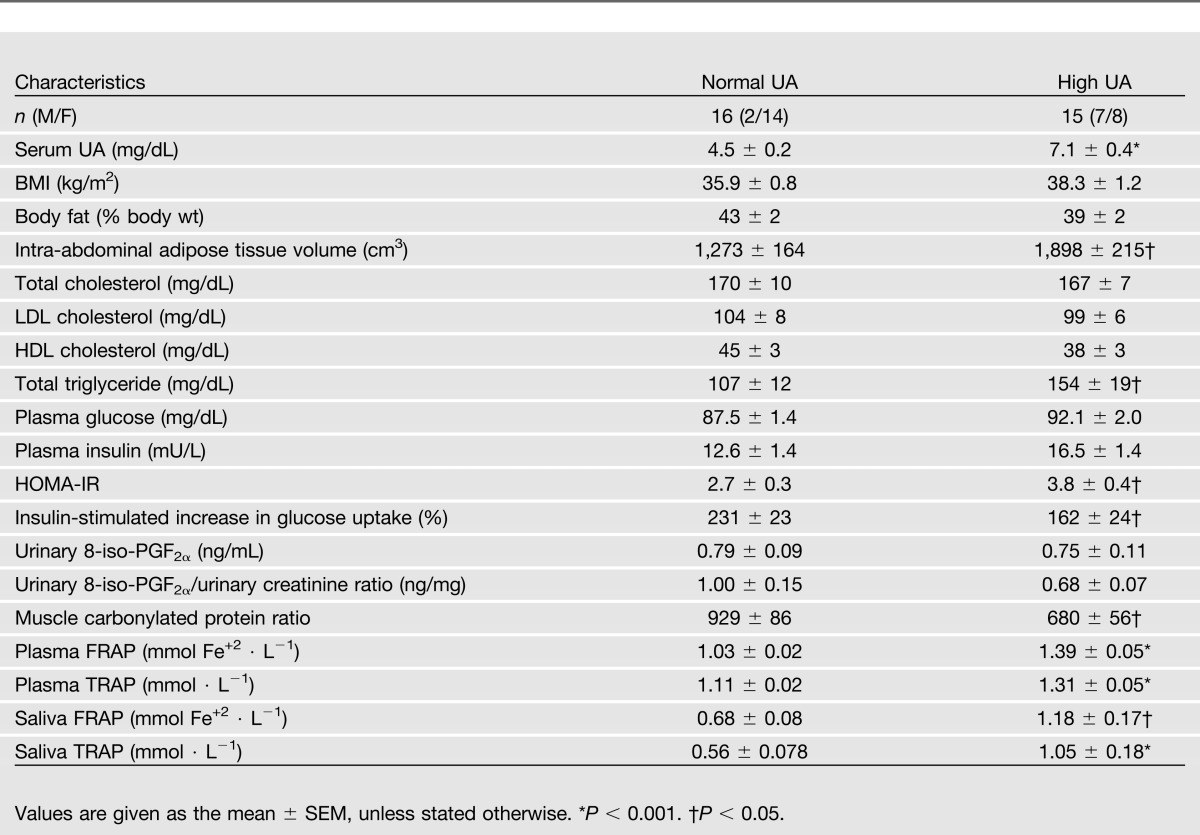
Although BMI and percentage of body fat in the high-UA group were not different than in the normal UA group, intra-abdominal adipose tissue volume was greater in the high-UA group than in the normal UA group (Table 1). Insulin sensitivity, assessed by HOMA-IR (Table 1) and by insulin-stimulated glucose disposal (Table 1), was greater in subjects with normal than in those with high serum UA concentrations. In the high-UA group, treatment with rasburicase caused a marked decrease in serum UA concentrations (from 7.1 ± 0.5 to 0.0 ± 0.0 mg/dL, P < 0.001) but did not affect HOMA-IR (from 3.7 ± 0.4 to 3.6 ± 0.3, P = 0.828) or insulin-stimulated glucose disposal (from 168 ± 25% to 156 ± 17%, P = 0.468).
Table 1.
Body composition and metabolic characteristics of subjects with normal or high serum UA concentrations
Antioxidant Capacity and Oxidative Stress
Baseline plasma and saliva NEAC, assessed as TRAP and FRAP, were greater in subjects with high UA than those with normal serum UA concentrations (Table 1). Rasburicase treatment caused a marked decrease in both plasma and saliva NEAC values (Fig. 1). Baseline urinary 8-iso-PGF2α levels and urinary 8-iso-PGF2α/urinary creatinine ratio in the high-UA group were not significantly different than those in the normal UA group (Table 1). Rasburicase treatment caused a significant (P < 0.05) increase in urinary 8-iso-PGF2α/creatinine ratio and a trend toward an increase in urinary 8-iso-PGF2α levels (P = 0.072) (Fig. 2A and B). Skeletal muscle protein carbonylation was lower in subjects who had high UA compared with those who had normal serum UA concentration (Table 1) and increased significantly after rasburicase infusion (Fig. 2C).
Figure 1.

Effect of lowering UA levels with rasburicase infusion on plasma and saliva antioxidant capacity, measured as the FRAP and the TRAP, in subjects with high basal serum UA concentrations. Values are the mean ± SEM. *P < 0.05.
Figure 2.

Effect of lowering UA concentrations with rasburicase infusion on markers of systemic (A and B) and skeletal muscle (C) oxidative stress in subjects with high basal serum UA concentration. Values are the mean ± SEM. *P < 0.05, †P = 0.072.
Discussion
Oxidative stress is purported to be involved in the pathogenesis of insulin resistance associated with obesity (1–3). Therefore, we hypothesized that an experimentally induced reduction in levels of serum UA, which is a major circulating antioxidant, would affect antioxidant capacity, markers of oxidative stress, and insulin sensitivity. We found that a marked decrease in serum UA levels caused a decrease in serum and saliva antioxidant capacity, assessed by TRAP and FRAP assays, and an increase in oxidative stress, assessed by measuring urinary isoprostanes and skeletal muscle protein carbonylation. However, the increase in oxidative stress did not have a significant effect on insulin sensitivity. These data demonstrate that circulating UA is a potent antioxidant, but oxidative stress is not a major cause of insulin resistance in obese people.
Obesity and the metabolic syndrome are associated with elevated serum UA concentrations (7–9). Data from a series of studies have shown a strong and independent correlation between serum UA and insulin resistance in subjects with the metabolic syndrome (8,18), and that serum UA concentration is a strong predictor of the future development of diabetes (19). We also found that subjects with high serum UA concentrations were more insulin-resistant than those with normal serum UA concentrations. However, our data suggest that the association between circulating UA and insulin resistance does not represent a causal relationship, because a marked reduction in serum UA levels did not significantly affect whole-body insulin sensitivity. The reason for the increase in serum UA levels associated with obesity and metabolic dysfunction is not known, but several mechanisms could be responsible, including the following: 1) increased intake of dietary purines, alcohol, and fructose, which produce UA (20,21); 2) impaired renal function and renal microvascular disease, which can increase UA production and/or decrease UA clearance (22); and 3) hyperinsulinemia, which increases renal UA reabsorption (23). It is possible that an increase in UA concentration is a protective mechanism to attenuate the adverse effects of an increase in oxidative stress (1,4,24). It has been hypothesized that the evolutionary loss of the uricase gene and a concomitant increase in serum UA concentrations in nonhuman and human primates provide a mechanism for increasing antioxidant capacity to compensate for the lost ability to synthesize ascorbic acid, a potent antioxidant (6,25). We found that our insulin-resistant subjects who had high serum UA concentrations had greater antioxidant capacity, but similar or lower markers of oxidative stress, than our obese and more insulin-sensitive subjects with normal serum UA concentrations. Moreover, the experimentally induced decrease in serum UA concentration resulted in a decrease in antioxidant capacity with a concomitant increase in markers of oxidative stress. These data suggest that high serum UA concentration prevents excessive oxidative stress in obese insulin-resistant people.
Our data shed light on whether oxidative stress is a clinically relevant cause of insulin resistance in obese people. We found that an acute increase in oxidative stress, induced by a marked reduction in circulating UA concentration, did not cause a significant change in insulin sensitivity. Although these data suggest that oxidative stress is not an important factor in the pathogenesis of insulin resistance, we cannot exclude the possibility that we were unable to detect small changes in insulin sensitivity or that a chronic reduction in UA concentration and an increase in oxidative stress might have a more profound effect on insulin action. Moreover, we cannot exclude the possibility that rasburicase might have adverse UA-independent effects on both oxidative stress and insulin sensitivity. In addition, although our data demonstrate that short-term reduction of serum UA in obese subjects with high, but normal, UA concentrations does not have a therapeutic effect on insulin action, this does not mean that pharmacological reduction of hyperuricemia does not have clinical benefits in specific patient populations (26,27).
The results from the current study demonstrate that UA is a major circulating antioxidant in obese people and might provide a protective mechanism to prevent systemic oxidative damage by free radicals. Although an increase in oxidative stress is associated with insulin resistance in obese people, our data suggest that oxidative stress does not have important effects on insulin action in vivo.
Article Information
Acknowledgments. The authors thank Melisa Moore and Martha Hessler of the Center for Human Nutrition, Washington University School of Medicine, for help with subject recruitment; Freida Custodio and Jennifer Shew of the Center for Human Nutrition, Washington University School of Medicine, for technical assistance; and the staff of the Clinical Research Unit, Washington University School of Medicine, for help in performing the studies.
Funding. This study was supported by National Institutes of Health grants DK-37948 and DK-56341 (Nutrition Obesity Research Center), UL1 RR024992 (Clinical and Translational Science Award), RR-00954 (Biomedical Mass Spectrometry Resource), and PO1 DK-58398.
Duality of Interest. This study was also supported by a grant from Sanofi. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. E.F. contributed to the design of the study, performed the metabolic studies, collated and analyzed data, and wrote the manuscript. M.S. contributed to the design and discussion of the study, performed the antioxidant capacity analyses, and reviewed and edited the manuscript. I.C.B. performed the antioxidant capacity analyses, and reviewed and edited the manuscript. S.L.H. performed the oxidative stress analyses, and reviewed and edited the manuscript. S.K. contributed to the design of the study, obtained funding, reviewed the data, contributed to the discussion, and reviewed and edited the manuscript. S.K. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Al-Aubaidy HA, Jelinek HF. Oxidative DNA damage and obesity in type 2 diabetes mellitus. Eur J Endocrinol 2011;164:899–904 [DOI] [PubMed] [Google Scholar]
- 2.Maddux BA, See W, Lawrence JC, Jr, Goldfine AL, Goldfine ID, Evans JL. Protection against oxidative stress-induced insulin resistance in rat L6 muscle cells by mircomolar concentrations of α-lipoic acid. Diabetes 2001;50:404–410 [DOI] [PubMed] [Google Scholar]
- 3.Rudich A, Tirosh A, Potashnik R, Hemi R, Kanety H, Bashan N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes 1998;47:1562–1569 [DOI] [PubMed] [Google Scholar]
- 4.Furukawa S, Fujita T, Shimabukuro M, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 2004;114:1752–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsuoka T, Kajimoto Y, Watada H, et al. Glycation-dependent, reactive oxygen species-mediated suppression of the insulin gene promoter activity in HIT cells. J Clin Invest 1997;99:144–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A 1981;78:6858–6862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonora E, Targher G, Zenere MB, et al. Relationship of uric acid concentration to cardiovascular risk factors in young men. Role of obesity and central fat distribution. The Verona Young Men Atherosclerosis Risk Factors Study. Int J Obes Relat Metab Disord 1996;20:975–980 [PubMed] [Google Scholar]
- 8.Yoo TW, Sung KC, Shin HS, et al. Relationship between serum uric acid concentration and insulin resistance and metabolic syndrome. Circ J 2005;69:928–933 [DOI] [PubMed] [Google Scholar]
- 9.Klein BE, Klein R, Lee KE. Components of the metabolic syndrome and risk of cardiovascular disease and diabetes in Beaver Dam. Diabetes Care 2002;25:1790–1794 [DOI] [PubMed] [Google Scholar]
- 10.McGuire EA, Helderman JH, Tobin JD, Andres R, Berman M. Effects of arterial versus venous sampling on analysis of glucose kinetics in man. J Appl Physiol 1976;41:565–573 [DOI] [PubMed] [Google Scholar]
- 11.Fabbrini E, Cella M, McCartney SA, et al. Association between specific adipose tissue CD4(+) T-cell populations and insulin resistance in obese individuals. Gastroenterology 2013;145:366–374.e1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pui CH, Mahmoud HH, Wiley JM, et al. Recombinant urate oxidase for the prophylaxis or treatment of hyperuricemia in patients with leukemia or lymphoma. J Clin Oncol 2001;19:697–704 [DOI] [PubMed] [Google Scholar]
- 13.Patterson BW, Zhao G, Klein S. Improved accuracy and precision of gas chromatography/mass spectrometry measurements for metabolic tracers. Metabolism 1998;47:706–712 [DOI] [PubMed] [Google Scholar]
- 14.Serafini M, Del Rio D. Understanding the association between dietary antioxidants, redox status and disease: is the total antioxidant capacity the right tool? Redox Rep 2004;9:145–152 [DOI] [PubMed] [Google Scholar]
- 15.Morrow JD. Quantification of isoprostanes as indices of oxidant stress and the risk of atherosclerosis in humans. Arterioscler Thromb Vasc Biol 2005;25:279–286 [DOI] [PubMed] [Google Scholar]
- 16.Curtis JM, Grimsrud PA, Wright WS, et al. Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes 2010;59:1132–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magkos F, Fabbrini E, Korenblat K, Okunade AL, Patterson BW, Klein S. Reproducibility of glucose, fatty acid and VLDL kinetics and multi-organ insulin sensitivity in obese subjects with non-alcoholic fatty liver disease. Int J Obes (Lond) 2011;35:1233–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vuorinen-Markkola H, Yki-Järvinen H. Hyperuricemia and insulin resistance. J Clin Endocrinol Metab 1994;78:25–29 [DOI] [PubMed] [Google Scholar]
- 19.Bhole V, Choi JW, Kim SW, de Vera M, Choi H. Serum uric acid levels and the risk of type 2 diabetes: a prospective study. Am J Med 2010;123:957–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emmerson BT. The management of gout. N Engl J Med 1996;334:445–451 [DOI] [PubMed] [Google Scholar]
- 21.Johnson RJ, Andrews P, Benner SA, Oliver W. Theodore E. Woodward award. The evolution of obesity: insights from the mid-Miocene. Trans Am Clin Climatol Assoc 2010;121:295–305 [PMC free article] [PubMed] [Google Scholar]
- 22.Messerli FH, Frohlich ED, Dreslinski GR, Suarez DH, Aristimuno GG. Serum uric acid in essential hypertension: an indicator of renal vascular involvement. Ann Intern Med 1980;93:817–821 [DOI] [PubMed] [Google Scholar]
- 23.Quiñones Galvan A, Natali A, Baldi S, et al. Effect of insulin on uric acid excretion in humans. Am J Physiol 1995;268:E1–E5 [DOI] [PubMed] [Google Scholar]
- 24.Miglio C, Peluso I, Raguzzini A, et al. Fruit juice drinks prevent endogenous antioxidant response to high-fat meal ingestion. Br J Nutr. 12 August 2013. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 25.Sevanian A, Davies KJ, Hochstein P. Serum urate as an antioxidant for ascorbic acid. Am J Clin Nutr 1991;54(Suppl.):1129S–1134S [DOI] [PubMed] [Google Scholar]
- 26.Fink HA, Wilt TJ, Eidman KE, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med 2013;158:535–543 [DOI] [PubMed] [Google Scholar]
- 27.Wei L, Mackenzie IS, Chen Y, Struthers AD, MacDonald TM. Impact of allopurinol use on urate concentration and cardiovascular outcome. Br J Clin Pharmacol 2011;71:600–607 [DOI] [PMC free article] [PubMed] [Google Scholar]