

Abstract
Attention deficit hyperactivity disorder (ADHD) is a neurobehavioral disorder affecting children and adults. Genetic and environmental factors are associated with the etiology of ADHD. Among the environmental factors, exposure of the developing brain to nicotine is considered a major risk factor. Recent evidence suggests that environmental influences on the brain and behavior may be transmitted from one generation to the next. We used a prenatal nicotine exposure (PNE) mouse model of ADHD to test the hypothesis that PNE-induced hyperactivity, a proxy for human ADHD phenotype, is transmitted from one generation to the next. Our data reveal transgenerational transmission of PNE-induced hyperactivity in mice via the maternal but not the paternal line of descent. We suggest that transgenerational transmission is a plausible mechanism for propagation of environmentally induced ADHD phenotypes in the population.
Keywords: ADHD, dopamine, hyperactivity, methylphenidate, nicotine, transgenerational transmission
Introduction
Attention deficit hyperactivity disorder (ADHD) is a neurobehavioral disorder characterized by hyperactivity, inattention, and impulsivity. ADHD affects ∼10% of children and 5% of adults in the United States (Faraone et al., 2003; Barbaresi et al., 2004; Kessler et al., 2006). Genetic and environmental factors contribute to the etiology of ADHD (Biederman et al., 2006; Swanson et al., 2007). Exposure to nicotine during prenatal and early postnatal periods is a major environmental contributor (Milberger et al., 1996; Milberger et al., 1998; Ernst et al., 2001). Children born to mothers who smoked cigarettes before, during, or immediately after pregnancy have a twofold higher risk of developing ADHD (Milberger et al., 1996; Milberger et al., 1998). We showed previously that ADHD associated with prenatal nicotine exposure is symptomatically indistinguishable from ADHD due to other causes (Biederman et al., 2012). In another recent study (Zhu et al., 2012), we showed that a mouse model of prenatal nicotine exposure (PNE) shares anatomical, biochemical, and behavioral features with human ADHD. Although maternal cigarette smoking is a significant contributor to ADHD in the exposed children, it should be emphasized that some studies report no major adverse consequences of maternal cigarette smoking on cognitive abilities of the children (Thapar et al., 2009; Obel et al., 2011).
In the present study, we show that PNE-induced hyperactivity, a proxy for the human ADHD phenotype, is transmitted in the mouse model from one generation to the next. Our findings may have significant implications for transgenerational transmission of environmentally induced ADHD in human populations.
Materials and Methods
Animals.
We used C57BL/6 mice (Charles River Laboratories) to create a PNE mouse model (Zhu et al., 2012). Female mice (F0 generation) were administered (−)-nicotine (0.1 mg/ml) dissolved in saccharin (2%; both from Sigma) in drinking water starting 3 weeks before mating and throughout pregnancy. The saccharin renders the water palatable. The male breeders were not exposed to nicotine or saccharin. Control groups only received 2% saccharin in the drinking water (SAC group). Nicotine administration in drinking water to pregnant dams is widely used for modeling fetal nicotine exposure caused by maternal tobacco use during pregnancy (Sparks and Pauly, 1999; Paz et al., 2007; Zhu et al., 2012). On the day of birth (postnatal day 0; P0), offspring in the PNE and SAC groups (F1 generation) were cross-fostered to drug-naive nursing mothers. At ∼6–8 weeks of age, the F1 females from each of the PNE and SAC groups were bred with wild-type, drug-naive males to generate the F2 generation (Fig. 1). Similarly, at ∼6–8 weeks of age, the F2 females from each of the PNE and SAC groups were bred with wild-type, drug-naive males to generate the F3 generation (Fig. 1). In a parallel set of experiments, F1 males from each of the PNE and SAC groups were bred with wild-type, drug-naive females to produce the F2 generation (Fig. 1). Neither the F2 nor the F3 generation was exposed to nicotine directly either during development or at adulthood. The only nicotine exposure was for the F0 females as adults in drinking water and the F1 mice (males and females) in utero due to exposure of the F0 mother during and before pregnancy. We used no more than one male and one female offspring from any given litter in our analyses to minimize the contribution of litter effects to the data.
Figure 1.

Generation of F1, F2, and F3 mice from the PNE dams. Dams in the founder (F0) generation were exposed to nicotine in drinking water beginning 3 weeks before they were crossed with a drug-naive sire. The nicotine exposure continued throughout pregnancy. At ∼6–8 weeks of age, male and female offspring (F1) born to the PNE dams were crossed with drug-naive male and female mice, respectively, to produce the F2 generations. At ∼6–8 weeks of age, PNE-F2 female mice were crossed with drug-naive males to produce the PNE-female-derived F3 generation. PNE-F2 male mice were not used for further breeding. From each generation, 6- to 8-week-old mice were used for experimental analysis. An identical breeding plan was used to generate F1, F2, and F3 mice from the prenatally saccharin-exposed dams (data not shown).
We showed in an earlier study that spontaneous locomotor activity is significantly elevated in 6-week-old F1 male and female mice in the PNE group compared with their counterparts in the SAC group (Zhu et al., 2012). Moreover, locomotor activity was not significantly different between mice in the SAC group and mice exposed to plain drinking water (without any additives), suggesting that saccharin alone did not influence locomotor activity (Zhu et al., 2012). Therefore, we excluded the plain water group and used the SAC group as controls in the present study.
Analysis of locomotor activity.
We examined spontaneous locomotor activity in 6- to 7-week-old F2 and F3 male and female mice derived from F1 and F2 male or female founders from the PNE or the SAC groups. The mice were placed individually in testing cages equipped with photobeam motion sensors (Photobeam Activity System; San Diego Instruments). Each instance in which consecutive breaks were recorded in adjacent photobeams (positioned 5.4 cm apart) was scored as an ambulatory event. Photobeam breaks were grouped into hourly activity measurements for statistical analysis. The analysis was conducted over a 17 h period from 15:00 to 08:00 h. Because locomotor activity increases transiently upon placement in the novel environment of the testing apparatus, we excluded from analysis the initial 4 h period from 15:00 to 19:00 h. Because the hyperactivity occurred in the F1 generation only during the lights-off period (Zhu et al., 2012), we performed the current analysis over a 12 h period from 19:00 to 07:00 h when the lights were off.
Statistics.
Differences between more than two experimental groups (prenatal treatment, gender, and generation) were compared for statistical significance using ANOVA. Differences between two groups were then analyzed using Tukey–Kramer pairwise comparisons. When only two experimental groups were involved, we used Student's t test.
Results
Locomotor activity was significantly increased in the F2 male and female mice derived from F1 PNE females (Fig. 2A,B) and in F3 male and female mice derived from F2 PNE females (Fig. 2C,D) compared with control SAC counterparts. In contrast, the activity in F2 mice derived from male F1 PNE mice was not significantly different from their SAC counterparts (Fig. 2E,F).
Figure 2.

Transgenerational transmission of hyperactivity induced by PNE. Spontaneous locomotor activity was analyzed between 17:00 and 09:00 h in the F2 (A, B) and F3 (C, D) generations. The founder mice for each generation were females from the PNE or SAC group and drug-naive males. Hourly (A, C) and cumulative (B, D) locomotor activity measurements are shown (mean ± SEM values). The cumulative activity represents locomotor activity during the lights-off period (19:00–07:00 h). Hourly measurements showed that both PNE male and female F2 (A) and F3 (C) mice showed significantly higher activity compared with the prenatal SAC counterparts. Cumulative activity showed significant effects of prenatal treatment in F2 (F(3,44) = 19.7; p = 0.0001) and F3 generations (F(3,44) = 14.3; p = 0.0001). Tukey–Kramer multiple-comparison tests showed significantly higher cumulative activity in both male and female PNE F2 and F3 mice compared with their SAC counterparts (p < 0.001). There was no significant difference between male and female mice in either the prenatal treatment group or either generation (p > 0.05). n = 12–13 for each group. PNE-induced hyperactivity is not transmitted from one generation to the next via male founders (E–F). Spontaneous locomotor activity was analyzed from 17:00–09:00 h in male F2 mice produced by breeding male PNE or SAC mice with drug-naive females. The cumulative activity represents locomotor activity during the lights-off period (19:00–07:00 h). Hourly (E) and cumulative (F) locomotor activity measurements are shown (mean ± SEM values). Hourly activity was similar between the PNE and SAC groups (E). Cumulative activity did not show significant differences between the two groups (t test, p = 0.13, n = 9 per group).
In an earlier study, we showed that a single oral administration of 0.75 mg/kg methylphenidate (MPH) reduces locomotor activity in PNE F1 male and female mice (Zhu et al., 2012). This oral MPH dose is equivalent to therapeutic doses for ADHD (Balcioglu et al., 2009). We administered MPH or saline by oral gavage (0.75 mg/kg) at 19:00 h, when the lights were turned off, to F2 mice. The MPH decreased locomotor activity in the F2 PNE male and female mice (Fig. 3), as well as in the F3 PNE male and female mice (Fig. 4) in the same manner as it did in F1 mice in our earlier study (Zhu et al., 2012).
Figure 3.

Effects of a single oral dose (0.75 mg/kg) of MPH on PNE induced spontaneous locomotor activity in male and female F2 mice derived from female founders. Locomotor activity was assayed from 17:00–09:00 h (A–D). MPH or saline (SAL) was gavaged at 19:00 h. Cumulative activity in the PNE (E) and SAC (F) groups was analyzed for the 12 h lights-off period from 19:00–07:00 h. Data are shown as means ± SEM. Hourly measurements showed that both male (A) and female (B) mice derived from PNE founders showed lower activity upon MPH administration compared with SAL administration. In the SAC group, MPH administration did not produce significant effects on the hourly activity compared with SAL administration in male (C) or female (D) mice. Cumulative activity over the entire 12 h lights-off period showed significant decreases in the PNE male and female mice (E) receiving MPH compared with SAL, whereas MPH had no significant effect on male or female mice in the SAC group (F). Cumulative activity showed significant effects of drug treatment in the PNE group (ANOVA: F(3,28) = 6.3; p = 0.003). Tukey–Kramer multiple-comparison test revealed significant differences in cumulative activity between SAL and MPH treatments for male and female mice (p < 0.05). ANOVA did not reveal significant changes in cumulative activity in the SAC group (F(3,37) = 1.02, p = 0.39).
Figure 4.
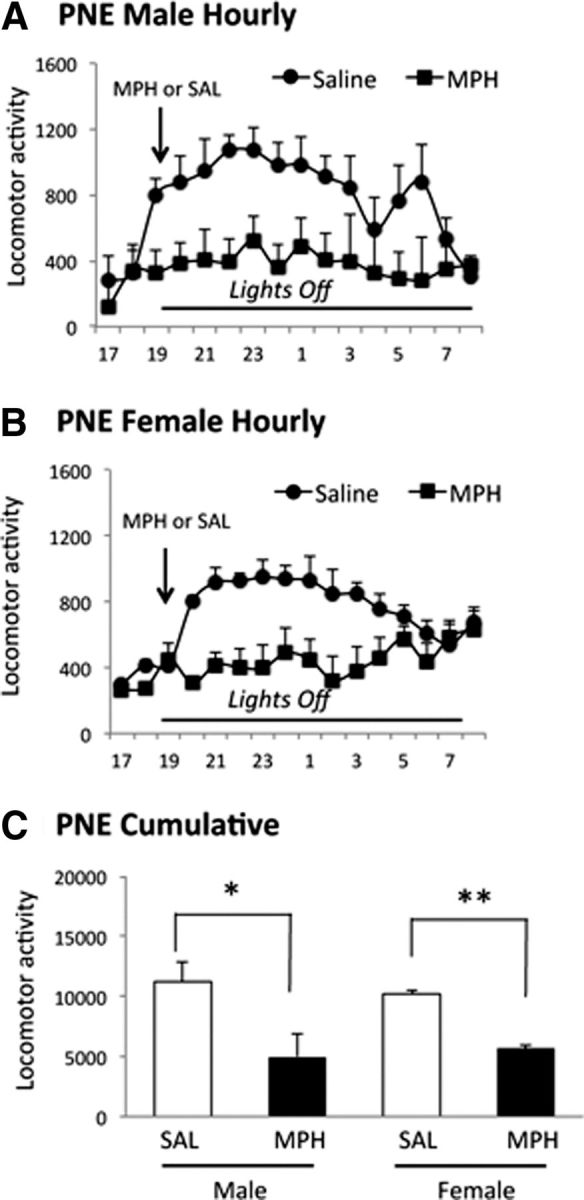
Effects of a single oral dose (0.75 mg/kg) of MPH on PNE induced spontaneous locomotor activity in male and female F3 mice derived from female F2 founders. Locomotor activity was assayed from 17:00–09:00 h (A, B). MPH or saline (SAL) was gavaged at 19:00 h. Cumulative activity (C) was analyzed for the 12 h lights-off period from 19:00 to 07:00 h. Data are shown as mean ± SEM values. Hourly measurements showed that both male (A) and female (B) F3 mice showed lower activity upon MPH administration compared with SAL administration. Cumulative activity over the 12 h lights-off period showed significant decreases in the PNE male and female mice (C) receiving MPH compared with SAL. Statistical analysis (t test) showed significant decreases in cumulative activity in male and female mice administered MPH compared with their counterparts receiving SAL (male: t = 3.39, p = 0.04; female: t = 6.09, p = 0.01; n = 4 per group).
Our data demonstrate that spontaneous locomotor activity is transmitted from F1 to F2 and F2 to F3 generations even though nicotine exposure occurred only for the F1 (in utero exposure) and F2 (in utero germ cell exposure) generations. Moreover, the transmission occurred via the maternal and not the paternal line, although in each generation both male and female offspring were hyperactive. The hyperactivity was associated with exposure to an environmental agent and not likely due to genetic causes, because every offspring from every PNE litter (F1) and maternally derived F2 and F3 generations showed hyperactivity. The hyperactivity did not show classical Mendelian inheritance.
Discussion
Our data show that PNE-induced hyperactivity is transmitted from one generation to the next, although only the founder generations are exposed to nicotine. The transmission occurs via the maternal and not the paternal line.
We have used a mouse model of PNE in which nicotine is administered to the pregnant dams via drinking water. We chose this model because of a number of advantages. For example, administration of nicotine in drinking water avoids the stress associated with systemic nicotine administration (e.g., intraperitoneal injection or osmotic pump; Rowell et al., 1983; Sparks and Pauly, 1999; Vaglenova et al., 2004; Paz et al., 2007). Prenatal stress significantly influences behavior and brain structure (Muhammad and Kolb, 2011). Nicotine administered via drinking water or non-oral routes produces comparable changes in locomotor activity and nicotinic acetylcholine receptor activity (Sparks and Pauly, 1999). A criticism of the oral nicotine paradigm is that it does not mimic the peaks and valleys of plasma nicotine levels associated with cigarette smoking (Benowitz et al., 1982). However, nicotine pharmacokinetics in the fetus are quite different from those in adults, and whether the peaks and valleys occur in the fetal blood and brain as a result of maternal cigarette smoking remains unclear (Wickstrom, 2007). Moreover, nicotine patch or smokeless tobacco use by pregnant women, which remains a significant public health concern (for review, see Wickstrom, 2007), is associated with sustained plasma nicotine concentrations (Benowitz et al., 1982; Hukkanen et al., 2005). Another criticism of experimental models of nicotine exposure is that cigarette smoke contains thousands of unique ingredients, many of which are harmful, and the adverse consequences of cigarette smoke may not be due to nicotine alone. However, the behavioral effects of cigarette smoke exposure can be recapitulated by exposure of the developing and mature brain to nicotine (Slotkin et al., 1987; Navarro et al., 1989a; Navarro et al., 1989b). Therefore, our PNE mouse model recapitulates the consequences of cigarette smoking during pregnancy for the developing brain.
Our finding of transgenerational transmission of hyperactivity in the PNE mouse model is uniquely relevant to ADHD for the following reasons. Prenatally nicotine exposed rodents not only show hyperactivity, but also attention deficits (Schneider et al., 2011). Hyperactivity and attention deficit constitute major domains of ADHD symptomatology. Unlike other ADHD animal models (Sagvolden et al., 2005), the PNE model carries ecological validity because cigarette smoking during pregnancy is associated with a nearly twofold increase in ADHD risk in the offspring (Milberger et al., 1996; Milberger et al., 1998; Biederman et al., 2012). The PNE mouse model also shows a frontal cortical hypodopaminergic state and reduced cingulate cortex volume (Zhu et al., 2012), cardinal features of untreated ADHD (Makris et al., 2007; Volkow et al., 2009). Finally, the hyperactivity in the PNE mouse model is significantly reduced and the frontal cortical dopamine levels are significantly elevated after oral MPH administration (Zhu et al., 2012). Therefore, the PNE mouse model shows face validity, construct validity, and predictive validity fulfilling the criteria for a valid animal model of human ADHD (Sagvolden et al., 2005). Therefore, the transgenerational transmission of hyperactivity in this model bears considerable significance and relevance for ADHD.
The hyperactivity in the F2 and F3 generations was responsive to oral MPH, suggesting that MPH-induced increases in dopamine content of the brain may have contributed to the reductions in hyperactivity. This was the case for the F1 generation in our earlier study (Zhu et al., 2012). Therefore, the CNS hypodopaminergic state in the F1 generation was likely transmitted to the F2 and F3 generation. In other words, the neurobiological mechanisms and the behavioral phenotypes appear to be transmissible from one generation to the next.
Our findings are also significant from another public health perspective. The rates of ADHD diagnosis increased by 24% over a 10-year period between 2001 and 2010 (Getahun et al., 2013). Although revised diagnostic criteria, sophisticated diagnostic tools, and increased awareness among parents, patients, and the general public alike may contribute to the increase in ADHD incidence, whether those factors alone can cause the increase remains unclear. Although genetic factors are associated with ADHD (Biederman et al., 1986; Wallis et al., 2008), a decade may not be long enough for newly acquired mutations to contribute to the dramatic rise. Increasing exposure to harmful environmental factors may be another potential culprit (Banerjee et al., 2007; Environmental Protection Agency, 2007). However, increasing public awareness and stringent public safety measures in recent decades may be expected to decrease the risk of harmful environmental exposures (Environmental Protection Agency, 2007). Therefore, in addition to the potential causes above, some other, as yet unknown factors or mechanisms may contribute to the increased incidence of ADHD.
Our findings suggest that the adverse effects of environmental factors on one generation can be transmitted to subsequent generations to amplify the impact of a temporally distal and discrete insult over time. In other words, we suggest that transgenerational transmission may be one mechanism contributing to the apparent increases in ADHD diagnosis over time. Our data do not reveal whether the transgenerational transmission would occur if the ADHD were treated successfully in a given generation. Future studies will test this interesting possibility.
A plausible mechanism for the transgenerational transmission of the PNE-induced brain and behavioral changes is heritable epigenetic modifications of the germ cell genome (for review, see Daxinger and Whitelaw, 2012). Nicotine is known to produce DNA methylation in a number of genes, including the gene coding for monoamine oxidase, a key enzyme in the metabolism of dopamine and other monoamines (Philibert et al., 2008; Philibert et al., 2010). Therefore, nicotine-induced epigenetic changes emerge as a potential mechanism for transgenerational heritability. Because the transmission of hyperactivity occurred only via the mother and not the father, mitochondrial genes, which are inherited exclusively from the mother, may be significant contributors. Alternatively, male germ cells may be somehow resistant to the effects of prenatal nicotine exposure, preventing transmission via the father. However, the male germ cells do acquire and transmit environment-induced epigenetic changes. For example, cocaine produces epigenetic changes in the sperm DNA (Vassoler et al., 2013) and these changes are transmitted to subsequent generations. Genomic imprinting is another plausible mechanism that may promote the heritability of hyperactivity in the PNE model. Recent evidence indicates that there are imprints/parent-of-origin effects on transcription at >1300 loci (Gregg et al., 2010b) and that ∼350 autosomal genes with sex-specific parent-of-origin effects may exist in the mouse brain (Gregg et al., 2010a). Pedigree analysis coupled with epigenetic techniques will be required to elucidate fully the mechanism(s) mediating heritable and sex-linked patterns, be it X-linkage, maternal effect, or parental imprinting.
In summary, our data show that the hyperactivity produced by PNE is transmitted via mothers from one generation to the next. A PNE-induced hypodopaminergic state in the brain may also be transmitted in the same manner because MPH administration ameliorates hyperactivity in multiple generations.
Footnotes
This work was supported by the United States Public Health Service (Grant R21DA027358). We thank Dr. Michael Schwarzschild for access to equipment and resources in his laboratory at the Massachusetts General Hospital, Boston, Massachusetts.
References
- Balcioglu A, Ren JQ, McCarthy D, Spencer TJ, Biederman J, Bhide PG. Plasma and brain concentrations of oral therapeutic doses of methylphenidate and their impact on brain monoamine content in mice. Neuropharmacology. 2009;57:687–693. doi: 10.1016/j.neuropharm.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee TD, Middleton F, Faraone SV. Environmental risk factors for attention-deficit hyperactivity disorder. Acta Paediatr. 2007;96:1269–1274. doi: 10.1111/j.1651-2227.2007.00430.x. [DOI] [PubMed] [Google Scholar]
- Barbaresi W, Katusic S, Colligan R, Weaver A, Pankratz V, Mrazek D, Jacobsen S. How common is attention-deficit/hyperactivity disorder? Towards resolution of the controversy: results from a population-based study. Acta Paediatr Suppl. 2004;93:55–59. doi: 10.1111/j.1651-2227.2004.tb03058.x. [DOI] [PubMed] [Google Scholar]
- Benowitz NL, Kuyt F, Jacob P., 3rd Circadian blood nicotine concentrations during cigarette smoking. Clin Pharmacol Ther. 1982;32:758–764. doi: 10.1038/clpt.1982.233. [DOI] [PubMed] [Google Scholar]
- Biederman J, Munir K, Knee D, Habelow W, Armentano M, Autor S, Hoge SK, Waternaux C. A family study of patients with attention deficit disorder and normal controls. J Psychiatr Res. 1986;20:263–274. doi: 10.1016/0022-3956(86)90030-0. [DOI] [PubMed] [Google Scholar]
- Biederman J, Monuteaux MC, Mick E, Wilens TE, Fontanella JA, Poetzl KM, Kirk T, Masse J, Faraone SV. Is cigarette smoking a gateway to alcohol and illicit drug use disorders? A study of youths with and without attention deficit hyperactivity disorder. Biol Psychiatry. 2006;59:258–264. doi: 10.1016/j.biopsych.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Biederman J, Petty CR, Bhide PG, Woodworth KY, Faraone S. Does exposure to maternal smoking during pregnancy affect the clinical features of ADHD? Results from a controlled study. World J Biol Psychiatry. 2012;13:60–64. doi: 10.3109/15622975.2011.562243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daxinger L, Whitelaw E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat Rev Genet. 2012;13:153–162. doi: 10.1038/nrg3188. [DOI] [PubMed] [Google Scholar]
- Environmental Protection Agency. EPA Office of Research and Development (8101R) (ICF International of Fairfax, Virginia, ed.) Washington, DC: EPA; 2007. A decade of children's environmental health research: highlights from EPA's Science to Achieve Results program. [Google Scholar]
- Ernst M, Moolchan ET, Robinson ML. Behavioral and neural consequences of prenatal exposure to nicotine. J Am Acad Child Adolesc Psychiatry. 2001;40:630–641. doi: 10.1097/00004583-200106000-00007. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Sergeant J, Gillberg C, Biederman J. The worldwide prevalence of ADHD: is it an American condition? World Psychiatry. 2003;2:104–113. [PMC free article] [PubMed] [Google Scholar]
- Getahun D, Jacobsen SJ, Fassett MJ, Chen W, Demissie K, Rhoads GG. Recent trends in childhood attention-deficit/hyperactivity disorder. JAMA Pediatr. 2013;167:282–288. doi: 10.1001/2013.jamapediatrics.401. [DOI] [PubMed] [Google Scholar]
- Gregg C, Zhang J, Butler JE, Haig D, Dulac C. Sex-specific parent-of-origin allelic expression in the mouse brain. Science. 2010a;329:682–685. doi: 10.1126/science.1190831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg C, Zhang J, Weissbourd B, Luo S, Schroth GP, Haig D, Dulac C. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science. 2010b;329:643–648. doi: 10.1126/science.1190830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukkanen J, Jacob P, 3rd, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 2005;57:79–115. doi: 10.1124/pr.57.1.3. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Adler L, Barkley R, Biederman J, Conners CK, Demler O, Faraone SV, Greenhill LL, Howes MJ, Secnik K, Spencer T, Ustun TB, Walters EE, Zaslavsky AM. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry. 2006;163:716–723. doi: 10.1176/appi.ajp.163.4.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris N, Biederman J, Valera EM, Bush G, Kaiser J, Kennedy DN, Caviness VS, Faraone SV, Seidman LJ. Cortical thinning of the attention and executive function networks in adults with attention-deficit/hyperactivity disorder. Cereb Cortex. 2007;17:1364–1375. doi: 10.1093/cercor/bhl047. [DOI] [PubMed] [Google Scholar]
- Milberger S, Biederman J, Faraone SV, Chen L, Jones J. Is maternal smoking during pregnancy a risk factor for attention deficit hyperactivity disorder in children? Am J Psychiatry. 1996;153:1138–1142. doi: 10.1176/ajp.153.9.1138. [DOI] [PubMed] [Google Scholar]
- Milberger S, Biederman J, Faraone SV, Jones J. Further evidence of an association between maternal smoking during pregnancy and attention deficit hyperactivity disorder: findings from a high-risk sample of siblings. J Clin Child Psychol. 1998;27:352–358. doi: 10.1207/s15374424jccp2703_11. [DOI] [PubMed] [Google Scholar]
- Muhammad A, Kolb B. Mild prenatal stress-modulated behavior and neuronal spine density without affecting amphetamine sensitization. Dev Neurosci. 2011;33:85–98. doi: 10.1159/000324744. [DOI] [PubMed] [Google Scholar]
- Navarro HA, Seidler FJ, Schwartz RD, Baker FE, Dobbins SS, Slotkin TA. Prenatal exposure to nicotine impairs nervous system development at a dose which does not affect viability or growth. Brain Res Bull. 1989a;23:187–192. doi: 10.1016/0361-9230(89)90146-9. [DOI] [PubMed] [Google Scholar]
- Navarro HA, Seidler FJ, Eylers JP, Baker FE, Dobbins SS, Lappi SE, Slotkin TA. Effects of prenatal nicotine exposure on development of central and peripheral cholinergic neurotransmitter systems: evidence for cholinergic trophic influences in developing brain. J Pharmacol Exp Ther. 1989b;251:894–900. [PubMed] [Google Scholar]
- Obel C, Olsen J, Henriksen TB, Rodriguez A, Järvelin MR, Moilanen I, Parner E, Linnet KM, Taanila A, Ebeling H, Heiervang E, Gissler M. Is maternal smoking during pregnancy a risk factor for hyperkinetic disorder? Findings from a sibling design. Int J Epidemiol. 2011;40:338–345. doi: 10.1093/ije/dyq185. [DOI] [PubMed] [Google Scholar]
- Paz R, Barsness B, Martenson T, Tanner D, Allan AM. Behavioral teratogenicity induced by nonforced maternal nicotine consumption. Neuropsychopharmacology. 2007;32:693–699. doi: 10.1038/sj.npp.1301066. [DOI] [PubMed] [Google Scholar]
- Philibert RA, Gunter TD, Beach SR, Brody GH, Madan A. MAOA methylation is associated with nicotine and alcohol dependence in women. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:565–570. doi: 10.1002/ajmg.b.30778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert RA, Beach SR, Gunter TD, Brody GH, Madan A, Gerrard M. The effect of smoking on MAOA promoter methylation in DNA prepared from lymphoblasts and whole blood. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:619–628. doi: 10.1002/ajmg.b.31031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowell PP, Hurst HE, Marlowe C, Bennett BD. Oral administration of nicotine: its uptake and distribution after chronic administration to mice. J Pharmacol Methods. 1983;9:249–261. doi: 10.1016/0160-5402(83)90064-5. [DOI] [PubMed] [Google Scholar]
- Sagvolden T, Russell VA, Aase H, Johansen EB, Farshbaf M. Rodent models of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57:1239–1247. doi: 10.1016/j.biopsych.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Schneider T, Ilott N, Brolese G, Bizarro L, Asherson PJ, Stolerman IP. Prenatal exposure to nicotine impairs performance of the 5-choice serial reaction time task in adult rats. Neuropsychopharmacology. 2011;36:1114–1125. doi: 10.1038/npp.2010.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin TA, Cho H, Whitmore WL. Effects of prenatal nicotine exposure on neuronal development: selective actions on central and peripheral catecholaminergic pathways. Brain Res Bull. 1987;18:601–611. doi: 10.1016/0361-9230(87)90130-4. [DOI] [PubMed] [Google Scholar]
- Sparks JA, Pauly JR. Effects of continuous oral nicotine administration on brain nicotinic receptors and responsiveness to nicotine in C57BL/6 mice. Psychopharmacology (Berl) 1999;141:145–153. doi: 10.1007/s002130050818. [DOI] [PubMed] [Google Scholar]
- Swanson JM, Kinsbourne M, Nigg J, Lanphear B, Stefanatos GA, Volkow N, Taylor E, Casey BJ, Castellanos FX, Wadhwa PD. Etiologic subtypes of attention-deficit/hyperactivity disorder: brain imaging, molecular genetic and environmental factors and the dopamine hypothesis. Neuropsychol Rev. 2007;17:39–59. doi: 10.1007/s11065-007-9019-9. [DOI] [PubMed] [Google Scholar]
- Thapar A, Rice F, Hay D, Boivin J, Langley K, van den Bree M, Rutter M, Harold G. Prenatal smoking might not cause attention-deficit/hyperactivity disorder: evidence from a novel design. Biol Psychiatry. 2009;66:722–727. doi: 10.1016/j.biopsych.2009.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaglenova J, Birru S, Pandiella NM, Breese CR. An assessment of the long-term developmental and behavioral teratogenicity of prenatal nicotine exposure. Behav Brain Res. 2004;150:159–170. doi: 10.1016/j.bbr.2003.07.005. [DOI] [PubMed] [Google Scholar]
- Vassoler FM, White SL, Schmidt HD, Sadri-Vakili G, Pierce RC. Epigenetic inheritance of a cocaine-resistance phenotype. Nat Neurosci. 2013;16:42–47. doi: 10.1038/nn.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Kollins SH, Wigal TL, Newcorn JH, Telang F, Fowler JS, Zhu W, Logan J, Ma Y, Pradhan K, Wong C, Swanson JM. Evaluating dopamine reward pathway in ADHD: clinical implications. JAMA. 2009;302:1084–1091. doi: 10.1001/jama.2009.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis D, Russell HF, Muenke M. Review: genetics of attention deficit/hyperactivity disorder. J Pediatr Psychol. 2008;33:1085–1099. doi: 10.1093/jpepsy/jsn049. [DOI] [PubMed] [Google Scholar]
- Wickström R. Effects of nicotine during pregnancy: human and experimental evidence. Curr Neuropharmacol. 2007;5:213–222. doi: 10.2174/157015907781695955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhang X, Xu Y, Spencer TJ, Biederman J, Bhide PG. Prenatal nicotine exposure mouse model showing hyperactivity, reduced cingulate cortex volume, reduced dopamine turnover, and responsiveness to oral methylphenidate treatment. J Neurosci. 2012;32:9410–9418. doi: 10.1523/JNEUROSCI.1041-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]