

Abstract

The l-arabinose isomerase (l-AI) and the d-xylose isomerase (d-XI) encoding genes from Lactobacillus reuteri (DSMZ 17509) were cloned and overexpressed in Escherichia coli BL21 (DE3). The proteins were purified to homogeneity by one-step affinity chromatography and characterized biochemically. l-AI displayed maximum activity at 65 °C and pH 6.0, whereas d-XI showed maximum activity at 65 °C and pH 5.0. Both enzymes require divalent metal ions. The genes were also ligated into the inducible lactobacillal expression vectors pSIP409 and pSIP609, the latter containing a food grade auxotrophy marker instead of an antibiotic resistance marker, and the l-AI- and d-XI-encoding sequences/genes were coexpressed in the food grade host Lactobacillus plantarum. The recombinant enzymes were tested for applications in carbohydrate conversion reactions of industrial relevance. The purified l-AI converted d-galactose to d-tagatose with a maximum conversion rate of 35%, and the d-XI isomerized d-glucose to d-fructose with a maximum conversion rate of 48% at 60 °C.
Keywords: l-arabinose isomerase, d-xylose (glucose) isomerase, d-tagatose, d-fructose, food grade, Lactobacillus
Introduction
l-Arabinose isomerase (l-AI; EC 5.3.1.4) catalyzes the reversible isomerization of l-arabinose into l-ribulose. Due to the ability to convert d-galactose into d-tagatose, this enzyme is also referred to as d-galactose isomerase.1
d-Tagatose, an isomer of d-galactose, has physical properties similar to those of sucrose.2 This low-calorie sweetener has numerous health and medical benefits, including prevention of dental caries, regulation of intestinal flora, and reduction of symptoms of type 2 diabetes.1 The sweetness of d-tagatose is comparable to that of sucrose, and it has received “generally recognized as safe” (GRAS) status from the U.S. Food and Drug Administration.3d-Tagatose can be produced from d-galactose by a chemical method using a calcium catalyst, but this process has some disadvantages including complex purification steps and the formation of chemical waste and byproducts. Therefore, biological production of d-tagatose using l-AI has been studied intensively in recent years.4 Several enzyme sources have been reported or patented including the l-AI from Escherichia coli,5G. stearothermophilus,6 and L. pentosus(7,8) High temperature improves the enzymatic isomerization because the reaction equilibrium is shifted toward d-tagatose. Acidic conditions have the advantage that the formation of undesirable byproducts is prevented.9,10
d-Xylose isomerase (d-xylose ketol-isomerase; d-XI; EC 5.3.1.5) catalyzes the reversible isomerization of d-xylose into d-xylulose. This enzyme is also referred to as glucose isomerase (GI) due to its ability to convert d-glucose to d-fructose, a reaction of commercial importance in the production of high- fructose corn syrup (HFCS). The chemical conversion (Lobry de Bruyn-Alberda van Ekenstein transformation) is nonspecific and leads to the formation of nonmetabolizable sugars (e.g., psicose) and other undesirable colored products. These disadvantages can be overcome by enzymatic isomerization.11 Several enzyme sources have been reported and patented, including Actinoplanes missouriensis,12Bacillus licheniformis,13 and S. rubiginosus(11,14) HFCS is used in the food industry because it does not cause a crystallization problem like sucrose. Further advantages are a lower price (10–20%) and higher sweetness (1.3 times) compared to sucrose.
An enzymatic procedure for the direct conversion of lactose into d-tagatose and d-glucose was described by Jørgensen et al.15 In this reaction the enzyme β-glycosidase catalyzes the hydrolysis of lactoserum, a byproduct of the manufacture of cheese and therefore a cheap source for d-galactose production at industrial scale. The resulting d-galactose is further isomerized to d-tagatose by l-AI and the residual d-glucose has to be removed. Alternatively, the enzyme d-XI could catalyze the conversion of d-glucose to d-fructose.2
Lactic acid bacteria (LAB) are extensively used in the food industry16 and are generally harmless to human beings.16,17 These Gram-positive bacteria are used in dairy production, wineries, and other industrial food fermentation processes. Furthermore, they are extensively used in the animal feed industry.18
Members of the genera Lactobacillus and Lactococcus are ideal candidates as safe cell factories.19,20Lactobacillus plantarum, a promising host for heterologous gene expression, was already successfully used for the production of β-galactosidase from Lactobacillus delbrueckii(21) and chitinase from Bacillus licheniformis.22 Available inducible expression systems23 are based on quorum-sensing mechanisms involved in the production of sakacins A and P.24−26 More recently, the erythromycin antibiotic resistance gene (erm) was replaced by the homologous alanine racemase gene (alr), and an alanine racemase-deficient host strain was constructed that obviates the undesirable fermentation in the presence of antibiotics and is suitable for applications in the food industry.19
In this study we present the heterologous expression of the l-AI and d-XI-encoding genes from the food grade microorganism L. reuteri in E. coli, the most commonly used organism for heterologous protein production, and the characterization of these novel enzymes.27L. reuteri, a heterofermentative and symbiotic species, is well adapted to colonize human and animal gastrointestinal tracts. Among probiotic microorganisms L. reuteri is unique due to its ability to produce and secrete the antimicrobial substance reuterin.28 Furthermore, we coexpressed the genes in the food grade host L. plantarum using two different expression systems and performed comprehensive studies on the conversion of d-glucose and d-galactose, respectively.
Materials and Methods
Chemicals and Enzymes
All chemicals were of the highest purity available and purchased from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise stated. Restriction endonucleases, T4 DNA ligase, and Phusion High-Fidelity DNA polymerase were obtained from Thermo Fisher Scientific Biosciences (St. Leon-Rot, Germany), whereas GoTaq DNA polymerase was from Promega (Madison, WI, USA). Phenylmethanesulfonyl fluoride (PMSF) was obtained from Fluka (Buchs, Switzerland), and chromatographic materials were obtained from GE Healthcare (Chalfont St. Giles, UK).
Bacterial Strains and Culture Conditions
The bacterial strains and plasmids used in this study are listed in Table 1. Lactobacillus reuteri DSMZ 17509 (strain designation 100-23) was obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). Lactobacillus strains were cultivated in deMan–Rogosa–Sharpe (MRS) medium at 30 °C without agitation. E. coli strain NEB5α (New England Biolabs, Ipswich, MA, USA), MB2159 (d-alanine auxotrophe), and BL21 Star (DE3) (Invitrogen, Carlsbad, CA, USA) were grown in Luria–Bertani (LB) medium at 37 °C with shaking at 125 rpm. When needed, media were supplemented with ampicillin at a concentration of 100 μg/mL and erythromycin at concentrations of 5 and 200 μg/mL for L. plantarum and E. coli, respectively. E. coli MB2159 and L. plantarum strain TLG02 were cultivated by adding 200 μg/mL d-alanine to the respective media. Solid media were prepared by adding 1.5% agar to the respective media.
Table 1. Strains and Plasmids Used in This Worka.
| strain or plasmid | relevant characteristics | ref |
|---|---|---|
| strains | ||
| L. reuteri | ||
| DSMZ 17509 | source for l-AI and d-XI encoding genes | DSMZ |
| L. plantarum | ||
| WCFS1 | wild type | (49) |
| TLG02 | Δalr, d-alanine auxotroph | (19) |
| E. coli | ||
| MB2159 | d-alanine auxotroph, cloning host | (50) |
| NEB5α | cloning host | New England Biolabs |
| BL21 Star (DE3) | expression host | Invitrogen |
| plasmids | ||
| pET-21a(+) | Ampr, T7 promoter, C-terminal His-Tag | Novagen |
| pSIP409 | Ermr, pSIP401 derivative, PsppQ | (25) |
| pSIP609 | pSIP409 derivative, alr replaced with erm | (19) |
| pPW002 | l-AI from L. reuteri in pET-21a(+) | this work |
| pPW004 | d-XI from L. reuteri in pET-21a(+) | this work |
| pPS1 | pSIP609 derivative, NcoI replaced with NheI | this work |
| pPW-015 | pSIP409 derivative, NcoI replaced with NheI | this work |
| pPS2 | l-AI and d-XI from L. reuteri in pPS1 | this work |
| pPS3 | l-AI and d-XI from L. reuteri in pPW-015 | this work |
| pPW116 | l-AI from L. reuteri in pJET1.2 | this work |
| pPW114 | d-XI from L. reuteri in pJET1.2 | this work |
Ampr, ampicillin resistance; Ermr, erythromycin resistance; alr, alanine racemase encoding gene; erm, erythromycin resistance gene.
DNA Amplification and Subcloning
DNA amplification was performed with Phusion High-Fidelity DNA Polymerase according to the manufacturer’s instructions. The oligonucleotide primers were obtained from VBC Biotech (Vienna, Austria) and are listed in Table 2. PCR products and restriction enzyme digested DNA were purified with GFX PCR DNA and a Gel Band Purification Kit (GE Healthcare) as recommended by the supplier. When needed, the PCR fragments were subcloned into the pJET1.2 vector (CloneJET PCR cloning kit, Thermo Fisher Scientific Biosciences), and E. coli was used as host for plasmid propagation before transformation into Lactobacillus. The amplified sequences were verified by a commercial sequencing service (LGC Genomics, Berlin, Germany).
Table 2. Primers Used in This Studya.
| primer | sequence (5′–3′) |
|---|---|
| araA_LbReu_fwd1 | ATGGATATTAAGAATTACGAATTTTGGTTT |
| araA_LbReu_rev1 | CTATTCAATAAAATCAACTTTAACA |
| xylA_LbReu_fwd1 | TCATCATGATAAGGATAAAGTTTTGG |
| xylA_LbReu_rev1 | ATTGCAATCGTTGTTCCATAAAGCCAATC |
| pSIP_SD_NheI_fwd1 | TACGTCCTGTAGAAACCCCAAC |
| pSIP409_SD_NheI_rev1 | CTACAGGACGTAGCTAGCCTAAAATCTCCTTGT |
| AraReuNheI_fwd1 | TTTTCGCTAGCATGGATATTAAGAATTACG |
| AraReuEcoRI_His6 | AACGCGAATTCGTTTCAATAAAATCAACTT |
| XylReuNheI_fwd1 | ATTTTGCTAGCATGGCTGATTTATGGA |
| XylReuEcoRI_His6 | CCGTGGAATTCGTCTTAGCAAGCGTTTC |
| AraReu_OE_rev1 | TAAAACTCCTGAACTAGTGGTGGTGGTGGTGGTGTTCAATAAAATCAAC |
| XylReu_OE_fwd1 | TTCAGGAGATTTTAATGGCTGATTTATGGAATAA |
| XylReu_OE_rev1 | CGAATGAATTCTTAGTGGTGGTGGTGGTGGTGCTTAGCAAGCGTTTC |
The restriction sites are underlined.
The araA gene (NCBI reference sequence for the genome: NZ_AAPZ02000002.1) was amplified by colony-PCR using the primers araA_LbReu_fwd1 and araA_LbReu_rev1. The xylA gene was amplified with the primer pair xylA_LbReu_fwd1 and xylA_LbReu_rev1. Each PCR product was ligated into the pJET1.2 vector as recommended by the supplier, resulting in plasmids pPW116 and pPW114, respectively. Each plasmid was transformed into chemically competent E. coli NEB5α cells according to the manufacturer’s instructions.
Plasmid Construction and Expression in E. coli
The araA and xylA genes were amplified using the primer pairs AraReuNheI_fwd1/AraReuEcoRI_His6 and XylReuNheI_fwd1/XylReuEcoRI_His6 as well as pPW116 and pPW114 as templates, respectively. The resulting PCR fragments were cut with the restriction enzymes NheI and EcoRI and ligated into the equally treated expression vector pET21-a(+) (Novagen, Darmstadt, Germany) in frame with the C-terminal His6-tag encoded by the vector. The resulting plasmids pPW002 and pPW004 were transformed into electrocompetent E. coli BL21 Star (DE3) according to the method of Inoue et al.29
Plasmid Construction and Expression in L. plantarum
The NcoI restriction site on the pSIP609 and pSIP409 vector, respectively, was replaced by a NheI restriction site by site-directed mutagenesis.30 A PCR was peformed using the primer pair pSIP_SD_NheI_fwd1 and pSIP409_SD_NheI_rev1. Subsequent digestion with DpnI and transformation into E. coli resulted in the plasmids pPS1 (pSIP609 derivative) and pPW015 (pSIP409 derivative), respectively.30 The fusion of the araA and the xylA genes was performed according to the overlap extension method.31 PCR reactions were done with the primer pairs XylReu_OE_fwd1/XylReu_OE_rev1 (pPW114 as template) and AraReuNheI_fwd1/AraReu_OE_rev1 (pPW116 as template). The resulting PCR products were purified and used as templates for a third reaction with the primers AraReuNheI_fwd1 and XylReu_OE_rev1. The resulting amplicon was digested with NheI and EcoRI and ligated into the equally treated vectors pPS1 and pPW-015, resulting in plasmids pPS2 and pPS3, respectively. These plasmids were transformed into E. coli MB2159 and NEB5α, respectively, for plasmid propagation. Competent cells of E. coli MB2159 were prepared and transformed according to the method of Inoue et al.29 The isolated plasmids were transformed into electrocompetent L. plantarum TLG02 and WCFS1 cells, respectively, according to the method of Josson et al.32
Sequence Analysis
The translated amino acid sequences were analyzed using the programs Translate and Compute pI/MW at http://www.expasy.org/.33
Expression in E. coli.
E. coli BL21 Star (DE3) carrying the plasmids pPW002 and pPW004, respectively, was grown overnight at 37 °C and 125 rpm in 3 mL of LB medium containing 100 μg/mL ampicillin. Two hundred milliliters of Terrific-Broth (TB) medium supplemented with 100 μg/mL in 1 L baffled shaking flasks were inoculated with 1 mL of the starter culture. The cells were incubated at 37 °C until an optical density at 600 nm (OD600) between 0.4 and 0.6 was reached. Lactose in a final concentration of 5 g/L was added to the culture for induction of the heterologous expression, and the cultures were incubated further at 25 °C for 20 h. Cells were then harvested by centrifugation (4000g, 20 min, 4 °C), resuspended in buffer A (50 mM sodium phosphate buffer, 500 mM NaCl, 5 mM imidazole, pH 6.0) supplemented with 5.7 mM PMSF, and disrupted by using a French press (Aminco, Silver Spring, MD, USA). Cell debris was removed by centrifugation (30000g, 30 min, 4 °C), and the supernatant was recovered as crude extract.
Expression in L. plantarum
L. plantarum carrying the plasmids pPS2 and pPS3, respectively, was grown overnight at 37 °C in 10 mL of MRS medium containing 5 μg/mL erythromycin in the case of the latter. These starter cultures were used to inoculate 1 L of the same media. The cultures were incubated at 30 °C until an OD600 between 0.3 and 0.4 was reached. Heterologous expression was induced by adding 25 ng/mL of the peptide pheromone IP-673 (supplied by the Molecular Biology Unit, University of Newcastle-upon-Tyne, UK). The cultures were incubated further at 30 °C for 20 h. Cells were washed twice by buffer A, and the cell pellet was resuspended in the same buffer containing 5.7 mM PMSF. The cells were homogenized by using a French press, and the cell debris was removed by centrifugation to obtain the crude extract.
Protein Purification
The crude extract was purified by one-step immobilized metal affinity chromatography (IMAC) on a prepacked 20 mL HisPrep FF 16/10 column (GE Healthcare). The column was pre-equilibrated with buffer A (see above), and the crude extract was applied at a rate of 2.5 mL/min. After an additional washing step (3 column volumes), the enzyme was eluted at the same rate by using a linear gradient of 5 mM–1 M imidazole in 10 column volumes. Active fractions were pooled, desalted, and concentrated in 50 mM sodium phosphate buffer (pH 6.0) using Amicon Ultra-15 centrifugal filter units (30 kDa cutoff; Millipore, Billerica, MA, USA) and stored at 4 °C. d-XI containing fractions were concentrated in 50 mM sodium acetate buffer (pH 5.0).
Protein Quantification, Electrophoresis, and Molecular Mass Determination
Protein concentration was determined according to the method of Bradford34 using a BSA standard curve and a prefabricated assay solution (Bio-Rad). SDS-PAGE was carried out using Mini-PROTEAN TGX precast gradient gels (4–15%) and Precision Plus Protein Unstained Standard. Bio-Safe Coomassie was used for visualization of the protein bands (Bio-Rad). The theoretical mass was derived from the ExPASy ProtParam tool (http://web.expasy.org/protparam/) and confirmed by size exclusion chromatography (SEC) using a 190 mL Sephacryl S300 column (GE Healthcare) equilibrated with 50 mM potassium phosphate buffer (pH 7.0) containing 150 mM NaCl and the molecular weight marker kit for gel filtration (Sigma-Aldrich).
Enzyme Assays and HPLC Analysis
l-AI activity was measured by determing the amount of formed l-ribulose or d-tagatose. The reaction mixture contained 50 mM l-arabinose or d-galactose, 1 mM CoCl2, 0.5 mM MnCl2, 50 μL of 0.5–1.0 mg/mL enzyme preparation, and 50 mM sodium phosphate buffer (pH 6.0) to bring the final volume to 0.5 mL. The reaction mixture was incubated at 65 °C for 10 min followed by heating the mixture to 95 °C for 5 min to stop the reaction. The amount of l-ribulose or d-tagatose was determined either by HPLC analysis or colorimetrically using the cysteine–carbazole–sulfuric-acid method35 with slight modifications. Fifty microliters of freshly prepared 1.5% (w/v) l-cysteine–HCl was added to 250 μL of reaction mixture (in a suitable dilution), 1.5 mL of ≈70% sulfuric acid and 50 μL of 0.12% (w/v) carbazole in ethanol were added, and the absorbance at 560 nm was measured after 1 h of incubation at room temperature. HPLC analysis was performed on a Dionex Summit HPLC system (Thermo Fisher Scientific) fitted with a Shodex RI-101 refractive index detector (Shoko Scientific, Yokohama, Japan) using an Aminex HPX 87-K column (Bio-Rad) with a guard column. Samples and standards were eluted at 80 °C with 1 g/L boric acid (pH 8.5, 0.5 mL/min). For calculation of the reaction product(s) l-arabinose, d-galactose, l-ribulose, and d-tagatose standards were included in the run. One unit of l-arabinose isomerase activity is defined as the amount of enzyme needed to produce 1 μmol of l-ribulose or d-tagatose per minute under the assay conditions.
d-XI activity was measured by the determination of the amount of formed d-xylulose (or d-fructose). The reaction mixture contained 50 mM d-xylose, 0.5 mM CoCl2, 0.5 mM MnCl2, 50 μL of 1 mg/mL enzyme preparation, and 50 mM sodium acetate buffer (pH 5.0) to bring the final volume to 0.5 mL. The reaction mixture was incubated at 65 °C for 10 min, and the reaction was stopped by heating the mixture at 95 °C for 5 min. For assaying the glucose isomerase activity, the reaction mixture contained 400 mM d-glucose, 5 mM CoCl2, 5 mM MnCl2, 5 mM MgSO4, 50 μL of 5 mg/mL enzyme preparation, and 50 mM sodium acetate buffer (pH 5.0) to bring the final volume to 0.5 mL. The reaction mixture was incubated at 60 °C for 10 min. The amount of d-xylulose or d-fructose was determined either by HPLC analysis or using the cysteine–carbazole–sulfuric-acid method35 with slight modifications (see above). For HPLC analysis and calculation of the reaction products d-xylose, d-glucose, d-xylulose, and d-fructose standards were included in the run. One unit of d-xylose isomerase activity is defined as the amount of enzyme needed to produce 1 μmol of d-xylulose or d-fructose per minute under the assay conditions.
Effect of Temperature, pH, and Divalent Metal Ions on Enzyme Activity
The temperature optimum was measured by assaying the enzyme samples over the range of 30–90 °C, at pH 5 (d-XI) and pH 6 (l-AI). Three buffer systems (sodium acetate/sodium phosphate/Tris-HCl) were used for measuring the pH optimum of enzyme activity at 65 °C with d-xylose (d-XI) and l-arabinose (l-AI) as substrate. The effects of various metal ions were determined by the addition of 1 mM MgSO4, CoCl2, or MnCl2 (or more than one in various concentrations) and assaying d-XI and l-AI activity, respectively, under standard conditions without 0.5 mM MnCl2 and 0.5 mM CoCl2 (d-XI) and without 0.5 mM MnCl2 and 1 mM CoCl2 (l-AI).
Determination of Kinetic Parameters
The kinetic paramters of l-AI were determined in 50 mM sodium phosphate buffer (pH 6.0) containing 0.5 mM MnCl2 and 1 mM CoCl2 and 5–800 mM l-arabinose (10–800 mM d-galactose). The reaction mixtures were incubated for 10 min at 65 °C. The kinetic parameters of d-XI were determined in 50 mM sodium acetate buffer (pH 5.0) containing 0.5 mM MnCl2, 0.5 mM CoCl2 (5 mM CoCl2, 5 mM MnCl2, 5 mM MgSO4), and 1–500 mM d-xylose (25–1500 mM d-glucose). The reaction mixtures were incubated for 10 min at 65 °C (60 °C). The observed data were fitted to the Michaelis–Menten equation, and kinetic constants were calculated by nonlinear least-squares regression. Using the molecular mass, turnover numbers (kcat) and catalytic efficiencies (kcat/Km) were calculated.
Enzymatic Conversion of d-Galactose to d-Tagatose
The conversion was carried out at 60 °C with 0.8 and 1.4 U of purified enzyme (i.e., 1.0 mg of l-AI produced in E. coli and 0.9 mg of l-AI expressed in L. plantarum), respectively, in a total volume of 1 mL. The conversion medium contained 500 mM d-galactose, 1 mM CoCl2, and 0.5 mM MnCl2 in 50 mM sodium phosphate buffer (pH 6.0). Samples were taken regularly for the analysis of d-galactose and d-tagatose by HPLC. The conversion rate represents the ratio between the formed d-tagatose and the initial d-galactose.
Enzymatic Conversion of d-Glucose to d-Fructose
The conversion was carried out at 60 °C with 2.1 and 1.1 U of purified enzyme (i.e., 1.9 mg of d-XI produced in E. coli and 0.9 mg of d-XI expressed in L. plantarum), respectively, in a total volume of 1 mL. The conversion media contained 500 mM d-glucose, 5 mM CoCl2, 5 mM MnCl2, and 5 mM MgSO4 in 50 mM sodium acetate buffer (pH 5.0). Samples were taken regularly for the analysis of d-glucose and d-fructose by HPLC. The conversion rate represents the ratio between the formed d-fructose and the initial d-glucose.
Results and Discussion
Heterologous Expression of the l-AI and d-XI Encoding Gene in E. coli
The l-AI encoding gene from L. reuteri DSMZ 17509 was successfully expressed in E. coli BL21(DE3). The nucleotide sequence contains an open reading frame of 1422 bp encoding a polypeptide of 473 amino acids. On the basis of the DNA sequence, two modified oligonucleotide primers containing restriction sites for in-frame ligation into the pET21-a(+) were designed and used to reamplify the araA sequence and to construct the expression vector pPW002. The l-AI encoding sequence was fused in frame with the C-terminal His6-tag encoded by the vector, and the construct was expressed under the control of the lactose- or IPTG-inducible T7 promoter. Approximately 137.9 U of l-AI activity/L fermentation medium was produced under the described conditions with a volumetric activity of 1.6 U/mL and a specific activity of 0.18 U/mg using d-galactose as substrate. The d-XI encoding gene from L. reuteri DSM 17509 contains an open reading frame of 1350 bp encoding a polypeptide of 449 amino acids. The xylA sequence was fused in frame with the C-terminal His6-tag encoded by the vector, and the heterologous expression resulted in approximately 780.5 U of d-XI activity/L fermentation medium with a volumetric activity of 8.9 U/mL and a specific activity of 0.57 U/mg using d-glucose as substrate.
Due to the use of an ampicillin resistance marker, the potential of the pET21-a(+) expression system is limited to food applications. Furthermore, the use of E. coli strains is not permitted in the food industry in most countries,36 due to possible endotoxin production.37
Coexpression of l-AI and d-XI Encoding Gene(s) in L. plantarum
The resulting expression vector contains the araA gene fused with a C-terminal His6-tag (CAC6) and a stop-codon (TAG). The following ribosomal binding site (RBS)38 was flanked downstream (5′) with TTC (three nucleotides downstream from the RBS) and upstream with the nucleotides ATTTTA (six nucleotides upstream of the RBS).26,39 The xylA gene was also fused with a His6-tag followed by the stop-codon (TAA). Approximately 28.6 U (18.3 U) of l-AI activity/L fermentation medium with a volumetric activity of 0.8 U/mL (0.5 U/mL) and a specific activity of 0.15 U/mg (0.08 U/mg) with d-galactose as substrate was produced in L. plantarum using the pSIP609 (pSIP409) vector. The coexpression of the xylA gene allowed production of approximately 32.3 U (55.9 U) of GI activity/L fermentation medium with a volumetric activity of 0.9 U/mL (1.6 U/mL) and a specific activity of 0.17 U/mg (0.23 U/mg) using the pSIP609 (pSIP409) expression system.
A food grade LAB strain was chosen as expression host as demonstrated by Rhimi et al.40 and Salonen et al.3 Although expression yields in E. coli are much higher, the combination of the (homologous) alanine racemase gene (alr) as selection marker and the safe cell factory L. plantarum has considerable potential for the production of ingredients and additives for the food industry.19 Compared to the erythromycin-dependent expression system pSIP409, the expression yield of the (first) araA gene was higher; in the case of the (second) xylA gene, the induction/expression worked less efficiently. However, the antibiotic-independent expression system pSIP609 allowed the successful coexpression of the genes araA and xylA from L. reuteri DSMZ 17509. The slightly higher yields with the pSIP609 system (alanine racemase as selection marker in combination with a suitable deficient host strain) are in agreement with previous data and can be explained by the avoidance of antibiotic detoxification and subsequently reduced plasmid loss.19
Purification of Recombinant l-AI and d-XI
The recombinant enzymes were purified to apparent homogeneity from cell extracts by a single-step purification protocol using immobilized metal affinity chromatography (Figure 1).
Figure 1.
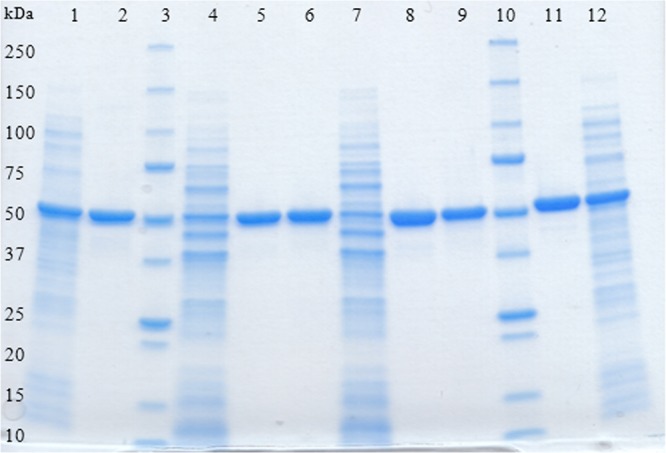
SDS-PAGE analysis of recombinant l-AI and d-XI from L. reuteri. Lanes: 1, l-AI-containing crude extract from E. coli; 2, purified l-AI (E. coli); 3, Precision Protein standard (Bio-Rad); 4, l-AI-containing crude extract from L. plantarum (pSIP609); 5, purified l-AI (pSIP609); 6, purified d-XI (pSIP609); 7, d-XI-containing crude extract from L. plantarum (pSIP409); 8, purified l-AI (pSIP409); 9, purified d-XI (pSIP409); 10, Precision Protein standard (Bio-Rad); 11, purified d-XI (E. coli); 12, d-XI-containing crude extract from E. coli.
The specific activities of the purified enzymes were 0.9 U/mg for l-AI and 1.1 U/mg for d-XI with E. coli as expression host and d-galactose and d-glucose, respectively, as substrate. The use of L. plantarum as expression host resulted in 1.6 U/mg (in the case of pSIP609 and 409) for l-AI and 1.2 U/mg (pSIP609) and 1.8 (pSIP409) for d-XI after purification. Although the coexpressed genes contained the same affinity tag, the resulting proteins are separated successfully and elute in different fractions.
Molecular masses of approximately 300 kDa (l-AI) and 190 kDa (d-XI) were determined using size exclusion chromatography. Considering the calculated molecular masses of 53.6 kDa (l-AI) and 51.04 kDa (d-XI), these data suggest that l-AI is naturally active as a homohexamer and d-XI is a tetramer. The hexameric quaternary structure of the l-AI from L. reuteri is also observed in the enzymes from E. coli(41) and L. plantarum NC8,42 whereas many other l-AIs exhibit tetrameric structures.9,42 The tetrameric quaternary structure of the d-XI from L. reuteri is the most common form of d-XIs,43 but dimeric and trimeric forms are also described.11
Effect of Temperature, pH, and Divalent Metal Ions on Enzyme Activity
The temperature profile of purified l-AI and d-XI (expressed in E. coli), respectively, was determined from 30 to 90 °C using the standard assay according to Dische and Borenfreund.35l-AI exhibited maximum activity at 65 °C, with 90 and 97% activity at 60 and 70 °C, respectively (Figure 2). The enzyme d-XI exhibited the same temperature optimum (65 °C) and retained 85% of its maximum activity at 60 and 70 °C, respectively (Figure 2). For industrial production of d-tagatose and d-fructose, respectively, an isomerization at elevated temperature of 60–65 °C resulted in higher conversion yields, better sugar solubility, and lower risk of microbial contamination. Higher temperatures above 70 °C resulted in the formation of undesired byproducts and browning effects.1,44
Figure 2.

Effect of temperature on l-AI (solid triangles) and d-XI (open squares) activity. Relative activities are presented as functions of temperature with l-arabinose and d-xylose, respectively, as substrate. The reaction mixtures were incubated for 10 min.
The standard assay was performed using different buffers with pH ranging from 4 to 10. The pH optimum of l-AI and d-XI was found in the slightly acidic pH range (6.0 and 5.0, respectively) (Figure 3). Acidic conditions avoid the formation of undesired byproducts, a great advantage for industrial purposes.3,44
Figure 3.

Effect of pH on l-AI (solid symbols) and d-XI (open symbols) activity. Relative activities are presented as functions of pH with l-arabinose and d-xylose, respectively, as substrate. The reaction mixtures were incubated for 10 min. The buffers used were acetate (circles), phosphate (squares), and tris (triangles).
The l-AI enzyme activity was assayed at 65 °C and pH 6.0 in the presence of various cations and d-galactose as substrate (Table 3). The activity was increased by the addition of 1 mM Co2+ (3.7-fold) and 1 mM Mn2+ (1.4-fold). A mixture of 1 mM Co2+ and 0.5 mM Mn2+ increased the enzyme activity 3.9-fold. The d-XI enzyme activity was assayed at 65 °C and pH 5.0 in the presence of various cations and d-xylose as substrate (Table 3). The activity was increased by the addition of 1 mM Co2+ (1.9-fold) and 1 mM Mn2+ (2.9-fold). A mixture of 0.5 mM Co2+ and 0.5 mM Mn2+ increased the enzyme activity 3.2-fold. Glucose isomerase activity was assayed at 60 °C and pH 5.0 and was maximally enhanced by the addition of 5 mM Co2+, 5 mM Mn2+, and 5 mM Mg2+ (data not shown).
Table 3. Effect of Metal Ions on l-AI and d-XI Activitya.
| metal ion | l-AI relative activity (%) | d-XI relative activity (%) |
|---|---|---|
| none | 26 | 32 |
| Co2+ | 94 | 62 |
| Mg2+ | 25 | 36 |
| Mn2+ | 41 | 93 |
| Co2+ + Mn2+ | 100 | 100 |
CoCl2, MgSO4, and MnCl2 were added to the enzyme reaction mixture at a final concentration of 1 mM. The mixture of CoCl2 and MnCl2 was added at a final concentration of 1 or 0.5 mM (in the case of l-AI) and 0.5 or 0.5 mM (in the case of d-XI), respectively.
A number of l-arabinose and d-xylose isomerases require metal ions for (maximal) activity (and stability).4,11 The use of metal cofactors, especially Co2+, increases downstream processing costs as they have to be removed from the final product.9 For industrial purposes, independence from or a very low requirement for metal ions is advantageous.3
Kinetic Parameters/Properties
The apparent Km, Vmax, kcat, and catalytic efficiency (kcat/Km) of l-AI using d-galactose as substrate were 647 ± 109 mM, 11 ± 1 U/mg, 59 ± 5 s–1, and 0.09 mM–1 s–1, respectively. For l-arabinose the apparent Km, Vmax, kcat, and catalytic efficiency (kcat/Km) were estimated to be 633 ± 69 mM, 179 ± 10 U/mg, 959 ± 55 s–1, and 1.5 mM–1 s–1, respectively.
The high Km value for d-galactose is comparable with that of the enzyme from Bifidobacterium longum(3) and L. pentosus,7,45 whereas enzymes from other Lactobacillus strains exhibit up to 10 times lower Km values.1,9,42 The low affinity for l-arabinose is comparable with the enzyme from Pediococcus pentosaceus PC-5, which shows no activity for this substrate.10
The apparent Km, Vmax, kcat, and catalytic efficiency (kcat/Km) of d-XI using d-glucose as substrate were 1099 ± 98 mM, 4.6 ± 0.2 U/mg, 15.5 ± 0.8 s–1, and 0.014 mM–1 s–1, respectively. For d-xylose the apparent Km, Vmax, kcat, and catalytic efficiency (kcat/Km) were estimated to be 177.4 ± 5.6 mM, 43.1 ± 0.6 U/mg, 146.6 ± 1.9 s–1, and 0.83 mM–1 s–1, respectively.
The lower Km value for d-xylose than for d-glucose is in agreement with the natural function of the d-XI to produce xylulose for the pentose phosphate or phosphoketolase pathway.46 The high Km value for d-glucose is compareable to that from L. brevis.47
Conversion Experiments
l-AI converted d-galactose to d-tagatose at a conversion ratio of approximately 35% after 25 h of incubation at 60 °C (Figure 4). The use of l-AI (120 U) from the related organism L. plantarum SK-2 resulted in a conversion rate of 39% after 96 h at 35 °C,45 whereas the enzyme from strain NC8 showed a yield of 30% after 6 h at 60 °C. A similar equilibrium was observed by using d-AI from L. sakei as biocatalyst (36% after 7 h at 40 °C),9 whereas a higher conversion yield (55% after 96 h at 65 °C) can be obtained by using the enzyme (9.98 U) from L. fermentum CGMCC2921 and 50 mM substrate.1
Figure 4.

Time course of d-tagatose (black symbols) and d-fructose (gray symbols) production during l-AI- and d-XI-catalyzed isomerization of d-galactose and d-glucose, respectively: black, open circle) conversion curve at 60 °C performed with 0.9 mg of l-AI (expressed in pSIP609 L. plantarum); (black, solid circle) conversion curve at 60 °C performed with 1.0 mg of l-AI (expressed in pET21-a(+) E. coli); (gray, open circle) conversion curve at 60 °C performed with 0.9 mg of d-XI (expressed in pSIP609 L. plantarum); (gray, solid circle) conversion curve at 60 °C performed with 1.9 mg of d-XI (expressed in pET21-a(+) E. coli).
The conversion of d-glucose resulted in approximately 48% d-fructose after 25 h of incubation and the use of d-XI produced in the expression host E. coli (Figure 4). Using d-XI produced in L. plantarum resulted in approximately 38% d-fructose after 25 h of incubation (Figure 4) when approximately 1 unit (approximately 1 mg) of purified protein was used for the conversion experiment. After an incubation of 3 h, conversion rates of approximately 42% (E. coli) and 30% (L. plantarum) were obtained; thus, the reaction reached its equilibrium after 3 h. A reaction temperature at 40 °C for 3 h resulted in conversion rates of approximately 30 and 9% (data not shown). For industrial purposes an operating temperature of 60 °C is favorable due to the reasons mentioned above (i.e., limited color formation and byproduct formation). HPLC analysis revealed that no byproducts were formed, either in the conversion of d-galactose or in the case of d-glucose (data not shown).
In summary, this study demonstrates the successful expression of the l-arabinose isomerase and d-xylose isomerase encoding genes from L. reuteri in E. coli as well as the coexpression of both genes in the food grade microorganism L. plantarum. Small-scale conversion experiments demonstrate the principal suitability of both enzymes for isomerization of d-galactose and d-glucose, the hydrolysis products of lactose, under preferred industrial conditions (acidic pH and 60 °C). Neither enzyme is ideally suited for the envisaged application due to their rather unfavorable Km values as well as their dependence on divalent metal cations. However, enzymes with more favorable properties can be screened from a large number of organisms that are considered safe, or an improvement of these properties by enzyme engineering methods48 may be an alternative. The expression system based on the pSIP vector series is suitable for other lactic acid bacteria as well, and construction of alanine racemase-deficient strains containing no foreign DNA has been shown to be straightforward for organisms that are amenable to transformation.19 Higher yields can be achieved by expression in E. coli and presumably also by expression of the single genes; however, the presented coexpression of two enzymes in a food grade host–vector system offers the possibility of cost-efficient production of enzyme preparations for the conversion of mixed substrates.
Acknowledgments
We thank Dr. Martin Tangney for kindly supplying E. coli MB2159 and Cindy Lorenz for technical assistance and performing the HPLC analysis.
Glossary
Abbreviations Used
- l-AI
l-arabinose isomerase
- d-XI
d-xylose isomerase
- GI
glucose isomerase
- L.
Lactobacillus
- HCFS
high-fructose corn syrup
Supporting Information Available
Additional figures. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by the Austrian Science Fund FWF (Grant P22094 to C.K.P.).
The authors declare no competing financial interest.
Supplementary Material
References
- Xu Z.; Qing Y.; Li S.; Feng X.; Xu H.; Ouyang P. A novel l-arabinose isomerase from Lactobacillus fermentum CGMCC2921 for d-tagatose production: gene cloning, purification and characterization. J. Mol. Catal. B: Enzymatic 2011, 701–21–7. [Google Scholar]
- Rhimi M.; Messaoud E. B.; Borgi M. A.; Khadra K. B.; Bejar S. Co-expression of l-arabinose isomerase and d-glucose isomerase in E. coli and development of an efficient process producing simultaneously d-tagatose and d-fructose. Enzyme Microb. Technol. 2007, 4061531–1537. [Google Scholar]
- Salonen N.; Nyyssölä A.; Salonen K.; Turunen O. Bifidobacterium longuml-arabinose isomerase overexpression in Lactococcus lactis, purification, and characterization. Appl. Biochem. Biotechnol. 2012, 1682392–405. [DOI] [PubMed] [Google Scholar]
- Oh D. K. Tagatose: properties, applications, and biotechnological processes. Appl. Microbiol. Biotechnol. 2007, 7611–8. [DOI] [PubMed] [Google Scholar]
- Roh H. J.; Yoon S. H.; Kim P. Preparation of l-arabinose isomerase originated from Escherichia coli as a biocatalyst for d-tagatose production. Biotechnol. Lett. 2000, 223197–199. [Google Scholar]
- Kim P.; Yoon S. H.; Seo M. J.; Oh D. K.; Choi J. H. Improvement of tagatose conversion rate by genetic evolution of thermostable galactose isomerase. Biotechnol. Appl. Biochem. 2001, 34299–102. [DOI] [PubMed] [Google Scholar]
- Ibrahim O. O.; Spradlin J. E.. Process for manufacturing d-tagatose USA. U.S. Patent 6,057,135, 2000.
- Kim P. Current studies on biological tagatose production using l-arabinose isomerase: a review and future perspective. Appl. Microbiol. Biotechnol. 2004, 653243–249. [DOI] [PubMed] [Google Scholar]
- Rhimi M.; Ilhammami R.; Bajic G.; Boudebbouze S.; Maguin E.; Haser R.; Aghajari N. The acid tolerant l-arabinose isomerase from the food grade Lactobacillus sakei 23K is an attractive d-tagatose producer. Bioresour. Technol. 2010, 101239171–9177. [DOI] [PubMed] [Google Scholar]
- Men Y.; Zhu Y.; Zhang L.; Kang Z.; Izumori K.; Sun Y.; Ma Y. Enzymatic conversion of d-galactose to d-tagatose: cloning, overexpression and characterization of l-arabinose isomerase from Pediococcus pentosaceus PC-5. Microbiol. Res. 2014, 169, 171–178. [DOI] [PubMed] [Google Scholar]
- Bhosale S. H.; Rao M. B.; Deshpande V. V. Molecular and industrial aspects of glucose isomerase. Microbiol. Rev. 1996, 602280–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrabet N. T.; Van Den Brande I.; Van Den Broeck A. One-step purification of Actinoplanes missouriensisd-sylose isomerase by high-performance immobilized copper-affinity chromatography: functional analysis of surface histidine residues by site-directed mutagenesis. Biochemistry 1992, 31102690–2702. [DOI] [PubMed] [Google Scholar]
- Boguslawski G.; Rynski M. J.. Novel strain of Bacillus licheniformis useful in production of glucose isomerase and method of screening Bacillus mutants for the ability to produce glucose isomerase in the absence of xylose. U.S. Patent 4,355,103, 1982.
- Vuolanto A.; Uotila S.; Leisola M.; Visuri K. Solubility and crystallization of xylose isomerase from Streptomyces rubiginosus. J. Cryst. Growth 2003, 2573–4403–411. [Google Scholar]
- Jørgensen F.; Hansen O. C.; Stougaard P. Enzymatic conversion of d-galactose to d-tagatose: heterologous expression and characterisation of a thermostable l-arabinose isomerase from Thermoanaerobacter mathranii. Appl. Microbiol. Biotechnol. 2004, 646816–822. [DOI] [PubMed] [Google Scholar]
- Peterbauer C.; Maischberger T.; Haltrich D. Food-grade gene expression in lactic acid bacteria. Biotechnol. J. 2011, 691147–1161. [DOI] [PubMed] [Google Scholar]
- Spath K.; Heinl S.; Egger E.; Grabherr R. Lactobacillus plantarum and Lactobacillus buchneri as expression systems: evaluation of different origins of replication for the design of suitable shuttle vectors. Mol. Biotechnol. 2012, 52140–48. [DOI] [PubMed] [Google Scholar]
- Giraffa G.; Chanishvili N.; Widyastuti Y. Importance of lactobacilli in food and feed biotechnology. Res. Microbiol. 2010, 1616480–487. [DOI] [PubMed] [Google Scholar]
- Nguyen T. T.; Mathiesen G.; Fredriksen L.; Kittl R.; Nguyen T. H.; Eijsink V. G. H.; Haltrich D.; Peterbauer C. K. A food-grade system for inducible gene expression in Lactobacillus plantarum using an alanine racemase-encoding selection marker. J. Agric. Food Chem. 2011, 59105617–5624. [DOI] [PubMed] [Google Scholar]
- Spath K.; Heinl S.; Grabherr R.. Direct cloning in Lactobacillus plantarum: electroporation with non-methylated plasmid DNA enhances transformation efficiency and makes shuttle vectors obsolete. Microb. Cell Factories 2012, 11, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. T.; Nguyen H. A.; Arreola S. L.; Mlynek G.; Djinović-Carugo K.; Mathiesen G.; Nguyen T. H.; Haltrich D. Homodimeric β-galactosidase from Lactobacillus delbrueckii subsp. bulgaricus DSM 20081: expression in Lactobacillus plantarum and biochemical characterization. J. Agric. Food Chem. 2012, 6071713–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H. A.; Nguyen T. H.; Nguyen T. T.; Peterbauer C. K.; Mathiesen G.; Haltrich D. Chitinase from Bacillus licheniformis DSM13: expression in Lactobacillus plantarum WCFS1 and biochemical characterisation. Protein Express. Purif. 2012, 812166–174. [DOI] [PubMed] [Google Scholar]
- Mathiesen G.; Sørvig E.; Blatny J.; Naterstad K.; Axelsson L.; Eijsink V. G. H. High-level gene expression in Lactobacillus plantarum using a pheromone-regulated bacteriocin promoter. Lett. Appl. Microbiol. 2004, 392137–143. [DOI] [PubMed] [Google Scholar]
- Sørvig E.; Grönqvist S.; Naterstad K.; Mathiesen G.; Eijsink V. G. H.; Axelsson L. Construction of vectors for inducible gene expression in Lactobacillus sakei and L. plantarum. FEMS Microbiol. Lett. 2003, 2291119–126. [DOI] [PubMed] [Google Scholar]
- Sørvig E.; Mathiesen G.; Naterstad K.; Eijsink V. G. H.; Axelsson L. High-level, inducible gene expression in Lactobacillus sakei and Lactobacillus plantarum using versatile expression vectors. Microbiology 2005, 15172439–2449. [DOI] [PubMed] [Google Scholar]
- Mathiesen G.; Huehne K.; Kroeckel L.; Axelsson L.; Eijsink V. G. H. Characterization of a new bacteriocin operon in sakacin P-producing Lactobacillus sakei, showing strong translational coupling between the bacteriocin and immunity genes. Appl. Environ. Microbiol. 2005, 7173565–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terpe K. Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2006, 722211–222. [DOI] [PubMed] [Google Scholar]
- Casas I. A.; Dobrogosz W. J. Validation of the probiotic concept: Lactobacillus reuteri confers broad-spectrum protection against disease in humans and animals. Microb. Ecol. Health Dis. 2000, 124247–285. [Google Scholar]
- Inoue H.; Nojima H.; Okayama H. High efficiency transformation of Escherichia coli with plasmids. Gene 1990, 96123–28. [DOI] [PubMed] [Google Scholar]
- Li S.; Wilkinson M. F. Site-directed mutagenesis: a two-step method using PCR and DpnI. BioTechniques 1997, 234588–590. [DOI] [PubMed] [Google Scholar]
- Ho S. N.; Hunt H. D.; Horton R. M.; Pullen J. K.; Pease L. R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989, 77151–59. [DOI] [PubMed] [Google Scholar]
- Josson K.; Scheirlinck T.; Michiels F.; Platteeuw C.; Stanssens P.; Joos H.; Dhaese P.; Zabeau M.; Mahillon J. Characterization of a Gram-positive broad-host-range plasmid isolated from Lactobacillus hilgardii. Plasmid 1989, 2119–20. [DOI] [PubMed] [Google Scholar]
- Artimo P.; Jonnalagedda M.; Arnold K.; Baratin D.; Csardi G.; De Castro E.; Duvaud S.; Flegel V.; Fortier A.; Gasteiger E.; Grosdidier A.; Hernandez C.; Ioannidis V.; Kuznetsov D.; Liechti R.; Moretti S.; Mostaguir K.; Redaschi N.; Rossier G.; Xenarios I.; Stockinger H. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40W1W597–W603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal. Biochem. 1976, 721–2248–254. [DOI] [PubMed] [Google Scholar]
- Dische Z.; Borenfreund E. A new spectrophotometric method for the detection and determination of keto sugars and trioses. J. Biol. Chem. 1951, 1922583–587. [PubMed] [Google Scholar]
- Fox P. F.; McSweeney P. L. H.. Advanced Dairy Chemistry: Vol. 3: Lactose, Water, Salts and Minor Constituents, 3rd ed.; Springer: New York, 2009. [Google Scholar]
- Kaswurm V.; Nguyen T. T.; Maischberger T.; Kulbe K. D.; Michlmayr H. Evaluation of the food grade expression systems NICE and pSIP for the production of 2,5-diketo-d-gluconic acid reductase from Corynebacterium glutamicum. AMB Express 2013, 311–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elagöz A.; Abdi A.; Hubert J. C.; Kammerer B. Structure and organisation of the pyrimidine biosynthesis pathway genes in Lactobacillus plantarum: a PCR strategy for sequencing without cloning. Gene 1996, 1821–237–43. [DOI] [PubMed] [Google Scholar]
- Pouwels P. H.; Leer R. J. Genetics of lactobacilli: plasmids and gene expression. Antonie van Leeuwenhoek, Int. J. Gen. Mol. Microbiol. 1993, 64285–107. [DOI] [PubMed] [Google Scholar]
- Rhimi M.; Chouayekh H.; Gouillouard I.; Maguin E.; Bejar S. Production of d-tagatose, a low caloric sweetener during milk fermentation using l-arabinose isomerase. Bioresour. Technol. 2011, 10233309–3315. [DOI] [PubMed] [Google Scholar]
- Manjasetty B. A.; Chance M. R. Crystal structure of Escherichia colil-arabinose isomerase (ECAI), the putative target of biological tagatose production. J. Mol. Biol. 2006, 3602297–309. [DOI] [PubMed] [Google Scholar]
- Chouayekh H.; Bejar W.; Rhimi M.; Jelleli K.; Mseddi M.; Bejar S. Characterization of an l-arabinose isomerase from the Lactobacillus plantarum NC8 strain showing pronounced stability at acidic pH. FEMS Microbiol. Lett. 2007, 2772260–267. [DOI] [PubMed] [Google Scholar]
- Umemoto Y.; Shibata T.; Araki T. d-Xylose isomerase from a marine bacterium, Vibrio sp. strain XY-214, and d-xylulose production from β-1,3-xylan. Mari. Biotechnol. 2012, 14110–20. [DOI] [PubMed] [Google Scholar]
- Liu S. Y.; Wiegel J.; Gherardini F. C. Purification and cloning of a thermostable xylose (glucose) isomerase with an acidic pH optimum from Thermoanaerobacterium strain JW/SL-YS 489. J. Bacteriol. 1996, 178205938–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Jiang B.; Pan B. Purification and characterization of l-arabinose isomerase from Lactobacillus plantarum producing d-tagatose. World J. Microbiol. Biotechnol. 2007, 235641–646. [Google Scholar]
- Ravikumar S.; Vikramathithan J.; Srikumar K. Purification and characterization of a novel thermostable xylose isomerase from Opuntia vulgaris Mill. Appl. Biochem. Biotechnol. 2011, 1645593–603. [DOI] [PubMed] [Google Scholar]
- Yamanaka K. Purification, crystallization and properties of the d-xylose isomerase from Lactobacillus brevis. Biochim. Biophys. Acta–Enzymol. 1968, 1513670–680. [DOI] [PubMed] [Google Scholar]
- Hong Y. H.; Lee D. W.; Pyun Y. R.; Lee S. H. Creation of metal-independent hyperthermophilic l-arabinose isomerase by homologous recombination. J. Agric. Food Chem. 2011, 592412939–12947. [DOI] [PubMed] [Google Scholar]
- Kleerebezem M.; Boekhorst J.; Van Kranenburg R.; Molenaar D.; Kuipers O. P.; Leer R.; Tarchini R.; Peters S. A.; Sandbrink H. M.; Fiers M. W. E. J.; Stiekema W.; Klein Lankhorst R. M.; Bron P. A.; Hoffer S. M.; Nierop Groot M. N.; Kerkhoven R.; De Vries M.; Ursing B.; De Vos W. M.; Siezen R. J. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Natl. Acad. Sci. U.S.A. 2003, 10041990–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strych U.; Penland R. L.; Jimenez M.; Krause K. L.; Benedik M. J. Characterization of the alanine racemases from two Mycobacteria. FEMS Microbiol. Lett. 2001, 196293–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.