

Abstract
Purpose
There are no effective therapies for patients with poorly differentiated papillary thyroid cancer (PTC) or anaplastic thyroid cancer (ATC), and metastasis to the bone represents a significantly worse prognosis. Src family kinases (SFKs) are overexpressed and activated in numerous tumor types and have emerged as a promising therapeutic target, especially in relation to metastasis. We recently showed that Src is overexpressed and activated in thyroid cancer. We therefore tested whether inhibition of Src with dasatinib (BMS-354825) blocks thyroid cancer growth and metastasis.
Experimental Design
The effects of dasatinib on thyroid cancer growth, signaling, cell cycle, and apoptosis were evaluated in vitro. The therapeutic efficacy of dasatinib was further tested in vivo using an orthotopic and a novel experimental metastasis model. Expression and activation of SFKs in thyroid cancer cells was characterized, and selectivity of dasatinib was determined using an Src gatekeeper mutant.
Results
Dasatinib treatment inhibited Src signaling, decreased growth, and induced cell-cycle arrest and apoptosis in a subset of thyroid cancer cells. Immunoblotting showed that c-Src and Lyn are expressed in thyroid cancer cells and that c-Src is the predominant SFK activated. Treatment with dasatinib blocked PTC tumor growth in an orthotopic model by more than 90% (P = 0.0014). Adjuvant and posttreatment approaches with dasatinib significantly inhibited metastasis (P = 0.016 and P = 0.004, respectively).
Conclusion
These data provide the first evidence that Src is a central mediator of thyroid cancer growth and metastasis, indicating that Src inhibitors may have a higher therapeutic efficacy in thyroid cancer, as both antitumor and antimetastatic agents.
Introduction
There are currently no effective therapies for patients with advanced thyroid cancer, which includes patients diagnosed with advanced papillary thyroid cancer (PTC) or anaplastic thyroid cancer (ATC; ref. 1). Notably, ATC is one of the most aggressive human cancers with greater than 95% mortality at 6 months. Extrathyroidal invasion and metastasis are the most common causes of thyroid cancer–related death, and metastasis to the bone predicts a significantly worse prognosis (2). Although much effort has been devoted to decipher the mechanisms involved in the progression of this cancer, little progress has been made in the development of new therapies (1, 3).
Src family kinases (herein referred to as SFKs or Src) are a multifunctional nonreceptor tyrosine kinase family that regulates a variety of cellular processes, including growth, survival, migration, and invasion (4). SFKs regulate these protumorigenic functions via activation of downstream signaling pathways, including mitogen-activated protein kinase/extracellular signal–regulated kinase (MAPK/ERK), phosphoinositide 3-kinase (PI3K), Stat3, p130Cas, paxillin, and focal adhesion kinase (FAK). SFKs are overexpressed and/or activated in many tumor types (5-10). Of the 9 SFK members, c-Src, Fyn, and Yes are most widely expressed, and c-Src itself has been most frequently implicated in tumorigenesis and metastasis (11). c-Src has been shown to play an important role in regulating osteoclast function and tumor colonization to the bone, making Src an attractive therapeutic target for the prevention and treatment of bone metastases (12, 13). While the remaining SFK members are expressed primarily in cells of hematopoietic origin, recent studies have shown that Lyn, Fyn, and Fgr are expressed and activated in epithelial-derived cancers (14-17).
Because SFKs plays a central role in the regulation of numerous protumorigenic pathways, the development of pharmacologic inhibitors targeting the Src pathway is an active area of investigation. Clinical trials are underway testing Src inhibitors in solid tumors, including BMS-354825 (dasatinib; Bristol-Myers Squibb), bosutinib (SKI-606, Wyeth; ref. 18), and AZD0530 (saracatinib; AstraZeneca; ref. 19). Dasatinib is approved by the U.S. Food and Drug Administration (FDA) for patients with imatinib-resistant chronic myelogenous leukemia (CML) and Philadelphia chromosome–positive acute lymphoblastic leukemia. Because inhibition of Src has the potential to inhibit the development and progression of metastases, Src inhibitors are being further investigated as antimetastatic agents in both adjuvant and treatment settings (20, 21).
Src signaling and the efficacy of SFK pathway inhibition has not been well studied in advanced thyroid cancer, and no studies have addressed the role of this pathway in thyroid cancer metastasis. In one previous study, FAK protein was overexpressed in a subset of PTC and ATC, but the phosphorylation status of FAK was not examined (22). We were the first to show that FAK is phosphorylated on a well-characterized Src-dependent site (Y861) in a subset of PTC patient tumor samples, and we predict that these tumors will be more aggressive and/or responsive to Src-directed therapies (23). The goals of this study were to evaluate the effects of Src inhibition using the clinically available Src inhibitor, dasatinib, on thyroid cancer growth and metastasis in vitro and in vivo.
Materials and Methods
Cell culture and generation of stable cell lines
Human thyroid cancer cell lines C643, TPC1, SW1736, BCPAP, K1, 8505C, HTh74, and HTh7 were authenticated by short tandem repeat profiling, as previously described (24). Cells were grown in RPMI (Invitrogen) containing 5% FBS (HyClone) and maintained at 37°C in 5% CO2 (24). BCPAP cells were transduced with pBABE-hygro, pBABE-WT-c-Src (Addgene plasmid 26983), or pBABE-c-Src-Dasatinib-Resistant-T338I (Addgene plasmid 26980) retrovirus, and selected with hygromycin (0.1 mg/mL; ref. 25).
Cellular growth assays
Cells (500/well for TPC1 and K1; 1,000/well for 8505C; 1,500/well for C643; and 2,000/well for BCPAP, SW1736, HTh74, and HTh7) were plated in triplicate in 96-well plates, and 24 hours later, cells were treated with increasing doses of dasatinib (BMS-354825; Bristol-Myers Squibb), as indicated. Cell growth was analyzed using the sulforhodamine B (SRB) assay (26). Briefly, after 72 hours of drug exposure, cells were fixed with 10% trichloroacetic acid at 4°C, and stained with 0.057% SRB (Sigma), after which unbound stain was removed by washing with 1% acetic acid. Protein-bound SRB was solubilized with 10 mmol/L unbuffered Tris base, and optical density was measured at absorbance wavelength of 570 nm.
Cell-cycle analysis
Subconfluent cells were treated with vehicle (dimethyl sulfoxide, DMSO), 10 or 50 nmol/L dasatinib in RPMI containing 5% FBS for 48 hours, as previously described (23, 27). Cell pellets were collected and stained with saponin/propidium iodide (PI), and cell-cycle distribution was determined as previously described (23, 27).
Apoptosis assays
Cells were plated in duplicate in white-walled 96-well plates, and 24 hours later treated with vehicle (DMSO), 10 or 50 nmol/L dasatinib for 24 hours in media supplemented with 0.1% FBS. Caspase-3/7 activities were measured using a luminescent CaspaseGlo-3/7 Assay (Promega).
Soft agar colony formation
Cells (104) were suspended in 0.3% agar with complete media and plated on a base layer of 0.64% agar (Difco Agar Noble, BD Biosciences) in 6-well dishes. Cells were treated with vehicle (DMS0), 10 or 50 nmol/L dasatinib, and media containing vehicle or drug was replenished every 3 to 4 days for a total of 20 days. Colonies were stained with nitroblue tetrazolium chloride (Amresco; 5 mg/mL) and incubated overnight at 37°C to develop the stain. Colonies were counted using ImageJ software.
Western blotting
Cells were treated with the indicated doses of dasatinib or vehicle (DMSO) and harvested in CHAPS lysis buffer containing 10 mmol/L CHAPs, 50 mmol/L Tris (pH 8.0), 150 mmol/L NaCl, and 2 mmol/L EDTA with 10 μmol/L Na3VO4 and 1× protease inhibitor cocktail (Roche). Protein extracts were resolved on 8% or 10% SDS-PAGE gels and transferred to Immobilon-P membranes (Millipore), as previously described (23, 27). Membranes were incubated at 4°C overnight with the following antibodies: p-Y416-SFK, SFK, Lyn, Fyn, Fgr, Yes, c-Src, p-Y710-Stat3, Stat3, p-Y410-p130Cas, p-Y118-paxillin, paxillin, pS473-Akt, Akt, FAK (Cell Signaling); p-Y861-FAK (Invitrogen); pp-ERK1/2, total ERK2 (Santa Cruz); or α-tubulin (CalBiochem) diluted in 5% nonfat dry milk or 5% bovine serum albumin in 20 mmol/L Tris, pH 7.4, 138 mmol/L NaCl, 0.1% Tween (TBST). Blots were incubated with secondary goat anti-rabbit or goat anti-mouse horseradish peroxidase–conjugated antibodies (GE Healthcare) and proteins were detected by enhanced chemiluminescence (ECL) detection (Pierce; refs. 23, 27).
Immunoprecipitation
Cells were lysed in RIPA buffer [10 mmol/L Tris (pH7.4), 100 mmol/L NaCl, 1 mmol/L NaF, 1 mmol/L EDTA, 1 mmol/L EGTA, 0.1% SDS, 10% glycerol, 0.5% sodoim deoxycholate, 1% Triton X-100, 1 mmol/L PMSF, 1× protease inhibitors, 2 mmol/L Na3VO4]. Equal amounts of protein (500 μg) were incubated with 100 ng of c-Src or Lyn antibody (Cell Signaling) or control IgG overnight at 4°C with 15 μL of beads. Immunoprecipitates were washed with RIPA, resolved on 10% SDS-PAGE gels, and detected using the ImmunoCruz IP/WB optima F system (Santa Cruz) with the indicated primary antibodies and ECL as described earlier.
Orthotopic murine model
BCPAP and 8505C cells (5 × 105) engineered to express a luciferase-IRES-GFP plasmid were injected into the right thyroid lobe of nude mice, as previously described (28-30). Briefly, male athymic nude mice (NCI; ~30 grams; 10–12 weeks old) were anesthetized with tribromoethanol (250 mg/kg). Thyroid cancer cells (5 μL cell suspension) were injected into the right thyroid gland with the aid of a dissecting microscope (Nikon SMZ645), and the skin closed with staples. Tumor establishment and progression was measured weekly by detection of bioluminescence with the Xenogen IVIS200 system (Caliper) in the UCCC Small Animal Imaging Core. Briefly, mice were injected with d-luciferin (3 mg in 200 μL), and bioluminescence activity (photons/s) was quantitated using the Living Image 2.60.1 software (Igor Corp). Minimum/maximum thresholds were normalized to compare images using the same scale. Final thyroid tumor size was measured with calipers and volume was calculated using the following formula: tumor volume = (length × width × height)/0.5236.
Experimental metastasis model
Male athymic nude mice (Harlan; 25 grams; 5-week old) received d-luciferin (3 mg) via intraperitoneal injection, and 5 minutes later each mouse was anesthetized using isofluorane. BCPAP-luc-IRES-GFP cells (105 cells in 100 μL PBS) were injected into the left ventricle of nude mice using a 26-gauge needle, as previously described (31). Successful left ventricle injection was monitored by the pulsatile flow of red blood into the needle hub indicating correct placement, and by whole body bioluminescence immediately following injection. Metastatic progression was monitored weekly by IVIS imaging. Mice were sacrificed if they lost more than 20% body weight and based on moribund criteria. All animal studies were conducted in accordance with the animal protocol procedures approved by the Institutional Animal Care and Use Committee at the University of Colorado Denver.
Drug preparation and administration
For in vivo studies, dasatinib (50 mg/kg) was prepared for daily oral gavage (5 d/wk) in 80 mmol/L sodium citrate buffer, pH 3.0. For the orthotopic murine model, mice were randomized on day 10 based on bioluminescence activity to receive drug or vehicle. In the metastatic murine model, mice received dasatinib or vehicle, as described earlier, starting 2 days before intracardiac injection (pretreatment), or on day 11 following randomization (posttreatment).
Results
Effects of dasatinib on thyroid cancer cell growth, apoptosis, and transformation
To study the role of SFK signaling in thyroid cancer, a panel of 8 thyroid cancer cell lines representing distinct thyroid tumor types (PTC and ATC) and clinically relevant oncogenic mutations (BRAF, RAS, RET/PTC1, PIK3CA; Table 1 and ref. 24) was used to study sensitivity to dasatinib (32). Figure 1A shows that treatment with increasing doses of dasatinib (0.019–1.25 μmol/L) for 3 days inhibited the growth of the C643, TPC1, BCPAP, and SW1736 cell lines by approximately 50% at low nanomolar concentrations, whereas higher concentrations were required to inhibit the growth of the K1 cell line. In contrast, the 8505C, HTh7, and HTh74 cell lines were not inhibited more than 50% by dasatinib treatment, even at doses greater than 1 μmol/L (Fig. 1A). IC50 values were calculated (Table 1), and cell lines exhibiting IC50 values of <100 nmol/L were considered sensitive to dasatinib, and include the TPC1, SW1736, BCPAP, and C643 cells with IC50 values between 30 and 80 nmol/L. The K1 cell line exhibited intermediate sensitivity (IC50 = 400 nmol/L), and the remaining cell lines (8505C, HTh74, HTh7) were resistant to dasatinib with IC50 values > 1.6 μmol/L (Table 1). The 100 nmol/L “sensitive” demarcation was chosen based on previous studies showing decreased selectivity of dasatinib at doses greater than 100 nmol/L (32, 33). Overall, we observed similar sensitivity to inhibition of Src with dasatinib and saracatinib in our previous study (23), indicating an important role for SFKs in thyroid cancer cell growth.
Table 1.
Antiproliferative effects of the Src inhibitor, dasatinib
| Cell line | Tumor type | Mutation | Dasatinib IC50, μmol/L |
|---|---|---|---|
| C643 | ATC | HRAS | 0.09 |
| TPC1 | PTC | RET/PTC1 | 0.03 |
| BCPAP | PTC | BRAF | 0.04 |
| SW1736 | ATC | BRAF | 0.08 |
| K1 | PTC | BRAF/PIK3CA | 0.4 |
| 8505C | ATC | BRAF | 2.7 |
| HTh74 | ATC | WT | >5 |
| HTh7 | ATC | NRAS | 1.6 |
NOTE: Thyroid cancer cells were treated with increasing doses of dasatinib for 3 days, as described in Fig. 1. IC50 values were calculated using nonlinear regression analysis with a slope sigmoidal dose–response curve in GraphPad Prism. No highlighting and dark gray highlighting indicates sensitive and resistant cell lines, respectively, and light gray highlighting indicates intermediate sensitivity.
Figure 1.
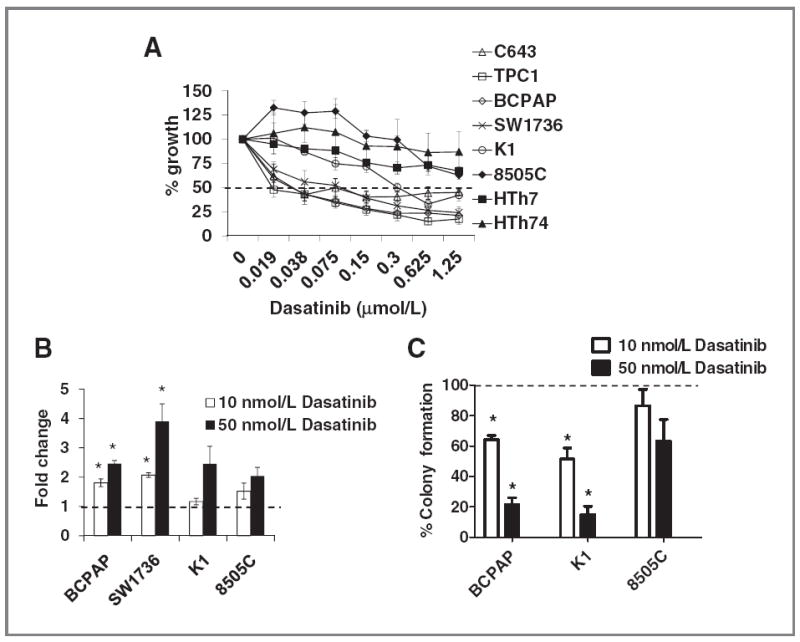
Effects of dasatinib on thyroid cancer growth, survival, and transformation. A, thyroid cancer cells were treated with the indicated doses of dasatinib or vehicle for 3 days and growth was measured using the SRB assay (26). Growth of the vehicle-treated cell was set to 100% (not shown). Results are the mean percent inhibition compared with vehicle-treated cells ± SEM of 3 experiments carried out in triplicate. The dashed line represents 50% growth inhibition. Significant inhibition of growth by one-way ANOVA was observed in the C643, TPC1, BCPAP, SW1736 (P < 0.0001), K1 (P = 0.001), and HTh7 (P = 0.01), although more than 50% growth inhibition was not observed in the HTh7 cells. B, cells were plated in RPMI containing 0.1% FBS with or without the indicated dose of dasatinib or vehicle for 24 hours, and caspase-3/7 activity was determined using the caspase-3/7 Glo assay, as described in Materials and Methods. Caspase-3/7 activity of the vehicle-treated cells was set to 1 and is represented by the dashed line. Results are the mean ± SEM of 3 experiments carried out in duplicate (*, P < 0.05). C, BCPAP, K1, and 8505C cells were plated in soft agar, and 1 day later, cells were treated with 10 or 50 nmol/L dasatinib or vehicle. Media with or without drug was replaced every 3 to 4 days for a total of 20 days. Number of colonies was counted using the ImageJ software. Colony formation of the vehicle-treated cells was set to 100% and is indicated by the dashed line. Percent colony formation of the dasatinib-treated cells compared with vehicle is shown. Results are the mean ± SEM of 3 experiments carried out in duplicate (*, P < 0.008).
To examine the mechanisms of inhibition of cell growth, cell-cycle progression was evaluated in representative dasatinib-sensitive and dasatinib-intermediate cell lines (BCPAP, SW1736, K1), as well as dasatinib-resistant cell lines (8505C, HTh7, HTh74). Table 2 shows that treatment with 10 or 50 nmol/L dasatinib resulted in an approximately 9% to 22% increase of cells in the G1 population in the BCPAP, SW1736, and K1 cells, and a corresponding 7% to 18% decrease in the percentage of cells in S-phase. In contrast, dasatinib treatment of the resistant 8505C, HTh7, and HTh74 cell lines resulted in a 0% to 10% increase in cells in the G1 population (Table 2), consistent with the resistant nature of these cells (Fig. 1A). To further explore the mechanism of growth inhibition by dasatinib, induction of apoptosis was also tested. Figure 1B shows a higher induction of caspase-3/7 activity in the dasatinib-sensitive cell lines (BCPAP and SW1736; ~2- to 4-fold; P < 0.05), whereas the dasatinib-intermediate (K1) and dasatinib-resistant (8505C) cells were less sensitive to dasatinib treatment, with a 1.2- to 2.5-fold induction of caspase-3/7 activity with the 10 and 50 nmol/L doses, respectively. Overall, these data indicate that cell cycle and apoptosis may be important mechanisms mediating growth inhibition in the sensitive cell lines but that different dominant mechanisms are present in different cell lines.
Table 2.
Effects of the Src inhibitor, dasatinib, on the cell cycle
| Vehicle (control)
|
Dasatinib, 10 nmol/L
|
Dasatinib, 50 nmol/L
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| G1 | S | G2–M | G1 | S | G2–M | G1 | S | G2–M | |
| BCPAP | 64.8 | 26.5 | 8.7 | 73.4 | 18.5 | 8.1 | 87.2 | 9.3 | 3.4 |
| SW1736 | 77.5 | 16.9 | 5.6 | 86.0 | 10.2 | 3.7 | 88.5 | 6.9 | 4.6 |
| K1 | 70.7 | 21.1 | 8.2 | 79.7 | 14.5 | 5.8 | 87.4 | 10.2 | 2.4 |
| 8505C | 58.6 | 28.9 | 12.5 | 58.5 | 30.1 | 11.4 | 65.5 | 23.1 | 11.4 |
| HTh7 | 61.1 | 26.8 | 12.2 | 60.8 | 27.0 | 12.2 | 67.2 | 23.9 | 8.9 |
| HTh74 | 65.3 | 22.5 | 12.2 | 71.1 | 19.0 | 9.9 | 75.1 | 17.3 | 7.6 |
NOTE: Cells were treated with the indicated dose of dasatinib or vehicle for 48 hours. Cell-cycle distribution was determined using the PI/saponin method. Results are the mean of 2 to 4 experiments.
To determine whether Src inhibition with dasatinib affects anchorage-independent growth, PTC and ATC cells exhibiting high (BCPAP), intermediate (K1), and low (8505C) sensitivity to dasatinib were tested. Figure 1C shows that treatment with 10 or 50 nmol/L dasatinib results in a dose-dependent decrease in colony formation in the dasatinib-sensitive BCPAP cells (36%–78% inhibition; P < 0.008), and dasatinib-intermediate K1 cells (49%–85% inhibition; P < 0.008). Colony formation of the 8505C cells, which were resistant to inhibition of growth with dasatinib (Fig. 1A; Table 1), were also not significantly inhibited under nonadherent conditions (Fig. 1C).
Dasatinib inhibition of SFK and FAK signaling in thyroid cancer cells
We next evaluated inhibition of activated SFKs and known downstream targets, in response to dasatinib treatment, in 4 PTC and ATC cell lines exhibiting high (BCPAP, SW1736), intermediate (K1), or low (8505C) sensitivity to dasatinib. Figure 2A shows that basal, endogenous SFK activity and expression is similar in all cell lines, and that treatment with dasatinib results in variable inhibition of SFK activity at the 1 to 10 nmol/L doses, and complete inhibition at 50 nmol/L dasatinib. Similar results were observed at earlier time points (4 hours, data not shown).
Figure 2.
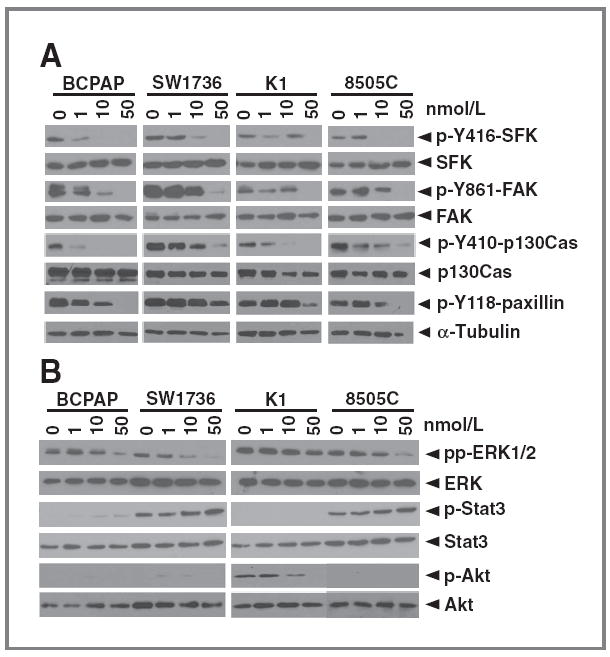
Effects of Src inhibition with dasatinib on cell signaling. Cells were treated with the indicated doses of dasatinib or vehicle for 24 hours. Cells were lysed in CHAPS buffer and equal protein (30 μg) was analyzed by Western blotting on 8% SDS-PAGE gels with the indicated antibodies. Blots shown are representative of at least 3 separate experiments.
FAK phosphorylation on the well-characterized Src-dependent site, tyrosine residue 861 (Y861), was also evaluated. Figure 2A shows similar levels of basal p-Y861-FAK in all 4 cell lines, with lower levels in the K1 and 8505C cells. Similar to p-Y416-SFK, dasatinib treatment resulted in decreased, but variable, p-Y861-FAK levels at 1 to 10 nmol/L doses of dasatinib, and complete inhibition at 50 nmol/L dasatinib in all cell lines (Fig. 2A).
p130Cas and paxillin are key downstream targets of the Src-FAK complex that function as adaptor proteins to promote tumor cell survival, proliferation, and migration (34-36). We therefore evaluated levels of p-Y410-p130Cas and p-Y118-paxillin and inhibition by dasatinib. Figure 2A shows elevated levels of p-Y410-p130Cas and p-Y118-paxillin are present in PTC and ATC cells and that treatment with increasing doses of dasatinib results in the inhibition of both p-p130Cas and p-paxillin. Taken together, these data indicate that dasatinib effectively inhibits the Src-FAK-p130Cas-paxillin pathway in thyroid cancer cells and that inhibition of this pathway does not distinguish sensitive from resistant thyroid cancer cells.
Effects of dasatinib treatment on ERK1/2, Stat3, and Akt signaling
Given the importance of the MAPK pathway in thyroid cancer, we evaluated the interaction of the Src pathway with MAPK in a panel of 4 thyroid cancer cell lines harboring oncogenic mutations in the MAPK pathway. Figure 2B shows that inhibition of Src with dasatinib does not inhibit MAPK (pp-ERK1/2) at low doses (1 or 10 mol/L), consistent with our previous studies using saracatinib (23). However, partial, but variable inhibition of pp-ERK1/2 was observed at higher doses of dasatinib (50 nmol/L; Fig. 2B and data not shown), suggesting that treatment with dasatinib may affect the oncogenic MAPK signaling in thyroid cancer.
We also evaluated the effects of SFK inhibition on the phosphorylation of Stat3 and Akt signaling, which have been associated with resistance to Src inhibitors, and promote survival in other tumor models (37-40). Figure 2B shows that p-Stat3 was not detected in the PTC-derived BCPAP and K1 cell lines, whereas elevated levels were detected in the ATC-derived SW1736 and 8505C cells. Interestingly, no significant effects of dasatinib were observed on p-Stat3 levels. Figure 2B shows that p-Akt levels are low in BRAF-mutant thyroid cancer cells (Fig. 2B; BCPAP, SW1736, 8505C), whereas elevated levels of p-Akt are present in the BRAF/PIK3CA-mutant K1 cells, where treatment with higher doses of dasatinib (50 nmol/L) were required to inhibit p-Akt (Fig. 2B). Taken together, these data indicate that Stat3 and Akt signaling are not major targets of Src in thyroid cancer.
Expression of Src family kinases in thyroid cancer cells
Because dasatinib has the potential to inhibit all SFKs (41), we evaluated expression of specific SFKs expressed in thyroid cancer cells, which has not been previously characterized. Figure 3A shows that c-Src and Lyn are the only family members expressed, whereas Fyn, Yes, Lck, and Fgr were not detected. To determine whether c-Src and Lyn are activated in thyroid cancer cells, extracts were prepared from cells treated with or without dasatinib, and c-Src or Lyn were immunoprecipitated using anti-c-Src or anti-Lyn–specific antibodies, followed by Western blotting to detect activation of c-Src and Lyn using the p-Y416SFK antibody, which cross-reacts with the activated and autophosphorylated form of c-Src (p-Y416) and Lyn (p-Y397). Figure 3B (top) shows that equivalent levels of total c-Src are immunoprecipitated in BCPAP cells treated with or without dasatinib. Activated p-Y416Src is specifically detected in the c-Src immunoprecipitates, and p-Y416Src is inhibited by dasatinib treatment (Fig. 3B, top). Lyn was also efficiently immunopreciptated, but phosphorylation of Lyn was not detected in the Lyn immunoprecipitations (Fig. 3B). Similar results were observed in the 8505C cells (Fig. 3B, bottom). Taken together, these data indicate that c-Src is the predominant SFK activated in representative sensitive (BCPAP) and resistant (8505C) thyroid cancer cells.
Figure 3.
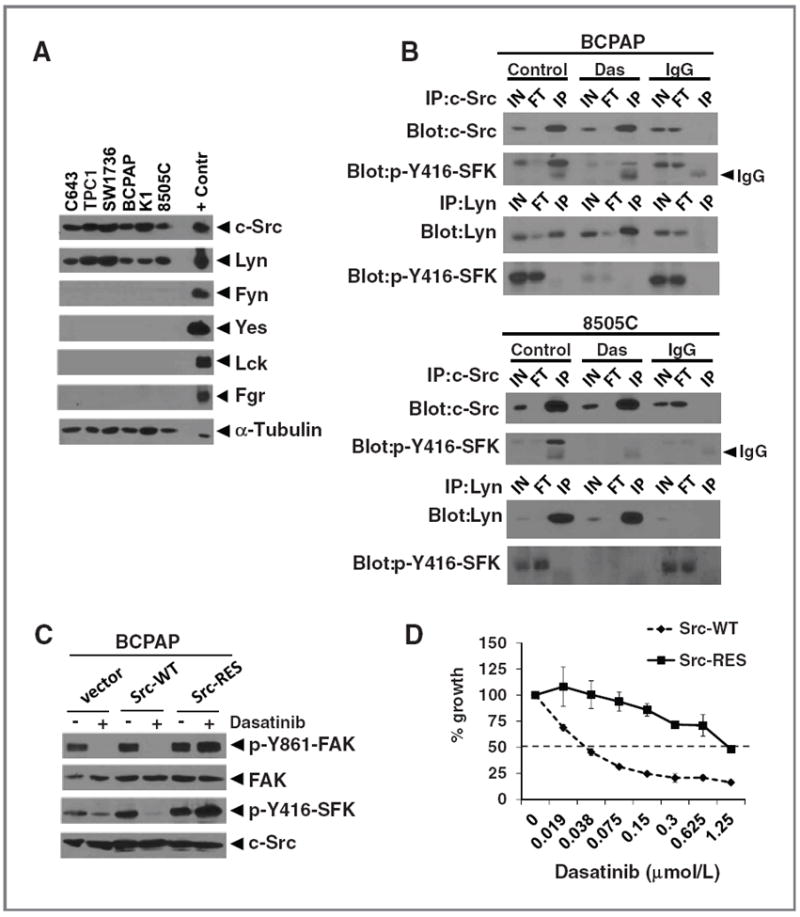
Src family kinase expression in thyroid cancer cells and effects of a Src gatekeeper mutant. A, expression of the indicated SFKs was measured by Western blot analysis in thyroid cancer cells (30 μg extract each), as described in Fig. 2. The following positive control extracts (10 μg each) were loaded for the indicated antibody: Jurkat (c-Src, Fyn, Lck), Raji (Lyn, Fgr), and U87MG(Yes). α-Tubulin was used as a loading control. B, cells were treated with or without dasatinib (Das, 50 nmol/L) for 4 hours and cell extracts (500 μg) were immunoprecipitated (IP) with anti-c-Src, anti-Lyn, or rabbit IgG. Immunoprecipitates were analyzed by Western blot analysis with the indicated antibodies. The p-Y416-SFK antibody cross-reacts with the activation site of c-Src (Y416) and Lyn (Y397). Results shown are representative blots of at least 3 experiments. C, BCPAP cells were transduced with retroviruses expressing wild-type Src (Src-WT), the T338I gatekeeper mutant Src (Src-RES), or empty vector (pBABE-hygro), and selected with hygromycin for 7 days before experimental manipulation. Cells were treated with dasatinib (50 nmol/L) or vehicle (DMSO) for 24 hours and cell extracts (30 μg) were analyzed by Western blot analysis with the indicated antibodies. D, BCPAP cells expressing Src-WT or Src-RES were plated in 96 wells. The following day, cells were treated with the indicated concentrations of dasatinib or vehicle (DMSO) for 3 days and growth was measured using the SRB assay as described earlier. The growth of the vehicle-treated cells was set to 100% (not shown). Results shown are the mean percent inhibition compared with vehicle-treated cells, ± SD from 3 experiments carried out in triplicate. Inhibition of growth by 50% is represented by the dashed line.
In addition to its role as a potent SFK inhibitor, dasatinib is a multikinase inhibitor (32). To test the specific role of c-Src in response to dasatinib treatment, we tested the ability of a c-Src inhibitor-resistant transgene to rescue the effects of dasatinib on signaling and cell growth. For these studies, we took advantage of the gatekeeper residue of Src (T338), which is required for dasatinib binding, to test whether expression of this dasatinib-resistant mutant rescues cells from dasatinib treatment (25, 42, 43). The c-Src dasatinib-resistant mutant (T338I, herein referred to as Src-RES), wild-type Src (Src-WT), or empty vector (pBABE-hygro) were stably expressed in the Src inhibitor–sensitive BCPAP cells at near physiologic levels (~1.5-fold above endogenous c-Src, Fig. 3C and data not shown). As expected, dasatinib treatment inhibits p-Y416SFK and p-Y861-FAK in cells expressing empty vector (pBABE-hygro) or Src-WT (Fig. 3C). In cells expressing Src-RES, p-Y416SFK levels were not inhibited by dasatinib treatment, indicating that the gatekeeper residue, Thr 338, is necessary for inhibition of Src by dasatinib. Similarly, inhibition of p-Y861-FAK by dasatinib was also blocked in cells expressing the Src-RES mutant, consistent with this residue being a major phosphorylation site targeted by Src (Fig. 3C).
We next tested the c-Src–dependent effects of dasatinib on growth of the BCPAP cells (Fig. 3D). As expected, we observed a dose-dependent decrease of growth in Src-WT expressing cells, with an IC50 value of 42 nmol/L, consistent with the response of parental (nontransduced) and empty vector–transduced cells (40 nmol/L; Table 1). Notably, expression of the Src-RES gatekeeper mutant blocked the growth inhibitory effects of dasatinib, shifting the IC50 value to 1,200 nmol/L (Fig. 3D). Similar results were observed in the SW1736 cells (data not shown). Thus, these data provide strong evidence that c-Src is the primary mediator of cell growth in response to Src inhibitor treatment in thyroid cancer cells.
Inhibition of Src with dasatinib inhibits PTC growth in a preclinical orthotopic mouse model
We next evaluated the in vivo antitumor effects of Src inhibition in an orthotopic thyroid cancer model using the BCPAP cells (sensitive to dasatinib in vitro) and 8505C cells (resistant to dasatinib in vitro). BCPAP or 8505C cells stably expressing a luciferase-IRES-GFP plasmid were injected into the right thyroid gland of athymic nude mice, and tumor establishment and progression were monitored by measuring luciferase activity using the Intravital Imaging System (IVIS). Daily treatment with dasatinib (50 mg/kg) was initiated on day 10. Using this approach, a significant inhibition of BCPAP orthotopic tumor growth was observed 6 days after treatment (day 16, P = 0.014), which was sustained through days 23 and 29 (P = 0.0003), compared with vehicle-treated mice (Fig. 4A, left). Representative bioluminescence images of orthotopic tumors are shown in Fig. 4B. The BCPAP orthotopic final tumor volumes were inhibited by more than 90% in response to dasatinib treatment (P = 0.0014; mean vehicle tumor volume = 289 mm3; mean dasatinib tumor volume = 11 mm3; Fig. 4C left). Strikingly, 3 of 8 dasatinib-treated BCPAP orthotopic mice did not have measurable tumor after therapy. In contrast, the growth of 8505C-derived tumors was not significantly inhibited by dasatinib treatment at any time point, as measured by bioluminescence imaging (Fig. 4A, right), and final tumor volumes (P = 0.8; Fig. 4C, right). Thus, these data validate our in vitro findings and show that inhibition of Src with dasatinib strongly inhibits thyroid tumor growth in vivo.
Figure 4.
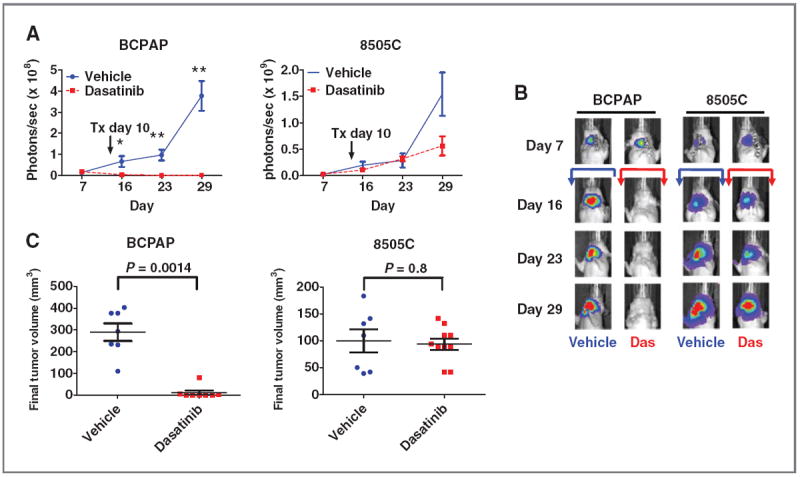
Inhibition of thyroid cancer growth by dasatinib in an orthotopic mouse model. A, BCPAP-luc PTC or 8505C-luc ATC cells (5 × 105) were injected into the right thyroid gland of nude mice and tumor establishment and growth were monitored by IVIS imaging. Dosing by daily oral gavage with dasatinib (50 mg/kg) or vehicle was started on day 10 and continued for the duration of the experiment. Data are expressed as the mean ± SEM area flux (photons/s) of the neck region at the indicated time points (*, P = 0.01; **, P = 0.0003). B, representative bioluminescence images are shown for each time point. The arrows indicate vehicle (blue) or dasatinib (red) treatment, which started at day 10. C, final tumor volume (mm3) on day 29 is shown. Data points represent tumor volumes of individual animals, and horizontal bars represent the mean tumor volume ± SEM (n = 7–8 animals per group for the BCPAP cells and 7–10 animals per group for the 8505C cells). BCPAP mean vehicle tumor volume = 289 mm3; BCPAP mean dasatinib tumor volume = 11 mm3. 8505C mean vehicle tumor volume = 98 mm3; 8505C mean dasatinib tumor volume = 94 mm3. Statistical analysis was conducted using the Student t test.
Inhibition of Src with dasatinib blocks PTC metastasis
One of the most lethal characteristics of thyroid cancer is the metastatic spread to distant sites, especially to the bone. The orthotopic model has resulted in limited distant metastases, and to date, there have been no bone metastasis models in thyroid cancer. We have therefore established an experimental metastasis model using an intracardiac injection approach, which allows for the widespread dissemination of tumor cells. Using this model system, we tested the Src inhibitor–sensitive BCPAP (PTC) cells, which have formed consistent distant metastases with a 70% to 90% take rate. For these studies, BCPAP-luc-IRES-GFP cells were injected into the left ventricle of athymic nude mice. Successful left ventricle injection was monitored by the spontaneous entrance of pulsatile blood into the hub of the syringe and by bioluminescence imaging immediately after injection to visualize the widespread distribution of luciferase expressing tumor cells (Fig. 5A). Representative histologic images of bone metastases are shown in Supplementary Fig. S1.
Figure 5.
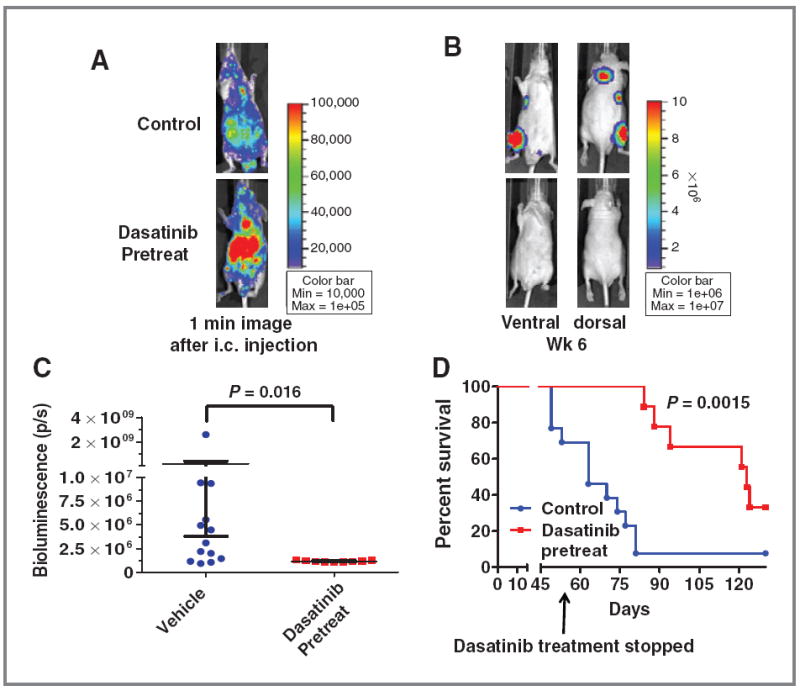
Inhibition of metastasis by adjuvant dasatinib treatment in an experimental metastasis model. Mice were pretreated with dasatinib (50 mg/kg) or vehicle by daily oral gavage 2 days before intracardiac (i.c.) injection of BCPAP cells (1 × 105/100 μL) into the left ventricle of athymic nude mice. A, representative images of vehicle or dasatinib-treated mice obtained by IVIS imaging 1 minute after i.c. injection shows widespread distribution of cancer cells. B, representative images of metastases 6 weeks post-i.c. injection. C, total bioluminescence activity (photons/s) at week 6 is shown for the vehicle and dasatinib-pretreated mice. Data points represent whole body bioluminescence activity of individual animals. Horizontal bars represent the mean ± SEM, n = 9–13 per group. Statistical analysis was conducted using the Student t test. D, Kaplan–Meier curve representing overall survival of vehicle versus dasatinib-pretreated mice. Dasatinib treatment was stopped at day 56, and the survival experiment was stopped at day 130. Statistical analysis was conducted using the log-rank test.
To determine the role of Src in metastasis, we first used an adjuvant treatment approach, in which mice were given vehicle or dasatinib (50 mg/kg) by daily oral gavage 48 hours before intracardiac injection of cancer cells. Using this approach, we observed an approximately 70% take rate in the vehicle-treated mice, compared with the pretreatment group where none of the mice developed distinct metastatic tumors, even after 6 weeks (Fig. 5C). Representative bioluminescence images of metastatic tumors are shown in Fig. 5B. Treatment with dasatinib was stopped on day 56, and Fig. 5D shows that dasatinib-treated mice had a significantly increased overall survival when compared with vehicle treated mice, with a mean survival of 63 days for the vehicle-treated mice versus 123 days for the dasatinib-treated mice (Fig. 5D; P = 0.0015). Next, to determine the effects of dasatinib treatment on established metastases, these studies were repeated and mice were treated with dasatinib (50 mg/kg) after the establishment of metastases on day 11 (Supplementary Fig. S2A). Supplementary Figure S2C shows that dasatinib treatment resulted in a sustained inhibition of metastatic tumor growth starting approximately 10 days after treatment (2-way ANOVA; P = 0.004). Representative images, showing inhibition of metastasis at week 7, are shown (Supplementary Fig. S2B). Overall, these results provide the first demonstration for Src signaling in thyroid cancer metastasis and indicate that inhibition of Src with dasatinib represents a promising therapeutic strategy to block both primary and metastatic tumor growth in patients with thyroid cancer.
Discussion
Little is known about the role of Src and FAK and the efficacy of SFK inhibitors in thyroid cancer. Our group was the first to show that elevated levels of phospho-SFK and FAK are present in thyroid cancer cell lines, and that phospho-FAK is present in a subset of PTC tumor samples (23). We further showed that inhibition of Src with the more selective Src inhibitor, saracatinib, inhibits the growth and invasion of thyroid cancer cell lines expressing elevated levels of phospho-FAK (23). In the current study, we have further tested the role of SFK signaling in thyroid cancer using dasatinib, which is a potent ATP-competitive inhibitor of Src, which is FDA-approved for the treatment of patients with leukemia, and is being tested in phase II clinical trials for several solid tumors (20). Here, we show that Src inhibition with dasatinib results in the inhibition of thyroid cancer cell growth and transformation in vitro, as well as primary and metastatic tumor progression in vivo, providing strong rationale for the use of Src inhibitors in patients with advanced thyroid cancer.
In this study, we identified cells with high, moderate, and low sensitivity to dasatinib in vitro and found that treatment with dasatinib blocks SFK and FAK signaling. Interestingly, we did not find a clear correlation between basal levels of these signaling molecules, or inhibition of these molecules by dasatinib with sensitivity to dasatinib, although phospho-FAK levels may be higher in the sensitive cell lines (Fig. 2A). The lack of correlation between phospho-SFK levels and sensitivity to dasatinib is similar to our previous studies using the Src inhibitor, saracatinib, as well studies in other tumor types, and suggest that even low levels of SFK activity are sufficient for these biologic responses (23, 44, 45). We also evaluated Stat3 and Akt signaling, which have been shown to promote survival and mediate resistance to Src inhibitor treatment in other tumor types (37-40). Importantly, we did not observe regulation of p-Stat3 in response to dasatinib treatment, and although p-Akt was inhibited with higher concentrations of dasatinib in the BRAF/PIK3CA-mutant K1 cells, low to undetectable levels of p-Stat3 and p-Akt were observed in both sensitive and resistant cell lines, indicating that these prosurvival pathways are likely not major targets of Src signaling in thyroid cancer. Finally, we found that dasatinib treatment inhibited pp-ERK1/2 at higher concentrations, but that this inhibition was variable, suggesting that the MAPK pathway is not a major target of Src in thyroid cancer cells. Accordingly, we did not identify any correlation between known oncogene mutations (BRAF, RAS, PIK3CA, RET/PTC1) and sensitivity to dasatinib (Table 1), consistent with our previous studies with the more selective Src inhibitor, saracatinib (23), as well as other tumor types (33, 40). Thus, additional studies are needed to identify predictive biomarkers of response to determine which patients will benefit the most from Src-directed therapies.
Although dasatinib is a multikinase inhibitor, in addition to a potent inhibitor of Src, our data show a strong correlation between the sensitivity of thyroid cancer cells to dasatinib (Fig. 1) and the more selective Src inhibitor, saracatinib (23), indicating an important role for Src in thyroid cancer cell growth, similar to one previous study in thyroid cancer (46). We have further shown the inhibitory effects of dasatinib are accompanied by inhibition of the Src pathway at low nanomolar concentrations (Fig. 2A), indicating these effects are likely mediated by Src and FAK signaling. To identify the specific SFK important in thyroid cancer, we characterized the expression of 6 SFK members in thyroid cancer cells, and show that c-Src and Lyn are the predominant members expressed (Fig. 3A). Interestingly, we found that whereas both c-Src and Lyn are expressed, c-Src is the predominant SFK phosphorylated (Fig. 3B). Because dasatinib can inhibit all Src family kinase members, as well as other kinases, we further tested the role of c-Src in the response to dasatinib using a gatekeeper approach. These studies showed expression of the c-Src gatekeeper mutant (Src-RES) rescues dasatinib-mediated inhibition of thyroid cancer cell growth, as well as inhibition of Src signaling, providing strong evidence that c-Src is the primary mediator of growth in response to dasatinib treatment (Fig. 3D). While these results do not prove Lyn has no effect, these data strongly support c-Src as the dominant if not only mediator of growth in response to Src inhibitors in thyroid cancer cells. These results are in contrast to recent studies in prostate and breast cancer, where both c-Src and Lyn are expressed and activated, and shown to mediate distinct biologic responses (15, 16, 47).
A number of studies in other tumor types, including colon cancer and NSCLC have shown that Src primarily regulates invasion and metastasis (33, 48, 49). Our studies show that treatment with dasatinib not only blocks transformation and metastatic progression, but also inhibits thyroid cancer cell growth. These results indicate that the use of Src inhibitors may have higher therapeutic efficacy in thyroid cancer, due to the potential to inhibit primary tumor growth (Fig. 4), in addition to metastatic outgrowth (Fig. 5; Supplementary Fig. S2).
Distant metastasis to the bone represents one of the most lethal characteristics of thyroid cancer, and there are currently no effective therapeutic options for these patients. Because of the role of Src signaling in bone metastases in other tumor types, and the importance of distant metastases in thyroid cancer, we developed an experimental metastasis model using an intracardiac injection approach. This is the first report describing the establishment of a thyroid cancer metastasis model to bone. Using a pretreatment, adjuvant approach, we found that while approximately 70% of mice developed metastases in the vehicle-treated control group, no mice developed metastases in the dasatinib-pretreated group after 6 weeks of treatment (Fig. 5). We found that although these mice exhibited a significantly longer survival of approximately 60 days than vehicle-treated mice, even after discontinuation of dasatinib (Fig. 5D), the majority of these mice developed tumors. To determine whether inhibition of Src could block the progression of established metastases, we used a post-treatment approach, where mice were treated with dasatinib after metastatic tumor establishment (Supplementary Fig. S2). Using this approach, dasatinib treatment significantly inhibited metastatic tumor progression by approximately 10-fold (Supplementary Fig. S2). Taken together, these results indicate that Src activity is important for tumor cell growth and/or survival, rather than early metastatic cell seeding. These results are consistent with our in vitro growth data (Fig. 1) as well as previous studies in prostate and breast cancer (16, 25). Thus, the precise role of Src in the progression of latent metastases, which is an important clinical problem in thyroid cancer (50), will be of particular interest to test in future studies. Collectively, these studies indicate that Src inhibitors will likely need to be used long-term or in combination with other therapies to achieve complete tumor remission, but nonetheless, these data provide a strong rationale for Src inhibitors as antitumor and antimetastatic agents in patients with advanced, metastatic disease.
In conclusion, our results show that inhibition of Src with dasatinib inhibits thyroid cancer cell growth and transformation in vitro and tumor growth and metastatic ougrowth in vivo. We show for the first time that c-Src and Lyn are the predominant SFK members expressed, and that c-Src is likely the major target of dasatinib in thyroid cancer cells. Finally, we provide the first evidence that dasatinib inhibits the progression of thyroid cancer metastasis in vivo. Thus, along with the growth inhibition observed in our orthotopic model, these results provide strong justification for the use of Src inhibitors as both antitumor and antimetastatic agents for advanced patients with thyroid cancer.
Supplementary Material
Translational Relevance.
New therapies with the potential to inhibit both tumor growth and metastasis are needed for patients with advanced thyroid cancer. The Src kinase pathway has emerged as a major player in cancer progression, especially relating to metastasis. We recently showed that the Src target, FAK, is overexpressed and phosphorylated in patient thyroid tumor samples, providing justification to pursue this pathway as a clinically relevant target. Here, we show that inhibition of Src with dasatinib blocks thyroid cancer growth and transformation in vitro. We further show that dasatinib has high therapeutic efficacy as an antitumor and antimetastatic agent in vivo, blocking tumor growth in an orthotopic model, and metastatic progression in an experimental metastasis model. Thus, Src plays a critical role in promoting both tumor growth and outgrowth of metastases, indicating that Src inhibitors may be of particular relevance in thyroid tumors with metastatic potential, as dual antitumor and antimetastatic agents.
Acknowledgments
The authors thank Drs. Jeffrey Myers and Maria Gule at MD Anderson Cancer Center for their guidance in establishing the orthotopic thyroid cancer model, and Dr. Carol Sartorius for her guidance, establishing the intracardiac injection model; Dr. Arthur Gutierrez-Hartmann for advice and critical reading of the manuscript, Dr. Jena French for histology expertise, and Dalan Jensen for statistical support; and Bristol-Myers Squibb for generously providing dasatinib for these studies.
Grant Support
This work was supported by National Cancer Institute grant K12-CA086913 (to R.E. Schweppe) and NIH 5 T32 DK063687 (to C.M. Chan). The UCCC DNA Sequencing and Analysis and Small Animal Imaging Cores are supported by NCI Cancer Center, grant P30 CA046934.
Footnotes
Authors’ Contributions
Conception and design: C.M. Chan, B.R. Haugen, R.E. Schweppe
Development of methodology: Q. Zhou, R.E. Schweppe
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): C.M. Chan, X. Jing, D.-J. Lim, G.S. Lund, V. Sharma, R.E. Schweppe
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): C.M. Chan, L.A. Pike, Q. Zhou, D.-J. Lim, S.B. Sams, G.S. Lund, B.R. Haugen, R.E. Schweppe
Writing, review, and/or revision of the manuscript: C.M. Chan, B.R. Haugen, R.E. Schweppe
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): C.M. Chan, L.A. Pike, B.R. Haugen, R.E. Schweppe
Study supervision: B.R. Haugen, R.E. Schweppe
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Pfister DG, Fagin JA. Refractory thyroid cancer: a paradigm shift in treatment is not far off. J Clin Oncol. 2008;26:4701–4. doi: 10.1200/JCO.2008.17.3682. [DOI] [PubMed] [Google Scholar]
- 2.Haugen BR, Kane MA. Approach to the thyroid cancer patient with extracervical metastases. J Clin Endocrinol Metab. 2010;95:987–93. doi: 10.1210/jc.2009-2305. [DOI] [PubMed] [Google Scholar]
- 3.Sherman SI. Tyrosine kinase inhibitors and the thyroid. Best Pract Res Clin Endocrinol Metab. 2009;23:713–22. doi: 10.1016/j.beem.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Kopetz S, Shah AN, Gallick GE. Src continues aging: current and future clinical directions. Clin Cancer Res. 2007;13:7232–6. doi: 10.1158/1078-0432.CCR-07-1902. [DOI] [PubMed] [Google Scholar]
- 5.Talamonti MS, Roh MS, Curley SA, Gallick GE. Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. J Clin Invest. 1993;91:53–60. doi: 10.1172/JCI116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636–42. doi: 10.1038/sj.onc.1203912. [DOI] [PubMed] [Google Scholar]
- 7.Bolen JB, Veillette A, Schwartz AM, Deseau V, Rosen N. Analysis of pp60c-src in human colon carcinoma and normal human colon mucosal cells. Oncogene Res. 1987;1:149–68. [PubMed] [Google Scholar]
- 8.Lutz MP, Esser IB, Flossmann-Kast BB, Vogelmann R, Luhrs H, Friess H, et al. Overexpression and activation of the tyrosine kinase Src in human pancreatic carcinoma. Biochem Biophys Res Commun. 1998;243:503–8. doi: 10.1006/bbrc.1997.8043. [DOI] [PubMed] [Google Scholar]
- 9.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–58. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 10.Cartwright CA, Meisler AI, Eckhart W. Activation of the pp60c-src protein kinase is an early event in colonic carcinogenesis. Proc Natl Acad Sci U S A. 1990;87:558–62. doi: 10.1073/pnas.87.2.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stein PL, Vogel H, Soriano P. Combined deficiencies of Src, Fyn, and Yes tyrosine kinases in mutant mice. Genes Dev. 1994;8:1999–2007. doi: 10.1101/gad.8.17.1999. [DOI] [PubMed] [Google Scholar]
- 12.Myoui A, Nishimura R, Williams PJ, Hiraga T, Tamura D, Michigami T, et al. C-SRC tyrosine kinase activity is associated with tumor colonization in bone and lung in an animal model of human breast cancer metastasis. Cancer Res. 2003;63:5028–33. [PubMed] [Google Scholar]
- 13.Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 14.Kim HS, Han HD, Armaiz-Pena GN, Stone RL, Nam EJ, Lee JW, et al. Functional roles of Src and Fgr in ovarian carcinoma. Clin Cancer Res. 2011;17:1713–21. doi: 10.1158/1078-0432.CCR-10-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi YL, Bocanegra M, Kwon MJ, Shin YK, Nam SJ, Yang JH, et al. LYN is a mediator of epithelial-mesenchymal transition and a target of dasatinib in breast cancer. Cancer Res. 2010;70:2296–306. doi: 10.1158/0008-5472.CAN-09-3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park SI, Zhang J, Phillips KA, Araujo JC, Najjar AM, Volgin AY, et al. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 2008;68:3323–33. doi: 10.1158/0008-5472.CAN-07-2997. [DOI] [PubMed] [Google Scholar]
- 17.Goldenberg-Furmanov M, Stein I, Pikarsky E, Rubin H, Kasem S, Wygoda M, et al. Lyn is a target gene for prostate cancer: sequence-based inhibition induces regression of human tumor xenografts. Cancer Res. 2004;64:1058–66. doi: 10.1158/0008-5472.can-03-2420. [DOI] [PubMed] [Google Scholar]
- 18.Quintas-Cardama A, Kantarjian H, Cortes J. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat Rev Drug Discov. 2007;6:834–48. doi: 10.1038/nrd2324. [DOI] [PubMed] [Google Scholar]
- 19.Hennequin LF, Allen J, Breed J, Curwen J, Fennell M, Green TP, et al. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl) ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem. 2006;49:6465–88. doi: 10.1021/jm060434q. [DOI] [PubMed] [Google Scholar]
- 20.Kim LC, Rix U, Haura EB. Dasatinib in solid tumors. Expert Opin Investig Drugs. 2010;19:415–25. doi: 10.1517/13543781003592097. [DOI] [PubMed] [Google Scholar]
- 21.Rucci N, Susa M, Teti A. Inhibition of protein kinase c-Src as a therapeutic approach for cancer and bone metastases. Anticancer Agents Med Chem. 2008;8:342–9. doi: 10.2174/187152008783961905. [DOI] [PubMed] [Google Scholar]
- 22.Kim SJ, Park JW, Yoon JS, Mok JO, Kim YJ, Park HK, et al. Increased expression of focal adhesion kinase in thyroid cancer: immunohistochemical study. J Korean Med Sci. 2004;19:710–5. doi: 10.3346/jkms.2004.19.5.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schweppe RE, Kerege AA, French JD, Sharma V, Grzywa R, Haugen BR. Inhibition of Src with AZD0530 reveals the Src-Focal Adhesion Kinase complex as a novel therapeutic target in papillary and anaplastic thyroid cancer. J Clin Endocrinol Metab. 2009;94:2199–203. doi: 10.1210/jc.2008-2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schweppe RE, Klopper JP, Korch C, Pugazhenthi U, Benezra M, Knauf JA, et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J Clin Endocrinol Metab. 2008;93:4331–41. doi: 10.1210/jc.2008-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16:67–78. doi: 10.1016/j.ccr.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–6. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 27.Schweppe R, Kerege A, Sharma V, Poczobutt J, Gutierrez-Hartmann A, Grzywa R, et al. Distinct genetic alterations in the MAPK pathway dictate sensitivity of thyroid cancer cells to MKK1/2 inhibition. Thyroid. 2009;19:825–35. doi: 10.1089/thy.2008.0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim S, Park YW, Schiff BA, Doan DD, Yazici Y, Jasser SA, et al. An orthotopic model of anaplastic thyroid carcinoma in athymic nude mice. Clin Cancer Res. 2005;11:1713–21. doi: 10.1158/1078-0432.CCR-04-1908. [DOI] [PubMed] [Google Scholar]
- 29.Ahn SH, Henderson Y, Kang Y, Chattopadhyay C, Holton P, Wang M, et al. An orthotopic model of papillary thyroid carcinoma in athymic nude mice. Arch Otolaryngol Head Neck Surg. 2008;134:190–7. doi: 10.1001/archoto.2007.36. [DOI] [PubMed] [Google Scholar]
- 30.Wood W, Sharma V, Bauerle K, Pike L, Zhou Q, Fretwell D, et al. PPARgamma promotes growth and invasion of thyroid cancer cells. PPAR Res. 2011;2011:171765. doi: 10.1155/2011/171765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang Y. Analysis of cancer stem cell metastasis in xenograft animal models. Methods Mol Biol. 2009;568:7–19. doi: 10.1007/978-1-59745-280-9_2. [DOI] [PubMed] [Google Scholar]
- 32.Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, et al. Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658–61. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 33.Johnson FM, Saigal B, Talpaz M, Donato NJ. Dasatinib (BMS-354825) tyrosine kinase inhibitor suppresses invasion and induces cell cycle arrest and apoptosis of head and neck squamous cell carcinoma and non-small cell lung cancer cells. Clin Cancer Res. 2005;11:6924–32. doi: 10.1158/1078-0432.CCR-05-0757. [DOI] [PubMed] [Google Scholar]
- 34.Tanjoni I, Walsh C, Uryu S, Tomar A, Nam JO, Mielgo A, et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol Ther. 2010;9:764–77. doi: 10.4161/cbt.9.10.11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J Clin Invest. 2009;119:252–66. doi: 10.1172/JCI37160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishibe S, Joly D, Liu ZX, Cantley LG. Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol Cell. 2004;16:257–67. doi: 10.1016/j.molcel.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 37.Byers LA, Sen B, Saigal B, Diao L, Wang J, Nanjundan M, et al. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin Cancer Res. 2009;15:6852–61. doi: 10.1158/1078-0432.CCR-09-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sen B, Saigal B, Parikh N, Gallick G, Johnson FM. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009;69:1958–65. doi: 10.1158/0008-5472.CAN-08-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsao AS, He D, Saigal B, Liu S, Lee JJ, Bakkannagari S, et al. Inhibition of c-Src expression and activation in malignant pleural mesothelioma tissues leads to apoptosis, cell cycle arrest, and decreased migration and invasion. Mol Cancer Ther. 2007;6:1962–72. doi: 10.1158/1535-7163.MCT-07-0052. [DOI] [PubMed] [Google Scholar]
- 40.Nagaraj NS, Smith JJ, Revetta F, Washington MK, Merchant NB. Targeted inhibition of SRC kinase signaling attenuates pancreatic tumorigenesis. Mol Cancer Ther. 2010;9:2322–32. doi: 10.1158/1535-7163.MCT-09-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–32. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 42.Du J, Bernasconi P, Clauser KR, Mani DR, Finn SP, Beroukhim R, et al. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat Biotechnol. 2009;27:77–83. doi: 10.1038/nbt.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, Rix U, Fang B, Bai Y, Edwards A, Colinge J, et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat Chem Biol. 2010;6:291–9. doi: 10.1038/nchembio.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emaduddin M, Bicknell DC, Bodmer WF, Feller SM. Cell growth, global phosphotyrosine elevation, and c-Met phosphorylation through Src family kinases in colorectal cancer cells. Proc Natl Acad Sci U S A. 2008;105:2358–62. doi: 10.1073/pnas.0712176105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shor AC, Keschman EA, Lee FY, Muro-Cacho C, Letson GD, Trent JC, et al. Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Cancer Res. 2007;67:2800–8. doi: 10.1158/0008-5472.CAN-06-3469. [DOI] [PubMed] [Google Scholar]
- 46.Chan D, Tyner JW, Chng WJ, BI C, Okamoto R, Said J, et al. Effect of dasatinib against thyroid cancer cell lines in vitro and a xenograft model in vivo. Oncol Lett. 2012;3:807–15. doi: 10.3892/ol.2012.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nam S, Kim D, Cheng JQ, Zhang S, Lee JH, Buettner R, et al. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 2005;65:9185–9. doi: 10.1158/0008-5472.CAN-05-1731. [DOI] [PubMed] [Google Scholar]
- 48.Serrels A, Macpherson IR, Evans TR, Lee FY, Clark EA, Sansom OJ, et al. Identification of potential biomarkers for measuring inhibition of Src kinase activity in colon cancer cells following treatment with dasatinib. Mol Cancer Ther. 2006;5:3014–22. doi: 10.1158/1535-7163.MCT-06-0382. [DOI] [PubMed] [Google Scholar]
- 49.Ammer AG, Kelley LC, Hayes KE, Evans JV, Lopez-Skinner LA, Martin KH, et al. Saracatinib impairs head and neck squamous cell carcinoma invasion by disrupting invadopodia function. J Cancer Sci Ther. 2009;1:52–61. doi: 10.4172/1948-5956.1000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ringel MD. Metastatic dormancy and progression in thyroid cancer: targeting cells in the metastatic frontier. Thyroid. 2011;21:487–92. doi: 10.1089/thy.2011.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.