

Abstract
The secretin-like (class B) family of G protein-coupled receptors (GPCRs) are key players in hormonal homeostasis and are interesting drug targets for the treatment of several metabolic disorders (type 2 diabetes, osteoporosis, obesity) and nervous system diseases (migraine, anxiety, depression). The recently solved crystal structures of the transmembrane domains of the human glucagon receptor and the human corticotropin-releasing factor receptor 1 have opened up new opportunities to study the structure and function of class B GPCRs. The current review shows how these structures offer more detailed explanations to previous biochemical and pharmacological studies of class B GPCRs, and provides new insights into their interactions with ligands.
Keywords: Class B G Protein-Coupled Receptor (GPCR), Glucagon receptor (GCGR), Corticotropin-releasing factor receptor 1 (CRF1), crystal structure, ligand binding
Understanding the function of class B GPCRs from their structure
Class B G protein-coupled receptors (GPCRs), also referred to as the secretin family of GPCRs, include receptors for 15 peptide hormones, which can be grouped into five subfamilies based on their physiological role (see Table 1 for an overview) [1]. These receptors are important drug targets in many human diseases including diabetes, osteoporosis, obesity, cancer, neurodegeneration, cardiovascular disease, headache and psychiatric disorders. However, the identification of small-molecule oral drugs for this family has proved extremely challenging (Table 1) [2–5]. Extensive high-throughput screening by the pharmaceutical industry has failed to identify chemical hits which mimic or block the large peptide ligands. An important reason for this is the lack of knowledge of the structure and location of druggable binding sites in class B GPCRs. Structurally, class B GPCRs consist of a large N-terminal extracellular domain (ECD) and a transmembrane domain (TMD) comprising the ‘GPCR’ signature of seven membrane spanning α-helices, which is involved in signalling via coupling to heterotrimeric G proteins that primarily activate adenylate cyclase to increase the levels of intracellular cyclic AMP, as well as those that increase inositol phosphate and intracellular calcium levels [2].
Table 1.
Human class B G protein coupled receptors.
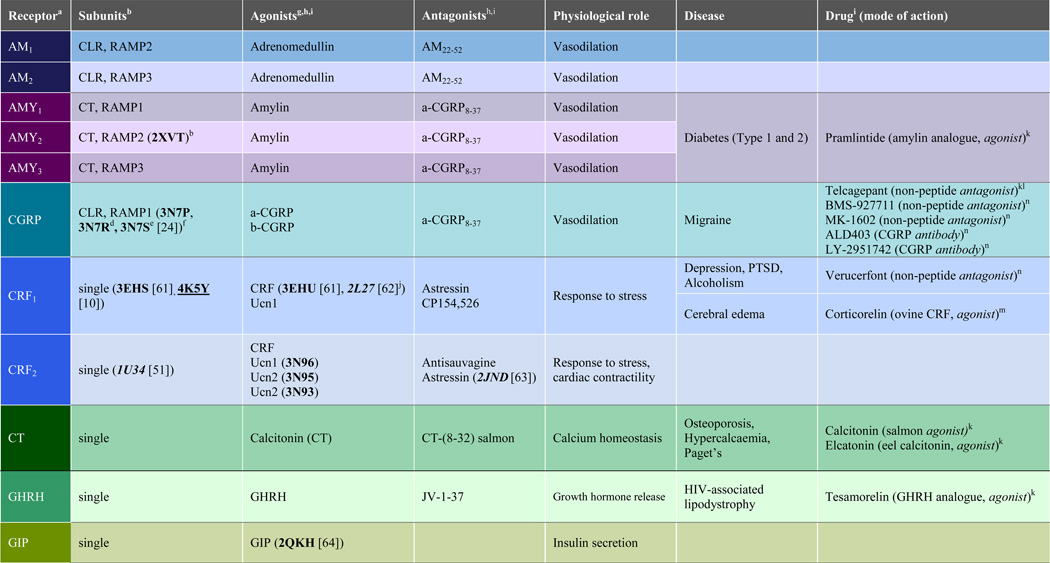 |
 |
The coloring scheme was chosen to give each subfamily of class B GPCRs a different color.
PDB IDs of corresponding crystal structures (bold) and NMR structures (italics) of apo ECD and TMD (underlined) are indicated between parentheses;
crystal structure of RAMP2 unit;
Complex with telcagepant;
Complex with olcegepant;
crystal structure of assembly of CLR and RAMP1 units;
The most potent endogenous agonists are given;
PDB IDs of corresponding crystal structures (bold) and NMR structures (Italics) of ECD-peptide agonist complexes are indicated between parentheses;
Amino acid sequences of peptide ligands and systematic names of non-peptide molecules are provided in Supporting Table S1;
α-helical cyclic CRF;
Drug on the market;
Reached clinical Phase 3 but discontinued;
In clinical Phase 3;
In clinical Phase 2;
In clinical Phase 1;
Abbreviations: AMY amylin, CRF corticotropin-releasing factor, CGRP calcitonin gene related peptide, CLR calcitonin like receptor, CT calcitonin, GHRH growth hormone releasing hormone, GIP glucose dependent insulinotropic peptide, GLP glucagon like peptide, PTH parathyroid hormone, PTHrP parathyroid hormone related protein, TIP tuberoinfundibular peptide, VIP vasoactive intestinal peptide, Ucn urocortin.
Structures of the ECDs of class B GPCRs in complex with their peptide ligands have been determined by X-ray crystallography and NMR (Table 1) and have provided information about structural mechanisms of ligand recognition and selectivity [4,6]. To date, crystal structures of more than 20 class A GPCRs have been determined [7,8]. While these structural studies have greatly increased our understanding of the molecular mechanisms of receptor ligand binding and activation [7] and have enabled structure based approaches to drug design [9], there has been a considerable delay in obtaining a structure of the TMD of any class B receptor, owing to the inherent instability of these receptors and the limited availability of high-affinity small-molecule agonists and antagonists. Recently, crystal structures of the TMDs of two class B receptors have been published, namely the human glucagon receptor (GCGR) and the human corticotropin-releasing factor receptor 1 (CRF1) [10,11]. This review compares the structures of these two class B GPCRs to each other as well as to class A receptors and discusses them in the context of previous pharmacological and biochemical data on class B receptors, furthering our understanding of receptor-ligand interactions.
Comparison of the CRF1 and GCGR structures
Using a mutagenesis approached called “conformational thermostabilization” described earlier [12], a crystallizable version of the TMD of the human CRF1 receptor (also known as CRHR1) with increased thermostability and bias towards the inactive conformation was generated and its structure has recently been determined by X-ray crystallography in complex with 2-aryloxy-4-alkylaminopyridine (CP-376395) [13], a small-molecule antagonist (Figure 1a) [10]. Simultaneously, the structure of the TMD of human GCGR, N-terminally fused to thermally stabilized apocytochrome b562RIL (referred to as BRIL) at residue 123 and likewise crystallized in the presence of a small-molecule antagonist, was reported (Figure 1b) [11]. With these two structures in hand, we can begin to identify structural similarities and differences within class B receptors. GCGR and CRF1 share approximately 30% sequence identity within the TMDs. Not surprisingly, their overall folds, including the extracellular and intracellular loops observed in the crystal structures, are similar. As expected, both TMDs consist of seven membrane-spanning α-helices that pack into the hallmark seven transmembrane (TM) helical bundle of GPCRs. Even though the general arrangement of the TM helices is similar to those observed in previously determined GPCR structures, the receptors assume conformations that are more open towards the extracellular side than any other GPCR of known structure. Both CRF1 and GCGR take on the shape of a letter V when viewed from within the membrane (Figures 1a and b). One arm of the V is made up by the extracellular halves of TM2-TM5, while the other comprises the extracellular portions of TM1, TM6, and TM7. As a result of this pronounced opening, both receptors feature a large solvent-filled cavity accessible from the extracellular side. Whilst the orthosteric binding site of class B receptors is, strictly speaking, composed of portions of both the ECD and the TMD, in the following discussions we will refer to this cavity as the putative peptide-agonist binding or orthosteric site.
Figure 1.

(a, b) Crystal structures of class B GPCRs CRF1 (PDB ID 4K5Y) and GCGR (PDB ID 4L6R) are shown in blue and orange ribbons, respectively, in two different views from within the membrane. TM helices and helix 8 are labeled. The disulfide bond tethering ECL2 to the tip of TM3 is shown as purple sticks. In CRF1 the small-molecule antagonist CP-376395 is shown in stick representation with carbon, nitrogen and oxygen atoms colored magenta, blue and red, respectively, and as skeletal formula in an inset. (c) Superposition of the two structures, with insets highlighting regions of particular interest. To highlight the structural differences in the extracellular halves of CRF1 and GCGR, the distance of approximately 11 Å between the Cα-atoms of residues 7.33b at the N-terminal end of TM7 is indicated with a red arrow. The small-molecule binding pocket is shown as a superposition of the two receptors around CP-376395, illustrating the antagonist binding mode and the substantial structural differences observed for TM6 of the two receptors.
Perhaps the most striking finding of the CRF1 crystal structure, and undoubtedly the most interesting from a drug discovery point of view, is the location of the small-molecule antagonist-binding site. The antagonist binds deep within the cytoplasmic half of the CRF1 receptor, far away from the orthosteric site (Figure 1a). Even though the GCGR structure did not identify an unambiguous binding site for the co-crystallized antagonist, it is clear that an antagonist binding pocket analogous to the one found in CRF1 is absent in this GCGR structure. Details of the small-molecule binding site are discussed below.
The GCGR structure revealed a potentially functionally important feature: the N-terminal end of TM1 extends far into the extracellular space (Figure 1b). This extension, dubbed the ‘stalk’, is thought to have a role in the positioning of the ECD (Figure 2a) relative to the helical bundle and may also interact with glucagon directly (see below). While we cannot rule out the possibility that the stalk region is an artifact of the construct or crystallization process, we believe it is real and plays an important role in the structure and function of GCGR. Even though no such extension was observed in the CRF1 structure, the ECD-TMD link might well take on a helical structure in the context of the full-length receptor. GCGR also features an unusually long helix 8 at the C-terminal end of the receptor (Figure 1b). Even though this helix is slightly bent out into the cytoplasm as compared to its orientation in class A GPCRs, probably a result of crystal lattice contacts, its N-terminal end is involved in ionic interactions at the cytoplasmic side of the receptor that seem conserved in class B GPCRs. The crystallized CRF1 construct was truncated after TM7, removing a potential helix 8. Superposition of CRF1 and GCGR shows that structural agreement is best in TM1–5 (Figure 1c). Even though their TM4 helices do not superimpose perfectly, probably a consequence of the disorder observed in this region in the GCGR crystal structure, the GWGxP motif, a highly conserved sequence motif in TM4 of class B receptors, appears to play an important structural role, creating a similar network of interactions in both receptors that non-covalently links TM2, TM3 and TM4. Another structural link observed in both structures is the interaction between TM1 and TM7. Ser1.50b (Wootten numbering in superscript [14], see Box 1 and Figure 3a) of TM1 interacts with the backbone of TM7 at Ser7.47b, Gly7.50b and Phe/Leu7.51b, thereby stabilizing the kink in this TM helix.
Figure 2.

(a) Overview of NMR and crystal structures of class B GPCR ECDs (magenta) and their complexes with peptide ligands (different colors) [18,19,24,34,39,51,61–68]. A complete overview of corresponding PDB IDs is presented in Table 1. (b) Structure-based sequence alignment of representative peptide ligands of class B GPCR, adopted from Parthier et al. [6]. The residues of the peptide ligands solved in ECD-ligand complex crystal structures [18,19,24,34,39,51,61–68] are marked using the same color as in Figure 2. The residues that are boxed black are found in an alpha helical conformation in the complex. Peptide ligand residues that covalently bind receptors in photo cross-linking or cysteine trapping studies [22,25,47,50,54,69,70] are colored and boxed green, while peptide ligand residues that have been mutated and studied in combination with receptor mutants [29,30,45,46,48] are colored and boxed red. Note that the first residue of GLP-1 is His7. A complete overview of all ECD structures and important peptide ligands for all class B GPCRs is presented in Table 1. Putative helix-capping residues [6] are colored blue and cysteines involved in a disulfide-bridge (calcitonin) are colored orange. D-phenylalanine (f), and norleucine (m) residues are indicated in stressin and asstressin. The last 41 and 99 residues of PTH and PTHrP, respectively, are not displayed.
BOX 1.
The Ballesteros–Weinstein (class A GPCRs) [60] and Wootten (class B GPCRs) [14] numbering schemes are based on the presence of several highly conserved residues in the TM helices of class A and class B GPCRs, respectively. The single most conserved residue in each TM helix is designated X.50 (class A Ballesteros-Weinstein number) or X.50b (class B Wootten number). X is the TM helix number, and all other residues in that helix are numbered relative to this conserved position. While the well-established Ballesteros-Weinstein numbering scheme serves well for analysis of sequence and structure similarities and differences across the whole family of GPCRs, the Wootten numbering scheme proves useful for the comparison of receptors within class B GPCRs. The spatial correspondence between residues in TM helices of class A and class B GPCRs makes it possible to align the class A Ballesteros-Weinstein and class B Wootten numbering schemes for comparisons between GPCR classes, as illustrated in Figure 3a.
Figure 3.

The spatial correspondence between residues in TM helices of class A and class B GPCRs makes it possible to align the most conserved residues in class A (designated X.50, Ballesteros-Weinstein numbering) and class B (designated X.50b, Wootten numbering) for comparisons between GPCR classes (Box 1). (b) Structural alignment of CRF1 (blue) and GCGR (orange) to two representative class A GPCRs, H1R (PDB ID 3RZE) and CXCR4 (PDB IDs 3ODU/3OE0) (in grey). Helices are depicted as cylinders, ligands glucagon (GCGR), CP-376395 (CRF1), doxepin (H1R), and IT1t and CVX15 (CXCR4) as sticks. The location of the Cα-atoms of the most conserved residues of TM1–3 and TM5 among class A and class B GPCRs (Box 1) are indicated by spheres (TM4 is not depicted for clarity).
In both receptors the extracellular halves of TM6 and TM7, and with them ECL3, point away from the center. In GCGR this region is closer to the long axis of the helical bundle than in CRF1 with a distance of approximately 11 Å between respective residues located at the extracellular end of TM7 (Figure 1c). In both structures these parts exhibit high crystallographic temperature factors, indicating high structural flexibility. The differences in the volumes of the orthosteric pockets are the direct consequence of the positions of these two helices relative to the rest of the helical bundle. The fact that this region possesses significant structural flexibility emphasizes the possible structural role of the ECD in class B GPCRs. Its influence on the shape and volume of the orthosteric pocket suggests a potential functional role in receptor activation, and the different orientations of TM6 and TM7 in the CRF1 and GCGR crystal structures might reflect different conformational/functional states.
On the intracellular side, the functionally relevant interaction between His2.50b and Glu3.50b [15,16] is present in both structures (Figure 1c). In the same region Tyr4007.57b of GCGR creates hydrogen bond interactions with Thr3516.42b and Glu2453.50b in a conformation that in class A GPCRs is linked to activation and interaction with the G protein [17]. In CRF1 the thermostabilizing mutation Tyr3637.57bAla contributes to the shift in the conformation of the receptor towards an inactive state. Helix 8 is not present in the CRF1 crystal structure while it is 26 residues long in the GCGR structure. In CRF1, the removal of helix 8 could have affected the orientation of the TM7 in the crystal structure, while in GCGR crystal contacts involving helix 8 may influence its relative orientation and length. Despite these possible experimental artifacts, Arg2.46b is in a comparable position in both structures and in GCGR creates a salt bridge with Glu406 located in helix 8 (Figure 1c). In GCGR there is an additional salt bridge between this conserved Glu and Arg3466.37b, which is a Lys in CRF1 (and Arg/Lys in other class B GPCRs).
Extracellular domain structures
The crystal structures and NMR structures of the ECDs of different class B GPCRs (Figure 2a) show that this domain has a conserved fold that includes two central antiparallel β-sheets and an N-terminal α-helix interconnected by several loops and stabilized by three conserved disulfide bonds. Ten of the 11 ECD-peptide ligand complexes show a similar binding mode in which the C-terminus of the peptide ligand adopts an α-helical conformation that binds between the two β-sheets of the ECD (Figure 2). The NMR structure of the complex between pituitary adenylate cyclase activating polypeptide (PACAP) and its receptor (PAC1) shows a somewhat distinct binding mode [18], but the ligand-free PAC1 crystal structure and mutation studies indicate that this receptor also adopts the same fold and accommodates the same ligand binding mode observed for the other ECDs [19]. This conserved fold and binding orientation suggests that common mechanisms underlie ligand recognition of class B GPCRs [4,6], and indicates that peptide ligand selectivity is in part determined by specific interactions between the two β-sheets and the connecting loops in the ECD. The ECD-peptide ligand structures and pharmacological studies with truncated and chimeric peptide ligands provide the basis for a ‘two domain’ binding model in which: i) the C-terminus of the hormone peptide forms an initial complex with the ECD, which ii) allows the N-terminus of the peptide ligand to interact with the TMD to activate the class B GPCR [4,6,20–22]. These insights into role of the ECD in the regulation of class B GPCRs have facilitated the development of peptide ligand antagonists and agonists (Table 1) [4,6]. For the calcitonin receptor subfamily, ligand selectivity is defined by the heterodimerization of the 7-TM proteins calcitonin (CT) or calcitonin receptor-like receptor (CLR) with one (or a combination) of three receptor-activity modifying proteins (RAMPs) [5,23]. RAMPs are essential for CT and CLR signaling as they play a role in ligand binding and are required for receptor translocation to the cell surface [23,24], and have been proposed to interact with other class B GPCRs (GCGR, PTH1, PTH2, VPAC1) as well [5]. The crystal structures of the ECDs of the calcitonin gene related peptide (CGRP) receptor comprising the CLR-RAMP1 complex show that the CLR-RAMP1 dimer is stabilized by hydrophobic and electrostatic interactions between the N-terminal helix of CLR, and helix 2 and 3 of RAMP1, and show how the small-molecule antagonists olcegepant and telcagepant can target the hetereodimer [24] (Figure 2a). In this family the 7-TM protein subunit is responsible for transmitting the signal of activation to the G protein whilst the RAMP complex interacts with the ECD of the 7-TM to alter the ligand specificity. In order to obtain small molecule drugs that are selective over the other family members these have been targeted to bind to the extracellular complex formed between the RAMP and 7-TM ECDs.
Peptide ligand recognition by the TMD of class B GPCRs
There has been no clear consensus on the binding site location of peptide ligands in the TMDs of class B GPCRs, which has been associated either with the extracellular loop regions [2,6], or with a pocket in the TMD [25,26]. Combination of previous structural information on ECD-ligand complexes (Table 1, Figure 2) with the recent GCGR and CRF1 TMD crystal structures allowed the construction of a full receptor-ligand model (Figure 4a) [11]. This model can account for the extensive interactions of the peptide ligand with extracellular loops, as well as residues deep in the helical bundle, as reflected by mutagenesis and photo-crosslinking studies in different class B GPCRs (Figure 4). The GCGR and CRF1 crystal structures reveal that some of the binding site residues previously thought to be positioned at the extracellular ends of the TM helices or in extracellular loops are in fact located deeper in the TMD. It has been proposed that an extended flexible conformation of the first residues allows the peptide ligands of class B GPCRs to reach deep into the pocket. This receptor-bound peptide conformation is proposed to be stabilized by conserved amino acid motifs that can induce an N-capping conformation (Figure 2b) [6,27] similar to the one observed in the receptor bound PACAP [28]. The extended stalk region (residues 125–136 in GCGR), which links the ECD and 7TM domain, may be involved in peptide ligand binding and help define the orientation of the ECD with respect to the TMD (Figure 4a). The α-helical structure of the stalk is stabilized by intrahelical interactions in the GCGR crystal structure (Glu 133–Lys 136) and GCGR-glucagon model (Glu127–Gln131 and Glu129–Lys132) and fits a GCGR-glucagon binding mode model supported by GCGR mutagenesis [11,29–35] and glucagon structure-activity relationship [36–38] studies in which: i) a hydrophobic region in the C-terminus of glucagon (residues Phe22, Val23, Trp25, and Met27) binds the ECD (as observed in the GLP-1 peptide bound GLP-1 ECD crystal structure [39]), ii) a second hydrophobic region in the middle region of glucagon (residues Tyr10, Tyr13, and Leu14 [38]) interacts with the stalk and extracellular loop 1, and iii) the N-terminus of glucagon (residues 1–6) interacts with the TMD (Figures 2 and 4). The helical structure of the stalk is further supported by a reduced glucagon affinity to a Ala135Pro mutant, which probably distorts the α-helical conformation [11]. Figure 4a shows how the mutations that affect glucagon binding [11,29–35] line the binding site within the TMD in the GCGR-glucagon model and include residues that are located deep in the pocket (Tyr1491.47b, Val1912.64b, Gln2323.37b, Glu3626.53b and Leu3867.43b) [11]. These results strongly support extension of the N-terminus of glucagon deep into the GCGR pocket, a region that could be equally important for ligand binding as in class A GPCRs. Figure 4b shows how the GCGR mutation data are paralleled by mutagenesis and photo-crosslinking studies of homologous residues in other class B GPCRs, including GLP-1 [14,21,26,39–42], GIP [43,44], secretin [45–47], VPAC1 [48,49], PTH1 [22,50], and CRF1 [10,51–54] receptors. For example, mutations of residues at positions 1.43b, 1.47b, 2.60b, 2.67b, 2.68b, 2.71b, 2.72b, 3.37b, 6.35b, 5.35b, 5.36b, 6.53b, 6.56b, and 7.43b have been shown to affect peptide ligand binding and/or potency in multiple class B GPCRs (Figure 4b) and line the proposed peptide ligand binding site in the TMD (Figure 4a). Interestingly, several N-terminally truncated forms of the CRF [20], GIP [55], GLP-1 [21], glucagon [56], and PTH [22] peptides are competitive antagonists for their corresponding receptor. C-terminally truncated ligands however remain active, indicating that interactions with peptide ligands in the TMD of class B GPCRs are required for receptor activation. Moreover, combined receptor and ligand mutation studies indicate that homologous residues in the peptide ligands of class B GPCRs (Figure 2b) interact with similar regions in this binding site (Figure 4a). For example, the homologous residues Gln3 of glucagon, Asp3 of secretin, and Asp3 of vasoactive intestinal polypeptide are located within the same vicinity in the TMD of GCGR (Lys1872.60b and Ile1942.67b) [29,30], rat secretin (Tyr1281.47b, Arg1662.60b, Lys1732.67b, and Asp1742.68b) [45,46], and VPAC1 (Arg1882.60b and Lys1952.67b) [48] receptors, respectively. It should be noted, however, that the N-terminal regions of peptide ligands that bind CRF1 and CRF2 are significantly longer than in the peptide ligands of other class B GPCRs (Figure 2b) [6] and probably adopt different binding modes. Similar regions in TM1 of GLP-1 (Leu1411.36b) and CRF1 (His1171.37b) receptors can for example be photo-cross-linked to residues at different positions in GLP (Val16) [25] and sauvagine (Met17) [54] peptides, respectively (Figure 2b and 4b). The wider pocket in the region around the top of TM6–7 and ECL3 in CRF1 compared to GCGR (Figure 1c) might explain how the receptor accommodates the larger N-terminal regions of its peptide ligands [6]. The distinct structural features and larger binding pocket of the GCGR and CRF1 compared to class A GPCR crystal structures provide new insights into the molecular details of peptide ligand binding and a more reliable structural template for the design of specific and potent small molecules to modulate class B GPCRs. Moreover, the apparent overlap of class B GPCR binding sites suggests that, despite possible structural differences between class B receptors, the GCGR and CRF1 TMD crystal structures may offer new opportunities to construct structural models to describe interactions between peptide ligands and other class B GPCRs.
Figure 4.
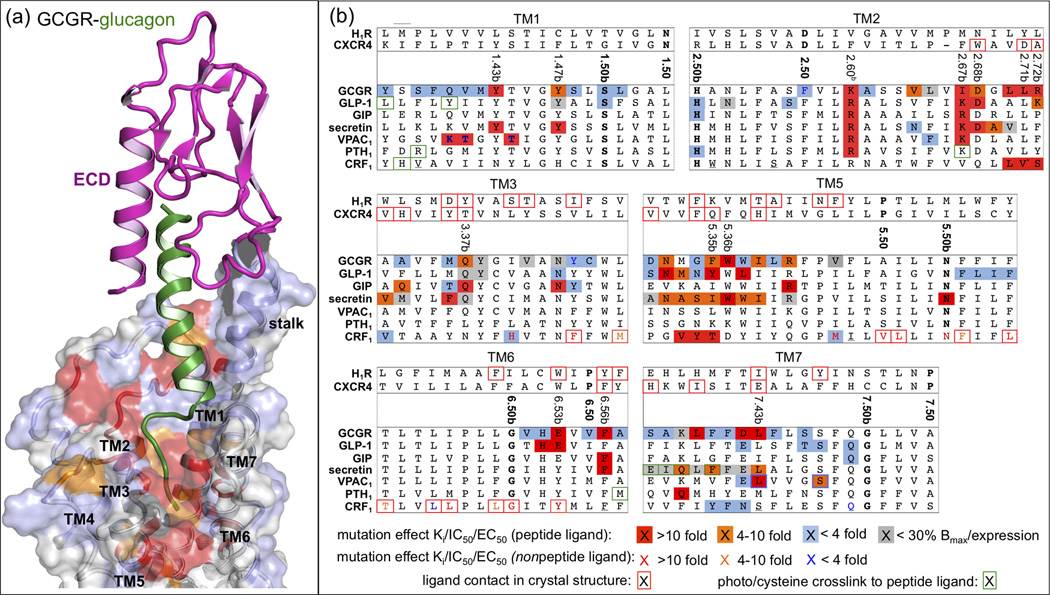
(a) Model of full GCGR-glucagon complex [11]. GCGR with the ECD (PDB ID 4ERS, in magenta) and TMD (PDB ID 4L6R, transparent surface) bound to glucagon (green). The effects of mutation studies of individual GCGR residues on glucagon binding are mapped onto the GCGR binding surface using the color coding presented in Figure 4b. (b) Effects of mutation studies in GCGR, GLP-1, GIP, secretin, VPAC1, PTH1, and CRF1 receptors [10,11,14,21,22,26,29–35,39–53] are mapped on a structure-based (Figure 3b) sequence alignment between representative class A GPCR crystal structures (H1R and CXCR4) and representative class B GPCRs. Only specific parts of TM1–3 and TM5–7 are shown, separated by grey dashed lines. Mutated residues that show <4 fold (blue), 4–10 fold (orange), and >10 fold (red) changes of Ki/IC50 values for ligand binding (or ligand potency/EC50 value if no Ki/IC50 value has been reported) are marked (peptide ligands) and colored (non-peptide ligands). Mutants that show receptor expression <30% of wild-type are marked grey. Ligand contact residues in CRF1 (Figure 1c), H1R, and CXCR4 crystal structures are boxed red. The most conserved residues in TM1–7 of class A (x.50) and class B (x.50b) GPCRs (Box 1, Figure 3a) are shown in bold. Receptor residues that covalently bind peptide ligands in photo cross-linking or cysteine trapping studies [22,25,47,50,54,69,70] are boxed green. Note that several mutation and photo cross-linking studies annotated for secretin and PTH receptors are performed with rat orthologs. Residues that are mutated in the conformationally thermostabilized [10] CRF1 crystal structure are underlined.
Druggability of the orthosteric and small-molecule binding sites
Despite the lack of sequence conservation, comparison of the CRF1 and GCGR structures with those of class A GPCR shows that the orientations and positions of TM helices are conserved between the two classes [10,11] (Figure 3b). This common GPCR fold is stabilized by similar regions of contacts between TM helices in both classes, but involves distinct patterns of conserved residues in class A [8] and class B [11] GPCRs (Figure 3a and 4b). The distances between the extracellular ends of TM2 and TM7 and TM3 and TM7 of CRF1 and GCGR are however (among) the largest observed in GPCR structures. Consequently, the orthosteric pockets of these two receptors are wider and deeper than those of any class A receptor [11] (Figure 5). In addition, several residues located deep in the orthosteric pocket between TM1 (1.47b) and TM2 (2.60b) play an important role in peptide ligand binding in class B GPCRs, while the corresponding structurally aligned residues (positions 1.43 and 2.53) are not involved in ligand binding in class A receptors. The crystal structures of the chemokine receptor CXCR4 and the histamine H1 receptor (H1R) (Figure 4b and 5) illustrate how the different binding pockets in class A GPCRs are targeted by peptide and non-peptide ligands. The CXCR4 structure is the only GPCR crystal structure in which a (non-peptide) ligand (1T1t) binds in the subpocket between TM1–3 and TM7 [57] (Figure 3b, 4b, and 5). The peptide ligand CVX15 targets an overlapping, but distinct binding site region in CXCR4 located between TM3–7 [57] (Figure 3b and 4a), which is occupied by non-peptide ligands in all other class A GPCR crystal structures [7]. The H1R ligand doxepin binds in a pocket that is located closest to the cytoplasm of all the ligand binding sites observed in class A GPCR crystal structures [58] (Figure 3b and 4a).
Figure 5.

Comparison of druggable binding sites of (a) GCGR, (c) CRF1, and the class A GPCRs (b) CXCR4 (PDB ID 3ODU/3EO0) and (d) H1R (PDB ID 3RZE). The surfaces of binding sites that are considered druggable [59] are colored orange (orthosteric pocket) and red (CP-376395 binding pocket). Of eight structurally aligned residues (see Figure 4b), the residues that line the binding pocket shown in the figures are labeled black; the residues that are not part of the depicted pockets are labeled light grey. Note that A6.56b and A7.43b are thermostabilizing mutations [10] in the CRF1 crystal structure (see also Figure 4b). The approximate position of the extracellular membrane boundary is shown as a dotted black line. Carbon atoms of peptide ligands (glucagon in GCGR and CVX15 in CXCR4) and non-peptide ligands (IT1t in CXCR4 and doxepin in H1R) are colored green and magenta, respectively.
The crystal structure of CRF1 revealed an unexpected small-molecule binding pocket located in the cytoplasmic half of the receptor (Figure 1c and 5). This position is more than 15 Å away from the putative peptide agonist binding site and more than 7 Å further down than the doxepin binding site in the class A GPCR H1R (Figure 3b). This is in contrast to the positions of the small-molecule antagonists and agonists found in the structures of class A GPCRs, which invariably bind close to or beyond the extracellular boundary of the membrane [8]. CP-376395 binds in this druggable site defined by residues of TM3, TM5 and TM6 showing a combination of hydrophobic and hydrophilic features compatible with drug-like small organic molecules. In this region, the sequence identity in class B GPCRs is remarkably high (Figure 4b). Of the 14 residues directly interacting with CP-376395, seven are identical in CRF1 and GCGR. Among them is Asn2835.50b, which forms an essential hydrogen bond with the ligand, while the other residues in common, Phe2845.51b, Ile2905.57b, Thr3166.42b, Leu3196.45b, Leu3236.49b and Gly3246.50b, provide hydrophobic interactions with the antagonist.
Compared to GCGR, in CRF1 TM6 in particular is shifted toward the membrane where CP-376395 is bound, adjusting for the presence of the antagonist (Figure 1c). A similar small-molecule binding pocket is not seen in the crystal structure of GCGR in which TM6 is closer to the center of the receptor: Ile3556.46b, Leu3586.49b and Leu2423.47b completely occupy this space (Figure 1c). In GCGR, one helical turn above in TM3, Tyr2393.44b creates hydrogen bonds with the backbone of Leu3586.49b holding TM3 and TM6 together. By contrast, in CRF1 residue 3.44b is a Phe and this interaction is therefore missing (Figure 1c). It is tempting to speculate that CP-376395 binds to CRF1 by ‘induced fit’, i.e. in the absence of the antagonist TM6 would be in a similar position as its equivalent in GCGR. Molecular motions or ‘breathing’ of the helical bundle then allow access for the antagonist to the binding pocket, which in turn keeps TM6 in the position observed in the crystal structure. CP-376395 is a selective CRF1 antagonist unable to inhibit CRF2. This difference seems linked to the nature of residues in position 3.40b and 5.43b [53]. While in CRF1 they are a His and a Met, respectively, a Val and Ile are present in CRF2. Interestingly, at these positions the two residues in GCGR are similar to CRF2 (Ile2353.40b and Val3115.43b). CP-376395 may act as an allosteric antagonist through stabilization of the inactive conformation of the receptor’s intracellular portion, probably preventing TM6 from moving toward the membrane and away from the helical bundle, which is necessary for docking of the G protein. This small-molecule binding pocket could also be exploited to design agonist ligands by stabilizing TM6 in such an open conformation as suggested by the mutation Thr4106.42Pro, which confers constitutive activity in the PTH1 receptor [15].
The analysis of the chemical and physical properties of a ligand binding site and the estimation of the water binding energies have proved a useful approach to understanding a receptor’s druggability [59]. Using these methods, we have compared the binding pockets of H1R, CXCR4, GCGR and CRF1 using GRID (Molecular Discovery) and WaterMap (Schrödinger) [59]. A druggable site is characterized by: (i) a combination of hydrophilic and hydrophobic regions compatible with balanced small molecule properties, and (ii) several clustered ‘unhappy’ (high energy compared to bulk solvent) water molecules, which are easily displaced by a ligand. The orthosteric site of H1R (Figure 5d) represents an excellent example of a druggable pocket, whereas the open binding region of CXCR4 (Figure 5b) with its limited number of unhappy waters is more challenging from a drug design perspective [59]. The orthosteric sites of both GCGR (Figure 5a) and CRF1 (Figure 5c) are similar to CXCR4 (Figure 5b): open and mostly occupied by bulk-like solvent, with only a single druggable hotspot at the bottom of the pocket. This hotspot in the orthosteric site of class B GPCRs is lined with residues that play an important role in peptide ligand binding (including the residues at positions 1.47b, 2.60b, 6.53b, and 7.43b) as summarized in Figures 4 and 5. The analysis of the CP-376395 binding site in CRF1 (Figure 1c and 5c) suggests instead a promising druggable region with features comparable to the H1R orthosteric site (Figure 5d). This unanticipated pocket opens new avenues for structure-based small-molecule drug discovery.
Concluding remarks
The GCGR and CRF1 crystal structures show distinct structural features and different binding pockets compared to class A GPCRs, and give new insights into the molecular details of peptide and small-molecule binding to class B GPCRs. The first two crystal structures of the TM domains of class B GPCRs provide a structural framework that will enable the design of biochemical and biophysical experiments detailing the complex structure of this class of receptors, and facilitate the design of stabilized constructs that may lead to solution of full-length class B GPCR–ligand complexes. The structures furthermore present more reliable structural templates for the design of specific and potent small molecules for the treatment of type 2 diabetes (GCGR) and depression (CRF1), in particular, and open new avenues for structure-based small-molecule drug discovery for class B GPCRs as a whole.
Supplementary Material
Highlights.
-
-
GCGR and CRF1 structures show different features compared to class A GPCRs
-
-
Class B structures and structure-based drug discovery for peptide hormone GPCRs
-
-
Glucagon, GLP1, and GIP peptide molecular recognition and diabetes
-
-
Corticotropin-releasing factor and stress
Acknowledgments
This work was supported by PSI:Biology grant U54 GM094618 (to R.C.S.); The Ministry of Health grants 2012ZX09304-011 and 2013ZX09507002 (M.-W.W.), Shanghai Science and Technology Development Fund 11DZ2292200 (M.-W.W.); Novo Nordisk-Chinese Academy of Sciences Research Fund NNCAS-2011-7 (M.-W.W.); Thousand Talents Program in China (M.-W.W.); COST Action CM1207, GLISTEN (C.d.G., K.H., A.B., F.M.). The authors are grateful to B. Tehan, J.S. Mason, M. Weir and other colleagues at Heptares Therapeutics, and V. Katritch, F.Y. Siu, and V. Cherezov, and other colleagues at The Scripps Research Institute for suggestions and comments. The authors thank A. Walker for comments and for assistance with manuscript preparation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lagerstrom MC, et al. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 2.Hoare SR. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov Today. 2005;10:417–427. doi: 10.1016/S1359-6446(05)03370-2. [DOI] [PubMed] [Google Scholar]
- 3.de Graaf C, et al. Structure-based discovery of allosteric modulators of two related class B G-protein-coupled receptors. ChemMedChem. 2011;6:2159–2169. doi: 10.1002/cmdc.201100317. [DOI] [PubMed] [Google Scholar]
- 4.Pal K, et al. Structure and mechanism for recognition of peptide hormones by Class B G-protein-coupled receptors. Acta Pharmacol Sin. 2012;33:300–311. doi: 10.1038/aps.2011.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Archbold JK, et al. Structural insights into RAMP modification of secretin family G protein-coupled receptors: implications for drug development. Trends Pharmacol Sci. 2011;32:591–600. doi: 10.1016/j.tips.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Parthier C, et al. Passing the baton in class B GPCRs: peptide hormone activation via helix induction? Trends Biochem Sci. 2009;34:303–310. doi: 10.1016/j.tibs.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Katritch V, et al. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venkatakrishnan AJ, et al. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 9.Congreve M, et al. The use of GPCR structures in drug design. Adv Pharmacol. 2011;62:1–36. doi: 10.1016/B978-0-12-385952-5.00011-7. [DOI] [PubMed] [Google Scholar]
- 10.Hollenstein K, et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–443. doi: 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- 11.Siu FY, et al. Structure of the human glucagon class B G-protein-coupled receptor. Nature. 2013;499:444–449. doi: 10.1038/nature12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robertson N, et al. The properties of thermostabilised G protein-coupled receptors (StaRs) and their use in drug discovery. Neuropharmacology. 2011;60:36–44. doi: 10.1016/j.neuropharm.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Chen YL, et al. 2-aryloxy-4-alkylaminopyridines: discovery of novel corticotropin-releasing factor 1 antagonists. J Med Chem. 2008;51:1385–1392. doi: 10.1021/jm070579c. [DOI] [PubMed] [Google Scholar]
- 14.Wootten D, et al. Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proc Natl Acad Sci U S A. 2013;110:5211–5216. doi: 10.1073/pnas.1221585110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schipani E, et al. Constitutive activation of the cyclic adenosine 3',5'-monophosphate signaling pathway by parathyroid hormone (PTH)/PTH-related peptide receptors mutated at the two loci for Jansen's metaphyseal chondrodysplasia. Mol Endocrinol. 1997;11:851–858. doi: 10.1210/mend.11.7.9934. [DOI] [PubMed] [Google Scholar]
- 16.Hjorth SA, et al. Constitutive activity of glucagon receptor mutants. Mol Endocrinol. 1998;12:78–86. doi: 10.1210/mend.12.1.0045. [DOI] [PubMed] [Google Scholar]
- 17.Rasmussen SG, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun C, et al. Solution structure and mutational analysis of pituitary adenylate cyclase-activating polypeptide binding to the extracellular domain of PAC1-RS. Proc Natl Acad Sci U S A. 2007;104:7875–7880. doi: 10.1073/pnas.0611397104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar S, et al. Crystal structure of the PAC1R extracellular domain unifies a consensus fold for hormone recognition by class B G-protein coupled receptors. PLoS One. 2011;6:e19682. doi: 10.1371/journal.pone.0019682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rivier J, et al. Synthetic competitive antagonists of corticotropin-releasing factor: effect on ACTH secretion in the rat. Science. 1984;224:889–891. doi: 10.1126/science.6326264. [DOI] [PubMed] [Google Scholar]
- 21.Donnelly D. The structure and function of the glucagon-like peptide-1 receptor and its ligands. Br J Pharmacol. 2012;166:27–41. doi: 10.1111/j.1476-5381.2011.01687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gardella TJ, et al. Molecular properties of the PTH/PTHrP receptor. Trends Endocrinol Metab. 2001;12:210–217. doi: 10.1016/s1043-2760(01)00409-x. [DOI] [PubMed] [Google Scholar]
- 23.Walker CS, et al. Regulation of signal transduction by calcitonin gene-related peptide receptors. Trends Pharmacol Sci. 2010;31:476–483. doi: 10.1016/j.tips.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 24.ter Haar E, et al. Crystal structure of the ectodomain complex of the CGRP receptor, a class-B GPCR, reveals the site of drug antagonism. Structure. 2010;18:1083–1093. doi: 10.1016/j.str.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 25.Miller LJ, et al. Refinement of glucagon-like peptide 1 docking to its intact receptor using mid-region photolabile probes and molecular modeling. J Biol Chem. 2011;286:15895–15907. doi: 10.1074/jbc.M110.217901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coopman K, et al. Residues within the transmembrane domain of the glucagon-like peptide-1 receptor involved in ligand binding and receptor activation: modelling the ligand-bound receptor. Mol Endocrinol. 2011;25:1804–1818. doi: 10.1210/me.2011-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neumann JM, et al. Class-B GPCR activation: is ligand helix-capping the key? Trends Biochem Sci. 2008;33:314–319. doi: 10.1016/j.tibs.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 28.Inooka H, et al. Conformation of a peptide ligand bound to its G-protein coupled receptor. Nat Struct Biol. 2001;8:161–165. doi: 10.1038/84159. [DOI] [PubMed] [Google Scholar]
- 29.Perret J, et al. Mutational analysis of the glucagon receptor: similarities with the vasoactive intestinal peptide (VIP)/pituitary adenylate cyclase-activating peptide (PACAP)/secretin receptors for recognition of the ligand's third residue. Biochem J. 2002;362:389–394. doi: 10.1042/0264-6021:3620389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Runge S, et al. Three distinct epitopes on the extracellular face of the glucagon receptor determine specificity for the glucagon amino terminus. J Biol Chem. 2003;278:28005–28010. doi: 10.1074/jbc.M301085200. [DOI] [PubMed] [Google Scholar]
- 31.Unson CG, et al. Roles of specific extracellular domains of the glucagon receptor in ligand binding and signaling. Biochemistry. 2002;41:11795–11803. doi: 10.1021/bi025711j. [DOI] [PubMed] [Google Scholar]
- 32.Roberts DJ, et al. Analysis of the glucagon receptor first extracellular loop by the substituted cysteine accessibility method. Peptides. 2011;32:1593–1599. doi: 10.1016/j.peptides.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 33.Prevost M, et al. Mutational and cysteine scanning analysis of the glucagon receptor N-terminal domain. J Biol Chem. 2010;285:30951–30958. doi: 10.1074/jbc.M110.102814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koth CM, et al. Molecular basis for negative regulation of the glucagon receptor. Proc Natl Acad Sci U S A. 2012;109:14393–14398. doi: 10.1073/pnas.1206734109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cascieri MA, et al. Characterization of a novel, non-peptidyl antagonist of the human glucagon receptor. J Biol Chem. 1999;274:8694–8697. doi: 10.1074/jbc.274.13.8694. [DOI] [PubMed] [Google Scholar]
- 36.Hruby VJ, et al. Conformational considerations in the design of glucagon agonists and antagonists: examination using synthetic analogs. Biopolymers. 1986;25(Suppl):S135–S155. [PubMed] [Google Scholar]
- 37.Unson CG, et al. Glucagon antagonists: contribution to binding and activity of the amino-terminal sequence 1–5, position 12, and the putative alpha-helical segment 19–27. J Biol Chem. 1989;264:789–794. [PubMed] [Google Scholar]
- 38.Ahn JM, et al. Development of potent truncated glucagon antagonists. J Med Chem. 2001;44:1372–1379. doi: 10.1021/jm000453e. [DOI] [PubMed] [Google Scholar]
- 39.Underwood CR, et al. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J Biol Chem. 2010;285:723–730. doi: 10.1074/jbc.M109.033829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mathi SK, et al. Scanning of the glucagon-like peptide-1 receptor localizes G protein-activating determinants primarily to the N terminus of the third intracellular loop. Mol Endocrinol. 1997;11:424–432. doi: 10.1210/mend.11.4.9913. [DOI] [PubMed] [Google Scholar]
- 41.Xiao Q, et al. Characterization of glucagon-like peptide-1 receptor-binding determinants. J Mol Endocrinol. 2000;25:321–335. doi: 10.1677/jme.0.0250321. [DOI] [PubMed] [Google Scholar]
- 42.Koole C, et al. Second extracellular loop of human glucagon-like peptide-1 receptor (GLP-1R) has a critical role in GLP-1 peptide binding and receptor activation. J Biol Chem. 2012;287:3642–3658. doi: 10.1074/jbc.M111.309328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tseng CC, et al. A point mutation in the glucose-dependent insulinotropic peptide receptor confers constitutive activity. Biochem Biophys Res Commun. 1997;232:96–100. doi: 10.1006/bbrc.1997.6231. [DOI] [PubMed] [Google Scholar]
- 44.Yaqub T, et al. Identification of determinants of glucose-dependent insulinotropic polypeptide receptor that interact with N-terminal biologically active region of the natural ligand. Mol Pharmacol. 2010;77:547–558. doi: 10.1124/mol.109.060111. [DOI] [PubMed] [Google Scholar]
- 45.Di Paolo E, et al. Mutations of aromatic residues in the first transmembrane helix impair signalling by the secretin receptor. Receptors Channels. 1999;6:309–315. [PubMed] [Google Scholar]
- 46.Di Paolo E, et al. Contribution of the second transmembrane helix of the secretin receptor to the positioning of secretin. FEBS Lett. 1998;424:207–210. doi: 10.1016/s0014-5793(98)00175-6. [DOI] [PubMed] [Google Scholar]
- 47.Dong M, et al. Mapping spatial approximations between the amino terminus of secretin and each of the extracellular loops of its receptor using cysteine trapping. FASEB J. 2012;26:5092–5105. doi: 10.1096/fj.12-212399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Solano RM, et al. Two basic residues of the h-VPAC1 receptor second transmembrane helix are essential for ligand binding and signal transduction. J Biol Chem. 2001;276:1084–1088. doi: 10.1074/jbc.M007696200. [DOI] [PubMed] [Google Scholar]
- 49.Ceraudo E, et al. Spatial proximity between the VPAC1 receptor and the amino terminus of agonist and antagonist peptides reveals distinct sites of interaction. FASEB J. 2012;26:2060–2071. doi: 10.1096/fj.11-196444. [DOI] [PubMed] [Google Scholar]
- 50.Gensure RC, et al. Identification of a contact site for residue 19 of parathyroid hormone (PTH) and PTH-related protein analogs in transmembrane domain two of the type-1 PTH receptor. Mol Endocrinol. 2003;17:2647–2658. doi: 10.1210/me.2003-0275. [DOI] [PubMed] [Google Scholar]
- 51.Grace CR, et al. NMR structure and peptide hormone binding site of the first extracellular domain of a type B1 G protein-coupled receptor. Proc Natl Acad Sci U S A. 2004;101:12836–12841. doi: 10.1073/pnas.0404702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liaw CW, et al. Localization of ligand-binding domains of human corticotropin-releasing factor receptor: a chimeric receptor approach. Mol Endocrinol. 1997;11:980–985. doi: 10.1210/mend.11.7.9946. [DOI] [PubMed] [Google Scholar]
- 53.Hoare SR, et al. Single amino acid residue determinants of non-peptide antagonist binding to the corticotropin-releasing factor1 (CRF1) receptor. Biochem Pharmacol. 2006;72:244–255. doi: 10.1016/j.bcp.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 54.Assil-Kishawi I, et al. Residue 17 of sauvagine cross-links to the first transmembrane domain of corticotropin-releasing factor receptor 1 (CRFR1) J Biol Chem. 2008;283:35644–35651. doi: 10.1074/jbc.M806351200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hinke SA, et al. Identification of a bioactive domain in the amino-terminus of glucose-dependent insulinotropic polypeptide (GIP) Biochim Biophys Acta. 2001;1547:143–155. doi: 10.1016/s0167-4838(01)00181-9. [DOI] [PubMed] [Google Scholar]
- 56.Unson CG, et al. Biological activities of des-His1[Glu9]glucagon amide, a glucagon antagonist. Peptides. 1989;10:1171–1177. doi: 10.1016/0196-9781(89)90010-7. [DOI] [PubMed] [Google Scholar]
- 57.Wu B, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shimamura T, et al. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mason JS, et al. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol Sci. 2012;33:249–260. doi: 10.1016/j.tips.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 60.Ballesteros JA, et al. Methods in Neurosciences. Vol. 25. Academic Press; 1995. pp. 366–428. [Google Scholar]
- 61.Pioszak AA, et al. Molecular recognition of corticotropin-releasing factor by its G-protein-coupled receptor CRFR1. J Biol Chem. 2008;283:32900–32912. doi: 10.1074/jbc.M805749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grace CR, et al. NMR structure of the first extracellular domain of corticotropin-releasing factor receptor 1 (ECD1-CRF-R1) complexed with a high affinity agonist. J Biol Chem. 2010;285:38580–38589. doi: 10.1074/jbc.M110.121897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grace CR, et al. Structure of the N-terminal domain of a type B1 G protein-coupled receptor in complex with a peptide ligand. Proc Natl Acad Sci U S A. 2007;104:4858–4863. doi: 10.1073/pnas.0700682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parthier C, et al. Crystal structure of the incretin-bound extracellular domain of a G protein-coupled receptor. Proc Natl Acad Sci U S A. 2007;104:13942–13947. doi: 10.1073/pnas.0706404104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pioszak AA, et al. Dimeric arrangement of the parathyroid hormone receptor and a structural mechanism for ligand-induced dissociation. J Biol Chem. 2010;285:12435–12444. doi: 10.1074/jbc.M109.093138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pioszak AA, et al. Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proc Natl Acad Sci U S A. 2008;105:5034–5039. doi: 10.1073/pnas.0801027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pioszak AA, et al. Structural basis for parathyroid hormone-related protein binding to the parathyroid hormone receptor and design of conformation-selective peptides. J Biol Chem. 2009;284:28382–28391. doi: 10.1074/jbc.M109.022905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Runge S, et al. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J Biol Chem. 2008;283:11340–11347. doi: 10.1074/jbc.M708740200. [DOI] [PubMed] [Google Scholar]
- 69.Dong M, et al. Spatial approximation between the amino terminus of a peptide agonist and the top of the sixth transmembrane segment of the secretin receptor. J Biol Chem. 2004;279:2894–2903. doi: 10.1074/jbc.M310407200. [DOI] [PubMed] [Google Scholar]
- 70.Ceraudo E, et al. The vasoactive intestinal peptide (VIP) alpha-Helix up to C terminus interacts with the N-terminal ectodomain of the human VIP/Pituitary adenylate cyclase-activating peptide 1 receptor: photoaffinity, molecular modeling, and dynamics. Mol Endocrinol. 2008;22:147–155. doi: 10.1210/me.2007-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.