

Abstract
Antigen binding to the B cell receptor (BCR) induces receptor desensitization, a condition characterized by cellular unresponsiveness to subsequent Ag stimulation despite the continued ability to bind Ag. To better understand the molecular mechanism of this unresponsiveness, we have used complementary lymphoma (K46 μ) and Ig transgenic (3-83 μδ) mouse models to study regulation of BCR signaling. Our findings in the lymphoma model show that an initial Ag encounter renders receptors unresponsive to subsequent Ag challenge, as measured by their inability to mobilize Ca2+ and to mediate phosphorylation of receptor-proximal kinases, including Lyn, Blk, and Syk. Most importantly, the Igα and Igβ components of desensitized receptors are not phosphorylated, and receptor-associated kinases are not activated upon Ag challenge. The molecular defect does not appear to result from Lyn inactivation, sequestration, or repression, since Lyn from desensitized cell lysates is activated in vitro by synthetic doubly phosphorylated immunoreceptor tyrosine-based activation motif peptides. A similar deficit in Ag-induced receptor phosphorylation was observed in desensitized B cells from 3-83 μδ transgenic mice. These studies indicate that Ag receptor desensitization reflects an inability to initiate activation of receptor-associated kinases that normally phosphorylate receptor Igαβ subunits, leading to signal propagation.
Desensitization of the B cell Ag receptor (BCR)3 follows binding of Ag to as few as 5% of receptors, leaving available receptors unresponsive to subsequent Ag binding (1–3). The molecular basis of receptor desensitization was initially studied in an in vitro system in which ligation of 5 to 10% of mIgM with anti-μ Abs desensitized the heterologous mIgD receptor (1, 4, 5). The cells did not mobilize Ca2+ and exhibited a block in signal transduction proximal to the receptor. Analysis of the involvement of protein kinase C (PKC) activation revealed that although a short-lived PKC-mediated BCR desensitization mechanism is operative, long term desensitization (>24 h) was mediated by a PKC-independent mechanism (1). In a more physiologically relevant setting, Ag-induced desensitization was studied by Lazarus et al., who extended these observations using a trinitro-phenol-specific B cell lymphoma and found receptor desensitization to be either upstream or independent of Ca2+ mobilization and PKC activation (6, 7). These studies defined the defect in desensitized cells as upstream of Ca2+ mobilization and proximal to the receptor. Finally, Cooke et al. have defined a signaling defect in anergic B cells characterized by failed Ca2+ mobilization and tyrosine phosphorylation of multiple undefined substrates following Ag stimulation. It is unknown whether the mechanisms involved in the unresponsiveness of anergic B cells are equivalent to those that mediate acute receptor desensitization.
Signal transduction through the BCR complex involves tyrosine phosphorylation of the mIg-associated transducing molecules, Igα and Igβ, (reviewed in Refs. 8–10). Within each of these molecules is found a conserved motif termed the ITAM (immunoreceptor tyrosine-based activation motif) that contains all the structural information necessary for transduction of signals through the receptor (10–12). Studies of signaling by ITAM-containing receptors are most consistent with the initial activation of Src family kinases through a receptor aggregation-dependent trans-activation mechanism, followed by ITAM phosphorylation (12–32). Syk activation is required for subsequent PLCγ phosphorylation and phosphoinositide hydrolysis leading to Ca2+ mobilization. The resting BCR is reportedly associated with the Src family members Lyn/Fyn/Blk through binding via their N-terminal unique regions to the nonphosphorylated ITAM sequence. Receptor aggregation, initial kinase activation, and ITAM phosphorylation lead to kinase reorientation, such that kinase binding now occurs via SH2 interaction with ITAM phosphotyrosine. This interaction leads to signal amplification through additional ITAM phosphorylation and kinase recruitment (17, 33). Full receptor activation appears to require the SH2-dependent recruitment of additional effector molecules, including Syk and Shc, that bind to ITAM phosphotyrosines (34–41).
In this report, we further define the molecular basis of BCR desensitization. Using a B lymphoma line specific for nitrophenol (NP) and NP-protein conjugates, we were able to show that ligation of the BCR by Ag leads to receptor desensitization. Our findings demonstrate that subsequent ligation of desensitized receptors does not lead to Ca2+ mobilization and fails to induce increased tyrosine phosphorylation of several key effectors. Most importantly, Igα, Igβ, Src family kinases, and Syk are not phosphorylated upon challenge, nor is receptor-associated kinase activity increased. However, Lyn from desensitized cells can bind doubly phosphorylated ITAM peptides and can be activated in vitro by doubly phosphorylated ITAM peptides. Examination of ex vivo B cells from 3-83 μδ Ig transgenic mice revealed that in this model, desensitization also leads to diminished Ag-induced receptor phosphorylation. These studies indicate that despite the ability of desensitized receptors to bind Ag, previous receptor ligation disrupts the earliest detectable events in receptor activation, Src family kinase activation and phosphorylation of the receptor Igα and Igβ subunits.
Materials and Methods
Cells
The K46 μ lymphoma line was (42, 43) cultured in Iscove’s modified Dulbecco’s medium (IMDM) supplemented with 5% FCS (HyClone, Logan, UT), 1 mM sodium pyruvate, 50 μg/ml gentamicin, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM glutamine, and 50 μM β-ME at 37°C in 7.5% CO2. The 3-83 μδ Ig transgenic mice express IgM and IgD specific for H-2Kk and have been previously described (44).
Isolation of primary B lymphocytes
Resting splenic B cells were prepared as previously described (1). Briefly, high density (ρ > 1.070) splenic B lymphocytes were isolated from heterozygous 3-83 μδ transgenic mice. Total splenic cell preparations were depleted of T lymphocytes by complement-mediated lysis, and resting cells were isolated by discontinuous density gradient centrifugation using Percoll.
Ag preparation and cell surface staining
NP-coupled BSA was prepared by mixing NP-CAP-OSucc/DMF (succinimidyl ester of nitrophenyl caproak) (Cambridge Research Biochemicals Ltd., Cambridge, MA) with BSA/3% NaHCO3. The mixture was incubated at room temperature for 2 h and then dialyzed against 3% NaHCO3, followed by dialysis against PBS. A typical reaction containing 4 mg of NP and 40 mg of BSA yielded a NP7BSA preparation with a valency of 7 to 10 nitrophenol groups/BSA molecule. The exact valency of each preparation was determined by calculating the molar ratio of NP to BSA. The molarity of NP was determined by OD430, and the molarity of BSA was determined by Bradford assay. NP-BSA and the BSA control were biotinylated according to the manufacturer’s instructions (EZ-link biotin, Cat # 21335, Pierce Chemical Co., Rockford, IL). Each preparation of Ag was tested by cell surface staining to determine the number of receptors occupied at any given dose and the valency of Ag. Briefly, cells were desensitized for 2 h at 37°C with nonbiotinylated NP7BSA, then 1 × 106 K46 μ cells/0.2 ml were washed three times with staining medium (balanced salt solution containing 2% FCS and 0.2% sodium azide). Untreated and treated cells were then stained with 20 μg/5 × 106 cells/ml of biotinylated NP7BSA. The cells were incubated on ice for 15 min, then washed three times with cold staining medium. Subsequently, phycoerythrin-conjugated avidin was added, and the suspension was incubated for an additional 20 min on ice, washed three times with staining medium, and analyzed by FACScan (Becton Dickinson, Mountain View, CA). Alternatively, the 3-83 reactive, H-2Kk Ag-mimetic sequence, CAHDWRSGFGGFQHLCCGAAGA, referred to as 3-83ag1, was defined by screening of phage display libraries as described by Sparks et al. (45). This reactive peptide insert was synthesized by the NJC Molecular Resource Center and coupled to N-ethylmaleimide-activated dexamine (100 kDa) to produce a hapten carrier ratio of 50 mol/mol. Characterization of the 3-83ag1 antigenic activity has been previously described (V. Kouskoff and D. Nemazee, manuscript in preparation). Receptor occupancy and doses required for optimal desensitization of the 3-83 μδ Ig transgenic B cells were determined as described, except that 3-83ag1dex was titrated as the Ag.
Measurement of Ca2+
Intracellular free calcium concentrations ([Ca2+]i) were determined as previously described (1). Briefly, K46 μ and ex vivo 3-83 μδ cells were washed once in buffer A (10 mM HEPES in HBSS, pH 6.9), then resuspended at 5 × 106 cells/ml in buffer A. Indo-1/AM in DMSO was added to the cell suspensions to a final concentration of 10 μM, and cells were incubated for 30 min at 37°C. The cells were diluted 1/2 with buffer B (10 mM HEPES and 3% FCS in HBSS, pH 7.4) and incubated for an additional 30 min at 37°C. The excess indo was removed by washing the cells three times with buffer C (IMDM containing 3% FCS). The cells were resuspended in buffer C at a concentration of 5 × 106/ml and maintained at room temperature until analysis. Flow cytometric analysis of [Ca2+]i was performed as previously described using the Ortho 50H flow cytometer (Ortho Diagnostics, Raritan, NJ) (46). Data were analyzed using MTime Software (Phoenix Flow Systems, San Diego, CA).
Induction of desensitization and preparation of cell lysates
Cells were harvested from log phase cultures by centrifugation and resuspended in fresh IMDM, then incubated at 37°C for approximately 2 h with the desensitizing dose of Ag (25 ng of NP7BSA/5 × 106 cells/ml for K46 μ, 300–500 ng of 3-83ag150Dex/5 × 106/ml for splenic B cells). Following this incubation, cells were again harvested by centrifugation and resuspended in IMDM, then treated with the challenge dose of Ag (20 μg of NP7BSA/50 × 106/ml for K46 μ, 1 μg of 3-83 μδ/50 × 106/ml splenic B cells). Treated cells were lysed in buffer containing 1% Nonidet P-40, 150 mM NaCl, 10 mM Tris (pH 7.5), 2 mM sodium o-vanadate, 1 mM PMSF, 0.4 mM EDTA, 10 mM NaF, and 1 μg/ml each of aprotinin, leupeptin and α1-antitrypsin. Lysates were incubated on ice for 5 min, then cleared of particulate nuclear/cytoskeletal components by centrifugation at 12,000 × g for 10 min. Alternatively, cells were lysed in 1% digitonin, 150 mM NaCl, 10 mM Tris (pH 7.5), 2 mM sodium o-vanadate, 1 mM PMSF, 0.4 mM EDTA, 10 mM NaF, and 1 μg/ml each of aprotinin, leupeptin, and α1-antitrypsin.
Ab preparations, immunoprecipitation, and immunoblot analysis
Rabbit antisera against Lyn residues 1–131 and Blk residues 1–218 (47) were prepared by immunization of rabbits with bacterially expressed, factor Xa-cleaved (Boehringer Mannheim, Indianapolis, IN) protein fragments generated as glutathione S-transferase fusion proteins. Anti-Syk antiserum was prepared against the Syk linker region (residues 150–215) as described by Couture et al. (38). The polyclonal anti-Igα was provided by Jan Jonstra (Toronto Western Hospital, Toronto, Canada). The Lyn, Igα, and Blk Abs were affinity purified using fusion proteins coupled to Sepharose. The Shc Ab (catalogue No. 06–203) was purchased from Upstate Biotechnology, Inc. (Lake Placid, NY), and anti-phosphotyrosine Ab, Ab-2, was purchased from Oncogene Science (Manhasset, NY). The monoclonal anti-μ Ab, b-7–6, was protein G purified from culture supernatants (48).
Immunoprecipitations of Lyn, Blk, Shc, Syk, and Igα were performed on lysates of 25 × 106 cell equivalents using 5 to 10 μg of affinity-purified Ab coupled to Sepharose beads, Immunoprecipitates were washed three times in lysis buffer, resuspended in 50 μl of reducing SDS-PAGE sample buffer, then fractionated using 10% SDS-PAGE gels. Electrophoretically fractionated proteins were transferred to polyvinylidene difluoride membranes using a semidry blotting apparatus following the conditions recommended by the manufacturer (Millipore Corp., Bedford, MA). Polyvinylidene difluoride membranes were blocked in Tris-buffered saline (TBS) containing 4% BSA (Boehringer Mannheim). For immunoblotting, membranes were incubated with primary Ab at the appropriate dilutions for 2 to 4 h, then washed three times with TBS containing 0.05% Triton X-100. The blots were subsequently incubated with secondary Ab, either protein A conjugated with horseradish peroxidase (Amersham, Arlington Heights, IL) or rat anti-mouse IgG1 (Zymed, South San Francisco, CA), for 30 min and washed three times with TBS/0.05% Triton. Immunoreactive proteins were detected by enhanced chemiluminescence (ECL, Amersham. Arlington Heights, IL). In some cases, membranes were stripped by incubation in buffer containing 100 mM β-ME, 2% SDS, and 62.5 mM Tris (pH 6.7) for 30 min at 56°C, then washed in TBS containing 0.05% Triton before reprobing with Abs.
In vitro kinase assay/Lyn activation with (p)2 ITAM peptide
To accomplish coprecipitation of receptor components, BCR was immunoprecipitated from digitonin lysates. Lyn was immunoprecipitated from Nonidet P-40 lysates of K46 μ cells as described above. Kinase activity was assayed essentially as previously described (49). Following three washes in lysis buffer, the BCR- or Lyn-adsorbed, Ab-coupled Sepharose beads were washed in kinase buffer (10 mM HEPES (pH 7.0), 10 mM MgCl2, 2 mM sodium o-vanadate, and 1 mM PMSF) and resuspended in 25 μl of kinase buffer containing 2 mM exogenous substrate (Lck auto-phosphorylation site RRLIEDAEYAARG), 10 μM ATP, and 10 μCi of [γ-32P]ATP (New England Nuclear-DuPont). Reactions were incubated for 10 min at 30°C, then quenched with trichloroacetic acid at a final concentration of 5%. The soluble portion of each reaction mixture was blotted to Whatman p81 phosphocellulose (Whatman, Clifton, NJ), which was then washed four times with a large volume of 75 mM phosphoric acid, dried with acetone, and subjected to liquid scintillation spectrometry.
Lyn activation by (p)2ITAM was assayed in vitro as previously described (28, 31). Immunoprecipitated Lyn (10 × 106 cell equivalents, adsorbed to 5 μg of anti-Lyn Sepharose) or receptor immunoprecipitates (25 × 106 cell equivalents, adsorbed to 20 μg b-7–6 · Sepharose) were first incubated for 1 h at 4°C with 0 to 500 μM (p)2 ITAM peptide or ITAM peptide containing a Y to F substitution of both conserved tyrosines (Y182, Y193) in a final volume of 15 μl. Following incubation, 10 μl of the reaction mix, containing the substrate and ATP, was added, and the reactions were conducted as described above.
Results
Desensitized K46 μ cells exhibit impaired Ca2+ mobilization and protein tyrosine phosphorylation in response to Ag stimulation
To address the molecular basis of receptor desensitization, we established an in vitro model to assess Ag regulation of receptor function using the B lymphoma, K46 (42). This lymphoma has been stably transfected with Ig heavy and light chain genes encoding an mIgM specific for NP (43). To establish the conditions under which Ag induces desensitization of K46 μ receptors, cells were exposed to various concentrations of Ag for 2 h before responsiveness was assessed by a second stimulation with Ag (challenge) and analysis of calcium mobilization. The 2 h point was chosen to avoid confusion with a short-lived, PKC-dependent mechanism of receptor desensitization (4). As shown in Figure 1, exposure of 5 × 106 cells to >1 ng of NP7BSA/5 × 106 cells/ml reduced responsiveness to challenge with 20 μg of NP7BSA/5 × 106 cells/ml. Complete unresponsiveness was seen at desensitizing ligand concentrations of >10 ng of NP7BSA/5 × 106 cells/1 ml. Therefore, 25 ng of NP7BSA/5 × 106 cells/ml was chosen as the desensitizing dose in subsequent experiments. As shown in Figure 2, 25 ng of NP7BSA/5 × 106/ml (middle panel) induced comparable [Ca2+]i mobilization as anti-receptor Ab (left panel) in non-desensitized, naive cells. In contrast, cells incubated for 2 h with 25 ng of NP7BSA/5 × 106/ml did not respond to challenge with 20 μg of NP7BSA/5 × 106/ml (right panel, closed arrow), although these cells remained responsive to ionomycin (right panel, open arrow).
FIGURE 1.
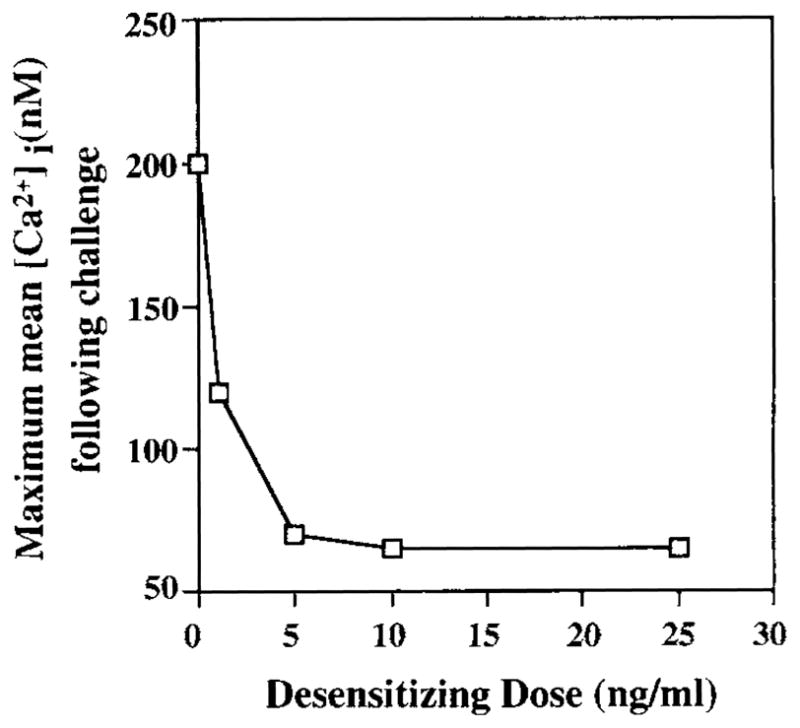
Ca2+ mobilization responses following challenge of K46 μ cells desensitized with increasing doses of Ag (NP7BSA). K46 μ cells were desensitized by incubation for 2 h with increasing doses (0–30 ng of NP7BSA/5 × 106 cells/ml) of Ag, then challenged with 20 μg of NP7BSA/5 × 106 cells/ml. Shown is the maximum mean [Ca2+]i achieved following challenge. The mean [Ca2+]i in resting unstimulated cells was 70 nM. Data were reproduced in two independent experiments.
FIGURE 2.
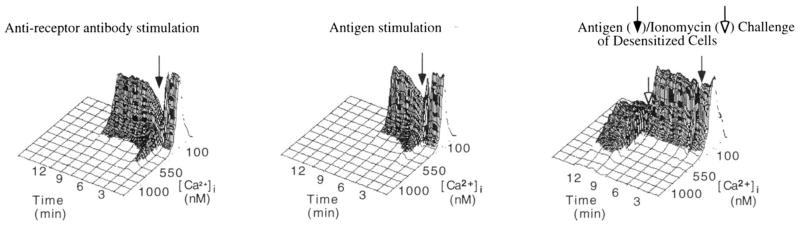
Isometric displays of [Ca2+]i in K46 μ cells responding to various stimuli. Left panel, Mean [Ca2+]i of untreated K46 μ cells before and after stimulation (stimulus added at arrow) with 10 μg anti-μ/ml. Middle panel, Mean [Ca2+]i of untreated K46 μ cells before and after stimulation with 25 ng of NP7BSA/5 × 106 cells/ml. Right panel, K46 μ cells that were desensitized with 25 ng of NP7BSA/5 × 106 cells/ml, then challenged with 20 μg of NP7BSA/5 × 106 cells/ml (added at filled arrow), followed by a subsequent challenge with ionomycin (1 μM; added at open arrow). Data were reproduced in three independent experiments and are expressed as cell number vs nanomolar [Ca2+)i over time.
To ensure that the failure of cells to respond to challenge was not simply due to loss or blockade of cell surface receptors, the approximate proportion of receptors occupied by the desensitizing dose and the proportion of receptors capable of binding Ag following the desensitizing period were determined compared with those on cells that had not encountered Ag. Although it would be more accurate to estimate receptor occupancy with a monovalent ligand, the monovalent Ags do not induce BCR signaling and receptor desensitization. Thus, to establish the relative receptor occupancy under the conditions in which receptor desensitization was induced, multivalent Ag (Biot-NP7BSA) was used to stain cells. First, to estimate the number of receptors occupied by low doses of Ag, including the dose used for desensitization, cells were stained with increasing concentrations of biotinylated NP7BSA. This staining revealed receptor occupancies of 14 to 28% at Ag doses ranging from 10 to 100 ng/ml, with the desensitizing dose of 25 ng occupying approximately 25% of the receptors (data not shown). To confirm the availability of Ag binding receptors following desensitization, biotinylated and nonbiotinylated forms of NP7BSA were prepared and used for staining. Cells were desensitized with nonbiotinylated Ag (25 ng of NP7BSA/5 × 106 cells/ml) for 2 h as shown in Figures 1 and 2, then stained with doses of biotinylated Ag (20 μg of Biot-NP7BSA/ml) and phycoerythrin-avidin that were shown in preliminary experiments to saturate receptors. Alternatively, cells were incubated for 2 h without Ag and stained with a dose of biotinylated Ag (20 μg of Biot-NP7BSA/ml) that saturated receptors to provide a control for the total number of receptors on naive cells. The data presented in Figure 3 show that, compared with control cells, cells desensitized for 2 h with 25 ng of NP7BSA/5 × 106 cells/ml had an estimated 70% of receptors still available for binding to Ag (control cell mean fluorescence of 170 compared with desensitized cell mean fluorescence of 122). This is consistent with the previous data indicating that 25% of receptors were occupied by this desensitizing dose. The availability of this proportion of receptors for Ag binding following desensitization with 25 ng/5 × 106 cells/ml was also confirmed using FITC-labeled NP1BSA (data not shown). Whether these remaining receptors had encountered Ag during the desensitizing period, and their Ag then dissociated cannot be determined. Thus, the lack of a response following challenge was not due to blockade or significant loss of receptors during the desensitizing period.
FIGURE 3.

Immunofluorescence histograms illustrating the relative numbers of receptors remaining following desensitization of K46 μ cells. The thick line reflects 0% receptor occupancy and was generated by analysis of cells stained with 20 μg of biotinylated BSA/ml followed by phycoerythrin-avidin. The black line was generated by analysis of cells that were desensitized for 2 h with 25 ng of nonbiotinylated NP7BSA/ml, then stained with 20 μg of biotinylated NP7BSA/ml and phycoerythrin-avidin. The thin gray line represents untreated cells, stained with 20 μg of biotinylated NP7BSA/ml and phycoerythrin-avidin to determine the mean fluorescence of cells at 100% receptor occupancy. Data are expressed as cell number vs log fluorescence intensity. Each histogram was generated based on analysis of 5000 cells.
To address the responsiveness of the desensitized cells at a more receptor-proximal point in the signaling pathway, patterns of protein tyrosine phosphorylation were compared in unstimulated, stimulated, desensitized, and desensitized/challenged K46 μ cells. As shown in Figure 4, cells stimulated for 1 min with the challenge dose of Ag (20 μg of NP7BSA/ml) exhibited a dramatic increase in tyrosine phosphorylation compared with unstimulated cells (lane 2 compared with lane 1). Cells that had been desensitized for 2 h showed essentially equivalent protein tyrosine phosphorylation as unstimulated cells. The level of tyrosine phosphorylation did not increase when the desensitized cells were challenged for 1 min (lane 4 compared with lane 3) in many experiments, although some experiments did show small amounts of inductive tyrosine phosphorylation (see WCL in Fig. 6). The Ag dose dependence of desensitization, as assessed by the tyrosine phosphorylation patterns, showed that cells desensitized for 2 h with 10, 25, or 50 ng/ml of Ag failed to respond to challenge with 20 μg/ml of Ag by protein phosphorylation, yet these same doses yielded maximal tyrosine phosphorylation when used to stimulate naive cells (data not shown). These results showed that the desensitized phenotype is evident based on the lack of induced protein tyrosine phosphorylation detectable in whole cell lysate immunoblots as well as by a failure to mobilize Ca2+ despite the availability of approximately 70% of receptors for binding Ag. Thus, the phenotype of desensitized cells appears equivalent to that of anergic cells from the anti-HEL transgenic model (50).
FIGURE 4.

Challenge of desensitized K46 μ cells does not lead to increased whole cell (WCL) protein tyrosine phosphorylation. Anti-phosphotyrosine immunoblot of whole cell lysates of 2.5 × 106 K46 μ equivalents. Lane 1, Unstimulated cells; lane 2, cells stimulated for 2 min with 20 μg of NP7BSA/50 × 106 cells/ml; lane 3, cells desensitized for 2 h with 25 ng of NP7BSA/5 × 106 cells/ml; lane 4, represents cells desensitized for 2 h with 25 ng of NP7BSA/5 × 106 cells/ml, then challenged for 2 min with 20 μg of NP7BSA/50 × 106 cells/ml. The experiment shown is representative of four independent experiments.
FIGURE 6.

Reduced tyrosine phosphorylation of Igα and Igβ in desensitized cells. Upper panel, Anti-phosphotyrosine immunoblot of SDS-PAGE-fractionated, nitrocellulose transfers of whole cell (WCL) lysates (lanes 1–4) from 2.5 × 106 K46 μ. or Igα IP from 25 × 106 cell equivalents (lanes 5–8). Lanes 1 and 5, Unstimulated cells; lanes 2 and 6, cells stimulated for 2 min with 20 μg of NP7BSA/50 × 106/ml; lanes 3 and 7, cells desensitized for 2 h with 25 ng of NP7BSA/5 × 106/ml; lanes 4 and 8, cells desensitized for 2 h with 25 ng of NP7BSA/5 × l06/ml, then restimulated for 2 min with 20 μg of NP7BSA/50 × 106/ml. Arrows depict the relative locations of Igα and lgβ. Lower panel, Anti-lgα immunoblot performed after stripping the upper blot. The arrow depicts the relative migration of Igα and corresponds to the band labeled Igα in the upper blot.
Ligation of desensitized receptors does not lead to phosphorylation of the receptor Igα and Igβ chains or associated kinases
To further address the molecular mechanism of receptor desensitization, we assessed the phosphorylation state of individual effectors known to be early intermediaries in the BCR signaling cascade. As shown in Figure 5A, immunoprecipitation of the Src family members, Lyn and Blk, followed by anti-phosphotyrosine immunoblotting revealed Ag-induced phosphorylation of both molecules (lane 1 compared with lane 2 in each panel). The phosphorylation state of these molecules did not completely returned to baseline after 2 h, but upon challenge, Lyn was not inducibly phosphorylated, and Blk showed only a slight increase in phosphorylation (lane 3 compared with lane 4 in each panel). Reprobing of the same blot with anti-Lyn and anti-Blk revealed that the differences in phosphotyrosine were indeed due to differences in kinase phosphorylation and not to differences in kinase recovery. A similar reduction in Shc and Syk tyrosine phosphorylation was seen following challenge of desensitized cells (Fig. 5B), suggesting that the ability to trigger phosphorylation of the kinases most proximal to the receptor is markedly diminished in desensitized cells.
FIGURE 5.

A, Reduced BCR-mediated tyrosine phosphorylation of Lyn and Blk in desensitized cells. Upper panel, Anti-phosphotyrosine immunoblot of SDS-PAGE-fractionated nitrocellulose transfers of anti-Lyn immunoprecipitates (IP) (upper left) and anti-Blk IP of 25 × 106 K46 μ cells (upper right). Lane 1 of each panel represents immunoprecipitates from unstimulated cells, lane 2 of each panel represents cells stimulated for 2 min with 20 μg of NP7BSA/50 × 106 cells/ml, lane 3 of each panel represents cells desensitized for 2 h with 25 ng of NP7BSA/5 × 106 cells/ml, lane 4 of each panel represents cells desensitized for 2 h with 25 ng of NP7BSA/5 × 106 cells/ml, then challenged for 2 min with 20 μg of NP7BSA/50 × 106 cells/ml. Lane 5 represents the proteins binding to an equivalent volume of protein A beads. The arrows beside each panel depict the positions of the 53- and 55-kDa forms of Lyn, and the 55-kDa form of Blk. Lower panel, Anti-Lyn or anti-Blk immunoblot performed after stripping the upper blots. Lyn and Blk bands correspond precisely to the annotated bands above. B, Reduced tyrosine phosphorylation of Syk and Shc in desensitized cells. Upper panel, Anti-phosphotyrosine immunoblots of SDS-PAGE-fractionated, nitrocellulose transfers of anti-Syk IP (upper left) and anti-Shc IP (upper right) from 25 × 106 K46 μ cells. Lane 1 of each panel represents immunoprecipitates from unstimulated cells, lane 2 of each panel represents cells stimulated for 2 min with 20 μg of NP7BSA/50 × 106/ml, lane 3 of each panel represents cells desensitized for 2 h with 25 ng of NP7BSA/5 × 106/ml, lane 4 of each panel represent cells desensitized for 2 h with 25 ng of NP7BSA/5 × 106/ml, then challenged for 2 min with 20 μg of NP7BSA/50 × 106/ml. Lane 5 represents the proteins from stimulated lysates that bind to an equivalent volume of protein A beads. The arrows beside each panel depict the 72-kDa form of Syk and the 46-and 52-kDa forms of Shc. Lower panel, Anti-Syk immunoblot or anti-Shc immunoblot performed after stripping the upper blots. Syk and Shc bands correspond precisely to the annotated bands above.
Since Src family kinases are physically associated with the BCR and are rapidly activated upon Ag binding, they represent strong candidates as mediators of ITAM phosphorylation. Src family kinases have been shown to mediate ITAM phosphorylation in BCR, TCR, and FcεRI reconstitution systems, and Lck expression is essential for TCR-mediated ITAM phosphorylation in Jurkat cells (16, 20, 51). Given the reduction of induced phosphorylation of Lyn and Blk, we assessed the effect of receptor desensitization on the phosphorylation state of the ITAM-containing receptor sub-units. Anti-phosphotyrosine immunoblotting of Igα immunoprecipitated from unstimulated, stimulated, desensitized, and desensitized/challenged cell lysates revealed that challenge-induced phosphorylation of Igα and Igβ was greatly reduced in desensitized cells (Fig. 6; inductive phosphorylation of lanes 5 and 6 compared with lanes 7 and 8). Based on previous demonstrations that Igβ occurs as a disulfide-linked heterodimer with Igα, is tyrosine phosphorylated, and has a relative molecular mass of 37 kDa, we conclude that the phosphotyrosine band at approximately 37 kDa in the anti-Igα precipitate is Igβ (52–54). Again, these differences reflected failed phosphorylation, since reprobing the same blot with anti-Igα revealed similar amounts of Ag in each sample. In all experiments performed, there was either failure to phosphorylate Igα and Igβ (Fig. 4, whole cell lysate) or a marked reduction in challenge-induced Igα and Igβ tyrosine phosphorylation (Fig. 6). In cases in which the reduction was not complete, anti-phosphotyrosine blotting of whole cell lysates (Fig. 6, lanes 1–4) indicated that desensitization was incomplete. These data reveal a defect at the level of receptor-mediated Igα and Igβ phosphorylation in desensitized cells and suggest that a failure to activate kinases that normally associate with the receptor leads to failed ITAM phosphorylation.
Activation of receptor-associated tyrosine kinase is markedly diminished in desensitized cells
Since phosphorylation of several receptor proximal kinases and the Igα/β transducing molecules was markedly diminished in desensitized cells, we assessed whether ligation of desensitized receptors leads to activation of receptor-associated tyrosine kinase. Receptor-associated kinase activity was measured by immunoprecipitating the BCR with an anti-IgM Ab, then assaying the kinase activity based on phosphorylation of an exogenous peptide substrate. As shown in Figure 7, in four independent experiments stimulated cells exhibited approximately threefold increased kinase activity compared with unstimulated cells. This receptor-associated kinase activity returned to the level found in unstimulated cells following incubation for 2 h and upon subsequent Ag challenge did not increase significantly. These results confirm a lack of ligand-induced increase in receptor-associated kinase activity in desensitized cells, consistent with the failure to phosphorylate receptor-associated Igα, Igβ, Lyn, and Syk.
FIGURE 7.

Ag does not induce activation of receptor-associated kinases in desensitized cells. Surface mlgM receptors were immunoprecipitated from digitonin lysates of 25 × 106 unstimulated, stimulated, desensitized, and desensitized/challenged K46 μ cells as described in Figure 5. The kinase activity of the immunoprecipitated receptor complexes was assessed using an exogenous Lck substrate. Data are shown as the fold increase over unstimulated vs the various cell treatments. The fold increase was calculated by assigning the counts per minute of the unstimulated sample a value of 1, then calculating the relative fold increase in the other cell treatments. The primary data (counts per minute of reaction – counts per minute of background) of unstimulated, stimulated, desensitized, and desensitized/challenged cells, respectively, are as follows: Expt. 1, 22,535, 57,881, 15,818, and 28,425; Expt. 2, 18,879, 51,463, 18,540, and 33,502; Expt. 3, 18,411, 35,716, 19,001, and 19,353; and Expt. 4, 16,002, 51,224, 26,499, and 11,479.
Lyn from desensitized cells binds to and is activated by doubly phosphorylated ITAM peptides
The results presented above clearly show a failure of Ag to induce phosphorylation and activation of receptor-associated kinases and phosphorylation of Igα and Igβ in desensitized cells. Whether this reflects a modification of Lyn, making it nonfunctional in signaling, or a modification of the receptor remained to be determined. To begin to address these possibilities we evaluated the ability of Lyn and Syk from normal and desensitized cells (Fig. 8A) to bind synthetic, phosphorylated and nonphosphorylated ITAM peptides. Previously it was shown that Lyn associates via its N-terminal unique region with the nonphosphorylated ITAM sequence, then reorients, undergoing an SH2-ITAM phosphotyrosine interaction upon ITAM phosphorylation (17, 33). As shown in Figure 8B, Syk and Lyn from desensitized cell lysates bound the doubly phosphorylated ITAM peptides ((p)2ITAM) similarly to Syk and Lyn from unstimulated cell lysates. The slight difference in the amount of Syk and Lyn binding the phosphorylated ITAM in unstimulated compared with desensitized lysates (Fig. 8B, lane 2 compared with lane 4) was not consistent in repeat experimentation. These results suggest that Lyn from desensitized cells is capable of binding a phosphorylated ITAM motif.
FIGURE 8.

Lyn and Syk from desensitized cells bind to phosphorylated and nonphosphorylated ITAMs. A, Anti-phosphotyrosine immunoblot of SDS-PAGE-fractionated, nitrocellulose transfers of 2.5 × 106 whole cell (WCL) lysate equivalents to show cells were desensitized. Lane 1, Unstimulated cells; lane 2, cells stimulated for 2 min with 20 μg of NP7BSA/50 × 106 cells/ml; lane 3, cells desensitized 2 h with 25 ng of NP7BSA/5 × 106 cells/ml; lane 4, cells desensitized 2 h with 25 ng of NP7BSA/5 × 106 cells/ml, then challenged for 2 min with 20 μg of NP7BSA/50 × 106 cells/ml. B, Anti-Syk (upper panel) and anti-Lyn (lower panel) immunoblots of SDS-PAGE-fractionated, nitrocellulose transfers of 25 × 106 cell equivalents binding (p)2ITAM peptides (20 μg of doubly phosphorylated ITAM) in unstimulated (lane 2), stimulated (lane 3), desensitized (lane 4), and desensitized/challenged (lane 5) lysates. Lane 1 depicts protein binding from stimulated lysates to an equivalent amount of underivatized Sepharose beads. C, Anti-Lyn immunoblot of SDS-PAGE-fractionated, nitrocellulose transfers of 25 × 106 cell equivalents binding the nonphosphorylated ITAM peptide (50 μg of ITAM) in unstimulated (lane 1) and desensitized (lane 2) lysates. Lane 3 represents protein binding an equivalent amount of underivatized Sepharose beads.
The available evidence indicates that Lyn normally associates with the resting receptor via Igα ITAM (17, 28, 33). This reservoir of receptor-associated kinase may be responsible for initial receptor phosphorylation (16, 20, 51). In an attempt to assess this aspect of Lyn function, we undertook quantitation of the amount of Lyn associated with the desensitized compared with resting receptors. Using in vitro kinase reaction labeling, we detected in both cells a receptor-associated phosphoprotein of an appropriate size to be Lyn (data not shown). Unfortunately, we were unable to confirm the identity of the molecule by immunoblotting with Src family kinase antisera, presumably because the amount of protein was below our limit of detection. As an alternative approach we assessed the ability of Lyn from desensitized and normal B cell lysates to bind nonphosphorylated ITAM peptides. Although much less Lyn binds the nonphosphorylated receptor compared with the phosphorylated receptor (exposure time in Fig. 8C approximately 5 times as long as that in Fig. 8B), the amount of Lyn competent to bind ITAM was similar in the unstimulated and desensitized cell lysates (Fig. 8, B and C). These results indicate that there is sufficient Lyn available to bind the ITAMs following desensitization and that Lyn in desensitized cells is normal in its capacity to bind phosphorylated and nonphosphorylated ITAMs.
As a final measure of Lyn functionality in desensitized cells, we tested whether Lyn could be activated in vitro by the doubly phosphorylated ITAM motif. Previously it has been shown that the doubly phosphorylated ITAM peptide can activate Lyn purified from unstimulated cells and baculovirus-expressed Lyn (31). Lyn was isolated by either receptor immunoprecipitation or anti-Lyn immunoprecipitation from unstimulated and desensitized K46 μ cells, and its activity was measured based on its ability to phosphorylate an exogenous peptide substrate following incubation with increasing doses of (p)2ITAM peptide. As shown in Figure 9A, (p)2ITAM peptide stimulated similar increases in receptor-associated Lyn activity whether the kinase was derived from unstimulated or desensitized cells indicating that receptor-associated Lyn from desensitized cells can be activated. Similarly, immunoprecipitated Lyn can be activated to the same level in unstimulated compared with desensitized cells (Fig. 9B). The activation of receptor-associated Lyn was quenched at a very high peptide concentration (Fig. 9A, 700 μM), while the activation of immunoprecipitated Lyn was slightly increased at a very high dose (Fig. 9B, 700 μM). Taken together, these results suggest that the defective signaling in desensitized cells does not reflect a modification of Lyn; rather, the defect appears to lie at the level of the receptor itself.
FIGURE 9.

Lyn from both desensitized and nondesensitized cells is activated by (p)2ITAM binding. Lyn was immunoprecipitated from NP7BSA desensitized (◆) and unstimulated (–□–) K46 μ cells by either anti-receptor (A) or anti-Lyn (B) immunoprecipitation. Immunoprecipitates from 10 × 106 cell equivalents were exposed to varied concentrations of (p)2ITAM for 1 h before kinase activity was assayed as described in Materials and Methods. Data are shown as fold activity plotted against the micromolar concentration of stimulating (p)2ITAM peptide. Fold stimulation was calculated by first subtracting the background counts per minute (reaction mix minus kinase) from the counts per minute at each data point, then dividing each corrected data point value by the value of the zero peptide control. The basal kinase activity of anti-Lyn immunoprecipitates from unstimulated cells was 81,335 cpm and that from desensitized cells was 49,625 cpm with a background counts per minute of 4,500; that from anti-receptor IP of unstimulated cells was 4,241 cpm, and that from desensitized cells was 3,577 cpm with a background counts per minute of 2,000.
Desensitized B cells from 3-83 μδIg transgenic mice show diminished Igα and Igβ phosphorylation following challenge with Ag
To assess whether the failure of Ag to induce phosphorylation of Igα and Igβ in desensitized K46 μ cells is also a feature of desensitized normal B cells, we assessed the phosphorylation state of Igα and Igβ in splenic B cells isolated from H-2Kk-specific 3-83 μδ Ig transgenic mice. To induce desensitization, we used an H-2Kk Ag mimetic epitope defined by screening a phage display library that exhibits binding specificity for this transgenic BCR. This 22-amino acid sequence (CAHDWRSGFGGFQHLCCGAAGA), which we henceforth refer to as 3-83ag1, mimics binding of MHC class I molecules to the 3-83 Id-positive mIg on transgenic B cells and stimulates the cells as efficiently as H-2Kb protein (data not shown; V. Kouskoff and D. Nemazee, manuscript in preparation). We generated a high valency Ag by coupling the peptide to a dextran backbone to obtain a hapten carrier ratio of 50 mol/mol of dextran (3-83ag150Dex). Desensitization was achieved by incubating the B cells with 500 ng/l × 106 cells/ml 3-83ag150Dex (desensitizing dose) for 2 h, then challenging the cells with 1 μg/ml of Ag. The dose of Ag used for desensitization (500 ng/1 × 106 cells/ml) left at least 50% of the receptors capable of binding Ag after the desensitization period, as determined by cell surface staining using biotinylated 3-83ag150Dex Ag (data not shown). The requirement for higher doses of Ag in desensitizing the 3-83 B cells is presumably a function of the Ag rather than the cell type, as optimal stimulation of the K46J lymphoma, which contains the 3-83 receptor, also required 500 ng of Ag.
To ensure that ligation of the remaining 50% of receptors following desensitization was capable of inducing a maximal phosphotyrosine response, we titrated the dose of Ag and compared the level of tyrosine phosphorylation to that achieved with the dose used to challenge desensitized cells. As shown in Figure 10A, doses of Ag >100 ng yielded tyrosine phosphorylation responses comparable to that produced when the 1 μg/ml dose was used to challenge desensitized cells. Thus, any diminution of phosphorylation seen in desensitized cells could not be the result of too few receptors ligated to yield an optimal phosphotyrosine response.
FIGURE 10.

A, Ca2+ mobilization responses of naive and desensitized 3-83 μδ transgenic B cells following stimulation with Ag (3-83ag150Dex). H-2Kk transgenic B cells were stimulated with 3-83ag150Dex (left panels; 1 μg/1 × 106 cells/ml) or desensitized for 2 h with 3-83ag150Dex (500 ng/1 × 106 cells/ml), then challenged with 1 μg/1 × 106 cells/ml of 3-83ag150Dex (right panels). Shown in the upper panels is maximum mean [Ca2+]i, and in the lower panels the percentage of cells responding is plotted as a function of time. The mean [Ca2+]i in resting unstimulated cells was 100 nM. The arrow depicts the time at which Ag was added. Data are representative of three replicate experiments. WCL, whole cell. B, The dose response of tyrosine phosphorylation following stimulation of transgenic 3-83 μδ cells. An anti-phosphotyrosine immunoblot of whole cell lysates of 3 × 106 splenic B cells was performed. Lane 1, Unstimulated cells; lane 2, cells stimulated for 2 min with 3-83ag150Dex (100 ng/1 × 106 cells/ml); lane 3, 250 ng/1 × 106 cells/ml of 3-83ag150Dex; lane 4, 500 ng/1 × 106 cells/ml of 3-83ag150Dex; lane 5, 750 ng/1 × 106 cells/ml of 3-83agl50Dex; lane 6, 1 μg/1 × 106 cells/ml of 3-83ag150Dex. Data are representative of two independent experiments. C, Reduced tyrosine phosphorylation of Igα and Igβ in desensitized 3-83 μδ splenic B cells. Upper panel, Anti-phosphotyrosine immunoblot of whole cell lysates (lanes 1–4) from 3 × 106 small resting B cells (ρ = >1.070) or Igα immunoprecipitate (IP) from 20 × 106 cell equivalents (lanes 5–8). Lanes 1 and 5, Unstimulated cells; lanes 2 and 6, cells stimulated for 2 min with 1 μg/1 × 106 cells/ml of 3-83ag150Dex; lanes 3 and 7, cells desensitized for 2 h with 500 ng/1 × 106 cells/ml of 3-83ag150Dex, then restimulated for 2 min with 1 μg/1 × 106 cells/0.05 ml of 3-83ag150Dex. The arrow beside the panel depicts the relative migration of Igα (see below). The faint band above the identified Igα band is Igβ. Lower panel, Anti-Igα immunoblot performed after stripping the upper blot. The arrow at the right depicts the relative migration of Igα and corresponds to the band labeled Igα in the upper blot.
To assess whether the failed BCR phosphorylation observed in the desensitized K46 μ system was also seen in Ag-specific primary B cells, we desensitized splenic B cells isolated from the 3-38 μδ transgenic mice using the 3-83ag150Dex Ag (500 ng/1 × 106 cells/ml) and assayed the ability of these cells to respond by Ca2+ mobilization to Ag challenge. As shown in Figure 10B, although basal [Ca2+] in desensitized cells was slightly elevated, the challenged cells did not show a further increase in [Ca2+], as clearly seen in the stimulated control cells. Similarly, analysis of tyrosine phosphorylation following desensitization showed a slight increase in basal phosphotyrosine following desensitization, but no inductive tyrosine phosphorylation upon challenge (Fig. 10C, lanes 1–4).
To further characterize the desensitized phenotype of transgenic B cells, we analyzed the phosphorylation state of Igα and Igβ following challenge of desensitized cells. As shown in Figure 10C, immunoprecipitation of Igα followed by anti-phosphotyrosine immunoblotting revealed a markedly diminished phosphorylation response similar to that observed in K46 μ cells (Fig. 10C, lanes 5–8). These differences in phosphorylation were not due to inefficient immunoprecipitation in desensitized cells or unequal sample loading, as revealed by anti-Igα immunoblotting analysis (Fig. 10C, lower panel). Thus, the phenotype of failed Igα phosphorylation when desensitized cells are challenge is seen in normal, Ag-specific B cells following receptor desensitization as well as in the K46 μ lymphoma model.
Discussion
We have established an in vitro model to define the molecular mechanisms underlying BCR desensitization. Using the B lymphoma, K46 μ, and splenic B cells from 3-83 μδ Ig transgenic mice, we induced receptor desensitization using a low dose of Ag, then measured the ability of cells to respond to challenge with a high dose of Ag. Unresponsiveness was measured by a decreased ability of cells to mobilize Ca2+ as well as diminished Ag-induced protein tyrosine phosphorylation. Our desensitization model uses a single receptor; hence, it is possible that during the 2-h desensitization period, Ag binding and dissociation (Kd of free NP for the receptor = 1.5 × 10−6; J. del Porto and G. Kelsoe, unpublished observations) may have resulted in Ag binding to all the cell surface receptors despite the fact that the desensitizing dose (25 ng/ml) occupied only about 25% of the receptors at any given time. Our model clearly shows that following Ag binding the receptors exhibit a desensitized phenotype, as indicated by their inability to signal upon receptor ligation. This clearly is not due to loss of receptors from the cell surface or the inability of the remaining receptors to bind Ag. The data presented in Figure 3 demonstrate that 70% of the total number of cell surface receptors are capable of binding Ag following the desensitization period, thus revealing a loss of only about 5% of receptors compared with cells that have not encountered Ag. We also show that ligation of just 70% of receptors is fully competent in eliciting a maximal phosphotyrosine response as clearly demonstrated in Figure 10A. Thus, the failure to initiate signal transduction following receptor ligation cannot be explained by the loss of surface receptor, the inability of the remaining receptors to bind Ag, or the inability of 70% of functional receptors to induce a response.
The studies described here address more precisely the mechanism of Ag-induced desensitization using a number of approaches. We assessed the phosphorylation state of receptor subunits and effectors that function proximal to the receptor and are not readily resolved in whole cell lysate phosphotyrosine blots. We observed reduced Ag-inducible phosphorylation of Lyn, Blk, Shc, and Syk from lysates of desensitized cells. This failed phosphorylation correlated with a lack of receptor-associated kinase activity in desensitized cells following challenge. Most interestingly, Igα/Igβ were not phosphorylated upon challenge of the desensitized K46 μ cells or desensitized splenic B cells from transgenic mice, suggesting that desensitized receptors fail to transduce signals leading to the earliest known events in BCR signal transduction. Finally, to address the possibility that inactivation of the Src family kinases is responsible for the desensitized phenotype, we assessed the functional state of the Src family member Lyn. We found that Lyn from desensitized lysates could bind phosphorylated and nonphosphorylated ITAM peptides as well as Lyn from unstimulated cell lysates. In addition, Lyn could be activated by exogenous phosphorylated ITAMs, revealing that the desensitized phenotype was not the result of Lyn kinase repression. Thus, we conclude that desensitization reflects the inability of Ag to initiate activation of receptor-associated kinases that normally phosphorylate the receptor Igαβ subunits. Unfortunately we were unable to detect the association of Lyn with either the desensitized or the stimulated BCR by immunoprecipitation; thus, the possibility remains that the defect in desensitized cells reflects inadequate association of Lyn with the desensitized receptor.
Signal transduction through Ag receptors is believed to occur via receptor aggregation mediated clustering, trans-phosphorylation, and activation of receptor-associated Src family members (16, 55). These kinases are thought to phosphorylate receptor-transducing subunits, in this case Igα and Igβ, which then recruit additional Src family kinases. Recent observations place activation of Src family members such as Lyn and Lck upstream of receptor phosphorylation during both B and T cell signaling (L. Pao and J. C. Cambier, unpublished observation) (35). Thus, failed phosphorylation of Igα and Igβ in desensitized cells may be secondary to failed Src family kinase phosphorylation and activation. This failed kinase activation could result from some modification of the kinase itself. For example, the negative regulatory C-terminal tyrosine might be phosphorylated because of inactivation of CD45, the phosphotyrosine phosphatase that dephosphorylates the C-terminal phosphotyrosine, or by activation of CSK, the kinase that phosphorylates the C-terminal tyrosine. Either of these mechanisms could keep the kinase in a closed or repressed conformation (56). Four pieces of evidence suggest that Lyn from desensitized cells is functionally normal and not repressed. Firstly, Src family kinases are not hyperphosphorylated in desensitized cells (Fig. 5A). Secondly, Lyn from these cells is equally competent to bind nonphosphorylated ITAMs (Fig. 8C). Thirdly, Lyn from desensitized and desensitized/challenged lysates binds the (p)2ITAM as efficiently as Lyn from unstimulated cells (Fig. 8B). These data suggest that the conformation of Lyn from desensitized cells is appropriate for binding to resting receptors via N-terminal unique regions and to phosphorylated receptors via SH2 domains as occurs during productive signaling (Fig. 9, A and B). It is noteworthy that C-terminally phosphorylated Lyn does not bind to phosphorylated ITAMs (57). Lastly, although both the receptor-associated and the cytoplasmic pools of Lyn from desensitized cells are inactive, as measured by tyrosine phosphorylation (Fig. 5A) and receptor-associated in vitro kinase activity (Fig. 7), they can be activated by the (p)2 ITAM peptide (Fig. 10). These findings support the idea that Lyn from desensitized cells is capable of normal function and is not repressed. Taken together, the data suggest that the defect that prevents activation of the desensitized receptors is not due to repression or other forms of negative regulation of Lyn, but, rather, indicates a defect at the level of the receptor and its coupling to Lyn.
Given our data that Lyn function in desensitized cells is normal, several alternative mechanisms can be envisioned to account for the desensitized phenotype. First, lack of receptor-mediated kinase phosphorylation and activation, and receptor phosphorylation could reflect a shift in equilibrium between receptor-associated tyrosine kinase and phosphotyrosine phosphatase activities. Excessive local phosphatase activity could maintain receptor-associated kinases in a nonphosphorylated state, thus preventing activation. Such a mechanism could also account for the ability to cross-desensitize unoccupied receptors. Likely candidates include CD45 and PTP1C/SHP-1. Although CD45’s role in BCR signaling has mainly been associated with the derepression of Src family kinases, CD45 has been shown to associate with the BCR, and it has been shown to dephosphorylate Igα and Igβ in vivo and in vitro (58–60). Recently, PTP1C/SHP-1 has been shown to associate with resting, but not activated, BCR, implicating the phosphatase in maintaining the resting BCR complex in the nonphosphorylated state (61). Using autoimmune/immunodeficient moth-eaten (me) and viable moth-eaten (meV) mice, Pani et al. further showed that cells that lack PTP1C/SHP-1 are hyper-responsive to submitogenic concentrations of F(ab′)2 anti-Ig Ab, and they exhibit reduced susceptibility to the inhibitory effects of FcγRIIB cross-linking on BCR-induced proliferation (61). The moth-eaten phenotype, when crossed with the HEL transgenic mice also showed a role for PTP1C/SHP-1 in BCR signaling. Cyster et al. showed that Ag triggered a greater and more rapid elevation of intracellular calcium in the PTP1C/SHP-1 deficient mice (62). Elimination of the autoreactive cells was triggered by a lower valency form of self Ag compared with HEL transgenics containing the PTP1C phosphatase. Thus, enhanced constitutive receptor-localized activity of these phosphatases could prevent activation of desensitized receptors.
Another possible mechanism for the observed unresponsiveness might involve an uncoupling of Lyn from an otherwise intact mIg/Igαβ complex due to impaired receptor aggregation. Since receptor cross-linking is essential for triggering the biochemical induction of signal transduction, receptor aggregation is believed to be essential for activation of T cell and B cell receptors. Aggregation has been shown to be important in FcεR1 activation through receptor clustering and trans-phosphorylation (18, 19). Receptor re-constitution studies of FcεR1 signaling have shown that receptor clustering controls Lyn-mediated FcεR1 tyrosine phosphorylation (20). Hence, defective BCR aggregation could account for the failure to activate the Src family kinases and subsequently the phosphorylation of Igα and Igβ. Our findings that desensitized cells contain competent, yet inactive, Lyn kinase is consistent with a possible mechanism involving a receptor aggregation defect.
Desensitization could also occur through modification, e.g., serine/threonine phosphorylation, of the Igα or Igβ, preventing Lyn association with the ITAM motif. Serine/threonine phosphorylation could directly effect the quality of the substrate or could signal the binding of some other blocking protein. Activation of such a kinase could result in modification of Igα or Igβ on both occupied and unoccupied receptors, explaining the ability of Ag binding to as few as 25% of receptors to desensitize the cell’s remaining receptors. In desensitization of the β-adrenergic receptor, serine/threonine phosphorylation by β-adrenergic receptor kinase leads to uncoupling of the receptor from Gs by a mechanism that may involve binding of arrestins (63). Consistent with a role for serine/threonine phosphorylation in desensitization, we have observed that PKC-activating phorbol diesters and diacylglycerol can induce desensitization, and van Noesel et al. have demonstrated PMA-induced serine phosphorylation of human Igα and Igβ (64). We believe that this is unlikely to be the mechanism of desensitization because we have shown that PMA-induced desensitization, unlike anti-receptor Ab or Ag-induced desensitization, is relatively short-lived (lasting <60 min), and we have been unable to document PMA or ligand induction of Igα or Igβ phosphorylation in murine B cells (K. Campbell and J. Cambier, unpublished observation) (4).
BCR desensitization may represent a normal physiologic consequence of Ag binding the BCR. Interestingly, the experiments from this model confirm the findings of Cooke et al., who showed that the BCR on anergic, anti-HE transgenic B cells are desensitized, as indicated by uncoupling of receptors from Ca2+ mobilization and tyrosine phosphorylation responses (50). Hence, receptor desensitization may represent an early step in the induction of anergy, as has been previously suggested (50, 65). Data from our two models, however, are inconsistent with the work of Brunswick et al., who showed that splenic B cells desensitized by anti-μ and challenged with anti-δ exhibit diminished Ca2+ mobilization but no detectable decrease in receptor-mediated tyrosine phosphorylation responses (5). This failure to detect defective, receptor-mediated tyrosine phosphorylation in desensitized cells is perhaps explained the by findings of Cooke et al., who showed that based on this parameter, anti-receptor Abs partially overcome desensitization induced by HEL Ag. Thus, due to unique specificity or increased cross-linking ability, anti-receptor Ab stimulation may lead to quantitatively distinct signaling not subject to normal physiologic regulation (50, 66, 67).
In conclusion, our data support a model in which engagement of the BCR on mature B cells by protein Ags leads to signaling events that set in motion pathways that have two functional consequences. The first is expression of B7.1 and B7.2 and increased expression of MHC class II, making the B cell competent to present Ag to T cells. This competent state is relatively short lived, being limited by the duration of up-regulation of B7 and MHC class II costimulatory molecules. The second consequence is receptor desensitization, a relatively long-lived condition, potentially lasting the life of the cell. Thus, in the absence of a properly timed T cell:B cell interaction, the B cell expression of T cell costimulatory molecules, e.g., B7.2 and B7.1, declines (this occurs in approximately 24–48 h; B. J. Vilen and J. C. Cambier, manuscript in preparation), and since renewed Ag stimulation does not induce re-expression of these molecules because of receptor desensitization, the B cell cannot participate in a thymus-dependent immune response. This B cell unresponsiveness is a manifestation of receptor desensitization that we have shown to occur through an uncoupling of the receptor from signal transduction pathways. Recently, it has been reported that mature peripheral B cells lacking mIg due to conditional gene ablation disappear from the periphery, suggesting the need for constitutive BCR signaling in maintaining cell survival (K. Rajewsky, unpublished observation). Thus, the eventual elimination of desensitized or anergic cells may result from the failure to sustain mIg signal transduction.
Acknowledgments
We thank Dr. Wayne Jensen for helpful discussion and critical reading of this manuscript. We also thank Dr. Jan Jonstra for the generous gift of anti-Igα, and Cortec, Inc. (Denver, CO), for the generous gift of dexamine.
Footnotes
This work was supported by U.S. Public Health Service Grants AI21768 and AI22295, an Ida and Cecil Green professorship of cell biology (to J.C.C.), and a National Institutes of Health-National Research Service Award and a Leukemia Society of America Fellow Award (to B.J.V.).
Abbreviations used in this paper: BCR, B cell antigen receptor; mlg, membrane immunoglobulin; PKC, protein kinase C; ITAM, immunoreceptor tyrosine-based activation motif; PLCγ, phospholipase Cγ; NP, nitrophenol; IMDM, Iscove’s modified Dulbecco’s medium; dex, dextran; [Ca2+]i, intracellular free calcium concentration; TBS, Tris-buffered saline; Id, idiotype; IP, immunoprecipitates.
References
- 1.Cambier J, Chen ZZ, Pasternak J, Ransom J, Sandoval V, Pickles H. Ligand-induced desensitization of B-cell membrane immunoglobulin-mediated Ca2+ mobilization and protein kinase C translocation. Proc Natl Acad Sci USA. 1988;85:6493. doi: 10.1073/pnas.85.17.6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goodnow CC. Transgenic mice and analysis of B-cell tolerance. Annu Rev Immunol. 1992;10:489. doi: 10.1146/annurev.iy.10.040192.002421. [DOI] [PubMed] [Google Scholar]
- 3.Goodnow CC, Crosbie J, Jorgensen H, Brink RA, Basten A. Induction of self tolerance in mature peripheral B lymphocytes. Nature. 1989;342:385. doi: 10.1038/342385a0. [DOI] [PubMed] [Google Scholar]
- 4.Cambier JC, Fisher CL, Pickles H, Morrison DC. Dual molecular mechanisms mediate ligand-induced membrane Ig desensitization. J Immunol. 1990;145:13. [PubMed] [Google Scholar]
- 5.Brunswick M, June CH, Mond JJ. B lymphocyte immunoglobulin receptor desensitization is downstream of tyrosine kinase activation. Cell Immunol. 1994;156:240. doi: 10.1006/cimm.1994.1168. [DOI] [PubMed] [Google Scholar]
- 6.Lazarus AH, Mills GB, Delovitch TL. Antigen-induced Ca2+ signaling and desensitization in B cells. J Immunol. 1990;144:4147. [PubMed] [Google Scholar]
- 7.Lazarus AH, Mills GB, Crow AR, Delovitch TL. Antigen-induced Fc receptor-dependent and -independent B cell desensitization. J Immunol. 1991;147:1739. [PubMed] [Google Scholar]
- 8.Cambier JC. Antigen and Fc receptor signaling The awesome power of the immunoreceptor tyrosine-based activation motif (ITAM) J Immunol. 1995;155: 3281. [PubMed] [Google Scholar]
- 9.Pleiman CM, D’Ambrosio D, Cambier JC. The B-cell antigen receptor complex: structure and signal transduction. Immunol Today. 1994;15:393. doi: 10.1016/0167-5699(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 10.Kim K-M, Alber G, Weiser P, Reth M. Signalling function of the B-cell antigen receptors. Immunol Rev. 1993;132:126. doi: 10.1111/j.1600-065x.1993.tb00840.x. [DOI] [PubMed] [Google Scholar]
- 11.Cambier JC. New nomenclature for the Reth motif (or ARH1/TAM/ARAM/YXXL) Immunol Today. 1995;16:110. doi: 10.1016/0167-5699(95)80105-7. [DOI] [PubMed] [Google Scholar]
- 12.Flaswinkel H, Reth M. Dual role of the tyrosine kinase activation motif of Ig-α protein during signal transduction via the B cell antigen receptor. EMBO J. 1994;13:83. doi: 10.1002/j.1460-2075.1994.tb06237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan AC, Iwashima M, Turck CW, Weiss A. ZAP-70: a 70 kd protein-tyrosine kinase that associates with the TCR ζ chain. Cell. 1992;71:649. doi: 10.1016/0092-8674(92)90598-7. [DOI] [PubMed] [Google Scholar]
- 14.Jouvin M-HE, Adamczewski M, Numerof R, Letourneur O, Valle A, Kinet J-P. Differential control of the tyrosine kinases lyn and syk by the two signaling chains of the high affinity immunoglobulin E receptor. J Bio Chem. 1994;269:5918. [PubMed] [Google Scholar]
- 15.Kihara H, Siraganian RP. Src homology 2 domains of syk and lyn bind to tyrosine-phosphorylated subunits of the high affinity IgE receptor. J Biol Chem. 1994;269:22427. [PubMed] [Google Scholar]
- 16.Straus DB, Weiss A. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell. 1992;70:585. doi: 10.1016/0092-8674(92)90428-f. [DOI] [PubMed] [Google Scholar]
- 17.Clark MR, Campbell KS, Kazlauskas SA, Johnson A, Hertz M, Potter TA, Pleiman C, Cambier JC. The B cell antigen receptor complex: association of Ig-α and Ig-β with distinct cytoplasmic effectors. Science. 1992;258:123. doi: 10.1126/science.1439759. [DOI] [PubMed] [Google Scholar]
- 18.Pribluda VS, Pribluda C, Metzger H. Transphosphorylation as the mechanism by which the high-affinity receptor for IgE is phosphorylated upon aggregation. Proc Nat Acad Sci USA. 1994;91:11246. doi: 10.1073/pnas.91.23.11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamashita T, Mao S-Y, Metzger H. Aggregation of the high-affinity IgE receptor and enhanced activity pf p53/p56lyn protein-tyrosine kinase. Proc Natl Acad Sci USA. 1994;91:11251. doi: 10.1073/pnas.91.23.11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scharenberg AM, Lin S, Cuenod B, Yamamura H, Kinet J-P. Reconstitution of interactions between tyrosine kinases and the high affinity IgE receptor which are controlled by receptor clustering. EMBO J. 1995;14:3385. doi: 10.1002/j.1460-2075.1995.tb07344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gold MR, Matsuuchi L, Kelly RB, DeFranco AL. Tyrosine phosphorylation of components of the B-cell antigen receptors following receptor crosslinking. Proc Natl Acad Sci USA. 1991;88:3436. doi: 10.1073/pnas.88.8.3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neuberger MS, Patel KJ, Dariavach P, Nelms K, Peaker CJG, Williams GT. The mouse B-cell antigen receptor: definition and assembly of the core receptor of the five immunoglobulin isotypes. Immunol Rev. 1993;132:147. doi: 10.1111/j.1600-065x.1993.tb00841.x. [DOI] [PubMed] [Google Scholar]
- 23.Campbell M-A, Sefton BM. Association between B-lymphocyte membrane immunoglobulin and multiple members of the src family of protein tyrosine kinases. Mol Cell Biol. 1992;12:2315. doi: 10.1128/mcb.12.5.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamada T, Taniguchi T, Yang C, Yasue S, Saito H, Yamamura H. Association with B-cell antigen receptor with protein-tyrosine kinase p72syk and activation by engagement of membrane IgM. Eur J Biochem. 1993;213:455. doi: 10.1111/j.1432-1033.1993.tb17781.x. [DOI] [PubMed] [Google Scholar]
- 25.Yamanashi Y, Kakiuchi T, Mizuguchi J, Yamamoto T, Toyoshima K. Association of B cell antigen receptor with protein tyrosine kinase Lyn. Science. 1991;251:192. doi: 10.1126/science.1702903. [DOI] [PubMed] [Google Scholar]
- 26.Yamanashi Y, Fukui Y, Wongsasant W, Kinoshita Y, Ichimori Y, Toyoshima K, Yamamoto T. Activation of Src-like protein-tyrosine kinase Lyn and its association with phosphatidylinositol 3-kinase upon B-cell antigen receptor-mediated signaling. Proc Natl Acad Sci USA. 1992;89:1118. doi: 10.1073/pnas.89.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burkhardt AL, Costa T, Misulovin Z, Stealy B, Bolen JB, Nussenzweig MC. Igα and Igβ are functionally homologous to the signaling proteins of the T-cell receptor. Mol Cell Biol. 1994;14:1095. doi: 10.1128/mcb.14.2.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clark MR, Johnson SA, Cambier JC. Analysis of Ig-α-tyrosine kinase interaction reveals two levels of binding specificity and tyrosine phosphorylated Ig-α stimulation of Fyn activity. EMBO J. 1994;13:1911. doi: 10.1002/j.1460-2075.1994.tb06460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurosaki T, Takata M, Yamanashi Y, Inazu T, Taniguchi T, Yamamoto T, Yamamura H. Syk activation by the src-family tyrosine kinase in the B cell receptor signaling. J Exp Med. 1994;179:1725. doi: 10.1084/jem.179.5.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurosaki T, Johnson SA, Pao L, Sada K, Yamamura H, Cambier JC. Role of the syk autophosphorylation site and SH2 domains in B cell antigen receptor signaling. J Exp Med. 1995;182:1815. doi: 10.1084/jem.182.6.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson SA, Pleiman CM, Pao L, Schneringer J, Hippen K, Cambier JC. Phosphorylated immunoreceptor signaling motifs (ITAMs) exhibit unique abilities to bind and activate Lyn and Syk tyrosine kinases. J Immunol. 1995;155:4596. [PubMed] [Google Scholar]
- 32.Weiss A, Littman D. Signal transduction by lymphocyte antigen receptors. Cell. 1994;76:263. doi: 10.1016/0092-8674(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 33.Pleiman CM, Abrams C, Timson-Gauen L, Bedzyk W, Jongstra J, Shaw AS, Cambier JC. Distinct domains within p53/56lyn and p59fyn bind nonphosphorylated and phosphorylated Ig-α. Proc Natl Acad Sci USA. 1994;91:4268. doi: 10.1073/pnas.91.10.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.D’Ambrosio D, Hippen KL, Cambier JC. Distinct mechanisms mediate SHC association with the activated and resting B cell antigen receptor. Eur J Immunol. 1996;26:1960. doi: 10.1002/eji.1830260842. [DOI] [PubMed] [Google Scholar]
- 35.Straus DB, Chan AC, Patai B, Weiss A. SH2 domain function is essential for the role of the Lck tyrosine kinase in T cell receptor signal transduction. J Biol Chem. 1996;271:9976. doi: 10.1074/jbc.271.17.9976. [DOI] [PubMed] [Google Scholar]
- 36.Shiue L, Zoller MJ, Brugge JS. Syk is activated by phosphotyrosine-containing peptides representing the tyrosine-based activation motifs of the high affinity receptor for IgE. J Biol Chem. 1995;270:10498. doi: 10.1074/jbc.270.18.10498. [DOI] [PubMed] [Google Scholar]
- 37.Law DA, Chan VWF, Datta SK, DeFranco AL. B-cell antigen receptor motifs have redundant signalling capabilities and bind the tyrosine kinases PTK72, Lyn and Fyn. Curr Biol. 1993;3:645. doi: 10.1016/0960-9822(93)90062-s. [DOI] [PubMed] [Google Scholar]
- 38.Couture C, Baier G, Atman A, Mustelin T. p56lck-independent activation and tyrosine phosphorylation of p72syk by T-cell antigen receptor/CD3 stimulation. Proc Natl Acad Sci USA. 1994;91:5301. doi: 10.1073/pnas.91.12.5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bauman G, Maier D, Freuler F, Tschopp C, Baudisch K, Wienands J. In vitro characterization of major ligands for Src homology 2 domains derived from protein tyrosine kinases, from the adaptor protein SHC and from GTPase-activating protein in Ramos B cells. Eur J Immunol. 1994;24:1799. doi: 10.1002/eji.1830240812. [DOI] [PubMed] [Google Scholar]
- 40.Smit L, de Vries-Smits AMM, Bos JL, Borst J. B cell antigen receptor stimulation induces formation of a Shc-Grb2 complex containing multiple tyrosine-phosphorylated proteins. J Biol Chem. 1994;269:20209. [PubMed] [Google Scholar]
- 41.Saxton TM, van Oostveen I, Bowtell D, Aebersold R, Gold MR. B cell antigen receptor cross-linking induces phosphorylation of the p21ras oncoprotein activators SHC and mSOS1 as well as assembly of complexes containing SHC, GRB-2, mSOS1, and a 145 kDa tyrosine phosphorylated protein. J Immunol. 1994;153:623. [PubMed] [Google Scholar]
- 42.Kim KJ, Kanellopoulos-Langevin C, Merwin RM, Sachs DH, Asofsky R. Establishment and characterization of BALB/c lymphoma lines with B cell properties. J Immunol. 1979;122:549. [PubMed] [Google Scholar]
- 43.Reth ME, Petrac P, Wiese L, Lobel L, Alt FW. Activation of V gene rearrangements in pre-B cells follows the expression of membrane-bound immunoglobulin heavy chains. EMBO J. 1987;6:3299. doi: 10.1002/j.1460-2075.1987.tb02649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nemazee D, Burki K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature. 1989;337:562. doi: 10.1038/337562a0. [DOI] [PubMed] [Google Scholar]
- 45.Sparks AB, Adey NB, Quilliam LA, Thorn JM, Kay BK. Screening phage-displayed random peptide libraries for SH3 ligands. Methods Enzymal. 1995;255:498. doi: 10.1016/s0076-6879(95)55052-6. [DOI] [PubMed] [Google Scholar]
- 46.Justement LB, Wienands J, Hombach J, Reth M, Cambier JC. Membrane IgM and IgD molecules fail to transduce Ca2+ mobilizing signals when expressed on differentiated B lineage cells. J Immunol. 1990;144:3272. [PubMed] [Google Scholar]
- 47.Pleiman CM, Clark MR, Timson Gauen LK, Winitz S, Coggeshall KM, Johnson GL, Shaw AS, Cambier JC. Mapping of sites on the src-family protein tyrosine kinases p55blk, p59fyn and p56lyn which interact with the effector molecules phospholipase C-γ2, microtubule-associated protein kinase, GTPase-activating protein. and phosphatidylinositol 3-kinase. Mol Cell Biol. 1993;13:5877. doi: 10.1128/mcb.13.9.5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Julius MH, Heusser CH, Hartmann K-U. Induction of resting B cells to DNA synthesis by soluble monoclonal anti-immunoglobulin. Eur J Immunol. 1984;14:753. doi: 10.1002/eji.1830140816. [DOI] [PubMed] [Google Scholar]
- 49.Goldman FG, Jensen WA, Johnson GL, Heasley L, Cambier JC. gp120 ligation of CD4 induces p56lck activation and TCR desensitization independent of TCR tyrosine phosphorylation. J Immunol. 1994;153:2905. [PubMed] [Google Scholar]
- 50.Cooke MP, Heath AW, Shokat KM, Zeng Y, Finkelman FD, Linsley PS, Howard M, Goodnow CC. Immunoglobulin signal transduction guides the specificity of B cell-T cell interactions and is blocked in tolerant self-reactive B cells. J Exp Med. 1994;179:425. doi: 10.1084/jem.179.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saouaf SJ, Kut SA, Fargnoli J, Rowley RB, Boen JB, Mahajan S. Reconstitution of the B cell antigen receptor signaling components in Cos cells. J Biol Chem. 1995;270:27072. doi: 10.1074/jbc.270.45.27072. [DOI] [PubMed] [Google Scholar]
- 52.Hombach J, Lottspeich F, Reth M. Identification of the genes encoding the IgM-α and Ig-β components of the IgM antigen receptor complex by amino terminal sequencing. Eur J Immunol. 1990;20:2765. doi: 10.1002/eji.1830201239. [DOI] [PubMed] [Google Scholar]
- 53.Campbell KS, Cambier JC. B lymphocyte antigen receptors (mIg) are non-covalently associated with a disulfide-linked, inducibly phosphorylated glycoprotein complex. EMBO J. 1990;9:441. doi: 10.1002/j.1460-2075.1990.tb08129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Campbell KS, Hager EJ, Friedrich J, Cambier J. The IgM antigen receptor complex is associated with phosphoprotein products of B29 and mb-1 genes. Proc Natl Acad Sci USA. 1991;88:3982. doi: 10.1073/pnas.88.9.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iwashima M, Irving BA, van Oers NSC, Chan AC, Weiss A. Sequential interactions of the TCR with two distinct cytoplasmic tyrosine kinases. Science. 1994;263:1136. doi: 10.1126/science.7509083. [DOI] [PubMed] [Google Scholar]
- 56.Cooper JA, Howell B. The when and how of Src regulation. Cell. 1993;73:1051. doi: 10.1016/0092-8674(93)90634-3. [DOI] [PubMed] [Google Scholar]
- 57.Pao LI, Cambier JC. Syk but not Lyn recruitment to BCR and activation following stimulation of CD45−/− B cells. J Immunol. 1997;158:2663. [PubMed] [Google Scholar]
- 58.Brown VK, Ogle EW, Burkhardt AL, Rowley RB, Bolen JB, Justement LB. Multiple components of the B cell antigen receptor complex associate with the protein tyrosine phosphatase, CD45. J Biol Chem. 1994;269:17238. [PubMed] [Google Scholar]
- 59.Justement LB, Campbell KS, Chien NC, Cambier JC. Regulation of B cell antigen receptor signal transduction and phosphorylation by CD45. Science. 1991;252:1839. doi: 10.1126/science.1648262. [DOI] [PubMed] [Google Scholar]
- 60.Lin J, Brown VK, Justement LB. Regulation of basal tryosine phosphorylation of the B cell antigen receptor complex by the protein tyrosine phosphatase CD45. J Immunol. 1992;149:3182. [PubMed] [Google Scholar]
- 61.Pani G, Kozlowski M, Cambier JC, Mills GB, Siminovitch KA. Identification of the tyrosine phosphatase PTP1C as a B cell antigen receptor-associated protein involved in the regulation of B cell signaling. J Exp Med. 1995;181:2077. doi: 10.1084/jem.181.6.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cyster JG, Goodnow CC. Protein tyrosine phosphatase 1C negatively regulates antigen receptor signaling in B lymphocytes and determines thresholds for negative selection. Immunity. 1995;2:13. doi: 10.1016/1074-7613(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 63.Benovic JL, Bouvier M, Caron MG, Lefkowitz RJ. Regulation of adenylyl cyclase-coupled β-adrenergic receptors. Annu Rev Cell Biol. 1988;4:405. doi: 10.1146/annurev.cb.04.110188.002201. [DOI] [PubMed] [Google Scholar]
- 64.Van Noesel CJM, Borst J, De Vries EFR, Van Lier RAW. Identification of two distinct phosphoproteins as components of the human B cell antigen receptor complex. Eur J Immunol. 1990;20:2789. doi: 10.1002/eji.1830201238. [DOI] [PubMed] [Google Scholar]
- 65.Rathmell JC, Townsend SE, Xu JC, Flavell RA, Goodnow CC. Expansion or elimination of B cell in vivo: dual roles for CD40- and Fas (CD95)-ligands modulated by the B cell antigen receptor. Cell. 1996;87:319. doi: 10.1016/s0092-8674(00)81349-5. [DOI] [PubMed] [Google Scholar]
- 66.Bishop GA, Pennell CA, Travis W, Haughton G, Frelinger JA. Antibodies specific for Ig idiotype, but not isotype, can substitute for antigen to induce IgM secretion by a B cell clone. Int Immunol. 1990;2:285. doi: 10.1093/intimm/2.4.285. [DOI] [PubMed] [Google Scholar]
- 67.Pleiman CM, Chien NC, Cambier JC. Point mutations define a mIgM transmembrane region motif that determines intersubunit signal transduction in the antigen receptor. J Immunol. 1994;152:2837. [PubMed] [Google Scholar]