

Abstract
Current imaging technology provides an experimental platform in which complex developmental processes can be observed at cellular resolution over an extended time frame. New computational tools are essential to achieve a systems-level understanding of this high-content information. We have devised a structured approach to systematically analyze complex in vivo phenotypes at cellular resolution, which divides the task into a panel of statistical measurements of each cell in terms of cell differentiation, proliferation and morphogenesis, followed by their spatial and temporal organization in groups and the cohesion within the whole specimen. We demonstrate the approach to C. elegans embryogenesis with in toto imaging and automated cell lineage tracing. We define statistical distributions of the wild-type developmental behaviors at single-cell resolution based on over 50 embryos, cumulating in over 4000 distinct, developmentally based measurements per embryo. These methods enable statistical quantification of abnormalities in mutant or RNAi-treated embryos and a rigorous comparison of embryos by testing each measurement for the probability that it would occur in a wild-type embryo. We demonstrate the power of this structured approach by uncovering quantitative properties including subtle phenotypes in both wild-type and perturbed embryos, transient behaviors that lead to new insights into gene function and a previously undetected source of developmental noise and its subsequent correction.
Keywords: Automated phenotyping, Cell migration, Cell tracking, Caenorhabditis elegans
INTRODUCTION
The interpretation of complex in vivo phenotypes is a crucial and challenging step required to achieve a functional understanding of molecular networks. Advances in imaging technology offer an unprecedented opportunity for systems-level studies with high spatial and temporal resolutions. Using automated image analysis, dozens to hundreds of measurements can be made to systematically and objectively extract and analyze complex information. In cell culture, such methods have enabled systems analysis and large-scale screens examining cell shapes, division, death and subcellular textures (Carpenter et al., 2006; Bakal et al., 2007; Neumann et al., 2010; Danuser, 2011). Meanwhile, 3D time-lapse imaging has allowed detailed, cellular resolution recording of extensive time windows of embryogenesis in organisms such as C. elegans (Schnabel et al., 1997; Bao et al., 2006), Drosophila (McMahon et al., 2008; Keller et al., 2010; Truong et al., 2011), zebrafish (Keller et al., 2008; Truong et al., 2011) and mouse (Kwon et al., 2008). Automated methods are needed to systematically dissect the intertwined in vivo processes of differentiation, proliferation and morphogenesis recorded at single-cell resolution (Megason and Fraser, 2007).
C. elegans has proven to be an effective model for systems-level analysis of in vivo phenotypes (Kamath et al., 2003; Rual et al., 2004; Fernandez et al., 2005; Gunsalus et al., 2005; Sönnichsen et al., 2005; Lehner et al., 2006; Piano et al., 2006; Byrne et al., 2007; Hunt-Newbury et al., 2007; Liu et al., 2009; Green et al., 2011). Automated image analysis and data mining methods are emerging to further facilitate such efforts (Dupuy et al., 2007; Long et al., 2009; White et al., 2010; Wählby et al., 2012). Early embryogenesis of C. elegans, where lineage patterning, gastrulation and extensive cell-cell signaling occur, is particularly amenable for systematic in vivo analysis because the entire cell lineage can be traced at minute-level resolution (Schnabel et al., 1997; Hamahashi et al., 2005; Bao et al., 2006; Dzyubachyk et al., 2009; Hench et al., 2009; Santella et al., 2010; Giurumescu et al., 2012). Genome-wide RNAi has been used to analyze behaviors of individual cells up to the 4-cell stage (Sönnichsen et al., 2005). Although this analysis was conducted during development, the effort was mainly focused on cell biological behaviors regarding different aspects of cell polarity and the cell cycle. The 46 behaviors were carefully chosen in cell-specific manners to be most biologically informative, but the manual effort required prohibits propagating the analysis beyond the 4-cell stage.
We present a structured approach to systematically quantify complex developmental phenotypes during C. elegans embryogenesis. We choose a small and uniform set of measurements per cell that can be automatically applied to any cell to systematically assay differentiation, proliferation and morphogenesis in the entire embryo at single-cell resolution. We have also developed methods to measure the organization of cellular behaviors at whole-organism and regional levels. We quantify the range of variability of these behaviors in 53 wild-type embryos to the 350-cell stage, when the embryo has gone through the first nine rounds of cell division and completed gastrulation (Sulston et al., 1983). This cumulates in over 4000 distributions of wild-type cellular behaviors. These distributions are used to calculate the probability that a particular quantity observed in a mutant or RNAi-treated embryo would be observed in a wild-type embryo. We demonstrate the power of this structured approach in detecting subtle phenotypes, transient behaviors and developmental noise and correction through analyses of wild-type and RNAi-treated embryos.
MATERIALS AND METHODS
Experimental methods
RNAi was administered by feeding as described (Fraser et al., 2000). Embryos were collected and imaged as previously described (Bao and Murray, 2011). Nuclei were segmented and tracked as described (Santella et al., 2010). Automated tracking results were edited by hand as described previously (Murray and Bao, 2012).
Software to quantify phenotypes was written using MATLAB (MathWorks). Morphogenesis visualizations were made using POV-Ray (POV-Ray 3.6, Persistence of Vision team) to generate images containing the desired shapes.
General framework
The developmental behaviors of each cell are measured, compared with the wild-type distribution and assigned P-values representing the likelihood that each measurement would be observed in a wild-type embryo. The statistics compiled for each cell’s measurements are available in supplementary material Appendix S1. Measurements in a pal-1(RNAi) embryo are also provided (supplementary material Appendix S2). The corresponding P-values for this pal-1(RNAi) embryo are displayed in a chart to visualize abnormalities (supplementary material Fig. S1). To guard against false positives as a result of the large number of statistical tests, we use the Benjamini and Hochberg method (Benjamini and Hochberg, 1995) on the entire P-value matrix. To be flagged as abnormal, we chose the arbitrary adjusted P-value cut-off of 0.01, which yields an average of 2.2 false positives per embryo. This cut-off can be tuned based on the subtlety of the phenotype of interest.
The method discussed here is available as a software package that we named Dev-scape. The source code is provided as supplementary material Appendix S3. Additionally, the source code can be downloaded from SourceForge (http://sourceforge.net/projects/devscape/files). Using Dev-scape from the source code requires MATLAB. Compiled versions for both PC and Mac, which allow users to run Dev-scape without access to MATLAB, are also available from SourceForge. Detailed instructions on installation and using Dev-scape are provided as part of the download. The PC version is recommended. Using the source code or the Mac version requires the user to compile POV-Ray.
When using Dev-scape, a graphical user interface guides users to specify the input, choose the types of analysis to be performed, and enter parameters about their data (supplementary material Fig. S2). When the analysis is completed, an output browser opens to facilitate examination of the results. The input file is a zip file as outputted by AceTree (Murray and Bao, 2012), which contains the result of automated cell lineage tracing. All results of Dev-scape analysis are written to text files to facilitate further analysis of data and comparison between embryos. A list of the output files is provided in supplementary material Table S1. Detailed instructions and explanations are provided when the user downloads Dev-scape.
Differentiation
We use tissue-specific markers to assay cell fate. Each of the five markers used is a well-studied master transcription factor in determining the corresponding tissue type (supplementary material Table S2). The reporter strains have been shown to have stable and reproducible expression that reliably reflects cell fate in both the wild type and known fate transformations (Mathies et al., 2003; Murray et al., 2008) (data not shown).
To make a binary classification of each cell as expressing or non-expressing, we first calculate each cell’s background corrected expression level at each time point as described (Murray et al., 2008). Next, we calculate the average expression level of each cell over time and perform a robust linear regression on all average expression levels using the MATLAB ‘robustfit’ function with weighting parameter ‘fair’ (supplementary material Fig. S3A). This method fits a line to the background population of non-expressing cells and identifies the expressing cells as outliers. To accomplish this, non-expressing cells are interspersed with expressing cells, which is achieved by ordering cells by lineage identity. Most expressing cells can be identified by the low weights they are assigned due to their not fitting the non-expressing line. Cells with positive expression values and a weight below 0.3 are classified as expressing. Cells that fit the line well are assigned weights between 0.7 and 1 and classified as not expressing. Many embryos contain a few cells with borderline expression levels; cells assigned weights between 0.3 and 0.7 are considered borderline expressing. In general, these cells are either not expressing or begin expression during the cell’s life. To differentiate between these two types, we examine the cell’s time series of expression levels with a Wilcoxon rank-sum test to identify cells with expression values that increase over time. The series of expression values is repeatedly split into early and late sections, which serve as separate distributions for the test. To search for any time point as the initiation of expression, the split progresses through the cell cycle and a cell is called expressing if any P-value below 0.001 is found (supplementary material Fig. S3B). This method is robust to both the number of low expressing cells and imaging noise that affects the perceived baseline level of expression. Examining 22 embryos, we found a false-positive rate of 0.02%.
Differentiation results are displayed in a lineage tree to allow users to compare various wild-type and mutant patterns. Trees can be displayed in the canonical way with branch length corresponding to time or with a uniform branch length to separate proliferation and differentiation data (supplementary material Fig. S3C).
Proliferation
Global clock and normalization of cell cycle lengths
Under normal experimental conditions, wild-type embryos exhibit a linear global clock in that individual cell cycle lengths in an embryo vary proportionally to each other (Bao et al., 2008) (supplementary material Fig. S4A). For each embryo examined, we calculate this clock by performing a robust linear regression (as above) on the wild-type average cell cycle lengths versus their length in the given embryo. We examine how well the linear regression fits the cell cycle lengths using root mean squared deviation (RMSD). A high RMSD (empirical cut-off of 7) means that the cell cycle lengths do not vary proportionally to each other. To choose an empirical RMSD cut-off, we examined the relationship between global clock linearity and RMSD in 91 embryos that were treated with RNAi for genes required for embryogenesis. In the case of linear changes of the global clock, we use the slope from the regression to scale each cell cycle length in the given embryo. For non-linear changes, we test for the pace of proliferation slowing down over time. This often occurs when an embryo is approaching arrest, as represented by cdt-1(RNAi) (supplementary material Fig. S4A). We look for changes in each lineage group’s proliferation rate over time by calculating the average cell cycle length for each generation of cells within a given lineage and measure the ratio between wild-type cells in consecutive generations. Results of all the calculations discussed above are outputted into a text file and most are displayed in graphs for the user to examine (supplementary material Table S1).
Synchrony and organization of proliferation
We cluster cells that divide at roughly the same time into groups based on the pairwise correlation of each cell’s history of division timing. Each cell’s history is represented by a vector where each element corresponds to a time point and is assigned a value representing the proportion of the cell cycle that the corresponding time point is away from a division. We use the MATLAB ‘clusterdata’ function to perform single linkage clustering based on the pairwise correlations.
We examine the synchrony of each wild-type group defined above in mutants/RNAi. For each cell within a given group, we determine whether the cell is still in sync with the group as follows. We measure the difference between the cell’s division time and the average division time of the group. This difference is then normalized to the cell’s cycle length and compared with the wild-type distribution of the normalized difference across embryos. We treat the wild-type distributions as normal because for 83% of the cells the distribution passes the D’Agostino’s K2 test for normality with an alpha value of 0.05 (D’Agostino et al., 1990).
Individual cell cycle length
Single cell-cycle length distributions were tested for normality using D’Agostino’s K2 test, which is suitable for our relatively small sample size (D’Agostino et al., 1990). We found that 81% of the distributions were normal with an alpha value of 0.05.
Morphogenesis
To properly characterize the spatial variation between embryos, we first align the embryo according to the body axes then normalize axes to a standard size. We detect the anterior-posterior axis by finding the largest axis of variation in cell positions at the final time point of a time-lapse series, typically with 100 to 350 cells. Because of the mounting technique, the left-right axis in the early embryo is aligned with the z-axis of the image stack (Bao et al., 2006). We determine the orientation of the left-right axis by examining the position of the left and right side AB cells at the AB4 stage. The length of each body axis is determined by finding the minimum and maximum cell position along that axis; for stability, we use the 99th percentile minimum and maximum. Each body axis is then linearly scaled to the wild-type average size compiled from the 53 embryos, normalizing the position of each cell in the process.
We measure each cell’s average position and displacement based on the normalized coordinates. Cell position is measured as the three coordinates (assumed to be independent) in the three body axes, 5 minutes into the cell’s life. This time point is chosen to capture the position near the beginning of the cell cycle while avoiding the variation associated with cell divisions due to cell collisions. Each covariance ellipsoid is calculated using principal component analysis to identify the directions of variation and the variance in those directions (supplementary material Fig. S5A). The displacement is measured as the magnitude of total displacement as well as the displacement along each axis between a cell’s position at birth and division or death. In total, we calculate seven quantities describing each cell’s behavior in embryonic morphogenesis. Each of these distributions was tested for normality using D’Agostino’s K2 test. On average, 89% of the distributions passed the test when an alpha value of 0.05 was used.
To identify groups of cells that migrate together, we use mean shift clustering on the cell positions and displacements, allowing the identification of cells that are near each other and moving together. We also quantify the coordination of cell movement by measuring the pairwise correlation of migration paths for each time point that both members of the pair are in the embryo (supplementary material Fig. S5B). The correlation is calculated in each body axis, and then weighted by the amount of displacement that occurs in that axis and then summed.
To quantify angular movement, we treat the center of the normalized embryo as the origin. We project all cells onto the center plane bisecting the dorsal-ventral axis and only examine cells beyond the center two-thirds. Displacement is calculated over 15-minute windows. Before performing the paired t-test comparing the correlation between C and non-C periphery cell movement in wild-type and pal-1 averages, we tested both distributions for normality using D’Agostino’s K2 test. Both were normally distributed with P-values of 0.37 and 0.39, respectively.
RESULTS
A statistical strategy to achieve single-cell phenotypic descriptions
As outlined in Fig. 1, we build upon the histone-fluorescent protein fusion-based automated segmentation (Santella et al., 2010), cell tracking (Bao et al., 2006) and lineage tracing (Bao et al., 2006) to characterize development. We use a collection of tissue-specific markers to measure differentiation. We characterize proliferation by measuring the temporal, spatial and lineage-based organization of cell divisions. To quantify morphogenesis, we measure cell position, displacement and organization of collective movement. We also provide visualization tools to aid the understanding of the phenotypes.
Fig. 1.

The process of converting time-lapse image data to a panel of probabilities. (A) Representative frames from a time series of C. elegans embryogenesis imaged with histone-fluorescent protein labels. Images are segmented and cells are tracked. (B) Each cell’s differentiation, proliferation and morphogenesis are measured. (C) Measurements are compared with the wild-type probability distributions to obtain P-values that represent the probability of a measurement as far from or further from the mean being present in a wild-type embryo. After adjusting for testing multiple hypotheses, measurements within the wild-type range (P>0.01) are shown in white and significantly different values (P<0.01) are in red (when measurements are smaller than the mean) and in blue (when measurements are larger than the mean).
We have not only developed measurements for behaviors of the whole embryo and coordinated cell groups, but also for each cell. The key to characterizing the distribution of developmental behaviors of a given cell is the ability to find the equivalent cells across embryos. The general approach to this alignment in developmental biology is anatomy-based registration between specimens; for example, a particular tissue layer in a particular organ occupying a particular part of the body. The invariant cell lineage of C. elegans (Sulston et al., 1983) offers a convenient approximation of this general rule: because of the stereotypical orientation of each mitosis, the name of a cell provides the equivalent information as its anatomical position. This allows us to automate single-cell analysis through automated cell lineage tracing without additional computational steps of anatomy-based registration.
For single-cell measurements, the probability that a value would be found in a wild-type embryo is calculated by determining the probability that a measurement this extreme or more extreme would appear in a wild-type embryo. Repeating this process for each measurement and each cell leads to the calculation of 4000 statistical quantifications per embryo (supplementary material Fig. S1). After correcting for multiple tests, this list of the statistical significance of phenotypes can be readily used to guide in-depth characterization of in vivo gene function and for comparisons between embryos to identify related gene functions or construct functional gene networks. We describe the individual methods below and provide the technical details of each method in the Materials and methods.
Differentiation
We use tissue-specific marker expression to assay when cells take on a fate. With two-color imaging, one color is used to trace the lineage and the other to assay the expression of a fate marker. We use markers that are transcription factors expressed in the major tissue types: gut, hypodermis, muscle, neuron and pharynx. We examine each cell to determine whether it is expressing the marker or not. As shown in Fig. 2A,B, this approach provides detailed dynamics of marker expression in individual cells.
Fig. 2.

Consistent wild-type differentiation patterns allow the discovery of perturbations at single-cell resolution. (A) Nuclear expression of HND-1::GFP in the MS lineage for each time point. Expression levels in cells marked by asterisks are shown in B. (B) Expression levels over time show that HND-1 is transiently expressed in MSxp cells, but not in MSxa cells. a.u., arbitrary units. (C) Summaries of expression across different markers, all at the 350-cell stage. Cells are represented by vertical lines displayed in lineage order and color coded by fate marker expression. Each color represents a combination of markers being expressed in that cell. The expression of each marker is observed in separate embryos. The first row shows markers that are expressed in every wild-type embryo examined. The second row shows markers that are expressed in at least 50% of wild-type embryos. The third row shows markers that are expressed in one or both of two pal-1(RNAi) replicates. In the case that only one replicate is expressing, we display whichever behavior occurs more commonly in wild-type embryos. The bottom row shows which of the pal-1(RNAi) expression patterns were abnormal.
To decide whether a cell expresses the marker, we apply an embryo-specific binary classification based on marker expression levels that are found in each segmented nucleus at each time point (Murray et al., 2008). The automated classification relies on the fact that the majority of cells do not express a given fate marker and performs an iterative multilinear regression to summarize the background fluorescence level, which can vary widely between embryos (supplementary material Fig. S3A). Cells with expression levels significantly higher than the background are classified as expressing. To better capture expression that is initially low, we use a Wilcoxon rank-sum test to identify cells with a time-dependent increase of expression level (supplementary material Fig. S3B) (Murray et al., 2008). Using these two statistical classifications, expressing cells match well with known fates (Fig. 2C; see Materials and methods).
To formulate a wild-type differentiation standard we examine the consistency of marker expression in multiple wild-type embryos. Owing to the invariant lineage of C. elegans, fates are also invariant at the single-cell level and any fluctuation in expression status is due to marker inconsistency. Because of this fundamental reliability, and the time-consuming nature of data collection, we examined four to ten embryos per marker. As the relatively small number of samples limits the statistical power, we formulated the standard to include only the consistently expressing and consistently non-expressing cells among all wild-type embryos. If, for a given marker, a cell sometimes expresses and sometimes does not in wild-type embryos, neither state can be considered abnormal in a mutant or RNAi-treated embryo.
To systematically assay cell fate across the lineage, one can use a panel of selected markers. We selected five well-studied tissue markers with consistent expression patterns corresponding to the five major tissue types, namely cnd-1 for neuronal, pha-4 for pharyngeal and endodermal, nhr-25 for hypodermal, elt-2 for endodermal and hnd-1 for muscular fate (Mathies et al., 2003; Murray et al., 2012). Between the five tissue-type markers, 40% of cells consistently express at least one marker and 100% of cells consistently do not express at least three markers. This means that loss of expression can be detected in 40% of cells and a gain of expression can be detected in all cells.
To test our approach, we examined the phenotype of pal-1(RNAi). PAL-1 is the ortholog of Caudal in Drosophila and is a transcription factor that is required to specify the fate of the posterior lineages, named C and D, that give rise to muscle and hypodermal cells (Hunter and Kenyon, 1996). Consistent with the known function of pal-1, we observed a loss of muscle and disruption of hypodermal fates in the C and D lineages and no other consistent deviation from wild type (n=2 embryos per marker; Fig. 2C) (Hunter and Kenyon, 1996; Edgar et al., 2001). PAL-1 also has a redundant role in specifying the E lineage, which gives rise to endoderm (Maduro et al., 2005). As expected, pal-1(RNAi) alone does not affect expression of the endoderm marker elt-2.
Proliferation
We measured proliferation at three levels. First, we examined the global pace of proliferation by counting the number of cells over time. To formulate a standard for how closely this number of cells matches the wild type, we plotted each average wild-type cell cycle length verses the cell cycle length in the embryo being examined. In wild-type embryos, this relationship is linear and we refer to the slope of the line on this plot as the global clock (supplementary material Fig. S4A) (Bao et al., 2008). The global clock is known to vary slightly between embryos due to external factors and we detect intra-embryo global clock changes as a phenotype, such as slowing toward arrest. pal-1(RNAi) does not lead to a significant change in the global clock (Fig. 3A; supplementary material Fig. S4A). By contrast, RNAi against cdt-1, a conserved DNA replication licensing factor (Zhong et al., 2003), dramatically slows the global clock.
Fig. 3.
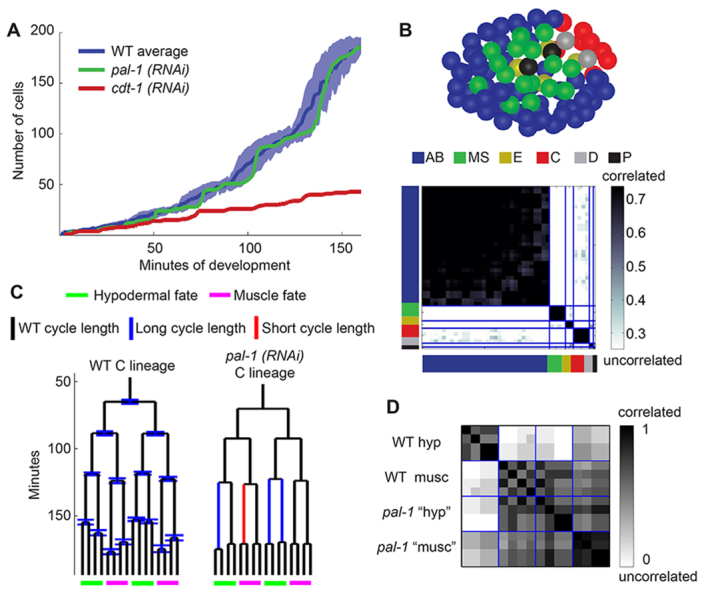
Analyzing proliferation at multiple scales can uncover a variety of phenotypes. (A) The number of cells in the embryo over time reflects the global proliferation rate. Blue represents the wild-type average plus and minus one standard deviation. Green and red each represent an RNAi-treated embryo. (B) (Top) The spatial distribution of cells that divide synchronously. Each synchronous group is marked by a different color. These groups correspond perfectly with lineage groups. The embryo is at the start of the AB64 stage, viewed ventrally. (Bottom) A heat map showing the degree of correlation between cells. The colors along the axes represent the lineage groups as in the top panel. Division timing is correlated within lineage groups, but not between them. (C) Wild-type average cell cycle lengths and standard deviations in the C lineage, and pal-1(RNAi) cell cycle lengths highlighting statistically significant deviations (P<0.01). (D) Correlation of the division timing histories of the C lineage in a wild-type embryo and pal-1(RNAi) embryo. The wild-type lineage forms two groups separated by cell fates, whereas all of the pal-1(RNAi) cells are correlated with the wild-type muscle division timing. hyp, hypodermal; musc, muscle.
Second, to assay the spatial organization of proliferation, such as mitotic waves, we examined groups of synchronized divisions. For de novo detection of these groups, we clustered cells based on pairwise correlations of their division histories (Fig. 3B; supplementary material Fig. S4C). In wild-type C. elegans embryos the clusters correspond to lineage groups, which are known to divide synchronously, validating the clustering method. We also measured the synchrony within each of these defined groups by examining the difference between the group’s average cell division time and individual cell division times (supplementary material Fig. S4B).
Third, we measured the pace of proliferation in individual cells in terms of cell cycle length. Across embryos, wild-type single cell-cycle length distributions were normal (see Materials and methods) and had an average percent coefficient of variation of 4.3. This level of consistency allows small aberrations to be detected as statistically significant (supplementary material Fig. S4C).
Our sensitive measurements uncovered new phenotypes of pal-1 loss of function and insights regarding how this conserved master transcription factor coordinates proliferation between different tissue types. Consistent with the knowledge that pal-1(RNAi) disrupts cell fate only in the C and D lineages (Hunter and Kenyon, 1996; Edgar et al., 2001), we found that pal-1(RNAi) only consistently affected cell cycle lengths in these two lineages (P<0.01). D lineage cell cycle length abnormalities are all in the form of increases in cycle length, appearing in seven out of ten pal-1(RNAi) embryos examined. The C lineage abnormalities comprised both increases and decreases in cell cycle length and occurred in nine out of ten pal-1(RNAi) embryos (Fig. 3C). In the C lineage, divisions of the presumptive hypoderm cells become synchronous with those of the presumptive muscle cells (Fig. 3D; supplementary material Fig. S4B), and the entire C lineage proliferates at the same pace as the wild-type muscle cells in the C lineage. These results indicate that the C lineage has a default proliferation rate that is independent of pal-1. This default rate is used in the muscle sublineages but is accelerated by pal-1 in the hypoderm.
Morphogenesis: measuring cellular position and migration
To characterize the variation in cell positions, we fit the distribution of the wild-type position of each cell across 53 wild-type embryos with a covariance ellipsoid (Fig. 4A; supplementary material Fig. S5A). Wild-type cell positions are consistent, as ellipsoids drawn with radii of one standard deviation in each component do not overlap (supplementary material Movie 1). For cell movement, we found that the trend of migration is consistent for a given cell between embryos, but that the path is usually noisy (supplementary material Fig. S6). To emphasize the movement instead of the noise, we quantify the amount of displacement. To measure the overall similarity of an embryo to the wild type, we calculate the RMSD between average positions and those in the embryo of interest (Schnabel et al., 2006). We also developed visualization techniques to allow users to better grasp the 3D cell migration dynamics, which are difficult to describe (Fig. 4C,F; supplementary material Fig. S5, Movie 2).
Fig. 4.

Morphogenesis quantification and visualization reveals disruption in pal-1(RNAi). (A) Dorsal view of covariance ellipsoids at the AB32 stage. The long axis of each ellipsoid is aligned with the direction of most variation of a cell’s position in 53 wild-type embryos examined 5 minutes after cell birth. Each radius shows one standard deviation in one principal component of the cell position distribution. (B) A pal-1(RNAi) embryo has an increasing number of cells with abnormal displacement as development progresses. (C,F) Cell displacement patterns are shown for wild type and pal-1(RNAi) during the AB64 stage. (D,G) Brown, light blue and dark blue cells are part of co-migrating groups identified by performing mean shift clustering on the positions and displacements in C and F. (E,H) Pairwise correlation coefficients of migration trajectories within the same embryos show high correlation in wild type and lack of correlation in pal-1(RNAi). A subset of the lineage is shown here; see supplementary material Fig. S5 for the complete correlation matrix of the wild-type embryo.
Furthermore, we use two methods to identify groups of cells that migrate together. The first is a weighted average of the correlation of migration along each developmental axis (Fig. 4E,H; supplementary material Fig. S5B). This allows cells to be grouped based on the dynamics of their migration. The second is mean shift clustering based on cell positions and displacements (Fig. 4D,G).
We examined the function of pal-1 in organizing posterior morphogenesis using the above methods. In pal-1(RNAi), displacement is largely diminished. This is first detected at the AB32 stage, with the cell named Cap being the only cell with an abnormal amount of displacement (Fig. 4B). Many more cells have an abnormal amount of displacement during AB64 and AB128. More specifically, the displacement is variably reduced among C8 cells (P<0.01) and the co-migration is diminished (Fig. 4C,F). Two other co-migrating groups adjacent to C8 (ABplp8 and ABprp8) are also diminished (Fig. 4H). Given the lineage-specific function of pal-1, these later migration abnormalities are likely to be secondary defects. We did not observe the early misplacement of the C granddaughters and their anterior neighbors (ABarppa/p) that was reported in the strong loss-of-function allele pal-1(ct224) (Edgar et al., 2001), probably owing to the weaker penetrance of RNAi.
Morphogenesis: developmental noise and correction
The covariance ellipsoid visualization revealed that, at the AB64 stage, many of the ellipsoids are aligned in a circular pattern when viewed dorsally (Fig. 5A). This pattern of directional variance is caused by a previously unobserved global cell movement, producing a rotation about the dorsal-ventral axis in a subset of embryos. This new rotation is perpendicular to a previously observed rotation about the long axis of the embryo that occurs one cell cycle later (Giurumescu et al., 2012). In the subset of embryos that undergo this rotation, cells move clockwise in a 15-minute time window beginning a few minutes after the C4 cells are born (Fig. 5A). This movement exaggerates the angle between the left-right midline and the long axis of the embryo (Pohl and Bao, 2010). Cells later move counterclockwise, so that by the start of the AB128 stage the exaggerated angle is reduced (Fig. 5A). Measurements of the angular displacements show that wild-type embryos undergo varying degrees of this shift and countershift (Fig. 5B).
Fig. 5.

Angular shift is initiated by C cells. (A) Mid AB32 shows clockwise motion in peripheral cells in an individual embryo. Early AB64 shows the covariance ellipsoids of cell positions over 53 wild-type embryos. Late AB64 shows counterclockwise motion in the same embryo as in mid AB32. Early AB128 shows the covariance ellipsoids of cell positions over the same set of wild-type embryos as in late AB64. (B) The maximum magnitude of the average clockwise angular movement (x-axis) plotted versus counterclockwise angular movement (y-axis) in wild-type and pal-1(RNAi) embryos. Average is calculated across C cell and non-C periphery cell groups in each embryo. Angular displacement is defined within a plane bisecting the dorsal-ventral axis. Only cells beyond the center two-thirds of the embryo are included (blue and red in A). (C) One embryo’s average angular displacement in C cells (red in A) and non-C periphery cells (blue in A) over time. (D) Time-shifted correlation between C and non-C periphery cells over a range of temporal shifts. Lines show average correlation values and range shows plus and minus one standard deviation. The wild-type curve shows that moving the non-C periphery cells back in time relative to the C cells leads to larger correlation, i.e. comparing C cell movement to the movement of the non-C periphery cells 4 minutes later results in the largest correlation.
Based on cell displacements over time, the C4 cells are the first to move in the initial clockwise shift (supplementary material Fig. S7). Time-shifted correlations of the angular displacement curves between C4 and other cells confirmed this observation (Fig. 5C). The wild-type embryos (n=53) showed a maximal correlation value around -4 minutes. We then used pal-1(RNAi) to perturb the C4 cells. In pal-1(RNAi) embryos (n=10) the shift-countershift was diminished, and there was no significant correlation between the residual C4 and non-C movements [Fig. 5D; paired t-test between wild-type and pal-1(RNAi) average correlation values, P=4×10-7]. The correlation between shift and countershift magnitudes is also diminished [Pearson’s correlation coefficient=0.56 in wild type and 0.14 in pal-1(RNAi)]. Taken together, our analyses suggest that variation in C4 movement leads to noise and correction in global cell positions.
Morphogenesis: emerging global patterns
We found that novel patterns of whole-embryo collective migrations can arise in RNAi knockdowns of specific genes (Fig. 6). For example, in RNAi of pie-1, which encodes a zinc-finger protein required for germline fate (Mello et al., 1996), bilateral cells in the ABp lineage form two half-circular flows in opposite directions that merge at the anterior end of the midline (Fig. 6A). Interestingly, apx-1(RNAi) produces strikingly similar morphogenetic patterns (Fig. 6B), which is reflected in the similarity in their P-value matrices (Fig. 6C,D). As a notch ligand required for fate induction of ABp in the 4-cell embryo, expression of apx-1 is dependent on pie-1 (Mango et al., 1994; Mickey et al., 1996). Therefore, it is most likely that the pattern that emerged in pie-1(RNAi) is a result of abolishing apx-1 expression.
Fig. 6.

Collective migration patterns emerge in RNAi treatments. (A,B) Each rod represents a cell’s displacement over a 15-minute period. (A) In an embryo treated with pie-1(RNAi), cells at the posterior and anterior extremes of the embryo migrate from left to right and meet at on the embryo’s right side. (B) An apx-1(RNAi) embryo shows cells producing a similar pattern to those in A. (C,D) The P-value chart for cell positions and movements in A (C) and in B (D). Measurements with P<0.01 are in red or blue. A-P, anterior-posterior; L-R, left-right; D-V, dorsal-ventral.
It has been proposed that cell movement during C. elegans embryogenesis is governed by systematic pairwise cell-cell interactions that guide cells to sort among themselves (Bischoff and Schnabel, 2006; Schnabel et al., 2006). The co-migrating groups and the new patterns of global organization apparent in the above experiments can assist investigations of how systematic local interactions can lead to the emergence of global patterns.
DISCUSSION
We have presented a structured approach to systematically quantify complex developmental phenotypes at cellular resolution based on long-term 3D time-lapse imaging. The measurements are designed to systematically capture three key aspects of development, namely differentiation, proliferation and morphogenesis, from single-cell behaviors to local and global patterns. This leads to over 4000 quantitative measurements in C. elegans embryogenesis through the first nine of ten total rounds of cell division. We tested these methods by systematically dissecting the phenotypes of pal-1(RNAi). In addition to recapitulating the known phenotypes, our approach detected subtle but significant changes in individual cells revealing a new function of this conserved transcription factor in coordinating proliferation between two tissue types.
Our work builds on several studies of C. elegans embryogenesis at single-cell resolution. Pioneering work using DIC imaging and manual lineaging documented the nature of the cell movements and the relatively high level of noise (Schnabel et al., 1997). Relative positions have also been characterized based on distance and Voronoi diagrams (Schnabel et al., 2006; Hench et al., 2009), which we did not address in this study. Based on the same imaging and cell-tracking platforms used in our study, computational methods and an extensive collection of transgenic strains have been developed to quantify single-cell marker expression (Murray et al., 2008; Murray et al., 2012). Most recently, cell tracking has been extended late into embryogenesis to quantitatively characterize highly coordinated cell movement patterns (Giurumescu et al., 2012). The statistical nature of our study, quantifying not only the average behaviors but the degree of variation in wild-type development, is a key addition to these previous studies of single-cell behavior in C. elegans embryogenesis.
Our results demonstrate the value of the structured approach in characterizing complex developmental processes. In particular, they show that a small and uniform set of measurements applied to each cell can effectively capture the key information of complex developmental processes. Although the invariant cell lineage of C. elegans plays an important technical role in our study, the conceptual framework of combining long-term 3D time-lapse imaging with a small but well-designed set of single-cell measurements can be applied to more complex organisms with variable development. As discussed above, this would require additional techniques of anatomy-based registration between specimens to identify equivalent cells. An additional consideration is that in C. elegans each cell is unique so that embryos can be compared on a cell-by-cell basis. In more complex organisms, the meaningful level of comparison would be each cell type, which may comprise multiple and variable numbers of cells that share the same properties. As shown in our study, clustering of cells based on single-cell measurements can be an effective approach to define cell groups with homogeneous behaviors, which can then be compared across specimens. As studies in developmental biology show, molecular markers can also be used to facilitate the detection of cell type to compare phenotypes.
More importantly, our approach enables systems-level analysis of in vivo development. The measurements constitute a panel of probabilities that summarizes multiple facets of development and readily supports quantitative measurement of functional similarity between genes and the construction of functional gene networks at single-cell resolution (Gunsalus et al., 2005). Furthermore, high temporal and spatial resolution allows the detection of transient and variable behaviors in a large set of embryos. This allowed us to capture and dissect noise and correction in the morphogenesis of an organism that is renowned for invariant and stereotypical development. Much of the current effort in studying developmental noise and robustness is devoted to molecular studies of gene expression (Balázsi et al., 2011). Our study suggests that systematic cell tracking and single-cell analysis will also open doors to study how noise and its control at the molecular level is transformed into cellular behaviors and how cellular-level noise and control affects the assembly of an organism.
Supplementary Material
Acknowledgments
We thank Christian Pohl, Anthony Santella, Robert Vogel and Jennifer Zallen for helpful discussions and critical reading of the manuscript; Anthony Santella for help with software packaging; and members of the Z.B. laboratory for software testing.
Footnotes
Funding
J.L.M. was supported by the Tri-Institutional Training Program in Computational Biology and Medicine. Work in the laboratory of Z.B. is supported by the National Institutes of Health [grants GM097576 and HD075602]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Author contributions
J.L.M. and Z.B. developed the method framework and wrote the manuscript. J.L.M. implemented the framework and created the visualizations. Z.D. performed experimental data collection and contributed to discussions.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.096040/-/DC1
References
- Bakal C., Aach J., Church G., Perrimon N. (2007). Quantitative morphological signatures define local signaling networks regulating cell morphology. Science 316, 1753–1756 [DOI] [PubMed] [Google Scholar]
- Balázsi G., van Oudenaarden A., Collins J. J. (2011). Cellular decision making and biological noise: from microbes to mammals. Cell 144, 910–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z., Murray J. I. (2011). Mounting Caenorhabditis elegans embryos for live imaging of embryogenesis. Cold Spring Harb. Protoc. 2011, pdb.prot065599 [DOI] [PubMed] [Google Scholar]
- Bao Z., Murray J. I., Boyle T., Ooi S. L., Sandel M. J., Waterston R. H. (2006). Automated cell lineage tracing in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 103, 2707–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z., Zhao Z., Boyle T. J., Murray J. I., Waterston R. H. (2008). Control of cell cycle timing during C. elegans embryogenesis. Dev. Biol. 318, 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 [Google Scholar]
- Bischoff M., Schnabel R. (2006). Global cell sorting is mediated by local cell-cell interactions in the C. elegans embryo. Dev. Biol. 294, 432–444 [DOI] [PubMed] [Google Scholar]
- Byrne A. B., Weirauch M. T., Wong V., Koeva M., Dixon S. J., Stuart J. M., Roy P. J. (2007). A global analysis of genetic interactions in Caenorhabditis elegans. J. Biol. 6, 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter A. E., Jones T. R., Lamprecht M. R., Clarke C., Kang I. H., Friman O., Guertin D. A., Chang J. H., Lindquist R. A., Moffat J., et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Agostino R. B., Belanger A., D’Agostino R. B. (1990). A suggestion for using powerful and informative tests of normality. Am. Stat. 44, 316–321 [Google Scholar]
- Danuser G. (2011). Computer vision in cell biology. Cell 147, 973–978 [DOI] [PubMed] [Google Scholar]
- Dupuy D., Bertin N., Hidalgo C. A., Venkatesan K., Tu D., Lee D., Rosenberg J., Svrzikapa N., Blanc A., Carnec A., et al. (2007). Genome-scale analysis of in vivo spatiotemporal promoter activity in Caenorhabditis elegans. Nat. Biotechnol. 25, 663–668 [DOI] [PubMed] [Google Scholar]
- Dzyubachyk O., Jelier R., Lehner B., Niessen W., Meijering E. (2009). Model-based approach for tracking embryogenesis in Caenorhabditis elegans fluorescence microscopy data. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2009, 5356–5359 [DOI] [PubMed] [Google Scholar]
- Edgar L. G., Carr S., Wang H., Wood W. B. (2001). Zygotic expression of the caudal homolog pal-1 is required for posterior patterning in Caenorhabditis elegans embryogenesis. Dev. Biol. 229, 71–88 [DOI] [PubMed] [Google Scholar]
- Fernandez A. G., Gunsalus K. C., Huang J., Chuang L. S., Ying N., Liang H. L., Tang C., Schetter A. J., Zegar C., Rual J. F., et al. (2005). New genes with roles in the C. elegans embryo revealed using RNAi of ovary-enriched ORFeome clones. Genome Res. 15, 250–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser A. G., Kamath R. S., Zipperlen P., Martinez-Campos M., Sohrmann M., Ahringer J. (2000). Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature 408, 325–330 [DOI] [PubMed] [Google Scholar]
- Giurumescu C. A., Kang S., Planchon T. A., Betzig E., Bloomekatz J., Yelon D., Cosman P., Chisholm A. D. (2012). Quantitative semi-automated analysis of morphogenesis with single-cell resolution in complex embryos. Development 139, 4271–4279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R. A., Kao H. L., Audhya A., Arur S., Mayers J. R., Fridolfsson H. N., Schulman M., Schloissnig S., Niessen S., Laband K., et al. (2011). A high-resolution C. elegans essential gene network based on phenotypic profiling of a complex tissue. Cell 145, 470–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunsalus K. C., Ge H., Schetter A. J., Goldberg D. S., Han J. D., Hao T., Berriz G. F., Bertin N., Huang J., Chuang L. S., et al. (2005). Predictive models of molecular machines involved in Caenorhabditis elegans early embryogenesis. Nature 436, 861–865 [DOI] [PubMed] [Google Scholar]
- Hamahashi S., Onami S., Kitano H. (2005). Detection of nuclei in 4D Nomarski DIC microscope images of early Caenorhabditis elegans embryos using local image entropy and object tracking. BMC Bioinformatics 6, 125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hench J., Henriksson J., Lüppert M., Bürglin T. R. (2009). Spatio-temporal reference model of Caenorhabditis elegans embryogenesis with cell contact maps. Dev. Biol. 333, 1–13 [DOI] [PubMed] [Google Scholar]
- Hunt-Newbury R., Viveiros R., Johnsen R., Mah A., Anastas D., Fang L., Halfnight E., Lee D., Lin J., Lorch A., et al. (2007). High-throughput in vivo analysis of gene expression in Caenorhabditis elegans. PLoS Biol. 5, e237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter C. P., Kenyon C. (1996). Spatial and temporal controls target pal-1 blastomere-specification activity to a single blastomere lineage in C. elegans embryos. Cell 87, 217–226 [DOI] [PubMed] [Google Scholar]
- Kamath R. S., Fraser A. G., Dong Y., Poulin G., Durbin R., Gotta M., Kanapin A., Le Bot N., Moreno S., Sohrmann M., et al. (2003). Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237 [DOI] [PubMed] [Google Scholar]
- Keller P. J., Schmidt A. D., Wittbrodt J., Stelzer E. H. (2008). Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science 322, 1065–1069 [DOI] [PubMed] [Google Scholar]
- Keller P. J., Schmidt A. D., Santella A., Khairy K., Bao Z., Wittbrodt J., Stelzer E. H. (2010). Fast, high-contrast imaging of animal development with scanned light sheet-based structured-illumination microscopy. Nat. Methods 7, 637–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon G. S., Viotti M., Hadjantonakis A. K. (2008). The endoderm of the mouse embryo arises by dynamic widespread intercalation of embryonic and extraembryonic lineages. Dev. Cell 15, 509–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner B., Crombie C., Tischler J., Fortunato A., Fraser A. G. (2006). Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat. Genet. 38, 896–903 [DOI] [PubMed] [Google Scholar]
- Liu X., Long F., Peng H., Aerni S. J., Jiang M., Sánchez-Blanco A., Murray J. I., Preston E., Mericle B., Batzoglou S., et al. (2009). Analysis of cell fate from single-cell gene expression profiles in C. elegans. Cell 139, 623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F., Peng H., Liu X., Kim S. K., Myers E. (2009). A 3D digital atlas of C. elegans and its application to single-cell analyses. Nat. Methods 6, 667–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro M. F., Kasmir J. J., Zhu J., Rothman J. H. (2005). The Wnt effector POP-1 and the PAL-1/Caudal homeoprotein collaborate with SKN-1 to activate C. elegans endoderm development. Dev. Biol. 285, 510–523 [DOI] [PubMed] [Google Scholar]
- Mango S. E., Thorpe C. J., Martin P. R., Chamberlain S. H., Bowerman B. (1994). Two maternal genes, apx-1 and pie-1, are required to distinguish the fates of equivalent blastomeres in the early Caenorhabditis elegans embryo. Development 120, 2305–2315 [DOI] [PubMed] [Google Scholar]
- Mathies L. D., Henderson S. T., Kimble J. (2003). The C. elegans Hand gene controls embryogenesis and early gonadogenesis. Development 130, 2881–2892 [DOI] [PubMed] [Google Scholar]
- McMahon A., Supatto W., Fraser S. E., Stathopoulos A. (2008). Dynamic analyses of Drosophila gastrulation provide insights into collective cell migration. Science 322, 1546–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megason S. G., Fraser S. E. (2007). Imaging in systems biology. Cell 130, 784–795 [DOI] [PubMed] [Google Scholar]
- Mello C. C., Schubert C., Draper B., Zhang W., Lobel R., Priess J. R. (1996). The PIE-1 protein and germline specification in C. elegans embryos. Nature 382, 710–712 [DOI] [PubMed] [Google Scholar]
- Mickey K. M., Mello C. C., Montgomery M. K., Fire A., Priess J. R. (1996). An inductive interaction in 4-cell stage C. elegans embryos involves APX-1 expression in the signalling cell. Development 122, 1791–1798 [DOI] [PubMed] [Google Scholar]
- Murray J. I., Bao Z. (2012). Automated lineage and expression profiling in live Caenorhabditis elegans embryos. Cold Spring Harb. Protoc. 2012, pdb.prot070615 [DOI] [PubMed] [Google Scholar]
- Murray J. I., Bao Z., Boyle T. J., Boeck M. E., Mericle B. L., Nicholas T. J., Zhao Z., Sandel M. J., Waterston R. H. (2008). Automated analysis of embryonic gene expression with cellular resolution in C. elegans. Nat. Methods 5, 703–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray J. I., Boyle T. J., Preston E., Vafeados D., Mericle B., Weisdepp P., Zhao Z., Bao Z., Boeck M., Waterston R. H. (2012). Multidimensional regulation of gene expression in the C. elegans embryo. Genome Res. 22, 1282–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann B., Walter T., Hériché J. K., Bulkescher J., Erfle H., Conrad C., Rogers P., Poser I., Held M., Liebel U., et al. (2010). Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464, 721–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piano F., Gunsalus K. C., Hill D. E., Vidal M. (2006). C. elegans network biology: a beginning. WormBook 2006, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl C., Bao Z. (2010). Chiral forces organize left-right patterning in C. elegans by uncoupling midline and anteroposterior axis. Dev. Cell 19, 402–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual J. F., Ceron J., Koreth J., Hao T., Nicot A. S., Hirozane-Kishikawa T., Vandenhaute J., Orkin S. H., Hill D. E., van den Heuvel S., et al. (2004). Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res. 14, 2162–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santella A., Du Z., Nowotschin S., Hadjantonakis A. K., Bao Z. (2010). A hybrid blob-slice model for accurate and efficient detection of fluorescence labeled nuclei in 3D. BMC Bioinformatics 11, 580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel R., Hutter H., Moerman D., Schnabel H. (1997). Assessing normal embryogenesis in Caenorhabditis elegans using a 4D microscope: variability of development and regional specification. Dev. Biol. 184, 234–265 [DOI] [PubMed] [Google Scholar]
- Schnabel R., Bischoff M., Hintze A., Schulz A. K., Hejnol A., Meinhardt H., Hutter H. (2006). Global cell sorting in the C. elegans embryo defines a new mechanism for pattern formation. Dev. Biol. 294, 418–431 [DOI] [PubMed] [Google Scholar]
- Sönnichsen B., Koski L. B., Walsh A., Marschall P., Neumann B., Brehm M., Alleaume A. M., Artelt J., Bettencourt P., Cassin E., et al. (2005). Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature 434, 462–469 [DOI] [PubMed] [Google Scholar]
- Sulston J. E., Schierenberg E., White J. G., Thomson J. N. (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64–119 [DOI] [PubMed] [Google Scholar]
- Truong T. V., Supatto W., Koos D. S., Choi J. M., Fraser S. E. (2011). Deep and fast live imaging with two-photon scanned light-sheet microscopy. Nat. Methods 8, 757–760 [DOI] [PubMed] [Google Scholar]
- Wählby C., Kamentsky L., Liu Z. H., Riklin-Raviv T., Conery A. L., O’Rourke E. J., Sokolnicki K. L., Visvikis O., Ljosa V., Irazoqui J. E., et al. (2012). An image analysis toolbox for high-throughput C. elegans assays. Nat. Methods 9, 714–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A. G., Cipriani P. G., Kao H. L., Lees B., Geiger D., Sontag E., Gunsalus K. C., Piano F. (2010). Rapid and accurate developmental stage recognition of C. elegans from high-throughput image data. Proc. IEEE Comput. Soc. Conf. Comput. Vis. Pattern Recognit. 2010, 3089–3096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W., Feng H., Santiago F. E., Kipreos E. T. (2003). CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature 423, 885–889 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.