

Abstract
A class I ribonucleotide reductase (RNR) uses either a tyrosyl radical (Y•) or a MnIV/FeIII cluster in its β subunit to oxidize a cysteine residue ∼ 35 Å away in its α subunit, generating a thiyl radical that abstracts hydrogen (H•) from the substrate. With either oxidant, the inter-subunit “hole transfer” or “radical translocation” (RT) process is thought to occur by a “hopping” mechanism involving multiple tyrosyl (and perhaps one tryptophanyl) radical intermediates along a specific pathway. The hopping intermediates have never been directly detected in a Mn/Fe-dependent (class Ic) RNR nor in any wild-type RNR. The MnIV/FeIII cofactor of Chlamydia trachomatis RNR assembles via a MnIV/FeIV intermediate. Here we show that this cofactor-assembly intermediate can propagate a hole into the RT pathway when α is present, accumulating radicals with EPR spectra characteristic of Y•s. The dependence of Y• accumulation on the presence of substrate suggests that RT within this “super-oxidized” enzyme form is gated by the protein, and the failure of a β variant having the subunit-interfacial pathway Y substituted by phenylalanine to support radical accumulation implies that the Y•(s) in the wild-type enzyme reside(s) within the RT pathway. Remarkably, two variant β proteins having pathway substitutions rendering them inactive in their MnIV/FeIII states can generate the pathway Y•s in their MnIV/FeIV states and also effect nucleotide reduction. Thus, the use of the more oxidized cofactor permits the accumulation of hopping intermediates and the “hurdling” of engineered defects in the RT pathway.
Keywords: Ribonucleotide reductase, tyrosyl radical, iron, manganese, electron transfer
Introduction
By catalyzing conversion of ribonucleotides to 2′-deoxyribonucleotides, ribonucleotide reductases (RNRs) provide all organisms with the precursors for the de novo synthesis and repair of DNA.1,2 All known RNRs use a free-radical mechanism that is initiated by the abstraction of the hydrogen atom (H•) from the 3′-position of the nucleotide substrate by a transient cysteine thiyl radical (C•).3,4 The mechanism by which the C• is generated is the primary basis for the division of RNRs into classes I–III.1,2
A class Ia RNR, such as the most extensively studied orthologue from aerobically-growing Escherichia coli (Ec), comprises two subunits, α and β.1,5 Both subunits are homodimers, and they form a 1:1 complex (α2•β2) in the active state.6-11 The α subunit binds substrates and effectors10 and contains the cysteine residue (C439 in the Ec enzyme discussed hereafter) that forms the H•-abstracting C• during turnover.5,12,13 The C439• is generated in situ by a stable oxidant in β. This potent oxidant, a μ-oxo-Fe2III/III/tyrosyl radical (Y•) cofactor,14-16 is the product of an auto-activation reaction, during which the reduced (Fe2II/II) form of the diiron cluster reacts with O2.14 The activation reaction proceeds via a μ-peroxo-Fe2III/III intermediate17,18 to a state containing both a Fe2III/IV complex, designated X, and a cation radical residing on the near-surface residue, tryptophan 48 (W48).19 The W48 cation radical (W48+•) can undergo rapid reduction by a number of natural compounds (e.g., ascorbate, thiols),19,20 leaving X to oxidize the nearby (∼5 Å away) Y122 to the Y122• as X is reduced to the μ-oxo-Fe2III/III cluster of the active β subunit.21,22 The net effect of the transient oxidation of W48 by the diiron–O2 adduct and the subsequent reduction of the W48+• from solution is the shuttling of a single electron to the buried diiron cofactor.19
The catalytic cycle of nucleotide reduction begins with the one-electron oxidation of C439 in α by the long-lived Y122• ∼ 35 Å away in β upon formation of the α2•β2 complex having the nucleoside diphosphate (NDP) substrate and (deoxy)nucleoside triphosphate allosteric effector bound in α. The long-distance, inter-subunit, radical translocation (RT) process that generates the C439• does not occur by a simple electron-transfer (ET) step via a tunneling mechanism. Rather, ET proceeds in multiple “hopping” steps involving a chain of aromatic amino acid residues (Y356 and perhaps W48 in β, and Y731 and Y730 in α), which are conserved across all class I RNRs.23-29 The stable Y122• is proposed to acquire an electron from the closest pathway residue (W48 or Y356), and the resultant “hole” is then propagated, residue-by-residue, via a series of similar hopping steps. It is thought that the individual ET steps are coupled to local proton transfers, and, consequently, the term “proton-coupled electron transfer” (PCET) has been used to describe either the overall RT process or its constituent steps.5,11 FOOTNOTE: In this manuscript, we denote the net transfer of the hole in β to the conserved C439 in α as “radical translocation” (RT). “Hole hopping,” “hole translocation,” “electron relay,” and “radical transfer,” which have previously been used by us and others, have the same meaning. We disfavor “radical transfer” because the process invariably involves multiple ET events that interconvert different oxidized species (in most cases pathway radicals) rather than actual transfer of a free radical per se. We also disfavor the acronym PCET to denote the entire RT process, because the process involves multiple discrete (PC)ET events. END FOOTNOTE It is likely that, through this coupling to proton transfer, the thermodynamics of the individual steps and the overall process are tuned for efficiency and reversibility. It is also likely that dynamic control of one or more of the required proton transfers is the mechanism by which the protein controls RT so that it occurs only in the “ready” β2•α2•substrate•effector complex.28-30
Although the mechanisms of cofactor generation and substrate reduction have been studied extensively, the crucial RT step that initiates and terminates each turnover had, until very recently, remained enigmatic, owing, in part, to the occurrence of a substrate-dependent conformational change that allows the RT process to occur along the mediating pathway.30 This substrate dependence has been termed conformational “gating,” and the change that occurs in the α2•β2 complex when substrate binds as “opening of the gate.” Because the gate-opening step is slow and subsequent events are fast, the wild-type enzyme does not accumulate RT intermediates during turnover.5,30 In the last several years, Stubbe and co-workers have elegantly unveiled these intermediates by rational alterations to either the RT pathway or the cofactor. They replaced key Y residues in Ec RNR with three different unnatural Y analogues, 3,4-dihydroxyphenylalanine (DOPA),8,28,31 3-aminotyrosine (NH2-Y),8,29,32-35 and 3-nitrotyrosine (NO2-Y).36-38 The two former analogues are more easily oxidized than Y (i.e., their radicals have reduction potentials less than that of Y•).8,28,29,31-35 Effectively, their inclusion in the pathway introduces a thermodynamic depression in the hopping pathway, and the radical can then reside on this residue to a significant extent. The latter analogue was incorporated at position 122, and the resultant NO2-Y122• has an elevated reduction potential relative to the natural Y122•.36,37 The presence of the more potently oxidizing initiator appeared to subvert the gating mechanism, permitting the much more rapid propagation of the radical into the pathway and the accumulation of hopping intermediates.37
The class Ic RNR from Chlamydia trachomatis (Ct) differs from Ec RNR in that its β subunit lacks the stable, radical-initiating Y•.39 The Y is replaced structurally by a redox-inert phenylalanine (F127),39,40 and the function of the Y• is assumed by the MnIV ion of a MnIV/FeIII cofactor.41-43 All other RT pathway residues are conserved,39 suggesting that, aside from the identity of the C•-generating cofactor, the mechanisms of RT and nucleotide reduction might also be conserved. Analogously to cofactor assembly in Ec RNR, the MnIV/FeIII cofactor in Ct RNR-β assembles in a reaction of the fully reduced cofactor form (MnII/FeII) with O2.44 The reaction proceeds via a MnIV/FeIV intermediate that accumulates to near-stoichiometric yield and is sufficiently stable to permit its efficient trapping even in manual-mixing experiments.45 The intermediate is subsequently reduced by one electron at the FeIV site. The electron is shuttled through an activation-specific electron-relay pathway46 (shown in Scheme 1) that is, to the best of our knowledge, unique to the Ct enzyme. This pathway comprises the surface-exposed Y222 (the cognate in Ec RNR-β is a redox-inert leucine) and W51 (the cognate of W48 in the Ec protein). As is the case with Ec RNR, no pathway RT intermediates are observed during turnover in Ct RNR. While efforts to incorporate unnatural amino acids into the Ct enzyme have thus far been unsuccessful,47 the longevity [t1/2 of tens of seconds at 5 °C45] of the MnIV/FeIV cofactor assembly-intermediate, itself expected to be a more potent oxidant than the native MnIV/FeIII cofactor, led us to consider the possibility that it could be used to alter the redox-potential landscape of the RT pathway to favor accumulation of radical intermediates.
Scheme 1.
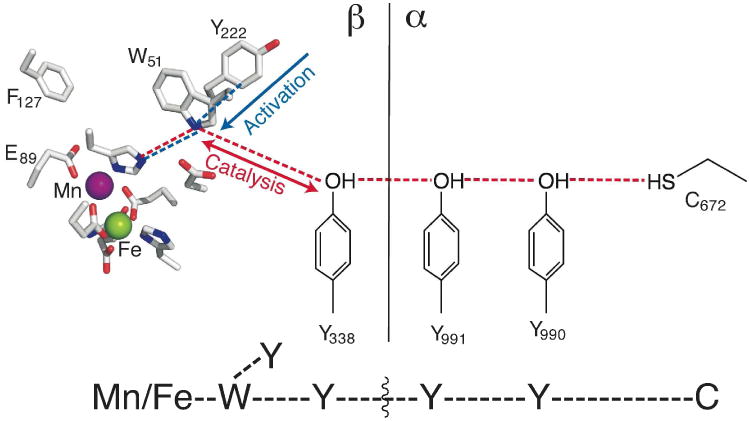
Orthogonal activation- and catalysis-specific electron-relay pathways in Ct RNR. At bottom is a simple schematic that is used throughout this work to assist in interpretation of the figures.
Indeed, we show herein that mixing the MnII/FeII-β complex of Ct RNR with an O2-containing solution of α, substrate (CDP), and allosteric effector (ATP) to form the MnIV/FeIV activation intermediate in the presence of all other reaction components results in accumulation of EPR-active species with hyperfine couplings characteristic of Y•s. The data imply the conversion of the MnIV/FeIV intermediate to a MnIV/FeIII-Y• species in a “super-oxidized” α2•β2 complex. Site-directed mutagenesis experiments demonstrate that the Y• resides within the RT pathway. Remarkably, two β variants with engineered RT pathway defects that render them inactive in their MnIV/FeIII states are, in their MnIV/FeIV states, capable of both generating Y•s and promoting a single turnover of the substrate. Thus, with the more oxidized cofactor, intermediates of the RT process can be accumulated in Ct RNR, and engineered defects in the RT pathway can effectively be “hurdled” to rescue nucleotide reduction in the disabled variants.
Experimental Procedures
Materials
Restriction enzymes and calf intestinal phosphatase were purchased from New England Biolabs (Ipswich, MA). 2-Methylbutane (iso-pentane), CDP, dCDP, ATP, DTT, MgSO4, Dowex-1 resin, and potassium tetraborate tetrahydrate (K2B4O7•4H2O) were purchased from Sigma-Aldrich (St. Louis, MO). (5-3H)-CDP was purchased from ViTrax (Placentia, CA). Ecoscint scintillation fluid was purchased from National Diagnostics (Atlanta, GA).
Over-expression and purification of Ct β and Ct α
Construction of the plasmid vectors pET28a-Ct α(Δ1-248) and pET28a-Ct β expressing the wild type subunits and the modified vectors expressing all the variants used in this study (except for the β-Y222F/Y338W variant) has been described elsewhere.41,46 The vector for β-Y222F/Y338W was constructed by applying a polymerase chain reaction (PCR) with the pET28a-Ct β-Y222F as the template and primers 1 (5′- TTA ACG CTT CAT ATG CAA GCA GAT ATT TTA GAT GG-3′; NdeI restriction site in bold) and 2 (5′-GGT GGT GCT CGA GCT ACC AAG TTA AGC TTG CTG CAT GTT GCC ATT CTA TAA CCC-3′; XhoI site in bold) to introduce the desired substitution (underlined). The PCR fragment was purified by agarose gel electrophoresis, restricted with NdeI and XhoI, repurified, and ligated with the larger (vector) fragment of NdeI- and XhoI-restricted pET28a-Ct β-wt. The sequence of the entire coding region of the vector was verified by ACGT, Inc. (Wheeling, IL). Purification of the N-terminally His6-tagged proteins has also been described.41,46
Synthesis of 2′-azido-2′-deoxyuridine-5′-diphosphate analogue (N3-UDP)
2′-Azido-2′-deoxyuridine was prepared as previously described.48 The 5′-diphosphate was appended by the method of Besada and coworkers.49 The quantity of tributylammonium phosphate was reduced from 6 to 3 equivalents to limit formation of the iso-triphosphate byproducts. The identity and purity of the product were verified by comparison of its 1H-, 13C-, and 31P-NMR spectra to published data for the compound.48-50
Preparation of samples for EPR spectroscopy
All samples (except in cases otherwise indicated) were prepared by adding 0.75 equiv of MnII and FeII to O2-free solutions of ∼ 2 mM apo-Ct β and subsequently rapidly adding an air-saturated solution (hereafter referred to as “α mix”) containing α, CDP (or dCDP or N3-UDP), ATP, DTT and MgSO4. Concentrations after mixing were 0.40 mM α, 0.20 mM β, 0.15 mM Mn, 0.15 mM Fe, 1 mM CDP (or dCDP or N3-UDP), 0.5 mM ATP, 10 mM DTT, and 10 mM MgSO4. Samples made with CDP and dCDP were frozen in cold (∼ –150 °C) 2-methylbutane 5–10 seconds after mixing (unless otherwise indicated), whereas samples with N3-UDP were frozen 1 minute after mixing.
EPR spectroscopy
EPR spectra were acquired on an ESP300 spectrometer equipped with a 4102ST X-band resonator and an ER 041 MR microwave bridge, all from Bruker (Billerica, MA) and described elsewhere.51 Cryogenic temperatures were maintained with an Oxford Instruments (Oxfordshire, UK) ESR 900 cryostat. Spectrometer conditions are provided in the figure legends. Simulation of EPR spectra of the Y•(s) was carried out with the program EasySpin.52 The simulation parameters were constrained as illustrated by Svistunenko53 and described in more detail in Supporting Information. The parameters are provided in Table S1.
“Spin” Quantification by EPR Spectroscopy
The extent of accumulation of the pathway Y•(s) was determined from analysis of the EPR spectra of duplicate samples prepared in a sequential-mix freeze-quench experiment. An O2-free solution of MnII/FeII-β was first mixed with an O2-saturated buffer solution, and this solution was allowed to react for 2 s to form the MnIV/FeIV intermediate. The pre-formed intermediate was mixed either with a solution of α, MgSO4, and DTT or with a solution of α, CDP, ATP, MgSO4, and DTT. The resultant solution was allowed to react for 1 s (ongoing studies suggest that the concentration of Y•(s) reaches its maximum value at this reaction time) before the reaction was terminated by freeze-quenching in cold (∼120 K) 2-methylbutane. Final concentrations after mixing were: 0.2 mM β, 0.15 mM Mn, 0.15 mM Fe, 0.3 mM α, 1 mM CDP (when present), 0.5 mM ATP (when present), 10 mM MgSO4, and 10 mM DTT. The signal intensities attributable to the MnIV/FeIV intermediate and the RT pathway Y•(s) were determined from the analysis of the EPR spectra as previously described.54 The results are summarized in Table S2.
Measurement of product formation with 3H-CDP
(5-3H)-CDP with specific activity >15 Ci/mmol was acquired from ViTrax (Placentia, CA). The assay procedure was adapted from that reported by Steeper and Steuart.55 Samples were prepared so as to be identical to those used for EPR spectroscopy, with 1 mM (5-3H)-CDP (∼5,000 cpm/nmol) substituted for CDP. The samples were incubated at 37 °C. Aliquots were removed at the desired reaction times, diluted 5-fold with reaction buffer (100 mM Na-HEPES, 10% v/v glycerol, pH 7.6), and quenched by incubation at 100 °C for 5 minutes. Quenched samples were centrifuged at 14,000 × g for 10 minutes. The cleared supernatant was transferred to a clean tube, and 1 μL of 10 U/μL calf intestinal phosphatase, 5 μL of 20 mM deoxycytidine, and 50 μL of 500 mM Tris-HCl (pH 8.5) were added. The resultant solutions were incubated for 1 hour at 37 °C, and each was then loaded onto a column containing 1.5 mL of Dowex-1 resin pretreated with 0.9 M potassium tetraborate (K2B4O7•4H2O). The dCDP product was eluted by washing with 9 mL distilled, deionized water. A 1 mL aliquot of the eluate was mixed with 5 mL of Ecoscint scintillation fluid; radioactivity was quantified by counting for 5 min per sample on a Beckman LS-6500 scintillation counter.
Results
Initial observation of a possible tyrosyl radical (Y•) and requirements for its formation
The well-characterized activation reaction of the Mn- and Fe-dependent Ct RNR β subunit begins with addition of O2 to its MnII/FeII cluster, which produces a long-lived, high-valent MnIV/FeIV intermediate that decays by reduction of the FeIV ion to FeIII.45 This intermediate has unique Mössbauer- and EPR-spectroscopic features (Figure 1A, spectrum i) reflecting, first, antiferromagnetic coupling of the S = 2 FeIV and the S = 3/2 MnIV to give STotal = 1/2 and, second, strong hyperfine coupling of the I = 5/2 55Mn nucleus to the electron spin.45 The spectra of samples prepared by mixing a solution of MnII/FeII-β with an air-saturated solution containing either α only (Figure 1A, ii) or α with the allosteric effector ATP (Figure 1A, iii) both reveal the presence of only the MnIV/FeIV intermediate. By contrast, when the activation reaction is initiated by mixing the MnII/FeII-β solution with an air-saturated solution of α and CDP substrate, an additional signal, which overlaps with the fourth line of the spectrum of the MnIV/FeIV intermediate at g = 2.00, also develops (Figure 1A, iv). The signal is not observed in samples having identical composition but prepared with pre-activated β harboring the stable MnIV/FeIII cofactor (Figure 3, ii). Analysis to resolve the new signal by subtraction of other spectral contributions shows that it is an organic radical that has hyperfine structure characteristic of a Y• (Figure 1B, iv). Although the presence of the substrate CDP is most crucial for the development of this signal, the effector ATP, while having negligible effect on its own (Figure 1, iii), appears to potentiate the effect of the substrate, resulting in greater intensity of the radical signal when both nucleotides are present (Figure 1, compare iv and v). Thus, accumulation of this radical is gated by binding of substrate and potentiated by binding of the allosteric effector in the same manner previously demonstrated for the RT step in both the Ec enzyme31 and the native form of the Ct enzyme.56 Interestingly, the product, dCDP, can replace the substrate in promoting radical accumulation (Figure 1, vi).
Figure 1.
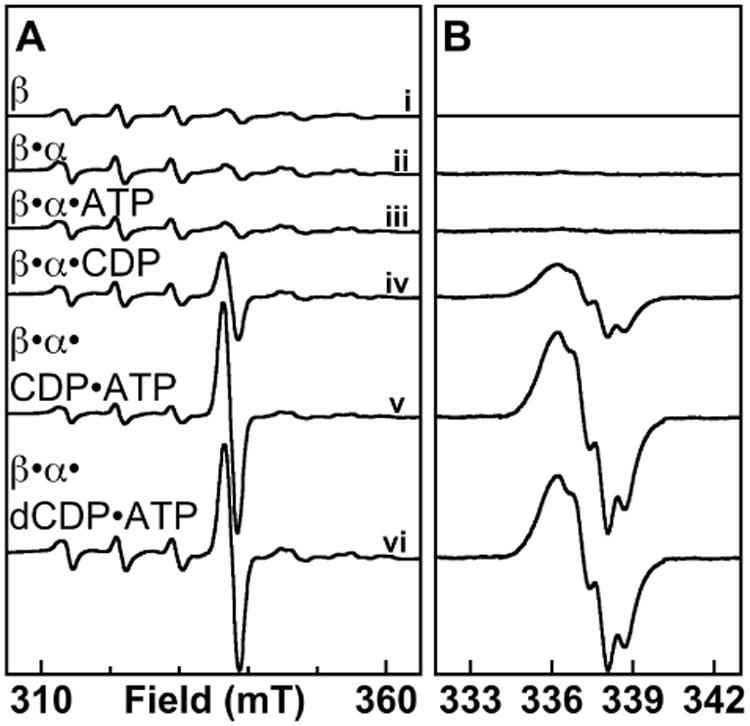
X-band EPR spectroscopic evidence for Y• accumulation in the super-oxidized Ct RNR holoenzyme complex. At ambient temperature (22 ± 2 °C), an O2-free solution of β (∼ 2 mM) containing 0.75 equiv each of MnII and FeII was mixed with a solution containing the other RNR reaction components to give final concentrations of: 0.2 mM β; 0 (i) or 0.4 mM (ii-vi) α; 0 (i-iii) or 1 mM (iv-vi) CDP; and 0 (i, ii, and iv) or 0.5 mM (iii, v, and vi) ATP. Reaction solutions were transferred to an EPR tube and frozen in cold iso-pentane (T∼ 125 K) 5 seconds after being thoroughly mixed. (A) Delineation of the requirements for radical formation, with spectra acquired over a wide range of the magnetic field. The spectrometer conditions were: T = 14 ± 0.2 K, microwave frequency = 9.47 GHz, microwave power = 20 μW, modulation frequency = 100 KHz, modulation amplitude = 10 G, time constant = 167 ms, scan time = 167 s. (B) Spectra acquired over a narrow range of the magnetic field with a smaller modulation amplitude (2 G) and 5 scans summed per spectrum to provide better resolution of the g ∼ 2 region. In B, the contribution of the spectrum of the MnIV/FeIV complex has been subtracted to resolve the signal of the organic radical component.
Figure 3.
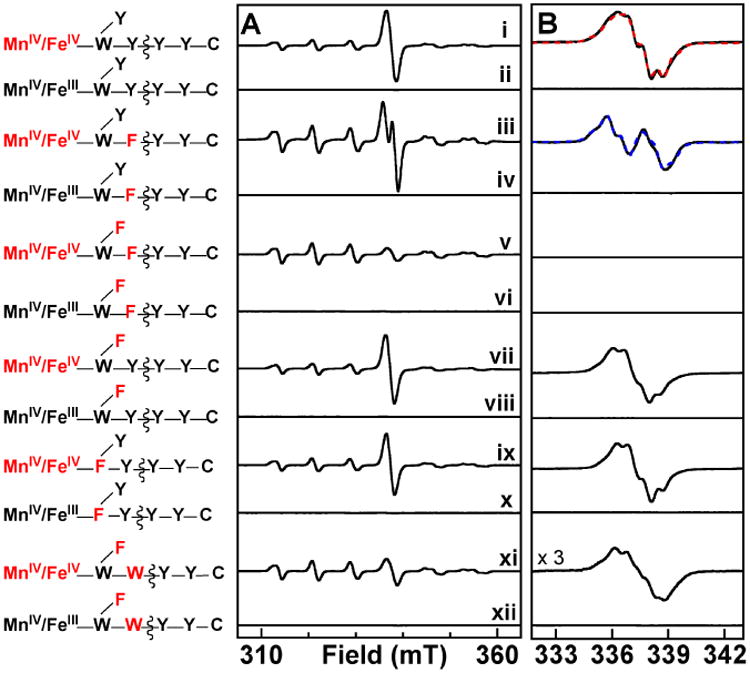
X-band EPR spectra illustrating the effects of substitutions of residues in the two orthogonal electron-relay pathways in Ct β on the nature of accumulating radical species in reactions of the MnII/FeII-β complexes with O2 in the presence of α, CDP, ATP, DTT, and MgSO4. A schematic of the electron-relay pathways is provided at the left, with the non-native cluster redox state and substituted amino acid residues highlighted in red. Sample preparation was as described in the legend of Figure 1. For samples containing the MnIV/FeIII states, each β protein was pre-treated by adding air-saturated 100 mM HEPES, 10% (v/v) glycerol buffer to the MnII/FeII-β complex, and incubating for 60 minutes at 5 °C. This solution was then mixed with the remaining components (α, CDP, ATP, MgSO4, and DTT) and frozen after ∼ 5 s. A displays the “raw” experimental spectra, and, in B, the contribution of the MnIV/FeIV intermediate has been removed to resolve the spectra of the radical components. The red and blue dashed spectra in B are simulations with parameters provided in Supporting Information. Spectrometer conditions are provided in the legend of Figure 1.
Evidence for “half-of-sites reactivity” in radical generation in the super-oxidized enzyme
The spectra of Figure 1 illustrate that accumulation of the putative Y•(s) is in no case associated with complete decay of the MnIV/FeIV activation intermediate. This is perhaps surprising, given that the latter species is expected to generate the former species. We formulated three alternative explanations for their simultaneous presence in samples of the super-oxidized enzyme complex: (1) an equilibrium between the MnIV/FeIV-containing and MnIV/FeIII–Y•-containing forms, reflecting reversibility of RT between the cluster and the Y residue(s); (2) incomplete complex formation between α and the MnIV/FeIV-containing β, which would prevent the fraction of the MnIV/FeIV cluster present in unbound β from generating a Y•; and (3) half-of-sites reactivity in RT, which would prevent one of the two MnIV/FeIV clusters present in each β2 subunit from generating a Y•. Explanation 1 was ruled out on the basis of the kinetics of MnIV/FeIV cluster and Y• decay following formation of the super-oxidized enzyme form (Figure 2A). Whereas two species in rapid equilibrium should develop and decay in constant proportion as a single kinetic entity, the residual MnIV/FeIV complex (squares) decays considerably (∼ 6-fold) faster than the Y• (circles) in these samples. As a test of explanation 2, the ratio of α:β was varied from 1:1 to 6:1, and the relative proportions of MnIV/FeIV cluster and Y• were assessed by EPR (Figure 2B). No significant change could be discerned with increasing α:β, implying that the β subunit is almost fully saturated by α even at a 1:1 molar ratio. The results of Figure 2 thus imply that explanation 3, half-of-sites reactivity within the α2•β2 complex, is the most likely reason for the incomplete decay of the MnIV/FeIV cluster in its generation of the putative Y•.
Figure 2.
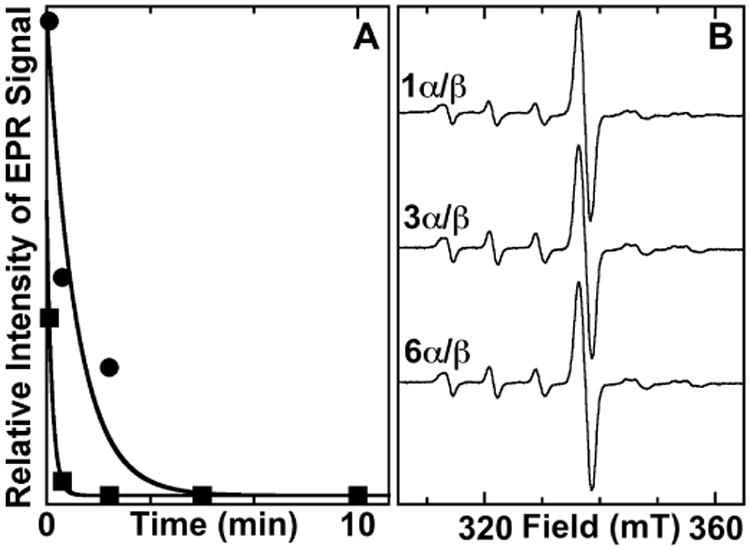
Tests of two possible explanations for the persistence of a fraction of the MnIV/FeIV complex even after maximal accumulation of the putative Y•(s). (A) Kinetics of decay of the putative Y•(s) (circles) and the MnIV/FeIV complex (squares). Traces were fit by the equation describing a single-exponential decay process, giving rate constants of 0.017 s-1 and 0.1 s-1, respectively. The difference in decay rate constants rules out the possibility that the two species are in rapid equilibrium. (B) X-band EPR of samples prepared with ratios of α:β ranging from 1:1 to 6:1. Samples were prepared as described for Figure 1, except they contained 0.14 mM β and 0.14 – 0.84 mM α. The failures of the radical signal to increase and the MnIV/FeIV signal to be diminished rules out the possibility that weak binding between the two subunits results in a fraction of the MnIV/FeIV-β remaining unbound and unable to generate the radical. Spectrometer conditions are the same as for Figure 1.
To test this hypothesis further, we prepared duplicate sets of freeze-quenched EPR samples by a double-mixing protocol. A solution of MnII/FeII-β was first mixed with O2-containing buffer to generate the MnIV/FeIV complex, and the intermediate was then reacted for 1 s with either α, MgSO4, DTT, CDP, and ATP in the experiment or only α, MgSO4, and DTT in the control. Comparison of the EPR spectra of the complete-reaction samples to those of the no-nucleotide control samples (Figure S2 and Table S2) reveals that the intensity of the EPR signature of the MnIV/FeIV intermediate decays by an average of 45%, very similar to the 50% loss expected under the assumption of “half-of-sites reactivity.”8,28,31,57,58 This lost intensity is fully accounted for by the new g = 2.00 signal attributed to the Y•(s). The spin quantification is thus completely consistent with the hypothesis that the Y•(s) is (are) generated by the MnIV/FeIV complex on one side of each α2•β2•ATP•CDP complex.
Evidence that the radical(s) reside within the RT pathway
Our previous work provided evidence that Ct RNR possesses two orthogonal pathways for reduction of the Mn/Fe cluster: the activation-specific pathway, comprising Y222 and W51 of β (blue lines in Scheme 1), mediates reduction of the FeIV of the MnIV/FeIV intermediate; the catalysis-specific RT pathway, comprising Y338 and W51 of β (in addition to Y991 and Y990 of α; red lines in Scheme 1), mediates propagation of the oxidizing equivalent from the MnIV ion of the functional MnIV/FeIII cofactor to the essential C672 in α during turnover.46 The primary basis for identification of the latter pathway was the undetectable catalytic activities of the W51F and Y338F variants, but the importance of Y338 is also strongly supported by its alignment with Y356 of Ec β, which the work of Stubbe and colleagues has convincingly shown to undergo substrate-dependent, protein-gated, transient oxidation during catalysis.8,27,31,33,34,36-38,59 To ascertain whether the putative Y• detected in Figure 1 resides within the catalytic RT pathway, we substituted Y338 with the redox-incompetent F and tested for loss of, or changes to, the signal of the Y•. The EPR spectrum of a sample prepared by mixing MnII/FeII-β-Y338F with an air-saturated solution containing α, CDP, and ATP again has a radical signal at g = 2.00 suggestive of a Y• (Figure 3, iii). The signal is, however, markedly different from that observed with wt β (Figure 3, i), suggesting that the radical resides, at least in part, on a different Y residue. Previous work examining reactions of the Ct holoenzyme with hydroxyurea (HU) had suggested that, with Y338 replaced by F, substrate-dependent propagation of the oxidizing equivalent from the MnIV/FeIII factor onto Y222 can occur, initiating HU reduction of the cofactor to the MnIII/FeIII state.56 We therefore suspected that the different signal observed in the super-oxidized complex with β-Y338F might result from propagation of the oxidizing equivalent into the activation-specific pathway and onto Y222. To test this possibility, we interrogated the double-variant β protein with substitutions of both Y338 and Y222 with F. The EPR spectrum of a sample prepared with this double variant lacks any signal for an organic radical (Figure 3, v). By contrast, the Y222F variant supports formation of a radical with an EPR spectrum very similar to that observed with wt β (Figure 3, vii). The simplest interpretation of the change in the radical EPR spectrum upon substitution of Y338 alone, abolition of the spectrum upon substitution of both electron-relaying Y residues in β, and absence of any effect of the Y222F substitution by itself is that the Y• observed with the wt subunit resides within the catalysis-specific RT pathway (on Y338β, Y991α, Y990α, or distributed among these residues) and that the Y338F substitution causes aberrant propagation onto Y222 of the activation-specific RT pathway. It is noteworthy that formation of this off-pathway (Y222) radical again requires the presence of substrate. Thus, even the aberrant propagation into the activation-specific pathway still behaves (as it did in the HU study56) as if gated by substrate binding. As previously noted, this conclusion has implications for the location and nature of the conformational gate of the RT process.
Hurdling engineered defects in the RT pathway
In comparison to Ec Y356 and Ct Y338, the roles of W48 of Ec β and W51 of Ct β in catalysis are less clear. It has been established that W48 functions in electron relay during activation of Ec β: it is oxidized to a cation radical concomitantly with formation of the Y122•-generating diiron intermediate, X.19 Moreover, the W48F substitution blocks electron transfer to the cofactor in multiple contexts.60,61 Analogously, the W51F substitution in Ct β slows reduction of the FeIV site of the MnIV/FeIV activation intermediate.46 Whether either W residue similarly undergoes transient oxidation during RT in catalysis is not yet known. To probe the role of W51 in the RT pathway, we tested for the ability of Ct β-W51F to support pathway-radical accumulation in its super-oxidized state. Remarkably, despite its incompetence for CDP reduction in the MnIV/FeIII state (most likely owing to blocked RT),46 the variant protein is fully competent for radical accumulation in its super-oxidized state (Figure 3, ix). The EPR spectrum in the g = 2.00 region is almost identical to that observed with wt Ct β. Accumulation of the radical is again dependent on the presence of the substrate CDP, suggesting proper gating of RT in the holoenzyme complex with β-W51F.
The inactivity of Ct β-W51F in the MnIV/FeIII state and competence of its MnIV/FeIV super-oxidized state to generate a pathway radical suggest that the stringency of RT-pathway structural requirements is relaxed in the super-oxidized state. As an additional test of this notion, the crucial, subunit-interfacial Y338 was substituted by W, a residue that might conceivably still function in RT but with an altered redox potential. This substitution was combined with Y222F in Ct β, so that the ability of W338 to support pathway radical accumulation could be assessed without interference from the signal of the off-pathway Y222•. Not surprisingly, the β-Y222F/Y338W protein lacks detectable catalytic activity in its MnIV/FeIII state (the double variant is fully competent to generate this form in the reaction of its reduced enzyme form with O2). Thus, the Y338W substitution results in loss of proper RT with the stable MnIV/FeIII cofactor. However, in the reaction of MnII/FeII -β-Y222F/Y338W with O2 in the presence of α, CDP, and ATP, a radical signal does indeed develop (Figure 3, xi). The shape of the spectrum is again suggestive of a Y•, which, if on the RT pathway, would in this case necessarily be located within α. The super-oxidized complex of this β double variant generated with the α-Y991F variant (which readily supports radical accumulation in complex with wt β; see Figure S1, black in the Supporting Information) lacks a significant radical signal (Figure S1, green), providing additional evidence that the radical in the β-Y222F/Y338W•α(wt) complex resides within α on either Y991 or Y990 (or perhaps distributed between them).
Functional competence of the Y•(s) in the super-oxidized Ct RNR complexes
The raison d'être of forward (β → α) RT within the RNR holoenzyme complex is to initiate nucleotide reduction by abstraction of H• from the 3′ carbon of the substrate. If the Y• or Y•s observed in super-oxidized Ct RNR do, as the above evidence suggests, reside within the RT pathway, they might be expected to function in initiation of turnover. The substrate analogue, 2′-azido-2′-deoxyuridine-5′-diphosphate (N3-UDP), can serve as a reporter of 3′-H• abstraction, because this step initiates a cascade of events leading to irreversible trapping of the oxidizing equivalent within α in the form of an analogue-derived radical, in which the unpaired spin resides primarily on a nitrogen atom derived from the azide moiety (this radical is hereafter abbreviated N•).6,62 It was previously shown that treatment of the stable, active form of β with N3-UDP in the presence of α and ATP converts the EPR-silent MnIV/FeIII cofactor to its EPR-active MnIII/FeIII form concomitantly with the accumulation of the N•,41 and this observation was the key evidence establishing that the MnIV/FeIII cofactor is functional in Ct RNR.41 The distinctive EPR signature of the N• (Figure 4, i) is partially resolved also from the spectra of the MnIV/FeIV activation intermediate and RT-pathway Y•(s) in the super-oxidized enzyme, making the analogue potentially useful as a probe of 3′-H• abstraction also in this state. For the wt enzyme, interpretation would be complicated by the fact that the super-oxidized MnIV/FeIV state decays to the active MnIV/FeIII form, which is itself known to support N• production. Thus, it would be ambiguous as to which cofactor form had been responsible for 3′-H• abstraction in the event that abstraction were found to occur. However, the β-W51F and β-Y222F/Y338W variants are both inactive in their MnIV/FeIII states yet capable of supporting RT-pathway Y• accumulation in their super-oxidized states, thereby potentially resolving this ambiguity. As expected on the basis of their incompetence for CDP reduction, neither variant supports accumulation of the N• when its stable MnIV/FeIII state is incubated with α, ATP, and N3-UDP (Figure 4, iii and v). Remarkably, samples prepared by mixing the MnII/FeII form of either variant with an O2-containing solution of α, ATP, and N3-UDP and freezing after one minute clearly do exhibit the signature of the N• (Figure 4, ii and iv), thus associating the radicals observed in the super-oxidized states of these RT-pathway-defective variants with successful 3′-H• abstraction. A control reaction employing the β-Y338F variant, which is incompetent for accumulation of RT-pathway Y•(s) but instead supports formation of the off-pathway Y222•, shows that N3-UDP can replace CDP in the gating function, thereby supporting development of the EPR signal assigned to Y222•, but, as expected, does not undergo conversion to the N• in the holoenzyme complex with this β variant (Figure 4, vi).
Figure 4.
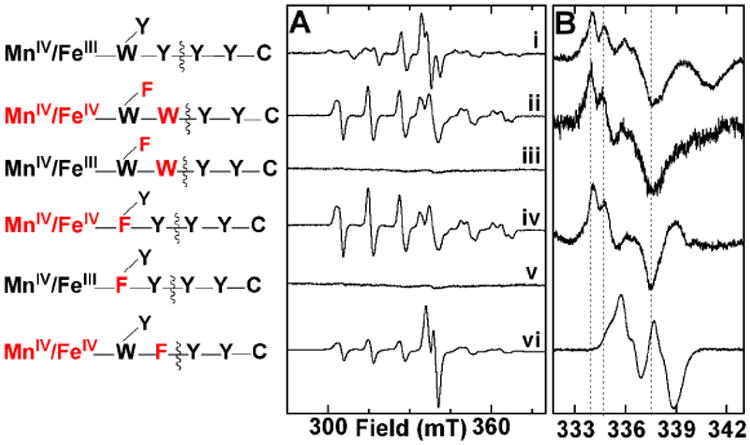
X-band EPR spectra of samples employing the radical-trapping substrate analogue, 2′-azido-2′-deoxyuridine-5′-diphosphate (N3-UDP), to provide evidence for the functional competence of the RT-pathway radicals in the super-oxidized enzyme for 3′-H• abstraction. (A) Raw experimental spectra acquired over a wide range of the magnetic field and (B) spectra acquired over a narrow range of the magnetic field and processed by subtraction of the contributions from either the MnIII/FeIII complex (i) or the MnIV/FeIV complex (ii, iv, and vi) to resolve the spectra of the organic-radical components. The method of sample preparation and the spectrometer conditions are the same as for Figures 1 and 2.
The functional competence of the RT-pathway Y•(s) was further assessed by testing directly for CDP reduction to dCDP. A published assay55 employing tritium-labeled CDP substrate (3H-CDP, labeled at C5 of cytidine) was used to quantify the product. Background levels of radioactivity were observed in the product fractions from reactions initiated by mixing the stable MnIV/FeIII forms of the β-Y338F, β-Y222F/Y338W, and β-W51F with α, ATP, and 3H-CDP (Figure 5, orange, black, gray, respectively). By contrast, reactions initiated by mixing the MnII/FeII forms of the variants with O2-containing solutions of the other reaction components (so as to generate the super-oxidized enzyme form) produced ∼ 0.5 equiv dCDP per β for the two variants [β-Y222F/Y338W (red) and β-W51F (blue)] shown above to support 3′-H• abstraction from N3-UDP, but gave no significant product-associated radioactivity in excess of the background for the uniformly inactive β-Y338F variant (green). The activity profile of the variants is entirely consistent with that deduced both by EPR [accumulation of pathway Y•(s) or absence thereof] and by use of the radical-trapping N3-UDP analogue (production of the N• or failure thereto). The correlation implies that the radicals observed in the super-oxidized enzyme forms are functionally competent. The quantity of dCDP observed with the conditionally active β-Y222F/Y338W and β-W51F variants suggests that only a single turnover can occur and that the known half-of-sites reactivity limits the super-oxidized forms to a single-turnover within just one side of the hetero-tetrameric α2•β2 complex. The conclusion of a single turnover suggests, in turn, that the bi-directionality of RT between β and α that is required for catalysis may be deranged as the enzyme operates with the more oxidized cofactor and RT pathway defects, thus precluding subsequent turnovers. The conclusion of a single turnover is also consistent with observations by Stubbe and co-workers on RT-pathway variants of the class Ia Ec RNR.28,37
Figure 5.
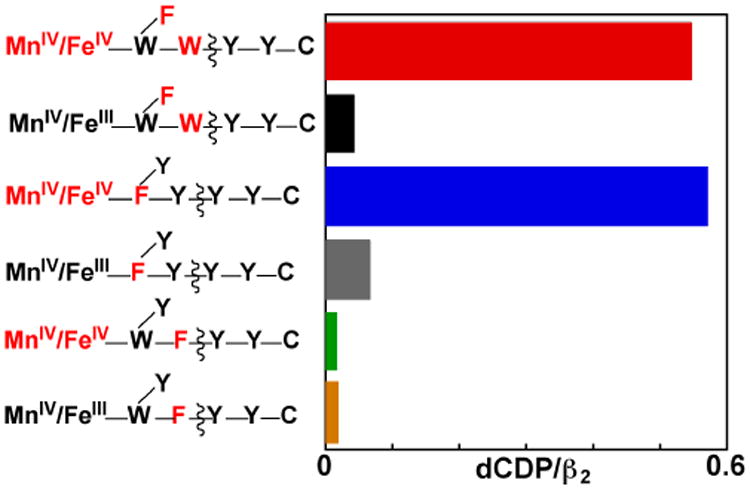
Product (dCDP) yields demonstrating the functional competence of RT-pathway radicals in the super-oxidized states of Ct β variants with pathway substitutions rendering them inactive for CDP reduction in their stable MnIV/FeIII states. Reactions were initiated by mixing the indicated MnII/FeII-β complex or the pre-formed MnIV/FeIII-β complex with an air-saturated solution containing the other reactions components, as in preparation of the EPR samples, but with [3H]-CDP (the site of labeling and specific activity are provided in the Experimental Procedures section) included as radioactive tracer for product quantification. After a one-minute reaction, samples were processed (as described in Procedures) based on a published method to separate substrate and product. The dCDP product was then quantified by scintillation counting.
Discussion
The characteristics of free-radical accumulation upon generation of the MnIV/FeIV activation intermediate in Ct RNR-β in the presence of α, a substrate (CDP), substrate analogue (N3-UDP), or product (dCDP), and an allosteric effector (ATP) provide convincing evidence for the location of the radical(s) within the RT pathway. Radical formation requires the substrate and is potentiated by the effector, implying gating in the same manner previously demonstrated for both Ct and Ec RNR.28-30,56 The line shape of the g = 2.00 EPR signal is consistent with that of a Y• (with appropriate dihedral angles between the ring and Cβ–H bonds),53,63 and Ct RNR has three Y residues (β-Y338, α-Y991, and α-Y990)39 that align in sequence with those shown by Stubbe and co-workers to undergo transient oxidation during catalysis by Ec RNR.27-31,37 The Y338F substitution in β changes the EPR spectrum and thus the identity of the accumulating radical (putatively to Y222•), and the combination of this substitution with the Y222F substitution in β, which disables the activation-specific electron-transfer pathway unique to the Ct enzyme,46 abolishes radical accumulation altogether. In the context of the β-Y222F/Y338X double variant, swapping the completely non-functional F for the potentially oxidizable W at position 338 restores radical accumulation, showing that a redox-active “stepping stone” is required at this position but need not be the native Y when the cofactor is in its super-oxidized state. By far the most compelling evidence for the location of the accumulating radicals within the RT pathway, however, is their association with 3′-H• abstraction: the variants that are competent for radical accumulation (β-W51F and β-Y222F/Y338W) also support both N• formation from the N3-UDP analog and a single turnover (reduction) of CDP substrate in their super-oxidized states, despite being essentially devoid of either activity in their stable MnIV/FeIII oxidation states.
The above observations on the super-oxidized state of Ct RNR have some analogy to recent studies by Stubbe and co-workers examining RT and turnover in the Ec RNR holoenzyme complex of a β variant with the radical harboring Y122 replaced by 3-nitrotyrosine (NO2-Y).37 The NO2-Y radical (NO2-Y•) is a more potent oxidant (by ∼ 200 mV) than Y•.37 As in our study, the “hot oxidant” (NO2-Y122•) was formed in situ in β, in this case immediately before mixing the subunits, because it is stable only for tens of seconds (half-life of ∼ 40 s at 25 °C). Whereas turnover by the wt Ec enzyme initiated by Y122• normally occurs at < 10 s-1,30 owing to a rate-limiting conformational change that is probably part of the opening of the gate for Y122• reduction and RT, both reduction of NO2-Y122• and dCDP production were found to be much faster in the variant [biphasic kinetics with observed first-order rate constants of ∼ 300 s-1 and ∼ 70 s-1 for NO2-Y122• reduction and ∼ 100 s-1 for dCDP production].37 The authors concluded that the more potent oxidant essentially permits hurdling of the conformational gate in a manner analogous to the hurdling of engineered pathway defects in the super-oxidized Ct enzyme we report in this manuscript. Their results are also consistent with the hypothesis that the gating mechanism involves dynamic control of proton transfer to Y122•, because the rapid, putatively “ungated” reduction of the NO2-Y122• in the variant is not associated with protonation of the radical (i.e., the 3-nitrophenolate form is produced). Apparently, even without coupled proton transfer, the initial electron transfer from W48 or Y356 to the more potently oxidizing NO2-Y122• radical is still favorable, permitting forward RT into α. In addition, one or more new Y• was shown to accumulate at nearly the same rate constant as for dCDP production. More recent electron double resonance spectroscopic experiments have provided evidence that the oxidizing equivalent actually becomes distributed over Y356 of β, Y731 of α, and Y730 of α,38 suggesting that the reduction potentials of the individual Y•s are similar. The kinetics of RT-pathway Y• accumulation and the location(s) of the radical(s) within the pathway in the Ct RNR super-oxidized state and variants thereof are actively being pursued and may shed light on the extent to which the class Ia and class Ic enzymes behave similarly in this key step.
Even without knowledge of the kinetics and precise location of the pathway radical(s), the observations on the class Ic enzyme and its super-oxidized state speak to two outstanding questions regarding the key RT step. One key question concerns the role of W48/W51. There is no evidence to date that either undergoes transient oxidation in RT during catalysis. The inactivity of the Ct β-W51F variant might be taken as evidence that W51 has such a role, but the absence of any evidence for a W51-derived radical in the super-oxidized holoenzyme complex with the β-Y222F/Y338F double variant, in which radical propagation beyond W51 is blocked, suggests that W51 is not readily oxidized, even by the more oxidized MnIV/FeIV cofactor form. Coupled with our earlier evidence that W51 is oxidized to a radical during the reaction of the Fe2II/II complex of the Y222F variant of Ct β with O2,46 the apparent absence of any W51 oxidation by the MnIV/FeIV activation intermediate can be viewed as argument against its transient oxidation during propagation of the oxidizing equivalent from the less potent MnIV/FeIII cluster toward α during catalysis. A caveat to this conclusion is the possibility that oxidation of W51 by the Mn/Fe cofactor (in either oxidation state) might be thermodynamically disfavored, causing very little W51-derived radical to accumulate, even in the super-oxidized complex of β-Y222F/Y338F. Indeed, recent work by Gray, Winkler, and coworkers has shown that electron hopping can proceed even with an element that is thermodynamically uphill by as much as 200 mV,64-67 which would correspond to an equilibrium between the MnIV/FeIV–W51 and MnIV/FeIII–W51• forms of the super-oxidized complex favoring the W51-reduced (non-radical) form by more than 1000-fold. Determination of the kinetics of Y• formation in super-oxidized Ct RNR and the impact of the W51F substitution thereupon might provide a quantitative assessment of the importance of this residue in the process.
A second outstanding question concerns the nature and location of the gate for RT. In previous work, we interpreted data on the reduction of the Ct RNR cofactor from the MnIV/FeIII state to the MnIII/FeIII state by HU and the dependence thereof on the presence of the substrate and certain key electron-relaying residues as implying that the gate is adjacent to the cofactor and largely independent of the aromatic residues of the RT pathway, involving, at most, W51 of the pathway.56 The observation here that the W51F variant supports radical accumulation, again dependent on the presence of the substrate, suggests that W51 is dispensable at least for the apparent gating being manifested by the super-oxidized enzyme. This observation would imply that the gating function is contained within the immediate vicinity of the cofactor. Intriguingly, in addition to signaling the cofactor-proximal physical gate to open, the conformational change upon substrate binding in α must have additional impact on the RT pathway in β that allows the hole to propagate outward from the cofactor into α once the gate has opened. In the absence of α, the MnIV/FeIV cluster in β must propagate a hole into the activation-specific pathway involving Y222 more efficiently than it does into the catalysis-specific pathway involving Y338, because replacement of the former residue by F slows reduction of the FeIV site by 10–65-fold,46 whereas substitution of the latter has no effect on the kinetics of FeIV-site reduction.56 Conversely, in the holoenzyme complex, it seems that Y338 is oxidized to a radical in great preference to Y222, and only with the former residue replaced by the redox-incompetent F does Y222 undergo oxidation. Thus, the conformational change in some way switches the relative reactivities of Y222 and Y338 toward one-electron oxidation.
On the basis of these observations and those by Stubbe and co-workers on their Ec β-Y122NO2-Y variant,37 our working hypothesis is that the key event in opening of the RT gate in both class Ia and class Ic RNRs is a conformational change, driven by binding of the substrate in α and propagated across the subunit interface into β, that engages the mechanism by which a proton is transferred to or within the initial oxidant (Y122• or the MnIV/Fe(III,IV) cluster). This proton transfer (PT) is coupled to ET from the cofactor proximal RT residue (W48/51 or Y356/338) and makes this redox step sufficiently favorable to initiate the cascade of hopping events. For the case of Ec RNR, our recent collaborative study with the Stubbe group has provided evidence in support of their prior suggestion that the water bound to Fe1 of the μ-oxo-Fe2III/III cluster is the proton donor to the Y122• oxidant.68 Thus, it is likely that the D84 ligand to Fe1 in Ec β, which is (i) completely conserved among class Ia orthologues but invariably replaced by E in the class Ic orthologues and (ii) in hydrogen-bonding distance from both Y122 and the Fe1 water ligand in crystal structures of Ec β15,69 mediates the crucial PT. Therefore, the gate-opening transition could well involve re-orientation of this carboxylate ligand or enhancement of its conformational dynamics in a manner that enables its PT mediation. In consideration of this hypothesis, the behavior of the Ec β-D84E variant, which we previously showed is fully competent for cofactor assembly,70 in the holoenzyme complex becomes an intriguing issue for future examination, because it is expected that even subtle re-orientation of the carboxylate group by the presence of the additional methylene unit in the E84 side chain could either completely disable the crucial PT pathway or derange its conformational modulation, rendering the variant protein inactive, ungated, or both. Additionally, we43 and others71 previously have speculated that the gate-opening step in Ct RNR may involve protonation of either the oxo- or hydroxo-bridge of the μ-oxo-μ-hydroxo-bridged MnIV/FeIII cofactor.72 Whether the cognate residue in Ct β, E89, might also mediate the gatekeeping proton transfer is an open question.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM-55365 to JMB and CK) and the Alfred P. Sloan Foundation Minority Ph.D. Scholarship Program (to LMKD). The authors thank the reviewers for insightful comments.
Abbreviations
- RNR
ribonucleotide reductase
- Ct
Chlamydia trachomatis
- Ec
Escherichia coli
- β
cofactor subunit of class I RNR
- α
catalytic subunit of class I RNR
- PCET
proton-coupled electron transfer
- Y•
tyrosyl radical
- RT
radical-translocation
- NDP
nucleoside 5′-diphosphate
- wt
wild-type
- CDP
cytidine 5′-diphosphate
- dCDP
deoxycytidine 5′-diphosphate
- H•
hydrogen atom
- C•
cysteine thiyl radical
- N3-UDP
2′-azido-2′-deoxyuridine-5′-diphosphate
- W•
tryptophan radical
- N•
nitrogen-centered radical
- PT
proton transfer
- ET
electron transfer
Footnotes
Supporting Information. Description of and parameters used in the simulation of the tyrosyl radicals reported in Figure 3, a schematic drawing of the phenol ring of a tyrosine residue with numerical assignments used for the simulation of the radicals, and a figure demonstrating the competence of the β-wt•α-Y991F complex but not the β-Y222F/Y338W•α-Y991F complex to form a radical are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nordlund P, Reichard P. Annu Rev Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 2.Stubbe J. Curr Opin Struct Biol. 2000;10:731–736. doi: 10.1016/s0959-440x(00)00153-6. [DOI] [PubMed] [Google Scholar]
- 3.Stubbe J, Ackles D. J Biol Chem. 1980;255:8027–8030. [PubMed] [Google Scholar]
- 4.Mao SS, Yu GX, Chalfoun D, Stubbe J. Biochemistry. 1992;31:9752–9759. doi: 10.1021/bi00155a031. [DOI] [PubMed] [Google Scholar]
- 5.Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem Rev. 2003;103:2167–2202. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 6.Thelander L, Larsson B, Hobbs J, Eckstein F. J Biol Chem. 1976;251:1398–405. [PubMed] [Google Scholar]
- 7.Brown NC, Reichard P. J Mol Biol. 1969;46:25–38. doi: 10.1016/0022-2836(69)90055-2. [DOI] [PubMed] [Google Scholar]
- 8.Seyedsayamdost MR, Chan CTY, Mugnaini V, Stubbe J, Bennati M. J Am Chem Soc. 2007;129:15748–15749. doi: 10.1021/ja076459b. [DOI] [PubMed] [Google Scholar]
- 9.Thelander L. J Biol Chem. 1973;248:4591–4601. [PubMed] [Google Scholar]
- 10.Uhlin U, Eklund H. Nature. 1994;370:533–539. doi: 10.1038/370533a0. [DOI] [PubMed] [Google Scholar]
- 11.Reece SY, Hodgkiss JM, Stubbe J, Nocera DG. Philos Trans Royal Soc B. 2006;361:1351–1364. doi: 10.1098/rstb.2006.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stubbe J, Riggs-Gelasco P. Trends Biochem Sci. 1998;23:438–443. doi: 10.1016/s0968-0004(98)01296-1. [DOI] [PubMed] [Google Scholar]
- 13.Licht S, Gerfen GJ, Stubbe J. Science. 1996;271:477–481. doi: 10.1126/science.271.5248.477. [DOI] [PubMed] [Google Scholar]
- 14.Atkin CL, Thelander L, Reichard P, Lang G. J Biol Chem. 1973;248:7464–7472. [PubMed] [Google Scholar]
- 15.Nordlund P, Sjöberg BM, Eklund H. Nature. 1990;345:593–598. doi: 10.1038/345593a0. [DOI] [PubMed] [Google Scholar]
- 16.Stubbe J. Curr Opin Chem Biol. 2003;7:183–188. doi: 10.1016/s1367-5931(03)00025-5. [DOI] [PubMed] [Google Scholar]
- 17.Tong WH, Chen S, Lloyd SG, Edmondson DE, Huynh BH, Stubbe J. J Am Chem Soc. 1996;118:2107–2108. [Google Scholar]
- 18.Yun D, Garcia-Serres R, Chicalese BM, An YH, Huynh BH, Bollinger JM., Jr Biochemistry. 2007;46:1925–1932. doi: 10.1021/bi061717n. [DOI] [PubMed] [Google Scholar]
- 19.Baldwin J, Krebs C, Ley BA, Edmondson DE, Huynh BH, Bollinger JM., Jr J Am Chem Soc. 2000;122:12195–12206. [Google Scholar]
- 20.Krebs C, Chen S, Baldwin J, Ley BA, Patel U, Edmondson DE, Huynh BH, Bollinger JM., Jr J Am Chem Soc. 2000;122:12207–12219. [Google Scholar]
- 21.Bollinger JM, Jr, Edmondson DE, Huynh BH, Filley J, Norton JR, Stubbe J. Science. 1991;253:292–298. doi: 10.1126/science.1650033. [DOI] [PubMed] [Google Scholar]
- 22.Bollinger JM, Jr, Stubbe J, Huynh BH, Edmondson DE. J Am Chem Soc. 1991;113:6289–91. [Google Scholar]
- 23.Ekberg M, Sahlin M, Eriksson M, Sjöberg BM. J Biol Chem. 1996;271:20655–20659. doi: 10.1074/jbc.271.34.20655. [DOI] [PubMed] [Google Scholar]
- 24.Ekberg M, Pötsch S, Sandin E, Thunnissen M, Nordlund P, Sahlin M, Sjöberg BM. J Biol Chem. 1998;273:21003–21008. doi: 10.1074/jbc.273.33.21003. [DOI] [PubMed] [Google Scholar]
- 25.Rova U, Goodtzova K, Ingemarson R, Behravan G, Gräslund A, Thelander L. Biochemistry. 1995;34:4267–4275. doi: 10.1021/bi00013a016. [DOI] [PubMed] [Google Scholar]
- 26.Rova U, Adrait A, Pötsch S, Gräslund A, Thelander L. J Biol Chem. 1999;274:23746–23751. doi: 10.1074/jbc.274.34.23746. [DOI] [PubMed] [Google Scholar]
- 27.Seyedsayamdost MR, Yee CS, Reece SY, Nocera DG, Stubbe J. J Am Chem Soc. 2006;128:1562–1568. doi: 10.1021/ja055927j. [DOI] [PubMed] [Google Scholar]
- 28.Seyedsayamdost MR, Stubbe J. J Am Chem Soc. 2007;129:2226–2227. doi: 10.1021/ja0685607. [DOI] [PubMed] [Google Scholar]
- 29.Seyedsayamdost MR, Xie J, Chan CTY, Schultz PG, Stubbe J. J Am Chem Soc. 2007;129:15060–15071. doi: 10.1021/ja076043y. [DOI] [PubMed] [Google Scholar]
- 30.Ge J, Yu G, Ator MA, Stubbe J. Biochemistry. 2003;42:10017–10083. doi: 10.1021/bi034374r. [DOI] [PubMed] [Google Scholar]
- 31.Seyedsayamdost MR, Stubbe J. J Am Chem Soc. 2006;128:2522–2523. doi: 10.1021/ja057776q. [DOI] [PubMed] [Google Scholar]
- 32.Minnihan EC, Seyedsayamdost MR, Stubbe J. Biochemistry. 2009;48:12125–12132. doi: 10.1021/bi901439w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seyedsayamdost MR, Argirević T, Minnihan EC, Stubbe J, Bennati M. J Am Chem Soc. 2009;131:15729–15738. doi: 10.1021/ja903879w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minnihan EC, Seyedsayamdost MR, Uhlin U, Stubbe J. J Am Chem Soc. 2011;133:9430–9440. doi: 10.1021/ja201640n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seyedsayamdost MR, Yee CS, Stubbe J. Biochemistry. 2010;50:1403–1411. doi: 10.1021/bi101319v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yokoyama K, Uhlin U, Stubbe J. J Am Chem Soc. 2010;132:8385–8397. doi: 10.1021/ja101097p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yokoyama K, Uhlin U, Stubbe J. J Am Chem Soc. 2010;132:15368–15379. doi: 10.1021/ja1069344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yokoyama K, Smith AA, Corzilius B, Griffin RG, Stubbe J. J Am Chem Soc. 2011;133:18420–18432. doi: 10.1021/ja207455k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roshick C, Iliffe-Lee ER, McClarty G. J Biol Chem. 2000;275:38111–38119. doi: 10.1074/jbc.M006367200. [DOI] [PubMed] [Google Scholar]
- 40.Högbom M, Stenmark P, Voevodskaya N, McClarty G, Gräslund A, Nordlund P. Science. 2004;305:245–248. doi: 10.1126/science.1098419. [DOI] [PubMed] [Google Scholar]
- 41.Jiang W, Yun D, Saleh L, Barr EW, Xing G, Hoffart LM, Maslak MA, Krebs C, Bollinger JM., Jr Science. 2007;316:1188–1191. doi: 10.1126/science.1141179. [DOI] [PubMed] [Google Scholar]
- 42.Jiang W, Bollinger JM, Jr, Krebs C. J Am Chem Soc. 2007;129:7504–7505. doi: 10.1021/ja072528a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bollinger JM, Jr, Jiang W, Green MT, Krebs C. Curr Opin Struct Biol. 2008;18:650–657. doi: 10.1016/j.sbi.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 44.Jiang W, Yun D, Saleh L, Bollinger JM., Jr Biochemistry. 2008;47:13736–13744. doi: 10.1021/bi8017625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang W, Hoffart LM, Krebs C, Bollinger JM., Jr Biochemistry. 2007;46:8709–8716. doi: 10.1021/bi700906g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang W, Saleh L, Barr EW, Xie J, Gardner MM, Krebs C, Bollinger JM., Jr Biochemistry. 2008:8477–8484. doi: 10.1021/bi800881m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stubbe J. Unpublished data [Google Scholar]
- 48.McGee DPC, Vargeese C, Zhai Y, Kirschenheuter GP, Settle A, Siedem CR, Pieken WA. Nucleosides & Nucleotides. 1995;14:1329–1339. [Google Scholar]
- 49.Besada P, Shin DH. J Med Chem. 2006;49:5532–5543. doi: 10.1021/jm060485n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verheyden JP, Wagner D, Moffatt JG. J Org Chem. 1971;36:250–254. doi: 10.1021/jo00801a002. [DOI] [PubMed] [Google Scholar]
- 51.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 52.Stoll S, Schweiger A. J Magn Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 53.Svistunenko DA, Cooper CE. Biophys J. 2004;87:582–595. doi: 10.1529/biophysj.104.041046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dassama LMK, Yosca TH, Conner DA, Lee MH, Blanc B, Streit BR, Green MT, DuBois JL, Krebs C, Bollinger JM. Biochemistry. 2012;51:1607–1616. doi: 10.1021/bi201906x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steeper JR, Steuart CD. Anal Biochem. 1970;34:123–130. doi: 10.1016/0003-2697(70)90092-8. [DOI] [PubMed] [Google Scholar]
- 56.Jiang W, Xie J, Varano PT, Krebs C, Bollinger JMJ. Biochemistry. 2010;49:5340–5349. doi: 10.1021/bi100037b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bennati M, Robblee JH, Mugnaini V, Stubbe J, Freed JH, Borbat P. J Am Chem Soc. 2005;127:15014–15015. doi: 10.1021/ja054991y. [DOI] [PubMed] [Google Scholar]
- 58.Uppsten M, Färnegårdh M, Domkin V, Uhlin U. J Mol Biol. 2006;359:365–377. doi: 10.1016/j.jmb.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 59.Yee CS, Seyedsayamdost MR, Chang MCY, Nocera DG, Stubbe J. Biochemistry. 2003;42:14541–14552. doi: 10.1021/bi0352365. [DOI] [PubMed] [Google Scholar]
- 60.Parkin SE, Chen S, Ley BA, Mangravite L, Edmondson DE, Huynh BH, Bollinger JM., Jr Biochemistry. 1998;37:1124–1130. doi: 10.1021/bi9723717. [DOI] [PubMed] [Google Scholar]
- 61.Baldwin J, Voegtli WC, Khidekel N, Moënne-Loccoz P, Krebs C, Pereira AS, Ley BA, Huynh BH, Loehr TM, Riggs-Gelasco PJ, Rosenzweig AC, Bollinger JM., Jr J Am Chem Soc. 2001;123:7017–7030. doi: 10.1021/ja002114g. [DOI] [PubMed] [Google Scholar]
- 62.Fritscher J, Artin E, Wnuk S, Bar G, Robblee JH, Kacprzak S, Kaupp M, Griffin RG, Bennati M, Stubbe J. J Am Chem Soc. 2005;127:7729–7738. doi: 10.1021/ja043111x. [DOI] [PubMed] [Google Scholar]
- 63.Bender CJ, Sahlin M, Babcock GT, Barry BA, Chandrashekar TK, Salowe SP, Stubbe J, Lindström B, Petersson L, Ehrenberg A, Sjöberg BM. J Am Chem Soc. 1989;111:8076–8083. [Google Scholar]
- 64.Gray HB, Winkler JRQ. Rev Biophys. 2003;36:341–376. doi: 10.1017/s0033583503003913. [DOI] [PubMed] [Google Scholar]
- 65.Gray HB, Winkler JR. J R Biochim Biophys Acta. 2010;1797:1563–1572. doi: 10.1016/j.bbabio.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 66.Shih C, Museth AK, Abrahamsson M, Blanco-Rodriguez AM, Di Bilio AJ, Sudhamsu J, Crane BR, Ronayne KL, Towrie M, Vlček BH, Jr, Richards JH, Winkler JR, Gray HB. Science. 2008;320:1760–1762. doi: 10.1126/science.1158241. [DOI] [PubMed] [Google Scholar]
- 67.Warren JJ, Winkler JR, Gray HB. Coordination Chemistry Reviews. 2012 doi: 10.1016/j.ccr.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wörsdörfer B, Conner DA, Yokoyama K, Seyedsayamdost MR, Jiang W, Stubbe J, Bollinger JMJ, Krebs C. For review only. 2012 [Google Scholar]
- 69.Högbom M, Galander M, Andersson M, Kolberg M, Hofbauer W, Lassmann G, Nordlund P, Lendzian F. Proc Natl Acad Sci U S A. 2003;100:3209–3214. doi: 10.1073/pnas.0536684100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bollinger JM, Jr, Krebs C, Vicol A, Chen S, Ley BA, Edmondson DE, Huynh BH. J Am Chem Soc. 1998;120:1094–1095. [Google Scholar]
- 71.Roos K, Siegbahn PEM. Biochemistry. 2009;48:1878–1887. doi: 10.1021/bi801695d. [DOI] [PubMed] [Google Scholar]
- 72.Younker JM, Krest CM, Jiang W, Krebs C, Bollinger JM, Jr, Green MT. J Am Chem Soc. 2008;130:15022–15027. doi: 10.1021/ja804365e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.