

Abstract
Adeno-associated virus (AAV) recombination can result in chimeric capsid protein subunits whose ability to assemble into an oligomeric capsid, package a genome, and transduce cells depends on the inheritance of sequence from different AAV parents. To develop quantitative design principles for guiding site-directed recombination of AAV capsids, we have examined how capsid structural perturbations predicted by the SCHEMA algorithm correlate with experimental measurements of disruption in seventeen chimeric capsid proteins. In our small chimera population, created by recombining AAV serotypes 2 and 4, we found that protection of viral genomes and cellular transduction were inversely related to calculated disruption of the capsid structure. Interestingly, however, we did not observe a correlation between genome packaging and calculated structural disruption; a majority of the chimeric capsid proteins formed at least partially assembled capsids and more than half packaged genomes, including those with the highest SCHEMA disruption. These results suggest that the sequence space accessed by recombination of divergent AAV serotypes is rich in capsid chimeras that assemble into 60-mer capsids and package viral genomes. Overall, the SCHEMA algorithm may be useful for delineating quantitative design principles to guide the creation of libraries enriched in genome-protecting virus nanoparticles that can effectively transduce cells. Such improvements to the virus design process may help advance not only gene therapy applications, but also other bionanotechnologies dependent upon the development of viruses with new sequences and functions.
Keywords: adeno-associated virus, capsid, chimera, protein engineering, recombination, SCHEMA
INTRODUCTION
Recombination represents one mechanism by which genetic variability is introduced into viruses during evolution as they traverse protein sequence space.1,2 Exchange of genetic information through recombination has been studied to the greatest extent within RNA viruses, although there is evidence that single-stranded DNA (ssDNA) viruses acquire sequence diversity through recombination.3–6 This mechanism of creating genetic diversity is thought to be vital for multiple aspects of viral population dynamics.2,6 Homologous recombination between closely related viruses has been proposed to help maintain genomic stability by avoiding the accumulation of detrimental mutations that arise when viral genomes are replicated by error-prone DNA and RNA polymerases.2,4 In addition, homologous recombination is thought to provide viruses with an efficient means of acquiring adaptive phenotypic traits that require multiple simultaneous residue changes.7 These traits, which include altered tropisms and abilities to escape neutralizing antibodies and host immunity, are thought to improve virus fitness by creating an evolutionary advantage.3
In the laboratory, evolution is used to create viruses with new properties that have not yet been discovered in nature.8–11 One ssDNA virus that has been the focus of many studies is adeno-associated virus (AAV), a non-pathogenic member of the Parvoviridae virus family that packages a linear single-stranded DNA genome (~4.7kb) and infects mammals.12 AAV is a promising gene delivery vector for human gene therapy due to its ability to efficiently infect human cells, low rate of random chromosomal integration, and relatively low immunogenicity compared to other viruses.13,14 Efforts to improve AAV for gene therapy have focused on modifying the AAV capsid (~25nm diameter), which is composed of three monomeric protein subunits (VP1, VP2, and VP3) that self-assemble into a T = 1 icosahedral 60-mer during viral replication.15 These subunits are transcribed from the same gene using alternative splicing and variable start sites and share a common structural domain.15,16 Laboratory evolution efforts employing random recombination (DNA shuffling) for directed evolution have yielded AAVs with improved transduction of glioma cells,17 myocardium,18 neural stem cells,19 and human pluripotent stem cells.20 While these studies highlight the potential for using recombination to alter AAV function for diverse applications, the annealing-based recombination methods used in these studies have not allowed engineers to establish quantitative design rules for constructing capsid chimeras with user-defined sequences and functions.21 DNA shuffling methods used to create AAV libraries are limited to recombining closely related homologs with high (>70%) sequence identity and typically constrain crossovers to regions of high identity. These limitations can highly bias chimeric libraries in their diversity, structural disruption, and the number (and kinds) of amino acid changes relative to their parents.
Site-directed protein recombination22 may be capable of accessing additional AAV capsid diversity by creating capsid protein chimeras from distantly-related serotypes that are difficult to recombine using DNA shuffling. Another benefit to this approach is that crossover sites that are least likely to disrupt virus capsid structure can be identified using algorithms, such as SCHEMA, that anticipate the effects of each possible crossover site on protein structure.23 Using sequence and structural information from parental proteins as inputs, SCHEMA calculates the number of residue-residue contacts broken upon recombination (defined as E) in chimeras. Laboratory evolution studies using multiple enzymes (lactamases, cytochromes P450, cellulases, and arginases) have shown that E is a reliable metric for anticipating structural conservation upon recombination.24–27 Among chimeras with the same number of amino acid substitutions, those with lower calculated disruption retain structure and function with a higher frequency.24 These studies have also demonstrated that proteins with highly divergent sequences (<50% identity) can be recombined using SCHEMA to create libraries enriched in functional proteins that share as little as 70% sequence identity with known sequences.28 Furthermore, SCHEMA-guided recombination can be used to diversify catalytic function,25 increase protein thermostability,26 and identify sequence elements that contribute to stability and catalysis.27 To date, SCHEMA has not been applied to proteins that form large, megadalton complexes like AAV capsids, and it remains unclear how its predictions correlate with structural and functional properties in virus capsids.
Here we explore the effect of recombination on structural and functional conservation within the AAV capsid upon recombining distantly related AAV serotypes (AAV2 and AAV4) whose capsid proteins display 58% sequence identity within the VP3 region of the cap gene. To establish how calculated SCHEMA disruption correlates with experimental capsid disruption, we created seventeen chimeras with a range of E values and calibrated how E scales with a variety of capsid properties. This information will be critical for future studies that use SCHEMA to build larger virus mutant libraries and explore functional variation within the AAV capsid.
RESULTS AND DISCUSSION
Estimating Capsid Structural Disruption Using SCHEMA
SCHEMA disruption values were calculated using AAV4 and AAV2 capsid sequences (Fig. S1) and the AAV4 structure.15,29 These viruses assemble into capsids with similar structures (0.5Å RMSD16) containing 60 viral protein (VP) subunits, even though they have divergent sequences (~58% sequence identity within the overlapping primary sequence that is shared by VP1, VP2, and VP3). In AAV capsid structures, there were two types of residue-residue contacts (Fig. 1a) considered by SCHEMA that are predicted to contribute to the structural disruption in megadalton capsid chimeras: intramolecular and intermolecular VP subunit contacts. Intramolecular SCHEMA disruption (Eintra) was calculated as the number of residue-residue contacts within each VP subunit that are broken by homologous recombination (Fig. 1b).30 Intermolecular SCHEMA disruption (Einter) was calculated as the number of residue-residue contacts broken between a single VP subunit in the capsid and all subunits that have at least one atom within 4.5Å of that subunit (Fig. 1c). Total disruption per subunit (Esubunit) was calculated as the sum of Eintra and 0.5 × Einter, and capsid chimera disruption (Ecapsid) was calculated as the total number of intramolecular and intermolecular residue-residue contacts broken on a per capsid level (Fig. 1d). Each broken contact involving VP residues i and j is counted once when that contact is made by a residue pair within the same subunit. However, each broken intermolecular residue-residue contact involving VP residues i and j get counted twice per subunit because residues i and j within a single subunit contribute to two distinct symmetry-related ij residue-residue contacts involving multiple subunits (Fig. S2). For this reason, the number of ij residue-residue pairs counted from the perspective of a single subunit must be divided by two to obtain the number of intermolecular contacts broken per subunit within the context of a capsid.
Figure 1. Calculating SCHEMA disruption values for virus capsids.

(a) For each chimera considered, SCHEMA was used to calculate the number of residue-residue contacts broken (E) within VP chimeras created by recombining sequence elements from AAV2 (red) and AAV4 (blue). The number of contacts broken (b) within each capsid subunit and (c) between one VP subunit and all contacting subunits (transparent subunits) were used to calculate (d) the number of contacts broken within a fully assembled capsid. The total disruption per subunit (Esubunit) is defined as the sum of Eintra and 0.5·Einter, and the total disruption per capsid (Ecapsid) is calculated as 60·Eintra + 30·Einter. The chimera shown, whose subunits were created using residues 1–474 from AAV4 and 475–734 from AAV2, was mapped onto the AAV4 capsid structure (PDB ID: 2G8G) using Pymol.
To identify simple chimeras to construct for calibrating SCHEMA predictions, we calculated disruption (E) and amino acid substitution (m) levels for all possible single-crossover and double-crossover chimeras that can arise from homologous recombination of AAV2 and AAV4 within the region of the VP sequence whose structure is known (Fig. 2). Within the AAV capsid sequences considered by SCHEMA, AAV2 differs in sequence from AAV4 at 223 positions within its primary sequence. The m for each chimera is reported relative to AAV4 (m = 0). Using this frame of reference, the m of AAV2 is 223 and chimeras that are most divergent from both parental proteins have an intermediate m value. These calculations revealed that, on average, E values correlate with the mutation distance from AAV2 and AAV4 (Figs. 2 and S3). However, in the case of multiple crossover events, chimeras can be generated at each value of m with a range of E values, which can differ by over an order of magnitude. For example, at an m = 128, Ecapsid can range from 630 to 6,420 for double-crossover chimeras. A large range of Eintra and Einter was also observed at each m for double-crossover chimeras (Fig. S4). Since it is unclear whether or not intramolecular and intermolecular contacts broken by recombination are equally disruptive, four single-crossover and thirteen double-crossover chimeras displaying a range of disruption and mutation values were chosen for construction and characterization (Table S1). A comparison of chimera gene sequences and parental AAV2 and AAV4 shows that a subset of these chimeras grafts the heparin-binding domain (HBD) from AAV2 into AAV4 (Fig. 3). Additionally, a subset of our chimeras recombine the assembly-activating protein (AAP),31 a protein that is required for efficient capsid assembly. The sequence identity between AAP from AAV2 and AAV4 (54%) is similar to that observed for the structural proteins that make up the capsid. The structure of AAP is not yet known, so it could not be considered by SCHEMA.
Figure 2. SCHEMA disruption in chimeric VP capsids.

Capsid disruption (Ecapsid) was calculated for all chimeras that can be created by recombining AAV2 and AAV4 using (a) single and (b) double crossovers. Capsid disruption is shown relative to the per subunit amino acid substitution level (msubunit), which was calculated relative to AAV4 (m = 0).
Figure 3. SCHEMA values and genome packaging properties of AAV2-AAV4 capsid chimeras.

AAV2 (red) and AAV4 (blue) genes were recombined to create capsid proteins with a range of sequence properties. Translation initiation sites (black dashes) are shown for each VP protein (VP1, VP2, and VP3), as well as the location of the AAV2 heparin-binding domain (HBD, yellow dashes) that is not present in AAV4. Locations of AAV4 and AAV2 assembly-activating proteins (AAP) are indicated under the AAV2 sequence. The effective level of mutation per chimeric subunit (msubunit) is the minimum number of amino acid mutations required to convert each chimera into AAV4, and Ecapsid represents the number of per capsid residue-residue contacts disrupted by recombination. The genomic titers of each chimera were measured by Q-PCR and scored on a scale of 1 to 4 (diamonds) relative to the detection limit and the titers obtained with AAV2 and AAV4. Each diamond represents approximately 1-log in titer. n.d. = not detected above background.
Analysis of Chimera Capsid Assembly and Genome Packaging
Upon homologous recombination, VP subunits can display a range of phenotypes. VP chimeras can: (i) misfold or aggregate during translation, (ii) fold into a native-like topology, but fail to assemble into a capsid, (iii) fold and self-assemble into a capsid-like structure but fail to package their DNA cargo, (iv) form capsids that contain DNA which is not well protected from degradation, (v) form capsids that contain DNA with native AAV stability but defective in cellular binding and transduction, (vi) form capsids with native AAV stability and cellular binding but defective in transduction, or (vii) form capsids with native AAV stability and transduction. To first assess the structural disruption of our chimeras, we examined whether they assembled into capsids and packaged a genome. This was accomplished by expressing VP chimeras in HEK293T cells and separating fully assembled capsids from incompletely assembled capsids using a discontinuous iodixanol density gradient. VP subunits sediment to different layers of the gradient during ultracentrifugation based on their ability to self-assemble into complete 60-mers and package genomes. Genome-containing capsids reside in the 40% iodixanol layer, whereas empty capsids and other capsid intermediates reside within the interface between the 25% and 40% iodixanol layers.32–35 Viral genome packaging was analyzed using quantitative polymerase chain reaction (Q-PCR) as previously described.10
Q-PCR analysis of virus genomic titers extracted from the 40% iodixanol gradient layer is shown in Figure 3. Of the seventeen VP chimeras generated, nine yielded genomic titers detectable above background with values varying by approximately four orders of magnitude (Fig. S5). The three chimeric capsids with the highest genomic titers differed by less than 1-log from the titers observed with AAV2 and AAV4. Genomic DNA packaging did not correlate strongly with m or E. Chimeras with detectable titers differed by an average of 65 residues per subunit from either parent, whereas chimeras that did not protect genomes differed by an average of 58 residues. In addition, the values of Ecapsid for chimeras that packaged genomes ranged from 60 to 4,800 (average = 1,786 ± 1,522), whereas Ecapsid for chimeras that did not package genomes ranged from 930 to 2,100 (average = 1,818 ± 351). This finding demonstrates that AAV capsid structures tolerate larger numbers of broken residue-residue contacts compared with smaller enzymes whose recombination tolerance has been calibrated against SCHEMA.24,27,30 Furthermore, this result suggests that a large fraction of the VP chimeras that can be created by recombining AAV2 and AAV4 (Fig. 2) will form capsids and package DNA, since a large fraction of the possible double-crossover chimeras have an Ecapsid within the range that we sampled.
To assess whether chimeras with virus titers below the limits of detection simply have defects in VP chimera expression or if they express VP chimeras and assemble oligomers to some extent, we performed western blot analysis on VP chimeras within the 40% iodixanol layer (data not shown) and 25–40% iodixanol interface (Fig. S6). Whereas the 40% layer contains full (genome-packaging) 60-mer capsids, empty capsids and VP protein oligomers that are smaller than a fully assembled capsid partition to the 25–40% iodixanol interface.32–35 Due to the relatively low genomic titers of the chimeras and sensitivity limitation of our antibody, VP expression was not detected for most chimeras in the 40% layer. VP subunits from all of the single-crossover chimeras could be detected in the 25–40% interface using the B1 monoclonal antibody, which binds to an epitope within the C-terminus of each AAV2 VP. Since the double-crossover chimeras lack the epitope recognized by the B1 antibody, we used the A1 monoclonal antibody that reacts with a conserved epitope in the VP1 subunit of AAV2 and AAV4. A majority of the chimeras with undetectable genomic titers expressed VP1 at detectable levels in the 25–40% interface, albeit many at lower levels than AAV2 and AAV4. Only the VP1 from 254–435 could not be readily detected. These observations suggest that most of our chimeric VP subunits associate together into capsid-like oligomers. The finding that a vast majority of our chimeras retain the ability to oligomerize suggests that VP subunit oligomerization does not depend on Ecapsid in the range that we sampled.
Using Genome Protection as Measure of Capsid Intactness
While several chimeric VP proteins assembled into oligomers stable enough to protect genomes through iodixanol gradient ultracentrifugation and extraction, it was unclear how well these protein subunits were assembled together. To test if these capsids had similar structural integrity as AAV2 and AAV4, we investigated the extent to which each chimeric capsid protected its genome from nuclease digestion after purification and used the level of protection as a measure of capsid intactness.36 Chimeras with genomic titers detectable above background were treated with benzonase, and the percentage of genomes remaining after nuclease treatment was measured using Q-PCR. The genomic titers of nuclease-treated AAV2 and AAV4 did not decrease significantly compared to sham buffer-treated samples (Fig. 4a). As negative control, unprotected DNA encoding green fluorescence protein (GFP) was degraded to undetectable levels by a similar treatment. We found that chimeras protected genomes to varying extents. Interestingly, capsid intactness did not correlate with the measured genomic titers of the chimeras. Chimeras 438–633 and 492–734 did not protect their genomes well (1.1 ± 0.2% and 54.3 ± 2.3%, respectively) despite having high titers. On the other hand, chimera 345–357 protected its genome (108.5 ± 5.0%) despite its titer being more than 1,000-fold lower than either parent. While the linear correlation between Ecapsid and the observed nuclease protection among our small sampling of chimeras is weak (r = −0.28), the three chimeras (655–670, 607–612, and 345–357) with the greatest resistance to benzonase (98.2 ± 9.4%, 101.6 ± 5.0%, 108.5 ± 5.0%, respectively) also have the lowest calculated Ecapsid values: 60, 90, and 180, respectively. The average Ecapsid for these variants (110 ± 62) is >20-fold lower than that observed with the benzonase-sensitive chimeras (2,388 ± 1,741). Thus, calculated structural disruption correlates with capsid intactness to a greater extent than genome packaging.
Figure 4. Chimeric capsid intactness determined by nuclease sensitivity and TEM imaging.

(a) Q-PCR was used to determine the percentage of viral genomes remaining after treatment with benzonase. Percentage protected represents benzonase treated samples normalized to a sham condition lacking nuclease. Chimeras are sorted horizontally based on their genomic titers compared to wt capsids; diamonds at the top represent relative titers of each chimera before nuclease treatment compared to parental capsids as in Fig. 3. Error bars indicate the standard error of the mean (SEM) calculated from three independent experiments. Transmission electron microscopy of (b) AAV2, (c) AAV4, and (d) chimera 438–633 show that AAV2 and AAV4 capsids exclude negative stain whereas the chimeric 438–633 capsids contain negative stain (dark centers). Images were taken at 40,000x magnification; insets were taken at 150,000x magnification.
To better understand the capsid structure of chimera 438–633, which had a high titer but low nuclease-protection ability, we imaged this chimera using transmission electron microscopy (TEM) and compared the results to that of AAV2 and AAV4. Q-PCR was used to verify high genomic titers (>1011 genomes/mL) of virus samples one day prior to negative staining in order to ensure the capsids being imaged still contained genomes after buffer exchange (Table S2). At both lower (40,000x) and higher magnifications (150,000x), phenotypic differences are observed between chimera 438–633 and the wild type capsids. Whereas AAV2 (Fig. 4b) and AAV4 (Fig. 4c) capsids have clearly defined straight edges that form hexagonal particles (due to the icosahedral symmetry of the virus), chimera 438–633 capsids are irregularly shaped (Fig. 4d), although they have a similar diameter as native AAV. In addition, the negative staining of AAV2 and AAV4 reveals mostly intact capsids, whereas all of the chimeric 438–633 capsids appear empty, as evidenced by their darkened, uranyl formate-stained capsid interiors. These findings suggest that the genomes protected by this chimera were sensitive to the sample preparation protocol prior to TEM analysis, which included buffer exchange, concentration, and uranyl formate staining. Collectively, our data suggest chimera 438–633 forms a genome-containing capsid but has a defect in capsid intactness.
Ability of Chimeras to Bind Heparin
The first step in the AAV infection process is binding to glycoproteins on cell surfaces. The two AAV serotypes recombined differ in the proteoglycans and co-receptors on cell surfaces that they bind as a means of transduction. AAV2 binds negatively charged heparan sulfate proteoglycan (HSPG) using a cluster of positively charged amino acids (R487, R585, R588) at the 3-fold axis of symmetry within the capsid.37,38 In contrast, AAV4 binds α-2,3 O-linked sialic acid.39 The exact AAV4 capsid residues critical for this binding event have yet to be determined.16 Previous studies have demonstrated that insertion of the AAV2 HSPG binding domain into AAV5, which naturally does not bind HSPG, confers HSPG binding to AAV5 with similar affinity as AAV2.38 This previous finding suggested that AAV2-AAV4 chimeras containing the HSPG binding might also bind HSPG.
To test whether chimeras containing or lacking the AAV2 HSPG binding domain (HBD) associate with heparin, we performed heparin affinity chromatography. This was achieved by incubating viruses with heparin-sepharose beads, washing the beads to remove non-specific binding, and eluting bound viruses using a series of buffers with increasing ionic strengths. To determine the relative fractions of heparin-bound virus after each step, genomic titers within each elution were measured using Q-PCR (Fig. 5). Due to Q-PCR sensitivity limitations, we were only able to characterize heparin binding of chimeras with titers >5×109 genomes/mL. Chimeras that lack the HBD uniformly did not bind heparin and displayed a similar elution profile as wild type AAV4 capsids (Fig. S7). In contrast, all chimeras containing the AAV2 HBD associated with heparin, albeit to varying degrees. Chimeras deriving a larger fraction of their primary sequence from AAV2 bound heparin stronger than those with a smaller amount of AAV2 sequence. These findings demonstrate that although the cluster of positively charged amino acids (R487, R585, R588) in AAV2 is required for heparin binding, other sequence elements in AAV2 contribute to the strength of the interaction with heparin.
Figure 5. Association of chimeric capsids with heparin.

Heparin binding of chimeras containing (black) and lacking (white) the AAV2 heparin-binding domain (HBD) was analyzed using a heparin affinity column. Q-PCR was used to quantify the number of viruses that did not bind to the column, the number that eluted when the column was washed with buffer, and the number that eluted when the column was washed with buffers containing 250 mM, 650 mM, and 5 M NaCl. For each virus, the number of genomes detected by Q-PCR in all of the salt elutions was divided by the total number of viruses collected from the initial unbound flow-through, buffer wash, and salt elutions. Error bars indicate the SEM calculated from three independent experiments.
Transduction of Cells with Chimeric Capsids
To determine whether chimeras retained the ability to transduce cells, we incubated capsid chimeras that contained the GFP gene with Cos-7 cells and measured GFP reporter expression 48 hrs after infection using flow cytometry. We chose Cos-7 cells for this transduction assay because this cell line has the cell-surface receptors (HSPG and sialic acid) required for transduction by both AAV2 and AAV4.39 Only chimeras with genomic titers >2×109 genomes/mL had their in vitro transduction efficiency analyzed because this level of virus was required for a strong reproducible signal from our positive controls under our experimental conditions. At a MOI of 5,000 viruses/cell, chimeras displayed a range of transduction efficiencies (Fig. 6). As previously reported, AAV2 and AAV4 both yielded high percentages of GFP-positive cells.39 While two of the chimeras tested (655–670 and 607–612) infected Cos-7 cells with similar efficiency as AAV4 (>30% GFP-positive cells), three chimeras produced a low percentage of GFP-positive cells (<2%). SCHEMA disruption calculations revealed the chimeras that transduced most efficiently were those with low Ecapsid (60 and 180, respectively). The Ecapsid for these chimeras are >24-fold lower than the average Ecapsid for the remaining chimeras whose transduction was assessed. While our sample size is small (n = 5), the linear correlation between Ecapsid and the transduction efficiency of our chimeras is relatively strong (r = −0.70). Thus, calculated structural disruption is inversely correlated to the functional conservation of cellular transduction.
Figure 6. Transduction of Cos-7 cells by chimeric capsids.

Chimeric capsid transduction efficiency was compared to that of AAV2 and AAV4 by analyzing the level of GFP expression in Cos-7 cells after incubation with viruses carrying the GFP transgene. Reporter expression was quantified by flow cytometry 48 hrs post-transduction. Data represent the percentage of cells (out of 10,000 cells per sample) that expressed the GFP reporter and is shown as the mean of three independent experiments with error bars corresponding to the SEM. *, p<0.05. NS = not significant.
Schema-based Design Rules
Within our small chimera sample (n = 17), we obtained evidence that SCHEMA disruption is a useful metric for anticipating structural conservation in virus capsid chimeras generated via recombination (Table S3). We found that both chimeric capsid intactness and transduction were inversely related to Ecapsid for those chimeras that packaged genomes (Fig. 7). These results can be used to inform future studies that seek to create larger libraries of AAV chimeras that are enriched in VP subunits that assemble into capsids, package genomes, and transduce cells. Such libraries can be identified using the RASPP algorithm, which finds crossovers that minimize the average Ecapsid of chimeras encoded within different libraries subject to constraints on the distance between crossovers.40 RASPP-designed libraries should be useful for building artificial families of capsid chimeras, and analysis of sequence-function relationships in such libraries should help identify sequence elements that contribute to different aspects of AAV structure and function.26,27,41
Figure 7. Calculated structural disruption in capsids is inversely correlated with genome-protection and transduction ability.
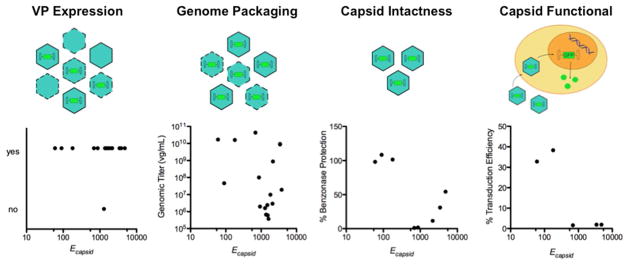
The sequence space for finding chimeric capsids depends on the definition of structural and functional tolerance to recombination, with high stringency definitions increasing from left to right. The top images illustrate how theoretical capsid populations (green) change with each tolerance definition (empty = no internal genomes, full = with genomes, incomplete and/or unstable capsids = dotted capsid outlines). The bottom graphs summarize the relationship between Ecapsid and capsid measurements, including (i) western immunoblot detection of VP subunit expression (ii) Q-PCR detection of genome packaging which requires capsids to associate with the genome through an iodixanol density gradient, (iii) benzonase protection of genomes after capsid purification, a more stringent definition of capsid intactness that requires capsids to be stably assembled around genomes, and (iv) cellular transduction. Among these four capsid properties, only capsid intactness and cellular transduction were inversely correlated with Ecapsid.
Previous studies have observed correlations between calculated SCHEMA disruption and molecular interactions that are dependent upon structural conservation like binding properties of proteins, such as heme binding to cytochromes P450.42 However, we did not observe a strong correlation between genome packaging by chimeric capsids and SCHEMA disruption. More than half of the chimeric VP proteins formed capsids that purified with viral genomes, including the three chimeras with the highest Ecapsid values. Surprisingly, the number of broken residue-residue contacts tolerated in these chimeras (>3,000 per capsid) is almost two orders of magnitude greater than the level that has been shown to disrupt the folding of monomeric enzymes.42 This finding indicates AAV capsid structures are robust to amino acid substitutions created by recombination,43 and it suggests that even the most distantly related AAV serotypes can be randomly recombined to create libraries that encode virus particles with genomes, albeit potentially deficient in transduction activity.
Among the double-crossover chimeras that did not protect viral genomes, more than half had crossovers in locations that resulted in recombination of AAP, the AAV protein required for efficient capsid assembly.31 Western blot analysis revealed that a majority of the chimeras with recombined AAP were expressed and soluble but unable to assemble into AAV-like genome-containing capsids, which may be because AAP activity is disrupted in these variants.31,44 This finding also suggests that one way to enrich the fraction of capsid forming chimeras in future recombination studies is to avoid crossover sites that recombine AAP or to co-express native AAP with VP chimeras. The former approach can be achieved by using SCHEMA to guide noncontiguous recombination of AAV VP proteins.45 Future selections of AAV libraries will be required to assess which approach is most effective for enriching libraries in capsid chimeras that package genomes. Studies involving libraries will also be useful for ascertaining whether each type of broken contact (intramolecular and intermolecular) is equally disruptive to capsid structure and function. If they are not equally disruptive, then a scaling factor will be needed to improve SCHEMA predictions of capsid structure and function.
Collectively, our results demonstrate the first application of the SCHEMA algorithm to the design of virus nanoparticles through site-directed recombination. Building and characterizing our small sample of virus chimeras have enabled us to calibrate SCHEMA predictions of different AAV capsid properties. Future work will involve building large chimera libraries and mining them for sequence-structure-function relationships with the goal of obtaining statistically rigorous quantitative design rules for virus capsid engineering. Virus nanoparticles engineered through site-directed recombination may find widespread use as supramolecular structures for drug delivery, bioimaging, tissue engineering, nanocatalysis, and nanotechnology.46–50
MATERIALS AND METHODS
SCHEMA Calculations
The crystallographic coordinates for the AAV4 capsid (PDB ID: 2G8G)29 and an AAV4 and AAV2 alignment of VP1 residues 211 to 734 were used with Equation 1 to calculate the number of residue-residue contacts (E) broken by recombination.
| (Equation 1) |
For these calculations, residues i and j were defined as contacting (cij = 1) if any atoms, excluding hydrogen and backbone carbon and nitrogen, were within 4.5 Å of one other within the capsid, otherwise cij = 0. If residues i and j formed an interacting residue-residue pair observed in either parent, Pij = 0, otherwise Pij = 1. Past studies observed a correlation between calculated and measured structural disruption when this distance cutoff was used for SCHEMA calculations.24–26 Contacts broken per subunit (Esubunit) were calculated as the sum of the number of contacts broken within one individual subunit (Eintra) and half of the number of contacts broken between that subunit and adjacent contacting subunits (Einter). Capsid chimera disruption (Ecapsid) was calculated as (subunits in a capsid)·Eintra + 1/2·(subunits in a capsid)·Einter. Intermolecular subunits are scaled differently because each residue-residue pair involves two subunits.
Cells
Escherichia coli XL1-Blue from Stratagene was used to amplify the plasmids built for this study, HEK293T cells were used to produce AAV capsids, and Cos-7 cells were used to analyze chimeric capsid transduction. HEK293T cells were maintained in Dulbecco’s modified Eagle Medium (DMEM, Life Technologies) supplemented with 10% fetal bovine serum (FBS, Atlanta-Biologicals) and 1% penicillin and streptomycin (Life Technologies). Cos-7 cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin and streptomycin. Cells were grown as adherent cultures in 5% CO2 at 37°C, sub-cultured after treatment with 0.25% trypsin-EDTA (Life Technologies) for 2–5 min at 37°C, and resuspended in complete medium.
AAV cap Gene Vectors
The vectors pXX252 and pXR453 were used to produce AAV2 and AAV4 viruses in HEK293T cells. To create a vector for expressing chimeric VP, pXR4 was PCR amplified to create pXR4-nocap, a vector that contains a unique SpeI restriction site at the codons encoding AAV4 VP1 residues 107 and 108 followed by unique NsiI and BmtI sites (Fig. S8). Each cap gene chimera was cloned into these unique restriction sites to create plasmids for expressing chimeric capsid subunits in HEK293T cells.
Capsid Gene Chimera Construction
To build single-crossover cap genes, Vent DNA polymerase was used to amplify AAV4 cap gene fragments using primers that amended 20 to 30 base pairs of the AAV2 cap gene to the 3’ end of the AAV4 amplicon, and AAV2 cap gene fragments were PCR amplified using primers that amended 20 to 30 base pairs of the AAV4 cap gene to the 5’ end of the AAV2 amplicon. Gibson assembly51 was used to splice each pair of fragments together to create an AAV4/AAV2 cap gene chimera, and these genes were cloned into pXR4-nocap using either Gibson assembly or standard cloning into the SpeI and BmtI restriction sites. A similar protocol was used to build and clone double-crossover cap genes. All plasmids were sequence verified. Chimera names indicate the first and last residue of a contiguous AAV2 fragment that was inserted into AAV4 according to the numbering of AAV4 (Fig. S9).
Virus Production
AAV4, AAV2, and chimeric virus capsids containing the GFP gene driven by cytomegalovirus (CMV) promoter were prepared by triple plasmid cotransfection of: (i) pITR-GFP (a plasmid containing a GFP transgene flanked by AAV inverted terminal repeats), (ii) pXX6 (a plasmid encoding adenoviral genes required for capsid assembly), and (iii) AAV4/AAV2 chimeric cap gene plasmid or wild type AAV2 (pXR2) or AAV4 (pXR4) cap gene plasmid into HEK293T cells using linear polyethylenimine with nitrogen:phosphate ratio of 20:1. Cells from a single 15-cm dish (BD Falcon) pre-coated with 0.001% poly-L-lysine (Sigma) were harvested and pelleted 48 hrs post-transfection, resuspended in 2 mL of gradient buffer (GB: 10 mM MgCl2, 150 mM NaCl, 10 mM Tris, pH 7.6), and lysed by three freeze-thaw cycles. Samples were treated with benzonase (Sigma) at 50 units/mL for 40 min at 37°C to remove nucleic acids. For TEM studies, 10 × 15-cm dishes were transfected. The rest of the protocol was the same as the single-dish preps, except cells were resuspended in 13 mL of GB prior to the freeze-thaw cycles. Cell lysate was collected after spinning at 3000 x g for 20 min at 4°C and purified by a 15–54% iodixanol step gradient in a Beckman quick-seal centrifuge tube. Sealed tubes were centrifuged at 48,000 rpm in a Beckman Type 70Ti rotor for 1.75 hrs at 18°C. Viruses were extracted from the 40% iodixanol layer using an 18-gauge needle and 3 mL syringe. 25–40% interface layers (containing empty capsids or virus-like particles) were also collected for further analysis. Viruses were stored in 3 mL cryovials (Biotix) at 4°C.
Quantifying Genomic Titers
Viruses were denatured by incubating them in 2 N NaOH at 75°C for 30 min. An equal volume of 2 N HCl was added prior to diluting samples using ultra-pure water containing sheared salmon sperm DNA (10 μg/mL, Life Technologies) as a carrier. Samples were titered by Q-PCR using SYBR green master mix (Applied Biosystems) and a C1000 thermal cycler (Bio-Rad) with primers against the CMV promoter (TACCGGGGATTTCCAAGTCTC and AATGGGGCGGAGTTGTTACGA). The virus preparation was applied to the pBlueScript II plasmid (lacking the AAV cap gene) as a negative control to determine the background signal.
Western Blotting
Virus samples were denatured by incubating at 75°C for 15 min after mixing with NuPAGE LDS Sample Buffer and NuPAGE Sample Reducing Agent (Life Technologies). Denatured samples were then loaded into the wells of NuPAGE Novex 7% Tris-Acetate gels. NuPAGE antioxidant was added to the running buffer used for gel electrophoresis. Gels were run at 10V for 10 min, 100V for 100 min, and transferred to nitrocellulose membranes at 30V for 90 min at 4°C. After a phosphate buffered saline with 0.1% Tween-20 (PBS-T) wash, nitrocellulose membranes were blocked using 5% milk in PBS-T for 1 hr at room temperature. After three PBS-T washes, membranes were incubated with the B1 monoclonal primary antibody (1:50, American Research Products) or A1 monoclonal primary antibody (1:100, American Research Products) in 3% BSA overnight at 4°C. Blots were washed using PBS-T (3x for 10 min each). Blots were then incubated with goat-anti-mouse HRP-conjugated secondary antibody (1:2000) in PBS-T containing 5% milk at room temperature for 90 min. After three sequential 10 min washes in PBS-T, blots were treated with Lumi-Light Western Blotting Substrate (Roche Applied Science) and imaged using a Fuji LAS-4000 imager.
Transmission Electron Microscopy
We generated 10-plate virus preparations of AAV2, AAV4, and 438–633 to obtain sufficient capsids for imaging. After iodixanol gradient separation, the virus extracted from the 40% layer was concentrated using an Amicon Ultra-4 centrifugal unit to increase the virus concentration and exchange the viruses into GB containing 0.001% Pluronic F-68 (Sigma). Continuous carbon sample grids (300 mesh; Ted Pella) were glow discharged, and each virus sample was applied to the grid and incubated for 5 min. Grids were wicked dry with filter paper and washed twice by immersion in separate drops of 50 μl of ultrapure water, wicking dry in between each wash. Each sample was negatively stained by immersion in 7.5 mg/mL uranyl formate (Ted Pella) that had passed through 0.2 μm filter. The sample was wicked dry and left to air dry for 15 min. TEM images were taken using a JEM FasTEM 2010 transmission electron microscope at 40,000x and 150,000x magnifications.
Nuclease Protection Assay
Viruses collected from the 40% iodixanol fraction (5 μL) were added to 45 μL of 1x Endo buffer (1.5 mM MgCl2, 0.5 mg/mL BSA, 50 mM Tris, pH 8.0). After mixing, samples were split into two PCR tubes (20 μL each) and incubated with 0.5 μL of benzonase nuclease (250 units/uL; Sigma) or 0.5 μL sham buffer (50% glycerol, 50 mM Tris-HCl, pH 8.0, 20 mM NaCl, 2 mM MgCl2) for 30 min at 37°C. Reactions were terminated by adding 0.5 M EDTA (0.5 μL). Samples (10 μL) were subsequently denatured by adding 2 N NaOH (10 μL), incubating the mixture at 75°C for 30 min, and adding 2 N HCl (10 μL) to neutralize the reaction. Titers were measured using Q-PCR. The percentage of protected genomes was calculated as 100x the titers of (benzonase treated) / (sham treated) samples.
Heparin Binding Assay
Only those viruses that could be purified to titers above 5×108 genomes/mL were tested for heparin binding. Heparin-agarose beads (100 μL, Sigma H-6508) were loaded into a low-retention 1.5 mL tube (Phenix Research Products) and washed twice with 200 μL of TD buffer (137 mM NaCl, 15 mM KCl, 10 mM Na2PO4, 5 mM MgCl2, 2 mM KH2PO4 pH 7.4). Approximately 5×108 to 1×109 viral genome-containing particles were added to TD to a final volume of 100 μL and were incubated with washed heparin-agarose beads for 15 min at room temperature with constant rotation. Samples were centrifuged for 5 min at 6,000 x g in an Eppendorf microcentrifuge, and the unbound supernatants were collected. The heparin-agarose beads were washed with 300 μL of TD. Bead-bound viruses were eluted using three buffers (200 μL each) containing increasing concentrations of NaCl (250 mM, 650 mM, and 5 M). Unbound, wash, and the three eluate fractions were collected separately, denatured by adding 2 N NaOH and incubating at 75°C for 30 min, neutralized by adding 2 N HCl, and titered with Q-PCR.
Transduction Assay
Cos-7 cells were seeded on 48-well plates at a density of 20,000 cells/well. After 20 h, cells were transduced with AAV2, AAV4, or chimeric viruses encoding GFP using an MOI of 5,000. Cells were incubated in the virus-containing, serum-free medium for 4 hr at 37°C. Media was replaced with complete serum-containing medium and incubated for 48 h. Cells were washed with PBS, detached from the plates by adding 0.25% trypsin-EDTA and incubating for 5 min at 37°C, and collected by spinning at 700 x g for 5 min at 4°C. Supernatant was discarded and cells were resuspended in 500 μL of 4°C PBS containing EDTA (5 mM). Transduction efficiency was quantified by analyzing the fraction of GFP-positive cells using a FACSCanto II flow cytometer (BD Biosciences) (10,000 total events collected).
Supplementary Material
Figure S1. Sequence alignment of the AAV capsid proteins recombined.
Figure S2. Scaling intermolecular contacts from subunits to capsids.
Figure S3. Eintra and Einter for single-crossover chimeras.
Figure S4. Eintra and Einter for double-crossover chimeras.
Figure S5. Genomic titers of chimeras.
Figure S6. Immunoblot analysis of chimeric VP expression.
Figure S7. Binding of chimeric capsids to heparin.
Figure S8. pXR4-nocap plasmid sequence.
Figure S9. AAV2-AAV4 chimera nomenclature.
Table S1. Subunit SCHEMA disruption values used to calculate Ecapsid.
Table S2. Genomic titer of viruses prepared for TEM before and after concentration.
Table S3. Summary of chimera characteristics.
Acknowledgments
Funding Sources: This research was supported by the Keck Center of the Gulf Coast Consortia Nanobiology Interdisciplinary Graduate Training Program (National Institute of Biomedical Imaging and Bioengineering T32EB009379) to MLH, Robert A. Welch Foundation (Grant No. C-1614) to JJS, National Science Foundation (Grant No. 1150138) to JJS, National Science Foundation Research Experiences for Undergraduates (Grant No. 1004853) to BAA.
The authors would like to thank Esteban Pimentel, Justin Chen, Ayesha Khan, and Shirley Liu for their assistance in the early phases of the study and Christopher Dempsey for help with electron microscopy.
ABBREVIATIONS
- AAP
assembly-activating protein
- AAV
adeno-associated virus
- CMV
cytomegalovirus
- GFP
green fluorescence protein
- HSPG
heparan sulfate proteoglycan
- HBD
HSPG binding domain
- MOI
multiplicity of infection
- Q-PCR
quantitative polymerase chain reaction
- ssDNA
single-stranded DNA
- TEM
transmission electron microscopy
- VNP
virus nanoparticle
Footnotes
Author Contributions: Conceived and designed experiments: MLH MLT JJS JS. Performed experiments: MLH BAA MLT. Analyzed data: MLH BAA JJS JS. Wrote manuscript: MLH BAA JJS JS.
Contributor Information
Michelle L. Ho, Email: michelle.liane.ho@gmail.com.
Benjamin A. Adler, Email: badler91@gmail.com.
Michael L. Torre, Email: mltorre3@gmail.com.
LITERATURE CITED
- 1.Negroni M, Buc H. Mechanisms of retroviral recombination. Annual review of genetics. 2001;35:275–302. doi: 10.1146/annurev.genet.35.102401.090551. [DOI] [PubMed] [Google Scholar]
- 2.Martin DP, Biagini P, Lefeuvre P, Golden M, Roumagnac P, Varsani A. Recombination in eukaryotic single stranded DNA viruses. Viruses. 2011;3:1699–738. doi: 10.3390/v3091699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao G, Alvira MR, Somanathan S, Lu Y, Vandenberghe LH, Rux JJ, Calcedo R, Sanmiguel J, Abbas Z, Wilson JM. Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:6081–6. doi: 10.1073/pnas.0937739100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lefeuvre P, Lett JM, Varsani a, Martin DP. Widely conserved recombination patterns among single-stranded DNA viruses. Journal of virology. 2009;83:2697–707. doi: 10.1128/JVI.02152-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lau SKP, Woo PCY, Yip CCY, Li KSM, Fu CTY, Huang Y, Chan KH, Yuen KY. Co-existence of multiple strains of two novel porcine bocaviruses in the same pig, a previously undescribed phenomenon in members of the family Parvoviridae, and evidence for inter- and intra-host genetic diversity and recombination. The Journal of general virology. 2011;92:2047–59. doi: 10.1099/vir.0.033688-0. [DOI] [PubMed] [Google Scholar]
- 6.Duffy S, Shackelton La, Holmes EC. Rates of evolutionary change in viruses: patterns and determinants. Nature reviews Genetics. 2008;9:267–76. doi: 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- 7.Felsenstein J. The evolutionary advantage of recombination. Genetics. 1974;78:737–56. doi: 10.1093/genetics/78.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nature Biotechnology. 2006;24:198–204. doi: 10.1038/nbt1182. [DOI] [PubMed] [Google Scholar]
- 9.Mateu MG. Virus engineering: functionalization and stabilization. Protein engineering, design & selection: PEDS. 2011;24:53–63. doi: 10.1093/protein/gzq069. [DOI] [PubMed] [Google Scholar]
- 10.Judd J, Wei F, Nguyen PQ, Tartaglia LJ, Agbandje-McKenna M, Silberg JJ, Suh J. Random Insertion of mCherry Into VP3 Domain of Adeno-associated Virus Yields Fluorescent Capsids With no Loss of Infectivity. Molecular therapy Nucleic acids. 2012;1:e54. doi: 10.1038/mtna.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powell SK, Kaloss Ma, Pinkstaff a, McKee R, Burimski I, Pensiero M, Otto E, Stemmer WP, Soong NW. Breeding of retroviruses by DNA shuffling for improved stability and processing yields. Nature biotechnology. 2000;18:1279–82. doi: 10.1038/82391. [DOI] [PubMed] [Google Scholar]
- 12.Muzyczka Nicholas, KIB . Parvoviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields virology. 4. Lippincott, Williams and Wilkins; New York, NY: 2001. pp. 2327–2360. [Google Scholar]
- 13.Kotin RM. Prospects for the use of adeno-associated virus as a vector for human gene therapy. Human gene therapy. 1994;5:793–801. doi: 10.1089/hum.1994.5.7-793. [DOI] [PubMed] [Google Scholar]
- 14.Daya S, Berns KI. Gene therapy using adeno-associated virus vectors. Clinical microbiology reviews. 2008;21:583–93. doi: 10.1128/CMR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie Q, Bu W, Bhatia S, Hare J, Somasundaram T, Azzi A, Chapman MS. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:10405–10. doi: 10.1073/pnas.162250899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Padron E, Bowman V, Kaludov N, Govindasamy L, Levy H, Nick P, McKenna R, Muzyczka N, Chiorini JA, Baker TS, Agbandje-McKenna M. Structure of adeno-associated virus type 4. Journal of virology. 2005;79:5047–58. doi: 10.1128/JVI.79.8.5047-5058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maguire Ca, Gianni D, Meijer DH, Shaket La, Wakimoto H, Rabkin SD, Gao G, Sena-Esteves M. Directed evolution of adeno-associated virus for glioma cell transduction. Journal of neuro-oncology. 2010;96:337–47. doi: 10.1007/s11060-009-9972-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang L, Jiang J, Drouin LM, Agbandje-McKenna M, Chen C, Qiao C, Pu D, Hu X, Wang DZ, Li J, Xiao X. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and in vivo selection. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3946–51. doi: 10.1073/pnas.0813207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jang JH, Koerber JT, Kim JS, Asuri P, Vazin T, Bartel M, Keung A, Kwon I, Park KI, Schaffer DV. An evolved adeno-associated viral variant enhances gene delivery and gene targeting in neural stem cells. Molecular therapy: the journal of the American Society of Gene Therapy. 2011;19:667–75. doi: 10.1038/mt.2010.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Asuri P, Bartel Ma, Vazin T, Jang JH, Wong TB, Schaffer DV. Directed evolution of adeno-associated virus for enhanced gene delivery and gene targeting in human pluripotent stem cells. Molecular therapy: the journal of the American Society of Gene Therapy. 2012;20:329–38. doi: 10.1038/mt.2011.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartel Ma, Weinstein JR, Schaffer DV. Directed evolution of novel adeno-associated viruses for therapeutic gene delivery. Gene therapy. 2012;19:694–700. doi: 10.1038/gt.2012.20. [DOI] [PubMed] [Google Scholar]
- 22.Carbone MN, Arnold FH. Engineering by homologous recombination: exploring sequence and function within a conserved fold. Current opinion in structural biology. 2007;17:454–9. doi: 10.1016/j.sbi.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 23.Voigt Ca, Martinez C, Wang ZG, Mayo SL, Arnold FH. Protein building blocks preserved by recombination. Nature structural biology. 2002;9:553–8. doi: 10.1038/nsb805. [DOI] [PubMed] [Google Scholar]
- 24.Meyer MM, Silberg JJ, Voigt CA, Endelman JB, Mayo SL, Wang Z, Arnold FH. Library analysis of SCHEMA-guided protein recombination. Protein science: a publication of the Protein Society. 2003;12:1686–93. doi: 10.1110/ps.0306603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Landwehr M, Carbone M, Otey CR, Li Y, Arnold FH. Diversification of catalytic function in a synthetic family of chimeric cytochrome p450s. Chemistry & biology. 2007;14:269–78. doi: 10.1016/j.chembiol.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinzelman P, Snow CD, Wu I, Nguyen C, Villalobos A, Govindarajan S, Minshull J, Arnold FH. A family of thermostable fungal cellulases created by structure-guided recombination. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5610–5. doi: 10.1073/pnas.0901417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Romero Pa, Stone E, Lamb C, Chantranupong L, Krause A, Miklos A, Hughes Ra, Fechtel B, Ellington AD, Arnold FH, Georgiou G. SCHEMA Designed Variants of Human Arginase I & II Reveal Sequence Elements Important to Stability and Catalysis. ACS synthetic biology. 2012;1:221–228. doi: 10.1021/sb300014t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyer MM, Hochrein L, Arnold FH. Structure-guided SCHEMA recombination of distantly related beta-lactamases. Protein engineering, design & selection: PEDS. 2006;19:563–70. doi: 10.1093/protein/gzl045. [DOI] [PubMed] [Google Scholar]
- 29.Govindasamy L, Padron E, McKenna R, Muzyczka N, Kaludov N, Chiorini Ja, Agbandje-McKenna M. Structurally mapping the diverse phenotype of adeno-associated virus serotype 4. Journal of virology. 2006;80:11556–70. doi: 10.1128/JVI.01536-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otey CR, Landwehr M, Endelman JB, Hiraga K, Bloom JD, Arnold FH. Structure-guided recombination creates an artificial family of cytochromes P450. PLoS biology. 2006;4:e112. doi: 10.1371/journal.pbio.0040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sonntag F, Schmidt K, Kleinschmidt JA. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:10220–5. doi: 10.1073/pnas.1001673107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayuso E, Mingozzi F, Bosch F. Production, purification and characterization of adeno-associated vectors. Current gene therapy. 2010;10:423–36. doi: 10.2174/156652310793797685. [DOI] [PubMed] [Google Scholar]
- 33.Potter M, Chesnut K, Muzyczka N, Flotte T, Zolotukhin S. Streamlined large-scale production of recombinant adeno-associated virus (rAAV) vectors. Methods in enzymology. 2002;346:413–30. doi: 10.1016/s0076-6879(02)46069-7. [DOI] [PubMed] [Google Scholar]
- 34.Zolotukhin S, Potter M, Zolotukhin I, Sakai Y, Loiler S, Fraites TJ, Chiodo Va, Phillipsberg T, Muzyczka N, Hauswirth WW, Flotte TR, Byrne BJ, Snyder RO. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods (San Diego, Calif ) 2002;28:158–67. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
- 35.Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene therapy. 1999;6:973–85. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 36.Smith RH, Levy JR, Kotin RM. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Molecular therapy: the journal of the American Society of Gene Therapy. 2009;17:1888–96. doi: 10.1038/mt.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Donnell J, Taylor Ka, Chapman MS. Adeno-associated virus-2 and its primary cellular receptor--Cryo-EM structure of a heparin complex. Virology. 2009;385:434–43. doi: 10.1016/j.virol.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Opie SR, Warrington KH, Agbandje-McKenna M, Zolotukhin S, Muzyczka N. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. Journal of virology. 2003;77:6995–7006. doi: 10.1128/JVI.77.12.6995-7006.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaludov N, Brown KE, Walters RW, Zabner J, Chiorini JA. Adeno-associated virus serotype 4 (AAV4) and AAV5 both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. Journal of virology. 2001;75:6884–93. doi: 10.1128/JVI.75.15.6884-6893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Endelman JB, Silberg JJ, Wang ZG, Arnold FH. Site-directed protein recombination as a shortest-path problem. Protein engineering, design & selection: PEDS. 2004;17:589–94. doi: 10.1093/protein/gzh067. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Drummond DA, Sawayama AM, Snow CD, Bloom JD, Arnold FH. A diverse family of thermostable cytochrome P450s created by recombination of stabilizing fragments. Nature biotechnology. 2007;25:1051–6. doi: 10.1038/nbt1333. [DOI] [PubMed] [Google Scholar]
- 42.Otey CR, Silberg JJ, Voigt CA, Endelman JB, Bandara G, Arnold FH. Functional Evolution and Structural Conservation in Chimeric Cytochromes P4503: Calibrating a Structure-Guided Approach. 2004;11:309–318. doi: 10.1016/j.chembiol.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 43.Drummond DA, Silberg JJ, Meyer MM, Wilke CO, Arnold FH. On the conservative nature of intragenic recombination. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5380–5. doi: 10.1073/pnas.0500729102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naumer M, Sonntag F, Schmidt K, Nieto K, Panke C, Davey NE, Popa-Wagner R, Kleinschmidt Ja. Properties of the adeno-associated virus assembly-activating protein. Journal of virology. 2012;86:13038–48. doi: 10.1128/JVI.01675-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith Ma, Romero Pa, Wu T, Brustad EM, Arnold FH. Chimeragenesis of distantly-related proteins by noncontiguous recombination. Protein science: a publication of the Protein Society. 2013;22:231–8. doi: 10.1002/pro.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doll TAPF, Raman S, Dey R, Burkhard P. Nanoscale assemblies and their biomedical applications. Journal of the Royal Society, Interface / the Royal Society. 2013;10:20120740. doi: 10.1098/rsif.2012.0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Z, Qiao J, Niu Z, Wang Q. Natural supramolecular building blocks: from virus coat proteins to viral nanoparticles. Chemical Society reviews. 2012;41:6178–94. doi: 10.1039/c2cs35108k. [DOI] [PubMed] [Google Scholar]
- 48.Steinmetz NF. Viral nanoparticles as platforms for next-generation therapeutics and imaging devices. Nanomedicine: nanotechnology, biology, and medicine. 2010;6:634–41. doi: 10.1016/j.nano.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manchester M, Singh P. Virus-based nanoparticles (VNPs): platform technologies for diagnostic imaging. Advanced drug delivery reviews. 2006;58:1505–22. doi: 10.1016/j.addr.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 50.Merzlyak A, Lee SW. Phage as templates for hybrid materials and mediators for nanomaterial synthesis. Current opinion in chemical biology. 2006;10:246–52. doi: 10.1016/j.cbpa.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 51.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature methods. 2009;6:343–5. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 52.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. Journal of Virology. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rabinowitz JE, Rolling F, Li C, Conrath H, Xiao W, Xiao X, Samulski RJ. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. Journal of virology. 2002;76:791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sequence alignment of the AAV capsid proteins recombined.
Figure S2. Scaling intermolecular contacts from subunits to capsids.
Figure S3. Eintra and Einter for single-crossover chimeras.
Figure S4. Eintra and Einter for double-crossover chimeras.
Figure S5. Genomic titers of chimeras.
Figure S6. Immunoblot analysis of chimeric VP expression.
Figure S7. Binding of chimeric capsids to heparin.
Figure S8. pXR4-nocap plasmid sequence.
Figure S9. AAV2-AAV4 chimera nomenclature.
Table S1. Subunit SCHEMA disruption values used to calculate Ecapsid.
Table S2. Genomic titer of viruses prepared for TEM before and after concentration.
Table S3. Summary of chimera characteristics.