

Abstract
Autism spectrum disorder (ASD) is characterized by deficits in language development and social cognition and the manifestation of repetitive and restrictive behaviors. Despite recent major advances, our understanding of the pathophysiological mechanisms leading to ASD is limited. Although most ASD cases have unknown genetic underpinnings, animal and human cellular models of several rare, genetically defined syndromic forms of ASD have provided evidence for shared pathophysiological mechanisms that may extend to idiopathic cases. Here, we review our current knowledge of the genetic basis and molecular etiology of ASD and highlight how human pluripotent stem cell-based disease models have the potential to advance our understanding of molecular dysfunction. We summarize landmark studies in which neuronal cell populations generated from human embryonic stem cells and patient-derived induced pluripotent stem cells have served to model disease mechanisms, and we discuss recent technological advances that may ultimately allow in vitro modeling of specific human neuronal circuitry dysfunction in ASD. We propose that these advances now offer an unprecedented opportunity to help better understand ASD pathophysiology. This should ultimately enable the development of cellular models for ASD, allowing drug screening and the identification of molecular biomarkers for patient stratification.
Keywords: Autism, Autism spectrum disorder, Human pluripotent stem cells, Human embryonic stem cells, Disease modeling, Induced pluripotent stem cell, Neuronal differentiation, Neurogenesis
Introduction
Autism is a neurodevelopmental disorder defined by impairments in social interaction, deficits in verbal and nonverbal communication, and the display of stereotyped and repetitive behaviors. Comorbid symptoms include intellectual disability, seizures, anxiety, aggression, and sleep disorders. Typical autism is at the extreme end of the autism spectrum, which also includes Asperger's syndrome, childhood disintegrative disorder and a set of disorders referred to as pervasive developmental disorder (PDD) not otherwise specified. The prevalence of autism spectrum disorder (ASD) is estimated to range between ∼25 and ∼110 in every 10,000, placing a substantial socioeconomic burden on families and society (Baird et al. 2006; Kogan et al. 2009). Although pharmacotherapy can manage ASD comorbidities, it does not address the core deficits. Current treatments are focusing on educational and behavioral interventions aimed at ameliorating maladaptive behaviors and promoting social interactions (Myers and Johnson 2007).
A prerequisite for the development of pharmacological agents targeting the core symptoms of ASD is a deeper understanding of the underlying pathophysiology. ASD has a strong genetic component and a small fraction of ASD cases is caused by defined genetic aberrations. Several mouse models of these syndromic forms of autism have provided insight into dysregulated pathways. The advent of somatic cell reprogramming now enables the study of ASD neurobiology using patient-derived cells. Ectopic expression of the four transcription factors Klf-4, Oct4, Sox2, and c-Myc induces reprogramming of somatic human cells to a pluripotent state (Takahashi et al. 2007; Takahashi and Yamanaka 2006). These human induced pluripotent stem cells (hiPSCs) retain the genetic composition of the cells they have been derived from (with the exception of stably integrated transgenes in the case of retro- or lentiviral approaches) and, owing to their pluripotent state, can be differentiated into cell types of all three germ layers. hiPSCs have characteristics similar to those of human embryonic stem cells (hESCs), derived from the inner cell mass of the embryonic blastocyst, and together hiPSC and hESCs are referred to as human pluripotent stem cells (hPSCs) in this review. Although reprogramming was initially established using dermal fibroblasts and using integrating viral vectors, recent advances allow derivation of hiPSCs from cell types accessible by less invasive methods, such as from keratinocytes (Aasen et al. 2008), peripheral blood T lymphocytes (Brown et al. 2010; Seki et al. 2011), and exfoliated renal epithelial cells found in urine samples (Zhou et al. 2011, 2012). In combination with efficient transgene-free reprogramming (Fusaki et al. 2009), these improvements facilitate the generation of hiPSCs from patients enrolled in clinical trials or studies.
A major limitation of hiPSC-based disease models is the fact that they necessitate the use of hiPSC lines generated from unaffected individuals as controls. Differences in genetic makeup are likely to contribute to interclonal variability between lines, further adding to differences in their properties caused by the reprogramming procedure itself (Hu et al. 2010). While in practice, these issues are addressed by generating multiple hiPSC lines from multiple patients and control individuals, the effect size of the phenotypes to be observed is usually unknown, making it impossible to estimate the number of cell lines required a priori. These barriers inherent in hiPSC-based disease modeling can be circumvented by targeted genetic manipulation of hPSCs. Sequence-specific designed zinc finger nucleases (ZFNs) or transcription activator-like effector nucleases (TALENs) can be used to generate a double strand break at a specific locus in the genome of hPSCs, allowing gene targeting via homology-directed repair or non-homologous end joining (Ding et al. 2013a; Lombardo et al. 2007). ZFNs and TALENs are now readily available (Kim et al. 2013; Xiao et al. 2013), enabling the disruption or mutation of virtually any gene. Recently developed advanced sequence-specific nuclease systems show improved targeting efficiency, making deletion even of large genomic segments possible, and thus open the door to model diseases caused by copy number variations (CNVs) (Cong et al. 2013; Ding et al. 2013b).
In this review, we provide a summary of the potential application of hPSCs for the generation of cellular models of ASD. We illustrate how insights from human genetic and transcriptome pathway studies as well as from mouse models inform the design of relevant hPSC models and discuss recent advances towards the generation of hPSC-derived neuronal populations that may enable modeling of human circuitry dysfunction in ASD.
Human genetics, mouse models, and pathway analyses: guiding the selection of appropriate hPSC models
Human genetics
ASD is highly heritable, with concordance rates in monozygotic twins estimated to be as high as 90 % (Bailey et al. 1995; Steffenburg et al. 1989). Even though the causes of ASD are largely unknown, heritability provides a strong rationale for modeling ASD using genetically modified hPSCs and patient-derived hiPSCs, since neuronal populations derived from them will carry the causative genetic aberrations, which may lead to relevant neuronal dysfunction observable in vitro. Despite high heritability, the upper limit of the fraction of patients that harbor known genetic mutations likely causative for ASD is currently estimated at ∼30–45 % (Schaaf and Zoghbi 2011; Stein et al. 2013), indicating that the majority of genetic defects remain to be identified in idiopathic cases. Indeed, recent results from CNV (Gilman et al. 2011; Levy et al. 2011; Sanders et al. 2011) and exome sequencing (Lim et al. 2013; Michaelson et al. 2012; Yu et al. 2013) studies in large autism cohorts point to the involvement of hundreds of genes conferring ASD risk when mutated, disrupted, or duplicated. Overall, these genetic studies support a largely polygenic susceptibility model in which a minority of patients carries one of a few highly penetrant genetic aberrations, while the majority of patients harbor a small subset of hundreds of genetic aberrations that individually convey subthreshold risk (Neale et al. 2012). Models of genetically defined, syndromic forms of autism have provided critical insights into disease-causing mechanisms. They support the notion that despite the genetic heterogeneity, pathophysiological trajectories of ASD converge onto a limited number of affected pathways. Future research will address whether these emerging concepts will extend to idiopathic cases of ASD.
Many of these high-risk alleles are recurrent CNVs that affect large arrays of genes, while others are point mutations or small insertions and deletions (indels) which, although individually unique, collectively converge on a limited set of genes (Kelleher and Bear 2008). Intriguingly, most of these genes fall into two broad functional classes, namely, genes predominantly involved in regulating synapse structure and function (e.g., SHANK3, CNTNAP2, NRXN1, NLGN3/4X) and those involved in transcriptional and/or translational control (e.g., TSC1/2, MECP2, NF1, PTEN) (see review by Bourgeron et al. in this issue). The large numbers of genes affected in CNVs (which are readily detected using genome-wide microarrays and thus have been found more frequently than smaller indels or mutations, whose detection requires some form of targeted sequencing) present a challenge for the identification of causal genes. Network-based approaches for the analysis of genetic associations, using as input data genes affected by CNVs from ASD patients, may help address this challenge. Figure 1 shows a roadmap illustrating how insights from network-based analyses of genetic and transcriptomic data from ASD patients can inform the design of relevant ASD cellular models.
Fig. 1.

A roadmap towards ASD modeling in hPSCs. Genomic analysis from ASD patients identifies those who harbor genetic defects that might be causal and hiPSCs from these patients can be generated from biopsy material. Insight into shared pathophysiological trajectories comes from pathway analyses of genetic aberrations, as well as from network-based analyses of gene expression in postmortem brains of ASD patients. Guided by the identification of pathways commonly dysregulated in larger subsets of patients, this knowledge informs the selection of genes to be targeted to create additional ASD models. If present in the patient cohort, these can be generated by somatic cell reprogramming. Alternatively, additional genotypes can be obtained from normal hPSCs via targeted genetic manipulation using genome editing. Control hPSCs may also be generated from patient hPSCs by reverting the causative genetic aberration using genome editing
In one such network-based approach, termed NETBAG (Gilman et al. 2011), a large set of gene descriptors was first used to construct a background network of genes that are clustered based on their likelihood of contributing to a shared phenotype. When 433 candidate genes affected by 47 de novo CNVs from a large autism patient cohort were then mapped onto the network and interrogated for their capacity to support the preformed connections, several single genes within CNVs emerged as significantly associated (Gilman et al. 2011). HPSC lines with gene disruptions in these candidate genes, or in genes directly linked within the same cluster, will expand the range of human cellular ASD models and will enable direct probing of implicated pathways. In addition, such models will subsume genetic aberrations shared by several patients. Recent advances in genome editing technology now make it possible to construct such synthetic hPSC-based models, including those for which patient cells are not available for reprogramming (Fig. 1). Common molecular dysfunction observed in these models will also aid in patient stratification.
Despite the paucity of knowledge of “culprit” genes in CNVs, these disorders have advanced our understanding of autism pathophysiology. CNVs where deletions and reciprocal duplications cause largely overlapping clinical phenotypes, and those where the reciprocal change leads to opposing phenotypes, have been particularly instructive. For example, a CNV on 16p11.2 deleting ∼600 kb and encompassing ∼25 known coding genes is associated with autism, while duplication of the same region is associated with autism and schizophrenia (McCarthy et al. 2009). 16p11.2 deletions and duplications are also linked to macrocephaly and microcephaly, respectively (McCarthy et al. 2009). In contrast, deletion of a ∼1.5-Mb chromosomal segment on 7q11.23 containing ∼28 genes causes Williams syndrome (Merla et al. 2010). Individuals affected by this disorder have generally good verbal skills relative to their intelligence quotient and, strikingly, tend to be exceedingly sociable, approach strangers readily and hyperfocus on the eyes of others (Mervis and John 2010). In contrast, duplication of this region is associated with autism, mental retardation, language impairment, and epilepsy (Mervis and John 2010; Sanders et al. 2011). Lastly, loss of a ∼5-Mb region on 15q11-13, harboring 10–15 coding genes, on the maternal chromosome causes Angelman syndrome (Pembrey et al. 1989), characterized by developmental delay, speech impairment, ataxia, and a unique combination of behavioral phenotypes that include a generally happy disposition, frequent smiling and laughing, and excitability (Williams et al. 2006). In contrast, individuals with duplications or triplications encompassing this region are autistic, with penetrance of ASD phenotypes correlating with CNV dosage (Hogart et al. 2010). Thus, syndromes related to one another by reciprocal CNVs illustrate that opposing gene dosage can lead to either converging or opposing clinical phenotypes. The example of the 16p11.2 CNV suggests that the autism phenotype may not be linked to its specific copy number (and, consequently, to gene dosage). Analyses of CNVs in rodents and humans have shown that, instead, it may be the structural change in the genome caused by the CNV that contributes to the phenotype. Support for this hypothesis comes from rodent studies of CNV-dependent dosage sensitivity of gene expression. Here, an only modest correlation between copy number and gene dosage was observed in general, with particularly low correlation in the brain (Henrichsen et al. 2009a,b; Reymond et al. 2007). These observations have major implications for modeling functional consequences of CNVs in mouse models (see below).
Nevertheless, hiPSC models of these syndromes may help understand how and where on the pathophysiological trajectory these reciprocal CNV syndromes converge or diverge. Although knowledge of the critical genes in these CNVs is still limited, experiments in zebrafish have linked the head size phenotype in 16p11.2 CNVs to dysregulation of the neural progenitor cell (NPC) proliferation gene KCTD13, which codes for a protein that interacts with the proliferating cell nuclear antigen (Golzio et al. 2012). The finding that a subset of Angelman patients harbor point mutations in UBE3A within the 15q11-13CNV strongly suggests that dysfunction of this gene, coding for an E3 ubiquitin ligase, is primarily responsible for the phenotype (Kishino et al. 1997). Targeted genetic manipulation of these candidate genes in hPSCs will help identify the molecular underpinnings of ASD and may ultimately serve as cellular models (Fig. 1).
Further advances, alongside reductions in the costs associated with deep sequencing, will make whole genome sequencing of large patient cohorts feasible in the near future. These studies will undoubtedly reveal additional small risk-conferring genetic defects (indels and point mutations) outside of the coding regions of known genes, particularly in promoter and enhancer regions, where variation in human populations is higher than in coding regions, and within long non-coding ribonucleic acids (RNAs), which have recently come into focus as important players in neurogenesis and neuropsychiatric disorders (Lin et al. 2011). These advances will likely expand the set of informative genotypes, which can be exploited to model ASD with human hPSCs.
Insights from transcriptome analyses in human brain postmortem brain
Volumetric magnetic resonance imaging (MRI) studies, combined with structural analyses of postmortem brains of ASD patients, have identified associated neuroanatomical and cellular aberrations. Overall, these findings point to increased brain growth beginning in the first postnatal year, persisting at least into early childhood (Courchesne et al. 2007). More detailed analyses have revealed changes in neuronal size, number, and density, as well as defects in neuronal organization in frontal and temporal cortex, anterior cingulate, amygdala, and cerebellum (Amaral et al. 2008; Schumann and Nordahl 2011). Although these alterations are likely due to defects in regulation of neuronal differentiation, proliferation, and migration, their molecular underpinnings remain elusive. Transcriptome analyses of these brain regions in large cohorts of ASD patients may help identify commonly dysregulated pathways converging from the heterogeneous genetic background of the disorder (Fig. 1). Such studies, however, have only recently begun to emerge (Chow et al. 2012; Voineagu et al. 2011).
In a seminal paper, Geschwind and colleagues analyzed the transcriptomes of frontal and temporal cortex and cerebellum from 19 postmortem brains from idiopathic cases of ASD and from 17 control individuals (Voineagu et al. 2011). An analysis approach based on identification of gene coexpression networks revealed that the most significantly ASD-correlated module of functionally related genes was enriched for genes involved in synaptic function, as well as for known autism susceptibility genes. Thus, in addition to confirming involvement of synaptic dysfunction in ASD, these findings strongly suggest that transcriptomic approaches can identify molecular commonalities in ASD brains, and we can now address whether hiPSC-derived neurons can identify these molecular signatures. The dominant module was anchored by A2BP1, the gene coding for the neuron-selective splicing factor RBFOX1, thus implicating splicing dysregulation in ASD (Fogel et al. 2012). Since A2BP1 itself had previously been identified as an autism susceptibility gene in humans (Martin et al. 2007), these results provide a strong rationale for creating hPSC-based ASD models with defects in this gene. Intriguingly, the study also showed that differences in gene expression patterns between frontal and temporal cortex were attenuated in ASD brains, suggesting defects in cortical specification, patterning, and/or organization (Voineagu et al. 2011). Thus, similar to network-based analyses of de novo CNVs in ASD patients, postmortem transcriptome studies pinpoint pathways commonly dysregulated in ASD (Fig. 1). Transcriptome profiles will also help validate and benchmark hPSC-based models—either obtained by cellular reprogramming of patient-derived cells or generated by genome editing in normal hPSCs—of critical genes within these pathways and will be crucial to test the relevance of pathways implicated in ASD pathophysiology.
Mouse models
Mouse models of genetically defined, syndromic forms of ASD phenocopy core autism deficits and have provided critical insight into pathophysiological mechanisms. In mouse models of fragile X syndrome, Rett syndrome and tuberous sclerosis complex, reversal of autism phenotypes by pharmacological and/or genetic intervention has been achieved after brain development. These studies suggest that pharmacological intervention may be a viable disease-modifying approach in both children and adults. However, at least in part due to differences in trajectories of brain development and anatomy, translation from mouse models to the human condition has been particularly difficult for central nervous system (CNS) diseases (Dragunow 2008). Human cellular models that recapitulate human-specific aspects of brain development and anatomy, particularly cortical specification and organization, can help to bridge this gap in translational ASD research.
Differences in the genomic make-up and transcriptional regulation between mouse and human pose a challenge to model human genetic variants, particularly CNVs, in mouse. When genomic regions affected by CNVs are not syntenic between mouse and human, the mouse chromosomal region corresponding to its human counterpart will not contain the same set of genes. Since CNVs have been shown to affect expression patterns of genes distal to the CNV breakpoints (Reymond et al. 2007), this caveat also applies to chromosomal regions surrounding the CNV. Moreover, human-specific transcriptional processes, such as alternative promoter usage and alternative splicing, are also not replicated in a heterologous system. Thus, hPSC-based disease models of ASD caused by CNVs may be more likely to recapitulate human disease etiology.
The fact that some ASD patients carry two genetic defects suggests that these may act in concert, supporting a multi-hit model for ASD. For example, ASD patients with a deletion at 16p12.1 are more likely to harbor an additional CNV than would be expected by chance (Girirajan et al. 2010). In addition, three patients found to carry mutations in the synaptic scaffolding protein SHANK2 additionally harbored CNVs in 15q11-13 (Leblond et al. 2012), a region previously implicated in ASD (see above). Modeling these two-hit genotypes in mouse would require construction of double-knockout mice; a more direct path would therefore be the use of patient-derived hiPSCs for modeling of ASD caused by multiple genetic aberrations.
In the past, the vast majority of genetically defined ASD mouse models have been built to genocopy mutations found in the patient population. Our growing understanding of the common molecular pathways dysregulated in different forms of ASD has now begun to enable the design of novel mouse models (or the use of pre-existing models that have not previously been considered as ASD models) that may subsume aspects of ASD pathology arising through diverse etiologies. A first glimpse into this new direction in ASD research has been provided by the finding that RBFOX1 is downregulated in brains of idiopathic ASD patients, as described above (Voineagu et al. 2011). Excitingly, a CNS-specific Rbfox1 knockout mouse has been shown to have an increase in neuronal excitability and susceptibility to spontaneous seizures, a common ASD comorbidity (Gehman et al. 2011). It will be interesting to see whether this mouse also displays core behavioral phenotypes of autism.
One of the central hypotheses for molecular mechanisms shared among different forms of ASDs is aberrant regulation of translation causing increased protein synthesis (Kelleher and Bear 2008). For example, a positron emission spectroscopy imaging study of children with pervasive developmental disorder showed a positive correlation of autism index scores with protein synthesis rates in language brain regions (Shandal et al. 2011). In addition, translation is the final common mechanism regulated by the fragile X mental retardation protein (FMRP) and mTOR pathways, both strongly linked to autism (see below). Transgenic mice mildly overexpressing eIF4E, the rate-limiting factor in the assembly of the translation initiation complex, show autistic-like behaviors, increased miniature excitatory postsynaptic current (mEPSC) frequency and amplitude and enhanced hippocampal metabotropic glutamate receptor (mGluR)-mediated long-term depression (LTD) (Santini et al. 2013). Similarly, mice null for one of the three eIF4E-binding proteins (Eif4ebp2 knockout), which sequester eIF4E, display autism-related deficits and an increased ratio of excitatory to inhibitory synaptic inputs (Gkogkas et al. 2013). Strikingly, among 24 autism-relevant proteins examined in both models, only neuroligins (Nlgn1-4) showed enhanced translation, indicating strong selectivity in the genes affected by the loss of eIF4E-dependent translational control (Gkogkas et al. 2013). The ASD-like phenotypes could be reversed by Nlgn1 knockdown and by intracerebroventricular infusion of the small-molecule inhibitor of cap-dependent translation, 4-EGI (Santini et al. 2013). Translation dysfunction thus links upstream signaling defects to downstream synaptic dysfunction, strongly implicating translation hyperfunction as a defect common in both syndromic and idiopathic ASD. The advent of genome editing now makes it possible to similarly create hPSCs with translation dysregulation, paving the road towards human cellular models of idiopathic ASD (Fig. 1).
Existing hiPSC models of ASD: insights from genetically defined cases
Identification of phenotypes in hiPSC-based models of ASD has been achieved in four syndromic forms of ASD with known single gene defects: Rett syndrome (RTT), Phelan-McDermid Syndrome (PMDS), Timothy syndrome (TS), and fragile X syndrome (FXS). These studies have paved the way for the use of hiPSC-derived neuronal populations as cellular models of ASD. Electrophysiological analyses of hiPSC-derived neurons have demonstrated fundamental neuronal firing properties and spontaneous inhibitory and excitatory currents, demonstrating synaptic connectivity. Although the neurons of the middle to upper cortical layers of the human cortex provide the intracortical circuitry that is among those thought to be perturbed in ASD, these types of neurons are not generated in basic hPSC differentiation protocols. Nevertheless, since more recent differentiation protocols have begun to address neuronal region specificity, these hPSC lines can now be used for more detailed analyses of neuronal function at the network and circuit levels (see below).
Rett syndrome
RTT is a severe X-linked neurodevelopmental disorder that occurs in approximately 1 in 10,000 female births and confers a highly increased risk of developing autism-like behaviors and seizures (Chahrour and Zoghbi 2007). RTT was the first PDD for which a genetic cause could be identified (Amir et al. 1999) and, more than 10 years later, the first PDD for which hiPSC-based models were established (Marchetto et al. 2010). Thus, animal and cellular models of RTT have become prototypical for understanding the molecular pathophysiology of RTT and have nurtured the hope that insights gained from them may extend to other ASDs. Typical RTT is caused by defects in the gene encoding the transcriptional regulator, methyl CpG binding protein 2 (MECP2), involved in transcriptional regulation via alteration of chromatin structure (Baker et al. 2013). Clinical manifestations of RTT include severe intellectual disability, loss of language, stereotypic hand wringing, and motor abnormalities.
Even prior to the identification of MECP2 as the gene affected in RTT, morphological alterations to neuronal structure had been observed in human postmortem tissue, notably small neuronal size and reduced dendritic arbors in specific cortical areas and layers (Armstrong et al. 1995, 1998). Strikingly, these abnormalities were subsequently found in mice mutant for Mecp2 (Chen et al. 2001; Kishi and Macklis 2004). Although an in vitro approach using cultured dissociated neurons from these mice did not find any effects on dendritic branching or complexity (Chao et al. 2007), hiPSC-derived neurons from RTT patients and hESC-derived MECP2 hemizygous neurons, created by genome editing, also have significantly reduced cell soma size and dendritic branching (Cheung et al. 2011; Marchetto et al. 2010). MECP2 mutant neurons showed a global reduction in transcriptional output (Yazdani et al. 2012), with levels of actively transcribed genes being particularly affected (Li et al. 2013). Synaptic deficits have been observed in models spanning from human RTT hiPSC- (Marchetto et al. 2010) and hESC-derived neurons (Li et al. 2013), iPSC-derived neurons from mice mutant for Mecp2 and mouse mutant neuronal cultures (Chao et al. 2007), in vitro acute brain slices from mutant mice (Dani et al. 2005), and even, at least to a limited extent, to in vivo data from mutant mice (Chao et al. 2007). In the aggregate, these data suggest that, in the absence of MECP2, neurons fail to maintain gene expression at levels critical for neuron-specific functions, including activity-dependent transcription (Li et al. 2013).
Electrophysiological analysis of spontaneous miniature excitatory and inhibitory postsynaptic currents (mEPSCs and mIPSCs) can impart information on the number and type of synaptic connections in a neural network. HiPSC-derived neurons from patients with RTT show reduced frequency and amplitude of mEPSCs and, to a lesser extent, mIPSCs (Marchetto et al. 2010). Combined with data demonstrating fewer puncta of a presynaptic marker of excitatory synapses, the lower frequency indicates that RTT neurons produce fewer synapses. The reduction in amplitude suggests a concomitant reduction in postsynaptic receptors. While these abnormalities have been described in various rodent models of ASD (Chao et al. 2007; Peca et al. 2011), considerable care needs to be taken in their interpretation, given that in vivo developmental and in vitro cell line variability can be high. Moreover, the hiPSC-derived RTT neurons were a mixture of glutamatergic and GABAergic neurons with unknown regional identity. A detailed study of iPSC-derived neurons from Mecp2 mutant mice found reductions in mEPSC frequency in female heterozygotes, but no change in mEPSC amplitude or any abnormality in neurons from male heterozygotes (which have no MECP2 protein) (Farra et al. 2012). However, these studies did find consistent abnormalities in action potential amplitude and rise time, sodium currents, and the number of action potentials generated by a given stimulus (Farra et al. 2012). Although these detailed electrophysiological analyses have yet to be performed in hPSC-based models of RTT, analyses using multielectrode arrays (MEAs) unveiled reduced spontaneous action potential rates in the hESC-based RTT model (Li et al. 2013). Moreover, hiPSC-derived RTT neurons showed a reduced frequency of calcium transients under conditions selective for synaptically connected neurons (Marchetto et al. 2010). In the aggregate, these data point to alterations in network activity and connectivity in RTT neurons.
A candidate approach, informed by the knowledge of reduced excitatory drive and synaptic connections, led to the use of insulin-like growth factor 1 (IGF-1) in mouse models of RTT. Here, administration of IGF-1 to mice mutant for Mecp2 resulted in a partial reversal of many aspects of the phenotype, including the synaptic abnormalities (Tropea et al. 2009). Excitingly, IGF-1 applied to hiPSC-derived RTT neurons rescued the reduction in excitatory synapses (Marchetto et al. 2010). Similarly, the reduction in protein synthesis seen in hESC-derived MECP2 mutant neurons, created by genome editing, could be rescued by application of brain-derived neurotrophic factor (BDNF) or IGF-1 (Li et al. 2013). These observations provide a first glimpse of the possibility of using hPSC-derived RTT neurons as an ASD model for drug screening.
Phelan-McDermid Syndrome
Phelan-McDermid Syndrome (PMDS) is a neurodevelopmental disorder caused by deletion of of the SHANK3 gene on the terminal end of chromosome 22 (also known as 22q13 deletion syndrome). It causes a wide range of symptoms including seizures, intellectual disability and sleep disorders, as well as being strongly associated with ASD (Phelan and McDermid 2012). Recently, hiPSC-derived neurons have been generated from PMDS patients and were found to have significant deficits in excitatory synaptic transmission. Not only were these deficits rescuable by expression of SHANK3 itself, but they were also corrected by pharmacological treatment with IGF-1 (Shcheglovitov et al. 2013). These results add weight to the concept that synaptic abnormalities may be treatable by pharmacotherapy, and indeed that where diverse genetic defects converge on similar synaptic dysfunction there may be commonalities in treatment options.
Timothy syndrome
Timothy syndrome (TS) is an extremely rare autosomal dominant multisystem disorder caused by mutations in CACNA1C, coding for the alpha-1 (voltage sensor and pore-forming) subunit of the L-type voltage-gated calcium channel CaV1.2 (Splawski et al. 2004, 2005). A member of the long QT family of syndromes, TS causes prolongation of the cardiac QT interval in affected individuals. Patients with classic (type 1) TS harbor a recurrent de novo missense mutation (Gly→Arg) at position 406 in exon 8A. Approximately 25 patients with classic TS have been described, while two patients with a more severe variant of TS (atypical or type 2 TS) have a Gly→Arg mutation in the more highly expressed alternative exon 8 (Splawski et al. 2006). As CACNA1C is expressed in multiple tissues, mutation causes dysfunction in multiple organs, including syndactyly (in 100 % of type 1 cases), congenital heart disease (61 %), cardiac arrhythmia (84 %), immune deficiency (57 %), and autism (75 %). TS type 2 is characterized by the presence of extreme prolongation of the QT but absence of syndactyly. TS patients die at an average of 2.5 years, and the leading cause of death is ventricular tachycardia. Thus, as is common with syndromic forms of autism, morbidity caused by autistic behaviors is a relatively minor aspect within a large spectrum of severe clinical manifestations (Splawski et al. 2006).
Dolmetsch and colleagues generated cortical-enriched neuronal populations differentiated from several hiPSC lines derived from two type-1 TS patients and showed that mutant neurons exhibit action potentials wider than those of control cells, in agreement with a loss of CaV1.2 channel inactivation caused by the mutation (Pasca et al. 2011). Calcium imaging in cortical progenitors and in synapsin-positive mature neurons revealed an increase in the sustained intracellular calcium rise following membrane depolarization, strongly suggesting that TS NPCs and neurons have deficiencies in calcium signaling, with concomitant changes in activity-dependent gene expression. Remarkably, TS neuronal cultures contained more cells expressing tyrosine hydroxylase and produced more norepinephrine and dopamine, a phenotype also observed in the valproic acid model of ASD (D'Souza et al. 2009). Unexpectedly, of those neurons in the culture that expressed lower layer cortical markers, TS neurons also contained fewer cells expressing SATB2, critical for formation of corpus callosum projection neurons. This observation was confirmed in a transgenic mouse model expressing the mutant channel under the forebrain-specific FOXG1 promoter (Pasca et al. 2011). Thus, these results indicate that neuronal specification deficits caused by TS mutations can be recapitulated in both hiPSC-derived neurons and mouse models. The study also underscores the fact that differentiation of hiPSC-derived neurons of appropriate subtypes is critical to reveal alterations in neuronal specification, as such changes may contribute to neural circuitry dysfunction in ASD patients.
A mouse model of type 2 TS showed robust behavioral phenotypes reflecting the three core traits of autism, including restricted, repetitive, and perseverative behaviors, reduced socialization with other animals, and altered ultrasonic vocalization (Bader et al. 2011). Although analyses of alterations in neuron morphology and function or brain circuitry that may underlie these behaviors were not reported in this study, a similar mouse model was recently constructed and characterized by Dolmetsch and colleagues (Krey et al. 2013). CaV1.2 couples changes in membrane potential to dendritic arborization, possibly by recruiting small GTP-binding protein family members, which in turn regulate signaling controlling arborization. Frontal cortex layer 2/3 pyramidal neurons from TS mice showed a decrease in total basal dendritic length and the number of dendritic branches by postnatal day 14, indicating a net reduction in basal dendritic complexity in an early postnatal phase. Importantly, these effects on activity-dependent dendritic refinement were also observed in vitro, using both primary rat cortical neurons and several hiPSC neuronal lines from the two type-1 TS patients. Detailed analyses revealed that mutant CaV1.2 channels are deficient in recruiting sufficient levels of the small GTP-binding protein Gem, thus causing excessive activation of RhoA and activity-dependent dendritic retraction independent of calcium signaling (Krey et al. 2013). These insights highlight that hiPSC modeling of ASD requires assays tailored to the known function of the affected gene to unravel pathophysiological mechanisms. The study also illustrates the value of a complementary approach that includes evidence from rodent and human cellular models and in vivo rodent data.
Fragile X syndrome
Fragile X syndrome (FXS) is an X-linked dominant disorder and the most common form of inherited intellectual disability, affecting ∼1 in 4,000 newborns (Coffee et al. 2009). Clinically, FXS is characterized by mild to severe cognitive deficits and behavioral abnormalities that include hyperactivity and autism-like behaviors (Gallagher and Hallahan 2012). The syndrome is caused by the absence of expression of the fragile X mental retardation 1 (FMR1) gene, coding for the RNA binding protein FMRP. Typically, loss of FMRP in FXS is caused by abnormal CGG triplet repeat expansion in the 5′ untranslated region (5′ UTR) of the gene, leading to developmentally controlled CpG methylation in the promoter and 5′ UTR region and thus resulting in gene silencing (Feng et al. 1995). Repeat numbers >200 lead to complete silencing in neurons and cause FXS, while repeat expansions in the premutation range (55–200 repeats) are associated with upregulation of FMR1 expression and cause the late onset neurodegenerative disease, fragile X associated tremor ataxia syndrome, probably via a toxic gain of function of the CGG-rich RNA transcript (Gallagher and Hallahan 2012).
Although a mouse model carrying ∼230 repeats did not recapitulate the abnormal methylation of the promoter and 5′ UTR methylation seen in humans (Brouwer et al. 2008), Fmr1 knockout mice display many features of FXS, including autistic behaviors (Consortium TD-BFX 1994). FMRP, a suppressor of messenger RNA translation, is required for activity-dependent regulation of synaptic protein synthesis, which is stimulated by activation of group 1 mGluRs, including mGluR5. Since many of the long-term consequences of mGluR5-mediated signaling, including LTD, are dependent on protein translation, it has been hypothesized that mGluR5 inhibition could counteract neuronal dysfunction in FXS (Bear et al. 2004). Indeed, strong support for the “mGluR5 hypothesis for FXS” theory comes from several recent studies in which pharmacological or genetic intervention aimed at reducing mGluR5-dependent signaling prevented and/or reverted FXS phenotypes (Dolen et al. 2007; Michalon et al. 2012; Silverman et al. 2012).
Human ESCs derived from FXS-affected blastocyst-stage embryos express FMRP and gene silencing via promoter methylation occurs only upon differentiation (Eiges et al. 2007). In contrast, hiPSCs derived from fibroblasts of affected patients showed deoxyribonucleic acid (DNA) methylation and transcriptional silencing even in the pluripotent stage, indicating that somatic reprogramming did not erase these chromatin marks (Urbach et al. 2010). Thus, although these hiPSCs do not recapitulate developmentally regulated FMR1 silencing, they may nevertheless represent a useful model system for screening of compounds capable of reactivating the gene (Bar-Nur et al. 2012). Differentiation of hiPSCs towards neurons showed reduced neurite numbers and neurite lengths (Sheridan et al. 2011), phenotypes that resemble those seen in the Fmr1 knockout mouse (Braun and Segal 2000; Luo et al. 2010) and in human FXS postmortem tissue (Castren et al. 2005). In the future, it will be important to address whether FXS hiPSC-derived neurons display exaggerated mGluR5 signaling and defects in LTD and, if so, whether these phenotypes can be reverted with mGluR5 inhibitors.
ASD modeling in a dish: towards neuronal circuits
An outstanding question is whether hPSC-derived neurons will be useful for identifying new therapeutic targets and for developing disease-relevant phenotypes for high throughput screens. The ability to study diseases of the cerebral cortex in vitro and to observe disease-relevant phenotypes will depend on the ability to produce neuronal cultures of defined cellular composition.
Efficient neural specification using defined morphogens
Major advances in the establishment of human neuronal differentiation protocols now nurture the hope that we may be able to build specific cellular networks from hPSCs in the near future. A number of groups have established robust morphogen-based protocols to drive mouse embryonic stem cells into neurons of specific regional and functional identity, including dopaminergic, striatal, motor, and cortical neurons (Hansen et al. 2011). These morphogens, together with signaling through specific cell-to-cell contacts, dictate neuronal specification by inducing signal transduction cascades (Hansen et al. 2011). In contrast, early procedures to produce human neural cells from hPSCs either relied on coculture with stromal feeder cells, which induce neural conversion through the action of undefined factors, or depended on the generation of three-dimensional heterogeneous cellular aggregates (so-called embryoid bodies) from which neural precursor cells were subsequently isolated. Both approaches yielded neuronal cultures of variable cellular composition and quality. More recently, however, chemically defined protocols that induce rapid and virtually complete neural conversion have been developed; these should ultimately lead to standardized strategies for driving differentiation of hPSCs to relevant neuronal cell types.
A major breakthrough was the development of protocols that robustly, reproducibly, and efficiently convert hPSCs into neural ectoderm tissue. Noggin, a bone morphogenetic protein inhibitor, had previously been demonstrated to have robust neural-inducing properties (Pera et al. 2004). Similarly, the small molecule SB431542, which inhibits the Lefty/activin/transforming growth factor beta pathways by blocking phosphorylation of the ALK4, ALK5, and ALK7 receptors, enhances neural induction in hPSCs (Patani et al. 2009). The combination of noggin and SB431542 (dual SMAD inhibition), then enabled rapid and uniform neural induction of hPSCs under adherent culture conditions, and allowed the derivation of relevant neuron subtypes in a shorter time (∼19 days) (Chambers et al. 2009). Others have reported similarly efficient neural induction in both hESCs and hiPSCs with the small molecule dorsomorphin in place of noggin (Kim et al. 2010). Although the dual SMAD inhibition protocol in its original form gives rise to a heterogeneous population of neuronal cell types (both telencephalic and diencephalic), recent modifications have enabled the derivation of more specialized neuronal precursor and mature neuron populations, including floor plate precursors, midbrain dopaminergic neurons, and cortical neurons (Fasano et al. 2010; Kriks et al. 2011; Shi et al. 2012c).
Accelerating differentiation towards homogeneous neuronal populations
HPSC-derived neurons presently require long maturation times to reach a level of neuronal connectivity that allows the study of synaptic physiology (60–120 days). In order to facilitate adaptation of these cultures for drug screening, future improvements will therefore have to focus on assessing synaptic physiology and accelerating neuronal maturation. For example, in several studies, electrophysiologically active human neurons initially exhibit immature properties, including smaller, slower action potentials and the inability to fire repetitively, in contrast to more mature neurons (Shi et al. 2012c). Thus, baseline intrinsic membrane properties (e.g., resting membrane potential, input resistance) and action potential firing properties (e.g., threshold, amplitude, rise time), as well as percentages of neurons with differing firing rates, define the degree of differentiation, and should be systematically monitored while optimizing neuronal protocols to model synaptic dysfunction.
Modeling neuronal circuitry deficits and properties of specific synapses, such as cortico-striatal and excitatory/inhibitory connections, requires cultures that are highly enriched for specific neuronal subpopulations. Fluorescence-activated cell sorting (FACS) has been used to isolate hiPSC-derived neural crest cells (Lee et al. 2009), NPCs, and even mature neurons (Israel et al. 2012; Yuan et al. 2011). The use of specific combinations of neuronal cell surface markers may thus allow the prospective isolation of additional neuronal lineages. Significantly, in their study of hiPSC-derived neurons from patients with Alzheimer's disease, Goldstein and colleagues used FACS-based purification to reduce the variability in the efficiency of neuronal differentiation between hiPSC lines, providing increased power to identify disease-related changes (Israel et al. 2012). The identification of additional cell surface neuronal markers of specific subtypes, particularly middle and upper layer cortical neurons and inhibitory neurons, will facilitate progress towards pure populations of specific cortical subpopulations. Although not explored in hPSCs to date, reporter constructs expressing green fluorescent protein, under the control of a lineage-specific promoter, may be generated by genome editing, allowing the enrichment of additional neuronal lineages, including those for which cell surface markers are not known.
Figure 2 shows that using proliferating hPSC-derived NPCs as a starting point for differentiation dramatically shortens the time required to obtain mature neurons with gene expression profiles approximating that of human brain. Additional modulation of the composition of the differentiation factor cocktail can modestly accelerate maturation (Fig. 2). One exciting recent approach towards further speeding up differentiation and generating homogenous cell populations is direct lineage conversion from non-neuronal cells by the forced expression of neuronal transcription regulators (Pang et al. 2011; Vierbuchen et al. 2010). Overexpression of the forebrain transcription regulators ASCL1, BRN2, ZIC1, MYT1L, and OLIG2 directly converts human fibroblasts into neurons with forebrain glutamatergic properties within 21 days (Qiang et al. 2011). However, fibroblasts have limited proliferative capacity in vitro, thus limiting the number of cells available from a patient biopsy. Dopaminergic neurons (Sanchez-Danes et al. 2012a) and motor neurons (Hester et al. 2011) have been generated from hPSCs and NPCs by combining different sets of transcription factors, indicating that subtype-specific neuronal differentiation can be achieved. Such “induced neurons” from Alzheimer's and Parkinson's patients have shown disease-relevant cellular phenotypes (Qiang et al. 2011; Sanchez-Danes et al. 2012a,b). Although this approach may help reduce some of the variability in cellular homogeneity associated with current morphogen-based procedures, it is clear that differentiation by forced transgene expression may not allow the observation of subtle defects in neuronal specification and maturation, such as those observed in TS neurons (Pasca et al. 2011). Thus, although the usefulness of induced neurons in modeling the molecular and cellular defects of neurodegenerative diseases has been demonstrated, it remains to be seen whether they can also generate neurons capable of supporting a neuronal network relevant to neurodevelopmental disorders.
Fig. 2.

Transcriptome analyses validate the relevance of hPSC-based models of neuronal differentiation. Principal component analysis (PCA) of the expression profiles of 4,818 genes of postmortem tissue samples from human cerebellum and frontal and temporal cortex (Voineagu et al. 2011) are compared to the expression profiles of hESC-derived neural progenitor cells (NPCs) and their differentiated progeny (our unpublished work). NPCs were generated from hPSCs by dual SMAD inhibition and differentiated for 7–35 days (D) in medium containing BDNF, glial-derived neurotrophic factor, ascorbic acid and dibutyryl-cyclic adenosine monophosphate (+BGAA), or in basal medium (−BGAA). The analysis shows that transcriptomic profiles of neuronal cultures differentiated for 35 days more closely approximate that of human cortex than of cerebellum, suggesting cortical-like properties. Inclusion of BGAA increases the kinetics and/or extent of neuronal maturation
Cortical specification
Relatively simple differentiation protocols, such as the one used to generate the neurons analyzed in Fig. 2, can generate neurons that approximate the gene expression pattern of human cortex. However, these neurons do not recapitulate the elaborate cortical layer-specific specification seen in vivo. Particularly relevant to ASD disease modeling, inclusion of retinoic acid in the dual SMAD inhibition procedure leads to cortical neuron identity, based on the appearance of cortical projection markers in precursors, which subsequently give rise to upper and lower layer cortical projection neurons (Shi et al. 2012c). Remarkably, all classes of cortical projection neurons found in vivo are generated and the temporal order of their emergence recapitulates in vivo cortex development. Importantly, between 60 and 120 days after the onset of neuronal derivation, the retinoic acid/dual SMAD inhibition protocol yields mature cortical neurons with appropriate electrophysiological properties and functional synapses (Michaelson et al. 2012; Shi et al. 2012a,b). Prolonged exposure to noggin resulted in robust induction of telencephalic/cortical genes, while shorter treatments yield a mixture of diencephalon and telencephalon induction, but the in vitro temporal sequence of generation of cortical neuron types again followed in vivo development. When cortical pyramidal neurons generated in vitro were transplanted into mouse neonatal brain, they developed axonal projections and dendritic morphology which corresponded to endogenous cortical neurons. Thus, the fact that neurons generated in vitro develop properties of human cortical neurons in vivo offers an unprecedented opportunity to study ASD both in vitro and in animal xenograft models. These recent studies bring hope that specific cortical neuronal circuitry can be modeled using hPSCs relevant to ASD. Given that the expanded upper layers of the cortex are a cardinal feature of the human cortex, the specific generation of these neurons from hPSCs has potential for revealing specific aspects of cortical circuitry dysfunction in ASD.
Neuronal circuits
MRI studies and postmortem analyses of autistic individuals have consistently demonstrated anatomical abnormalities in several brain regions (including cortical frontal lobe, striatum, amygdala, and cerebellum), while functional MRI studies found differences in the activity of specific brain areas during cognitive tasks when compared to neurotypical controls (Amaral et al. 2008; Schumann and Nordahl 2011). Thus, meaningful relationships will need to be established between the cellular, molecular, and network-based properties of hPSC-based models of ASD, on the one hand, and the anatomical and functional endophenotypes of ASD patients' brains, on the other. In cellular models of ASD, disease-relevant dysfunction may reveal itself at distinct levels of functional complexity within the cell population (Fig. 3). The different phenotypic trajectories of different hPSC-based ASD models likely converge at several levels of functional complexity as well, illustrating the requirement for mature, synaptically connected neurons capable of sustaining a neuronal network (Fig. 3).
Fig. 3.
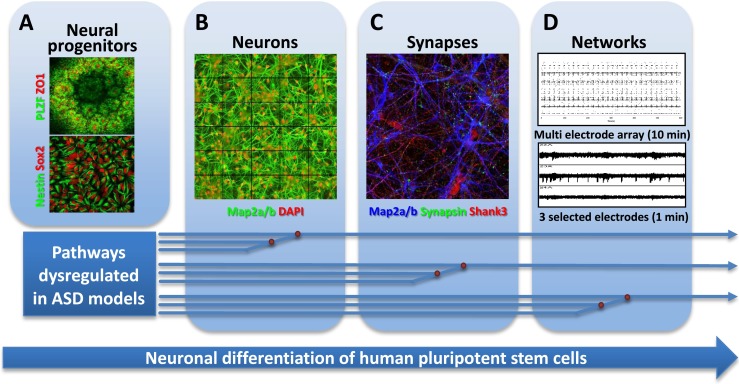
Towards cellular models of neuronal network dysfunction. Shown here are the principal stages of differentiation of hPSCs towards synaptically competent mature neurons. a First, neural ectoderm induction generates characteristic three-dimensional rosette-like structures expressing the transcription factor PLZF and the tight-junction protein ZO1 (upper). From these, neuroepithelial precursors expressing NPC markers Sox2 and nestin can be isolated and expanded (lower). b Further differentiation yields neurons extending MAP2a/b-positive dendrites. c Maturing neurons show colocalized synaptic markers, such as the presynaptic marker synapsin and the postsynaptic density protein SHANK3. d Mature neuronal cultures form functionally connected neuronal networks, characterized by synchronized spontaneous network activity seen in recordings from multi electrode arrays. Pathways dysregulated in different forms of ASD may converge on the cellular and molecular levels, allowing identification of early phenotypes in neuronal cultures from patient-derived hiPSCs. Such phenotypes include the identification of translational and transcriptional dysfunction which at the cellular level may be associated with abnormal proliferation and differences in extent and kinetics of early neuronal differentiation. Other pathways may converge at the synapse level, requiring mature neurons with synaptic connections. Dysregulation at the neuronal connectivity level due to synaptic dysfunction may only be identified in human neuronal cultures forming relevant neuronal circuits that resemble those of the human cortex
Aberrant neuronal network activity is strongly implicated as directly underlying many forms of brain dysfunction, including ASD. In particular, disruptions in the balance between excitatory/inhibitory (E/I) inputs have been observed in both ASD patients and mouse models of the disorder, pointing towards neural circuit dysfunction as the convergence point of pathophysiological trajectories that originate from diverse etiologies (Penzes et al. 2013). For example, Cntnap2 knockout mice, lacking a gene associated with ASD, show reduced numbers of GABAergic interneurons and decreased neuronal synchrony in the cortex (Penagarikano et al. 2011). Moreover, ASD mouse models engineered to display disinhibition of eIF4E-dependent control of protein synthesis have a decreased ratio between inhibitory and excitatory synaptic inputs (Gkogkas et al. 2013; Santini et al. 2013). Thus, the relevance of hPSC-based cellular models for ASD ultimately hinges on the successful in vitro generation of functional neuronal networks, characterized by synaptically connected excitatory and inhibitory neurons at physiologically relevant proportions. Given the different developmental origins and lineages of excitatory cortical pyramidal neurons and inhibitory interneurons, it remains a challenge to simultaneously produce both cell types in appropriate ratios from the same culture. Their separate generation by different protocols and subsequent coculture may provide a way forward. Indeed, protocols have recently been devised to direct the differentiation of hPSCs towards progenitors resembling those of the medial ganglionic eminence and further into forebrain interneurons (Nicholas et al. 2013) .
In the brain, inhibitory interneurons play a critical role in maintaining neural synchrony of oscillatory activity in local circuits (Huang et al. 2007). Since defects in synchrony are associated with neuropsychiatric disorders including ASD and schizophrenia (Penzes et al. 2013), there has been much interest in establishing neuronal in vitro cultures capable of supporting such synchronous activity. In their study of hiPSC-derived neurons from patients with schizophrenia, Brennand et al. (2011) observed reduced neuronal connectivity and glutamate receptor expression but, surprisingly, no abnormal synapse-dependent synchronous activity. Thus, it may be ambitious to expect that we can replicate complex brain network oscillations in an in vitro system. Nevertheless, synchronization is a sensitive measure of the maturity and connectivity of such in vitro neuronal networks, and so this functional readout may help reveal defects in network properties that are too subtle to be detected by cellular assays. Synchronous network activity can be monitored by culturing neurons on MEAs (Fig. 3) or by measuring intracellular calcium transients using Ca2+-sensitive dyes applied to neuronal cultures. MEAs allow repeated measurements of differentiation cultures over time, allowing investigation of maturation kinetics. In contrast to electrophysiological approaches, calcium imaging is amenable to higher-throughput assay formats, allowing the development of drug screens using hPSC-based cellular models of ASD. When combined with subsequent immunocytochemistry, specific neuronal subtypes (such as excitatory vs. inhibitory neurons) or subnetworks of cells that show aberrations in their network behavior can be identified. Owing to these advantages, it is therefore likely that measurement of network properties by calcium imaging will be one of the key technologies in translational ASD research.
Outlook
The key to understanding the pathophysiology of ASD and the development of therapeutics is the development of robust disease-relevant cellular models. These efforts comprise (1) establishing hiPSCs from biopsy material of ASD patients with diverse genetic etiologies and from neurotypical control individuals, (2) genetic engineering of normal hPSCs to generate matched isogenic sets of lines with targeted ASD-relevant genetic defects, (3) the development of robust, efficient and relevant neuronal differentiation protocols, and (4) the definition of ASD-related cellular phenotypes upon which corresponding cellular assays can be built. The design of these models and assays will need to be continuously informed and guided by insights from mouse models and from genetic, molecular, and functional studies in the ASD population. Given the diverse genetic and clinical landscape of ASD patients, it is evident that a single cellular model will not recapitulate all aspects of ASD pathology from all ASD patients. Nevertheless, our increased appreciation that different pathophysiological trajectories may converge on a limited set of molecular pathways fuels the hope that a limited number of cellular models may address a majority of relevant phenotypes. Major inroads into all these areas have been built in the recent past: the time is now ripe for hPSCs models to be implemented in ASD discovery research.
Acknowledgments
The authors have received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115300, resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007-2013) and EFPIA companies' in kind contribution. S.A. was supported by a Roche Postdoctoral Fellowship (RPF). We thank Veronica Costa for providing the cell cultures and Fred Knoflach for recording the multi-electrode array data used for Fig. 3.
Abbreviations
- ASD
Autism spectrum disorder
- BDNF
Brain-derived neurotrophic factor
- CNV
Copy number variation
- E/I
Excitatory/inhibitory
- FACS
Fluorescence-activated cell sorting
- FXS
Fragile X syndrome
- hESC
Human embryonic stem cell
- hiPSC
Human induced pluripotent stem cell
- hPSC
Human pluripotent stem cell
- IGF-1
Insulin-like growth factor 1
- Indel
Insertion/deletion
- iPSC
Induced pluripotent stem cell
- LTD
Long-term depression
- MEA
Multielectrode array
- mEPSC
Miniature excitatory postsynaptic current
- mGluR
Metabotropic glutamate receptor
- mIPSC
Miniature inhibitory postsynaptic current
- MRI
Magnetic resonance imaging
- NPC
Neural progenitor cell
- PCA
Principal component analysis
- PMDS
Phelan-McDermid Syndrome
- RTT
Rett syndrome
- TALEN
Transcription activator-like effector nuclease
- TS
Timothy syndrome
- UTR
Untranslated region
- ZFN
Zinc finger nuclease
Contributor Information
Laura C. Andreae, Email: laura.andreae@kcl.ac.uk
Ravi Jagasia, Email: ravi.jagasia@roche.com.
References
- Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, Vassena R, Bilic J, Pekarik V, Tiscornia G, Edel M, Boue S, Izpisua Belmonte JC. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends Neurosci. 2008;31:137–145. doi: 10.1016/j.tins.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Armstrong D, Dunn JK, Antalffy B, Trivedi R. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. 1995;54:195–201. doi: 10.1097/00005072-199503000-00006. [DOI] [PubMed] [Google Scholar]
- Armstrong DD, Dunn K, Antalffy B. Decreased dendritic branching in frontal, motor and limbic cortex in Rett syndrome compared with trisomy 21. J Neuropathol Exp Neurol. 1998;57:1013–1017. doi: 10.1097/00005072-199811000-00003. [DOI] [PubMed] [Google Scholar]
- Bader PL, Faizi M, Kim LH, Owen SF, Tadross MR, Alfa RW, Bett GC, Tsien RW, Rasmusson RL, Shamloo M. Mouse model of Timothy syndrome recapitulates triad of autistic traits. Proc Natl Acad Sci U S A. 2011;108:15432–15437. doi: 10.1073/pnas.1112667108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- Baird G, Simonoff E, Pickles A, Chandler S, Loucas T, Meldrum D, Charman T. Prevalence of disorders of the autism spectrum in a population cohort of children in South Thames: the Special Needs and Autism Project (SNAP) Lancet. 2006;368:210–215. doi: 10.1016/S0140-6736(06)69041-7. [DOI] [PubMed] [Google Scholar]
- Baker SA, Chen L, Wilkins AD, Yu P, Lichtarge O, Zoghbi HY. An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell. 2013;152:984–996. doi: 10.1016/j.cell.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Nur O, Caspi I, Benvenisty N. Molecular analysis of FMR1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J Mol Cell Biol. 2012;4:180–183. doi: 10.1093/jmcb/mjs007. [DOI] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Braun K, Segal M. FMRP involvement in formation of synapses among cultured hippocampal neurons. Cereb Cortex. 2000;10:1045–1052. doi: 10.1093/cercor/10.10.1045. [DOI] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, McCarthy S, Sebat J, Gage FH. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer JR, Huizer K, Severijnen LA, Hukema RK, Berman RF, Oostra BA, Willemsen R. CGG-repeat length and neuropathological and molecular correlates in a mouse model for fragile X-associated tremor/ataxia syndrome. J Neurochem. 2008;107:1671–1682. doi: 10.1111/j.1471-4159.2008.05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown ME, Rondon E, Rajesh D, Mack A, Lewis R, Feng X, Zitur LJ, Learish RD, Nuwaysir EF. Derivation of induced pluripotent stem cells from human peripheral blood T lymphocytes. PLoS One. 2010;5:e11373. doi: 10.1371/journal.pone.0011373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castren M, Tervonen T, Karkkainen V, Heinonen S, Castren E, Larsson K, Bakker CE, Oostra BA, Akerman K. Altered differentiation of neural stem cells in fragile X syndrome. Proc Natl Acad Sci U S A. 2005;102:17834–17839. doi: 10.1073/pnas.0508995102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Cheung AY, Horvath LM, Grafodatskaya D, Pasceri P, Weksberg R, Hotta A, Carrel L, Ellis J. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum Mol Genet. 2011;20:2103–2115. doi: 10.1093/hmg/ddr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow ML, Pramparo T, Winn ME, Barnes CC, Li HR, Weiss L, Fan JB, Murray S, April C, Belinson H, Fu XD, Wynshaw-Boris A, Schork NJ, Courchesne E. Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet. 2012;8:e1002592. doi: 10.1371/journal.pgen.1002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffee B, Keith K, Albizua I, Malone T, Mowrey J, Sherman SL, Warren ST. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. 2009;85:503–514. doi: 10.1016/j.ajhg.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium TD-BFX Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- Courchesne E, Pierce K, Schumann CM, Redcay E, Buckwalter JA, Kennedy DP, Morgan J. Mapping early brain development in autism. Neuron. 2007;56:399–413. doi: 10.1016/j.neuron.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Lee YK, Schaefer EA, Peters DT, Veres A, Kim K, Kuperwasser N, Motola DL, Meissner TB, Hendriks WT, Trevisan M, Gupta RM, Moisan A, Banks E, Friesen M, Schinzel RT, Xia F, Tang A, Xia Y, Figueroa E, Wann A, Ahfeldt T, Daheron L, Zhang F, Rubin LL, Peng LF, Chung RT, Musunuru K, Cowan CA. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12:238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, Musunuru K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 2013;12:393–394. doi: 10.1016/j.stem.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragunow M. The adult human brain in preclinical drug development. Nat Rev Drug Discov. 2008;7:659–666. doi: 10.1038/nrd2617. [DOI] [PubMed] [Google Scholar]
- D'Souza A, Onem E, Patel P, La Gamma EF, Nankova BB. Valproic acid regulates catecholaminergic pathways by concentration-dependent threshold effects on TH mRNA synthesis and degradation. Brain Res. 2009;1247:1–10. doi: 10.1016/j.brainres.2008.09.088. [DOI] [PubMed] [Google Scholar]
- Eiges R, Urbach A, Malcov M, Frumkin T, Schwartz T, Amit A, Yaron Y, Eden A, Yanuka O, Benvenisty N, Ben-Yosef D. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell. 2007;1:568–577. doi: 10.1016/j.stem.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Farra N, Zhang WB, Pasceri P, Eubanks JH, Salter MW, Ellis J. Rett syndrome induced pluripotent stem cell-derived neurons reveal novel neurophysiological alterations. Mol Psychiatry. 2012;17:1261–1271. doi: 10.1038/mp.2011.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasano CA, Chambers SM, Lee G, Tomishima MJ, Studer L. Efficient derivation of functional floor plate tissue from human embryonic stem cells. Cell Stem Cell. 2010;6:336–347. doi: 10.1016/j.stem.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Feng Y, Zhang F, Lokey LK, Chastain JL, Lakkis L, Eberhart D, Warren ST. Translational suppression by trinucleotide repeat expansion at FMR1. Science. 1995;268:731–734. doi: 10.1126/science.7732383. [DOI] [PubMed] [Google Scholar]
- Fogel BL, Wexler E, Wahnich A, Friedrich T, Vijayendran C, Gao F, Parikshak N, Konopka G, Geschwind DH. RBFOX1 regulates both splicing and transcriptional networks in human neuronal development. Hum Mol Genet. 2012;21:4171–4186. doi: 10.1093/hmg/dds240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348–362. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher A, Hallahan B. Fragile X-associated disorders: a clinical overview. J Neurol. 2012;259:401–413. doi: 10.1007/s00415-011-6161-3. [DOI] [PubMed] [Google Scholar]
- Gehman LT, Stoilov P, Maguire J, Damianov A, Lin CH, Shiue L, Ares M, Jr, Mody I, Black DL. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat Genet. 2011;43:706–711. doi: 10.1038/ng.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE, Baker C, Mefford HC, Kidd JM, Browning SR, Browning BL, Dickel DE, Levy DL, Ballif BC, Platky K, Farber DM, Gowans GC, Wetherbee JJ, Asamoah A, Weaver DD, Mark PR, Dickerson J, Garg BP, Ellingwood SA, Smith R, Banks VC, Smith W, McDonald MT, Hoo JJ, French BN, Hudson C, Johnson JP, Ozmore JR, Moeschler JB, Surti U, Escobar LF, El-Khechen D, Gorski JL, Kussmann J, Salbert B, Lacassie Y, Biser A, McDonald-McGinn DM, Zackai EH, Deardorff MA, Shaikh TH, Haan E, Friend KL, Fichera M, Romano C, Gecz J, DeLisi LE, Sebat J, King MC, Shaffer LG, Eichler EE. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42:203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, Major F, Lasko P, Ruggero D, Nader K, Lacaille JC, Sonenberg N. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature. 2013;493:371–377. doi: 10.1038/nature11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF, Kamiya A, Beckmann JS, Katsanis N. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485:363–367. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DV, Rubenstein JL, Kriegstein AR. Deriving excitatory neurons of the neocortex from pluripotent stem cells. Neuron. 2011;70:645–660. doi: 10.1016/j.neuron.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrichsen CN, Chaignat E, Reymond A. Copy number variants, diseases and gene expression. Hum Mol Genet. 2009;18:R1–R8. doi: 10.1093/hmg/ddp011. [DOI] [PubMed] [Google Scholar]
- Henrichsen CN, Vinckenbosch N, Zollner S, Chaignat E, Pradervand S, Schutz F, Ruedi M, Kaessmann H, Reymond A. Segmental copy number variation shapes tissue transcriptomes. Nat Genet. 2009;41:424–429. doi: 10.1038/ng.345. [DOI] [PubMed] [Google Scholar]
- Hester ME, Murtha MJ, Song S, Rao M, Miranda CJ, Meyer K, Tian J, Boulting G, Schaffer DV, Zhu MX, Pfaff SL, Gage FH, Kaspar BK. Rapid and efficient generation of functional motor neurons from human pluripotent stem cells using gene delivered transcription factor codes. Mol Ther. 2011;19:1905–1912. doi: 10.1038/mt.2011.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogart A, Wu D, LaSalle JM, Schanen NC. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol Dis. 2010;38:181–191. doi: 10.1016/j.nbd.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu BY, Weick JP, Yu J, Ma LX, Zhang XQ, Thomson JA, Zhang SC. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci U S A. 2010;107:4335–4340. doi: 10.1073/pnas.0910012107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Di Cristo G, Ango F. Development of GABA innervation in the cerebral and cerebellar cortices. Nat Rev Neurosci. 2007;8:673–686. doi: 10.1038/nrn2188. [DOI] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LS. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- Kim DS, Lee JS, Leem JW, Huh YJ, Kim JY, Kim HS, Park IH, Daley GQ, Hwang DY, Kim DW. Robust enhancement of neural differentiation from human ES and iPS cells regardless of their innate difference in differentiation propensity. Stem Cell Rev. 2010;6:270–281. doi: 10.1007/s12015-010-9138-1. [DOI] [PubMed] [Google Scholar]
- Kim Y, Kweon J, Kim A, Chon JK, Yoo JY, Kim HJ, Kim S, Lee C, Jeong E, Chung E, Kim D, Lee MS, Go EM, Song HJ, Kim H, Cho N, Bang D, Kim JS. A library of TAL effector nucleases spanning the human genome. Nat Biotechnol. 2013;31:251–258. doi: 10.1038/nbt.2517. [DOI] [PubMed] [Google Scholar]
- Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27:306–321. doi: 10.1016/j.mcn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- Kogan MD, Blumberg SJ, Schieve LA, Boyle CA, Perrin JM, Ghandour RM, Singh GK, Strickland BB, Trevathan E, van Dyck PC. Prevalence of parent-reported diagnosis of autism spectrum disorder among children in the US, 2007. Pediatrics. 2009;124:1395–1403. doi: 10.1542/peds.2009-1522. [DOI] [PubMed] [Google Scholar]
- Krey JF, Pasca SP, Shcheglovitov A, Yazawa M, Schwemberger R, Rasmusson R, Dolmetsch RE. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat Neurosci. 2013;16:201–209. doi: 10.1038/nn.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond CS, Heinrich J, Delorme R, Proepper C, Betancur C, Huguet G, Konyukh M, Chaste P, Ey E, Rastam M, Anckarsater H, Nygren G, Gillberg IC, Melke J, Toro R, Regnault B, Fauchereau F, Mercati O, Lemiere N, Skuse D, Poot M, Holt R, Monaco AP, Jarvela I, Kantojarvi K, Vanhala R, Curran S, Collier DA, Bolton P, Chiocchetti A, Klauck SM, Poustka F, Freitag CM, Waltes R, Kopp M, Duketis E, Bacchelli E, Minopoli F, Ruta L, Battaglia A, Mazzone L, Maestrini E, Sequeira AF, Oliveira B, Vicente A, Oliveira G, Pinto D, Scherer SW, Zelenika D, Delepine M, Lathrop M, Bonneau D, Guinchat V, Devillard F, Assouline B, Mouren MC, Leboyer M, Gillberg C, Boeckers TM, Bourgeron T. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012;8:e1002521. doi: 10.1371/journal.pgen.1002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, Tabar V, Sadelain M, Studer L. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461:402–406. doi: 10.1038/nature08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Ronemus M, Yamrom B, Lee YH, Leotta A, Kendall J, Marks S, Lakshmi B, Pai D, Ye K, Buja A, Krieger A, Yoon S, Troge J, Rodgers L, Iossifov I, Wigler M. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang H, Muffat J, Cheng AW, Orlando DA, Loven J, Kwok S, Feldman DA, Bateup HS, Gao Q, Vander Heiden MG, Sur M, Young RA, Jaenisch R. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell. 2013;13:446–458. doi: 10.1016/j.stem.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ET, Raychaudhuri S, Sanders SJ, Stevens C, Sabo A, MacArthur DG, Neale BM, Kirby A, Ruderfer DM, Fromer M, Lek M, Liu L, Flannick J, Ripke S, Nagaswamy U, Muzny D, Reid JG, Hawes A, Newsham I, Wu Y, Lewis L, Dinh H, Gross S, Wang LS, Lin CF, Valladares O, Gabriel SB, dePristo M, Altshuler DM, Purcell SM, State MW, Boerwinkle E, Buxbaum JD, Cook EH, Gibbs RA, Schellenberg GD, Sutcliffe JS, Devlin B, Roeder K, Daly MJ. Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron. 2013;77:235–242. doi: 10.1016/j.neuron.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Pedrosa E, Shah A, Hrabovsky A, Maqbool S, Zheng D, Lachman HM. RNA-Seq of human neurons derived from iPS cells reveals candidate long non-coding RNAs involved in neurogenesis and neuropsychiatric disorders. PLoS One. 2011;6:e23356. doi: 10.1371/journal.pone.0023356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, Ando D, Urnov FD, Galli C, Gregory PD, Holmes MC, Naldini L. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- Luo Y, Shan G, Guo W, Smrt RD, Johnson EB, Li X, Pfeiffer RL, Szulwach KE, Duan R, Barkho BZ, Li W, Liu C, Jin P, Zhao X. Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 2010;6:e1000898. doi: 10.1371/journal.pgen.1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, Muotri AR. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CL, Duvall JA, Ilkin Y, Simon JS, Arreaza MG, Wilkes K, Alvarez-Retuerto A, Whichello A, Powell CM, Rao K, Cook E, Geschwind DH. Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:869–876. doi: 10.1002/ajmg.b.30530. [DOI] [PubMed] [Google Scholar]
- McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE, Kusenda M, Krastoshevsky O, Krause V, Kumar RA, Grozeva D, Malhotra D, Walsh T, Zackai EH, Kaplan P, Ganesh J, Krantz ID, Spinner NB, Roccanova P, Bhandari A, Pavon K, Lakshmi B, Leotta A, Kendall J, Lee YH, Vacic V, Gary S, Iakoucheva LM, Crow TJ, Christian SL, Lieberman JA, Stroup TS, Lehtimaki T, Puura K, Haldeman-Englert C, Pearl J, Goodell M, Willour VL, Derosse P, Steele J, Kassem L, Wolff J, Chitkara N, McMahon FJ, Malhotra AK, Potash JB, Schulze TG, Nothen MM, Cichon S, Rietschel M, Leibenluft E, Kustanovich V, Lajonchere CM, Sutcliffe JS, Skuse D, Gill M, Gallagher L, Mendell NR, Craddock N, Owen MJ, O'Donovan MC, Shaikh TH, Susser E, Delisi LE, Sullivan PF, Deutsch CK, Rapoport J, Levy DL, King MC, Sebat J. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merla G, Brunetti-Pierri N, Micale L, Fusco C. Copy number variants at Williams-Beuren syndrome 7q11.23 region. Hum Genet. 2010;128:3–26. doi: 10.1007/s00439-010-0827-2. [DOI] [PubMed] [Google Scholar]
- Mervis CB, John AE. Cognitive and behavioral characteristics of children with Williams syndrome: implications for intervention approaches. Am J Med Genet C: Semin Med Genet. 2010;154C:229–248. doi: 10.1002/ajmg.c.30263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, Wu W, Corominas R, Peoples A, Koren A, Gore A, Kang S, Lin GN, Estabillo J, Gadomski T, Singh B, Zhang K, Akshoomoff N, Corsello C, McCarroll S, Iakoucheva LM, Li Y, Wang J, Sebat J. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 2012;151:1431–1442. doi: 10.1016/j.cell.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron. 2012;74:49–56. doi: 10.1016/j.neuron.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers SM, Johnson CP. Management of children with autism spectrum disorders. Pediatrics. 2007;120:1162–1182. doi: 10.1542/peds.2007-2362. [DOI] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, Cai G, Lihm J, Dannenfelser R, Jabado O, Peralta Z, Nagaswamy U, Muzny D, Reid JG, Newsham I, Wu Y, Lewis L, Han Y, Voight BF, Lim E, Rossin E, Kirby A, Flannick J, Fromer M, Shakir K, Fennell T, Garimella K, Banks E, Poplin R, Gabriel S, DePristo M, Wimbish JR, Boone BE, Levy SE, Betancur C, Sunyaev S, Boerwinkle E, Buxbaum JD, Cook EH, Jr, Devlin B, Gibbs RA, Roeder K, Schellenberg GD, Sutcliffe JS, Daly MJ. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas CR, Chen J, Tang Y, Southwell DG, Chalmers N, Vogt D, Arnold CM, Chen YJ, Stanley EG, Elefanty AG, Sasai Y, Alvarez-Buylla A, Rubenstein JL, Kriegstein AR. Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell. 2013;12:573–586. doi: 10.1016/j.stem.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, Citri A, Sebastiano V, Marro S, Sudhof TC, Wernig M. Induction of human neuronal cells by defined transcription factors. Nature. 2011;476:220–223. doi: 10.1038/nature10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, Bernstein JA, Hallmayer J, Geschwind DH, Dolmetsch RE. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17:1657–1662. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patani R, Compston A, Puddifoot CA, Wyllie DJ, Hardingham GE, Allen ND, Chandran S. Activin/Nodal inhibition alone accelerates highly efficient neural conversion from human embryonic stem cells and imposes a caudal positional identity. PLoS One. 2009;4:e7327. doi: 10.1371/journal.pone.0007327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pembrey M, Fennell SJ, van den Berghe J, Fitchett M, Summers D, Butler L, Clarke C, Griffiths M, Thompson E, Super M. The association of Angelman's syndrome with deletions within 15q11-13. J Med Genet. 1989;26:73–77. doi: 10.1136/jmg.26.2.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Sonnenblick LI, Gruver R, Almajano J, Bragin A, Golshani P, Trachtenberg JT, Peles E, Geschwind DH. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–246. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Buonanno A, Passafaro M, Sala C, Sweet RA. Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. J Neurochem. 2013;126:165–182. doi: 10.1111/jnc.12261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pera MF, Andrade J, Houssami S, Reubinoff B, Trounson A, Stanley EG, Ward-van Oostwaard D, Mummery C. Regulation of human embryonic stem cell differentiation by BMP-2 and its antagonist noggin. J Cell Sci. 2004;117:1269–1280. doi: 10.1242/jcs.00970. [DOI] [PubMed] [Google Scholar]
- Phelan K, McDermid HE (2012) The 22q13.3 deletion syndrome (Phelan-McDermid syndrome). Mol Syndromol (3–5):186–201 [DOI] [PMC free article] [PubMed]
- Qiang L, Fujita R, Yamashita T, Angulo S, Rhinn H, Rhee D, Doege C, Chau L, Aubry L, Vanti WB, Moreno H, Abeliovich A. Directed conversion of Alzheimer's disease patient skin fibroblasts into functional neurons. Cell. 2011;146:359–371. doi: 10.1016/j.cell.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Reymond A, Henrichsen CN, Harewood L, Merla G. Side effects of genome structural changes. Curr Opin Genet Dev. 2007;17:381–386. doi: 10.1016/j.gde.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Sanchez-Danes A, Consiglio A, Richaud Y, Rodriguez-Piza I, Dehay B, Edel M, Bove J, Memo M, Vila M, Raya A, Izpisua Belmonte JC. Efficient generation of A9 midbrain dopaminergic neurons by lentiviral delivery of LMX1A in human embryonic stem cells and induced pluripotent stem cells. Hum Gene Ther. 2012;23:56–69. doi: 10.1089/hum.2011.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, Jimenez-Delgado S, Caig C, Mora S, Di Guglielmo C, Ezquerra M, Patel B, Giralt A, Canals JM, Memo M, Alberch J, Lopez-Barneo J, Vila M, Cuervo AM, Tolosa E, Consiglio A, Raya A. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson's disease. EMBO Mol Med. 2012;4:380–395. doi: 10.1002/emmm.201200215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, Mason CE, Bilguvar K, Celestino-Soper PB, Choi M, Crawford EL, Davis L, Wright NR, Dhodapkar RM, DiCola M, DiLullo NM, Fernandez TV, Fielding-Singh V, Fishman DO, Frahm S, Garagaloyan R, Goh GS, Kammela S, Klei L, Lowe JK, Lund SC, McGrew AD, Meyer KA, Moffat WJ, Murdoch JD, O'Roak BJ, Ober GT, Pottenger RS, Raubeson MJ, Song Y, Wang Q, Yaspan BL, Yu TW, Yurkiewicz IR, Beaudet AL, Cantor RM, Curland M, Grice DE, Gunel M, Lifton RP, Mane SM, Martin DM, Shaw CA, Sheldon M, Tischfield JA, Walsh CA, Morrow EM, Ledbetter DH, Fombonne E, Lord C, Martin CL, Brooks AI, Sutcliffe JS, Cook EH, Jr, Geschwind D, Roeder K, Devlin B, State MW. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H, Klann E. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature. 2013;493:411–415. doi: 10.1038/nature11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CP, Zoghbi HY. Solving the autism puzzle a few pieces at a time. Neuron. 2011;70:806–808. doi: 10.1016/j.neuron.2011.05.025. [DOI] [PubMed] [Google Scholar]
- Schumann CM, Nordahl CW. Bridging the gap between MRI and postmortem research in autism. Brain Res. 2011;1380:175–186. doi: 10.1016/j.brainres.2010.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki T, Yuasa S, Fukuda K (2011) Derivation of induced pluripotent stem cells from human peripheral circulating T cells. Curr Protoc Stem Cell Biol Chapter 4: Unit4A 3 [DOI] [PubMed]
- Shandal V, Sundaram SK, Chugani DC, Kumar A, Behen ME, Chugani HT. Abnormal brain protein synthesis in language areas of children with pervasive developmental disorder: a L-[1-11C]-leucine PET study. J Child Neurol. 2011;26:1347–1354. doi: 10.1177/0883073811405200. [DOI] [PubMed] [Google Scholar]
- Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer JF, Dolmetsch RE (2013) SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. doi:10.1038/nature12618 [DOI] [PMC free article] [PubMed]
- Sheridan SD, Theriault KM, Reis SA, Zhou F, Madison JM, Daheron L, Loring JF, Haggarty SJ. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One. 2011;6:e26203. doi: 10.1371/journal.pone.0026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Livesey FJ. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat Protoc. 2012;7:1836–1846. doi: 10.1038/nprot.2012.116. [DOI] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Smith J, MacLean G, Orkin SH, Livesey FJ. A human stem cell model of early Alzheimer's disease pathology in Down syndrome. Sci Trans Med. 2012;4:124–ra129. doi: 10.1126/scitranslmed.3003771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Smith J, Robinson HP, Livesey FJ. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci. 2012;15:477–486. doi: 10.1038/nn.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Smith DG, Rizzo SJ, Karras MN, Turner SM, Tolu SS, Bryce DK, Smith DL, Fonseca K, Ring RH, Crawley JN. Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci Transl Med. 2012;4:131ra51. doi: 10.1126/scitranslmed.3003501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, Keating MT. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A. 2005;102:8089–8096. [Google Scholar]
- Splawski I, Timothy KW, Priori SG, Napolitano C, Bloise R. Timothy Syndrome. In: Pagon RABT, Dolan CR, Stephens K, Adam MP, editors. GeneReviews™ [Internet] Seattle: University of Washington; 2006. [Google Scholar]
- Steffenburg S, Gillberg C, Hellgren L, Andersson L, Gillberg IC, Jakobsson G, Bohman M. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry. 1989;30:405–416. doi: 10.1111/j.1469-7610.1989.tb00254.x. [DOI] [PubMed] [Google Scholar]
- Stein JL, Parikshak NN, Geschwind DH. Rare inherited variation in autism: beginning to see the forest and a few trees. Neuron. 2013;77:209–211. doi: 10.1016/j.neuron.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, Flannery R, Jaenisch R, Sur M. Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci U S A. 2009;106:2029–2034. doi: 10.1073/pnas.0812394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell. 2010;6:407–411. doi: 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA, Wagstaff J. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140:413–418. doi: 10.1002/ajmg.a.31074. [DOI] [PubMed] [Google Scholar]
- Xiao A, Wu Y, Yang Z, Hu Y, Wang W, Zhang Y, Kong L, Gao G, Zhu Z, Lin S, Zhang B. EENdb: a database and knowledge base of ZFNs and TALENs for endonuclease engineering. Nucleic Acids Res. 2013;41:D415–D422. doi: 10.1093/nar/gks1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani M, Deogracias R, Guy J, Poot RA, Bird A, Barde YA. Disease modeling using embryonic stem cells: MeCP2 regulates nuclear size and RNA synthesis in neurons. Stem Cells. 2012;30:2128–2139. doi: 10.1002/stem.1180. [DOI] [PubMed] [Google Scholar]
- Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, Ataman B, Schmitz-Abe K, Harmin DA, Adli M, Malik AN, D'Gama AM, Lim ET, Sanders SJ, Mochida GH, Partlow JN, Sunu CM, Felie JM, Rodriguez J, Nasir RH, Ware J, Joseph RM, Hill RS, Kwan BY, Al-Saffar M, Mukaddes NM, Hashmi A, Balkhy S, Gascon GG, Hisama FM, LeClair E, Poduri A, Oner O, Al-Saad S, Al-Awadi SA, Bastaki L, Ben-Omran T, Teebi AS, Al-Gazali L, Eapen V, Stevens CR, Rappaport L, Gabriel SB, Markianos K, State MW, Greenberg ME, Taniguchi H, Braverman NE, Morrow EM, Walsh CA. Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013;77:259–273. doi: 10.1016/j.neuron.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan SH, Martin J, Elia J, Flippin J, Paramban RI, Hefferan MP, Vidal JG, Mu Y, Killian RL, Israel MA, Emre N, Marsala S, Marsala M, Gage FH, Goldstein LS, Carson CT. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One. 2011;6:e17540. doi: 10.1371/journal.pone.0017540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Benda C, Duzinger S, Huang Y, Li X, Li Y, Guo X, Cao G, Chen S, Hao L, Chan YC, Ng KM, Ho JC, Wieser M, Wu J, Redl H, Tse HF, Grillari J, Grillari-Voglauer R, Pei D, Esteban MA. Generation of induced pluripotent stem cells from urine. J Am Soc Nephrol. 2011;22:1221–1228. doi: 10.1681/ASN.2011010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Benda C, Dunzinger S, Huang Y, Ho JC, Yang J, Wang Y, Zhang Y, Zhuang Q, Li Y, Bao X, Tse HF, Grillari J, Grillari-Voglauer R, Pei D, Esteban MA. Generation of human induced pluripotent stem cells from urine samples. Nat Protoc. 2012;7:2080–2089. doi: 10.1038/nprot.2012.115. [DOI] [PubMed] [Google Scholar]