

Abstract
Rationale
Advances in understanding the underlying mechanisms of conditions such as fragile X syndrome (FXS) and autism spectrum disorders have revealed heterogeneous populations. Recent trials of novel FXS therapies have highlighted several challenges including subpopulations with possibly differential therapeutic responses, the lack of specific outcome measures capturing the full range of improvements of patients with FXS, and a lack of biomarkers that can track whether a specific mechanism is responsive to a new drug and whether the response correlates with clinical improvement.
Objectives
We review the phenotypic heterogeneity of FXS and the implications for clinical research in FXS and other neurodevelopmental disorders.
Results
Residual levels of fragile X mental retardation protein (FMRP) expression explain in part the heterogeneity in the FXS phenotype; studies indicate a correlation with both cognitive and behavioral deficits. However, this does not fully explain the extent of phenotypic variance observed or the variability of drug response. Post hoc analyses of studies involving the selective mGluR5 antagonist mavoglurant and the GABAB agonist arbaclofen have uncovered significant therapeutic responses following patient stratification according to FMR1 promoter methylation patterns or baseline severity of social withdrawal, respectively. Future studies designed to quantify disease modification will need to develop new strategies to track changes effectively over time and in multiple symptom domains.
Conclusion
Appropriate selection of patients and outcome measures is central to optimizing future clinical investigations of these complex disorders.
Keywords: Fragile X syndrome, Autism spectrum disorder, FMRP, mGluR5, FMR1, Disease modification, Mavoglurant, AFQ056, GABA, Arbaclofen
Introduction
Based on advances in the understanding of the neurobiology of fragile X syndrome (FXS), targeted therapeutic agents designed to correct the underlying mechanisms of neural dysfunction have been assessed in patients. Investigating these new therapies for FXS in clinical trials has led to an increased understanding of the challenges involved in evaluating treatment efficacy in this complex condition. In particular, these early trials have highlighted several challenges: (a) heterogeneity, with the existence of subpopulations based on differing therapeutic response; (b) the lack of reliable biomarkers; and (c) the issue of specific and sensitive outcome measures, particularly in the context of future disease modification trials.
FXS is among the most common known inherited causes of intellectual disability and autism, typically caused by expansion of a cytosine–guanine–guanine (CGG) triplet repeat in the 5′ untranslated region of the fragile mental retardation 1 (FMR1) gene. The presence of >200 CGG repeats (full mutation), combined with extensive methylation of the FMR1 promoter, containing the repeat sequence and upstream CpG islands, leads to transcriptional silencing of FMR1 and a complete or partial absence of fragile mental retardation protein (FMRP) (Bell et al. 1991; Sutcliffe et al. 1992). FMRP is a dendritic RNA binding protein that modulates local translation of mRNA at the synapse and postsynaptic density. Dendritic translation, in turn, influences the morphology and functionality of the synapse (synaptic plasticity). Loss of FMRP leads to dysregulation of translation and abnormal neuronal signaling in specific pathways, culminating in the morphological defects and aberrant synaptic plasticity observed in Fmr1-knockout animal models and post mortem brain tissue from individuals with FXS.
In addition to cognitive deficits, individuals with FXS typically present behavioral problems which include a range of anxiety symptoms, attention deficits, hyperarousal, irritability, and autism or autistic-like symptoms including social deficits (Garber et al. 2008). Despite a common genetic etiology, there is a wide-ranging variability in clinical presentation of FXS.
Many of these symptoms are shared with autism spectrum disorders (ASD), and approximately 2–6 % of all cases of autism in males are caused by FXS (Hagerman et al. 2010), dependent on the intelligence quotient (IQ) level of the autism cohort. The prevalence of autism among individuals with FXS is approximately 30 % when using the Autism Diagnostic Observation Schedule/Autism Diagnostic Interview—Revised (Hall et al. 2008; Harris et al. 2008; Kaufmann et al. 2004; Rogers et al. 2001).
The mechanistic link underlying the high prevalence of autism in FXS may be mediated via FMRP and its role in regulating translation of a large number of proteins associated with autism: mGluR5, N-methyl-d-aspartate receptor subunits, mammalian target of rapamycin (mTOR), tuberous sclerosis complex 2, fragile-X-related protein 2, and neuroligin-3 (Ascano et al. 2012; Darnell et al. 2011; Iossifov et al. 2012). Many of these genes are associated with synaptic plasticity and function. The molecular pathways underlying the overlap between FXS and autism are complicated and poorly understood. A comprehensive review of the overlap between autism and FXS is beyond the scope of this paper, and several excellent reviews have been published on the topic.
In this review, we will discuss the clinical and molecular heterogeneity of FXS and the implications for clinical research. We will also highlight how future trials will need to carefully consider selection of the most appropriate patients and outcome measures, particularly in the context of evaluating disease modification. We will also consider the wider implications for other neurodevelopmental disorders, and whether what is learned from translational research in FXS can be extrapolated to other etiologies of ASD.
Fragile X syndrome—a clinically heterogeneous patient population
The significant heterogeneity in behavioral and cognitive deficits observed among individuals with FXS is explained in part by variations in residual levels of FMRP. The latter is determined by mosaicism of the CGG expansion size, methylation levels, and X chromosome inactivation. In females, inactivation of one of the two X chromosomes is random from one cell to the next, leading to a differential pattern of FMRP expression within tissues. As a result, the ratio of active normal and full mutation X chromosomes (X-activation ratio) significantly influences the extent of an individual’s symptoms; all females with FXS are mosaic by definition. Similarly, in males, variations in the pattern of methylation or size of CGG expansion can result in mosaicism, in which transcriptional silencing of FMR1 occurs in some but not all cells.
Several studies in both females and males have correlated the severity of intellectual disability with FMR1 activity and FMRP levels (Dyer-Friedman et al. 2002; Loesch et al. 2004; Reiss et al. 1995; Tassone et al. 1999). However, the variance reported in cognitive outcomes for males is also substantially influenced by the home environment, which is not the case for females (Dyer-Friedman et al. 2002; Hessl et al. 2001). Examination of the relationship between cognition and the molecular pathology of FXS has been limited by the sensitivity and floor effects of cognitive testing methods in very low functioning individuals. Only by developing new testing methods and/or algorithms for interpreting results from currently available cognitive measures can the extent of the link with FMRP levels be established. Table 1 provides details of the studies reviewed here. A study in 2009 sought to minimize the limitations associated with standard approaches to cognitive testing in the FXS population, by normalizing scores on the Wechsler Intelligence Scale for Children (WISC-III) (Hessl et al. 2009). Normalized WISC-III scores were based on raw score descriptive statistics from the publisher of the WISC-III (Psychological Corporation, San Antonio, TX, USA) and calculated using an age-dependent z-score transformation. The new scores and usual standardized scores were each correlated with the Vineland Adaptive Behavior Scales, and FMRP levels and the results compared. This analysis reported an enhanced correlation between cognition and residual FMRP expression for the new normalized scoring in contrast to standardized scoring, providing a more accurate assessment of the contribution of FMRP levels to cognitive skills in lower functioning individuals with FXS. Hessl et al. (2001) were among the first to show that behavioral effects correlated with FMRP expression levels. Correlations were the strongest for internalizing problems in females, including withdrawn and anxious/depressed behavior. In contrast, environmental factors were shown to play a significant role in behavioral problems for males (Hessl et al. 2001). In particular, non-pharmacological interventional therapy and the quality of the home environment were linked to fewer behavioral and autistic symptoms (Hessl et al. 2001).
Table 1.
Description of studies linking cognition to FMRP levels
| Study | Population | Cognitive measures | FMRP levels/FMR1 activity | Citation |
|---|---|---|---|---|
| Pedigree analysis of children with FXS and unaffected siblings | Aged 6–17 years | WISC-III | FMRP levels in peripheral blood determined by Immunocytochemistry as % of FMRP-positive lymphocytes | Dyer-Friedman et al. (2002); Hessl et al. (2001) |
| FXS: 80 M; 40 F | ||||
| Mosaicism: 9 M; 5 F | ||||
| Non-FXS siblings: 58 M; 62 F | ||||
| Pedigree analysis of 144 families with individuals affected by FXS | Aged 4–76 years | WAIS-III; WISC-III;WISC-R; WCST; RCFT; BDS | FMRP levels in peripheral blood determined by Immunocytochemistry as % of FMRP-positive lymphocytes | Loesch et al. (2004) |
| Full mutationa: 87 M; 58 F | ||||
| Premutationb: 32 M; 142 F | ||||
| Non-FXS relatives: 114 M; 57 F | ||||
| To specify and measure the relative contributions of genetics and epigenetic characteristics to variance in intellectual functioning | Aged 6–17 years | WISC-R | FMR1 activation ratio determined from southern blots of DNA extracted from peripheral lymphocytes | Reiss et al. (1995) |
| Full mutation: 29 F | ||||
| Non-FXS: 50 F | ||||
| Investigation of the relationship between degree of FMRP expression and deficits associated with FXS | Aged 2–60 | Leiter scale; WISC-R; WISC-III; WAIS-R; K-ABC; S-B; BSID; MDI; VABSc | FMRP levels in peripheral blood determined by Immunocytochemistry as % of FMRP-positive lymphocytes | Tassone et al. (1999) |
| Full mutation: 19 F | ||||
| Completely methylated: 36 M | ||||
| Partially methylated: 13 M | ||||
| Repeat size mosaicism: 12 M | ||||
| Examination of the sensitivity of the WISC-III in FXS | Aged 6–17 years | WISC-III | FMRP levels in peripheral blood determined by Immunocytochemistry as % of FMRP-positive lymphocytes | Hessl et al. (2009) |
| Full mutation: 134 M; 83 F | ||||
| Repeat size mosaicism: 44 M; 12 F | ||||
| Methylation mosaicism: 13 M; 1 F |
BDS Behavior Dyscontrol Scale, F females, M males, RCFT Rey Complex Figure Test, WAIS-III Weschler Adult Intelligence Scale—Third Edition, WCST Wisconsin Card Sorting Test, WISC-III Weschler Intelligence Scale for Children—Third Edition, WISC-R WISC—Revised, K-ABC Kaufman Assessment Battery for Children, S-B Stanford–Binet Intelligence test, MDI Mental Developmental Index, BSID Bayley Scales of Infant Development, VABS Vineland Adaptive Behavior Scale
aIncluded individuals with repeat size mosaicism, unmethylated full mutation
bIncluded two individuals with 40–49 CGG repeats
cUsed when standard IQ test could not be obtained
However, much of the variance in FXS phenotype remains unexplained, and most available studies are compounded by the methodology used to determine FMRP levels (Iwahashi et al. 2009; Willemsen et al. 1995, 1997). In the past, FMRP expression has been estimated indirectly using an immunocytochemical approach to calculate the percentage of lymphocytes expressing any FMRP, without quantifying the amount of FMRP expressed by the cell (Iwahashi et al. 2009; Willemsen et al. 1995, 1997). More quantitative methods capable of analyzing FMRP levels are required for improving our understanding of the relationship between the molecular pathology of FXS and the clinical phenotype. Furthermore, recent observations report a cross-reaction between the anti-FMRP monoclonal 7G1-1 and the RNA binding protein Caprin 1 (El et al. 2012), suggesting that anti-FMRP antibodies may not be as exclusive as previously thought. Other FMR1-related measures, such as DNA-methylation patterns and the characterization of additional genetic variants at the exome level, using next generation sequencing tools, may provide other means of explaining phenotypic heterogeneity in FXS.
Subgroups of FXS
Although FXS is usually associated with moderate to severe intellectual disability, the syndrome can present as learning difficulties in an individual with an IQ within the low normal or borderline range (70–90); this is most often observed in females. In one study, 50 % of females were placed in the normal or borderline range (i.e., IQ > 70) (de Vries et al. 1996). Residual FMRP levels are in part related to the X-activation ratio, potentially influencing the level of intellectual disability in females (Dyer-Friedman et al. 2002; Loesch et al. 2004; Reiss et al. 1995; Tassone et al. 1999). Clinical correlation of FMRP expression levels in lymphocytes and brain cells may be hampered by individual differences in the X-activation ratios for brain and blood.
Higher-functioning (IQ > 70) males with FXS also express greater levels of FMRP than those individuals with more pronounced deficits, due to mosaicism. Some of these mosaic males have a proportion of cells containing a premutation expansion (55–200 CGG repeats) and a proportion of cells with a full mutation (size mosaicism). Other individuals have the full mutation in both unmethylated and methylated forms (methylation mosaicism) (Hagerman et al. 1994; Tassone et al. 1999).
Further evidence of clinical subgroups is provided by the characterization of the Prader–Willi phenotype (PWP) (McLennan et al. 2011). PWP occurs in <10 % of individuals with FXS who present with hyperphagia, lack of satiation after meals, and hypogonadism or delayed puberty, but test negative for the 15q11–q13 deletion or uniparental maternal disomy associated with Prader–Willi syndrome. Instead, a study suggests that this subgroup has lowered expression of a gene located on chromosome 15 in the 15q11–q13 region, cytoplasmic FMR1 interacting protein 1 (CYFIP1) (Nowicki et al. 2007). CYFIP1 interacts with FMRP and Rac1, linking two processes which underlie synaptic remodeling—cytoskeletal reorganization and protein translation (Bardoni and Mandel 2002; Schenck et al. 2003; Zarnescu et al. 2005).
FXS has been described in association with other etiologies of intellectual disability, including autism, Down syndrome, Klinefelter syndrome, Turner syndrome, and trisomy X (Hagerman and Hagerman 2002). With the availability of comparative genomic hybridization and whole exome analyses, additional mutations may be uncovered in many more patients with a primary diagnosis of FXS. The heterogeneity in the FXS population and the varying treatment responses observed in recent trials now require new paradigms to design and implement future clinical trials for FXS.
Patient stratification for clinical trials
The appropriate selection of patients is crucial for any clinical research. In the case of FXS, patients have typically been selected based on a positive diagnostic test confirming the expansion of >200 CGG repeats in the FMR1 promoter. Treatments developed to target the underlying pathophysiology of the syndrome may address symptoms directly associated with FMRP deficits, and their therapeutic efficacy may be dependent largely on the extent of FMRP expression within the patient population. Conversely, with molecular stratification techniques, it may be possible to identify subpopulations of patients with FXS who are more likely to respond to certain treatments with specific molecular targets.
Molecular stratification of patients has been used in the clinical development of the selective mGluR5 antagonist, AFQ056 (mavoglurant). In a proof-of-concept crossover-design study, 30 male patients with FXS, aged 18–35 years, were treated with mavoglurant. A subgroup of patients with a completely methylated FMR1 promoter region were identified using a bisulfate-sequencing based method, more sensitive than the widely used Southern blot analysis. In a post hoc analysis, these individuals showed significant improvements in Aberrant Behavior Checklist—Community Edition (ABC-C) total score (−27.8 vs placebo; p < 0.001), despite no significant improvements in the overall population (Jacquemont et al. 2011).
Following these results, efficacy studies have been initiated in male and female adults and adolescents with FXS. Molecular profiling is used to enrich the population with completely methylated patients. Trials are designed with two strata: patients who have completely methylated FMR1 promoter regions and patients with a partially methylated promoter, as assessed using a newly developed DNA methylation assay based on DNA-methylation-specific restriction enzymes and real-time PCR. These trials are ongoing and no data are currently available.
The reasons for the significant response seen in the completely methylated population and the variable response among those in the partially methylated population in the initial study are unknown. Possible factors include the relationship between methylation status and FMR1 mRNA and FMRP expression (Jacquemont et al. 2011), resulting in a range of severity and varied behavioral and cognitive dysfunction exhibited by the completely methylated groups, relative to the partially methylated group.
As we understand and identify the additional genetic and environmental factors mediating phenotypic variability and therapeutic response to treatments (for example, using techniques such as array-comparative genomic hybridization, imaging genetics, whole genome, and exome sequencing), it may be possible to identify reliable biomarkers and use these to select patients most likely to benefit from specific treatments. This should lead to better clinical trial designs and lower attrition rates for FXS therapies.
Evaluating disease modification
Disease modification can be defined as a normalization or partial normalization of the core mechanism underlying FXS, which translates into a stabilization or improvement in symptoms. Therefore, improvements across multiple symptom domains in FXS (e.g., cognitive, behavioral, and neurological) could be considered as disease modifications. A key challenge to assessing disease modification is identifying an appropriate outcome measure, given the wide range of symptoms observed in this patient population. This is a critical factor that needs to be considered when designing future clinical trials evaluating disease modification.
Currently, it is unclear which measures are the most relevant for these studies. A report by the National Institutes of Health Working Outcome measures group concluded that there is currently no single measure that can effectively evaluate treatment for FXS, based on a review of published data (Berry-Kravis et al. 2013). They emphasized the need for greater consistency in the selection of outcome measures in clinical trials and the identification of a set of core measures for FXS. The group favored a single-composite approach grouping core features of FXS within each symptom domain. Although this would be useful in studies of drugs which target related symptoms, it might not detect improvements in a single sub-domain. Other challenges facing a single-composite approach include the variation in symptom presentation associated with age and level of impairment, plus the selection of features to be included in the measure. The ABC-C, expressive language sampling, prepulse inhibition, and neuroimaging were highlighted as promising measures in need of further development. Also, the group recommended testing outcome measures currently validated for other conditions that share core symptoms with FXS for their feasibility and validity in FXS.
As discussed in a recent review (Gross et al. 2012), any outcome measure selected for a clinical trial in FXS must be able to test a broad ability range, overcome problems of cooperation and variable performance, be reproducible and quantifiable, and show improvement in quality of life and function.
Recent trials of both mavoglurant (Jacquemont et al. 2011) and STX209 (arbaclofen) (Berry-Kravis et al. 2012) have looked at changes in the ABC-C and its subscales as clinical endpoints. The ABC-C was developed to assess problem behaviors in children and adults with intellectual disability (Aman et al. 1985) and has been effectively employed in trials for ASD treatments. However, it was unknown if the ABC-C is sensitive enough to detect disease modification in patients with FXS. A version of the ABC-C has been developed with enhanced specificity and sensitivity for FXS, ABC-C for FXS (Sansone et al. 2012) (Fig. 1). In post hoc analyses of the mavoglurant FXS trial, the responder subgroup (patients with a completely methylated FMR1 promoter region) showed similar significant improvements in both the ABC-C (−27.8 vs placebo; p < 0.001) (Jacquemont et al. 2011) and ABC-C for FXS (−25.61; p < 0.001) (Jaecklin et al., presented at the 13th International Fragile X Conference, July 25–29, 2012, Miami, FL, USA).
Fig. 1.
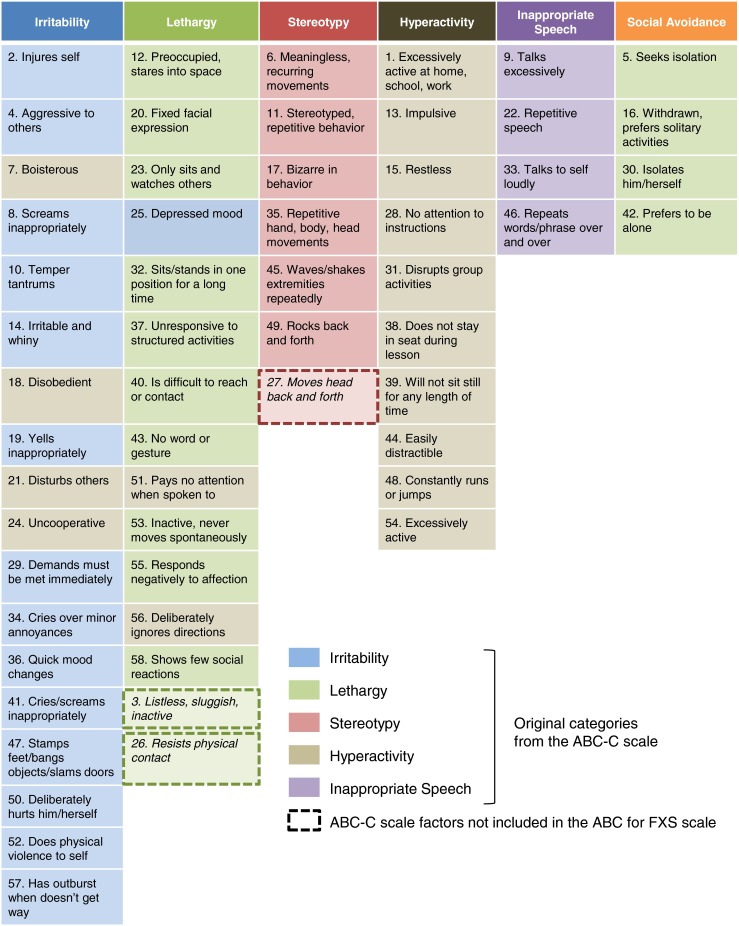
ABC-C for FXS scale. Development of the ABC-C for FXS scale led to the addition of the new Social Avoidance subscale containing specific factors from the ABC-C lethargy subscale
Post hoc analyses of the arbaclofen phase II trial in children and adults with FXS using the ABC-C for FXS scale reported significant improvements in the Social Avoidance subscale (−1.2 vs placebo; p = 0.01), despite no significant improvement in the other subscales of the ABC-C for FXS (Berry-Kravis et al. 2012). Additional benefit was found in a subgroup of patients with more severe social impairment at baseline (ABC-C Lethargy/Social Withdrawal ≥8). In these patients, there was a significant improvement in the average Social Avoidance subscale score (−2.2 vs placebo; p = 0.04).
Other behavioral rating scales which have been used to evaluate individuals with FXS and have shown sensitivity to change are summarized in Table 2. In the mavoglurant study, treatment benefits within the completely methylated population were captured using the following scales: Visual Analogue scale (VAS) ratings of parent-nominated behaviors, Clinical Global Impression—Severity (CGI-S) scale, Clinical Global Impression—Improvement (CGI-I), CGI efficacy index, Repetitive Behavior Scale—Revised, and Social Responsiveness Scale—Adult Research Version, despite no change in the primary endpoint (Jacquemont et al. 2011). Similarly, VAS ratings showed improvements following arbaclofen treatment in the entire per-protocol cohort, in the absence of an improvement in the primary endpoint, the ABC-C irritability subscale, or in other subscales of the ABC-C (Berry-Kravis et al. 2012). Generally, the scales listed in Table 2 are rarely used as primary outcome measures. However, a phase II trial of minocycline in children and adolescents with FXS used the CGI-I scale as the primary outcome measure, reporting a significant overall improvement (2.49 ± 0.13 vs 2.97 ± 0.13 in placebo; p = 0.02) (Leigh et al. 2013). Post hoc analyses of the VAS scores categorized according to behavior observed significant changes in VAS ratings of parent-nominated anxiety and mood-related behaviors (5.26 ± 0.46 vs 4.05 ± 0.46 in placebo; p = 0.05).
Table 2.
Other clinician/parental rating scales assessing behavioral symptoms which have shown sensitivity to change in FXS studies
| Rating scalesa | Study | |||||
|---|---|---|---|---|---|---|
| Lithium | Minocycline | Aripiprazole | Mavoglurant | Arbaclofen | Acamprosate | |
| Open label (n = 19) | ||||||
| Open label (n = 16) | Phase IIb (n = 66) | Open label (n = 12) | Phase II (n = 30) | Phase II (n = 63) | Open label (n = 12) | |
| CGI (National Institute of Mental Health 1970) | B 4.7 (0.9) | Open label | B 4.5 (0.5) | CM subgroup (n = 7) | CGI-S | CGI-I |
| B 5.1 (0.13) | E 1.9 | |||||
| E 4.5 (0.12) | № improved 9 | |||||
| p = 0.09 | CGI-S | |||||
| B 4.7 (0.5) | SSI subgroup (n = 27) | B 4.25 (0.45) | ||||
| C −1.6 (0.8) | E 3.5 (0.5) | D −1.78 (−2.34 to −1.22) | CGI-I | |||
| № improved 12 | E 2.5 (0.24) | E 3.33 (0.5) | ||||
| p = 0.003 | p = 0.02 | |||||
| Phase II | p = 0.008 | p < 0.001 | CGI-S | p < 0.0001 | ||
| C 1.3 (1.1) | CGI-I (n = 55) | B 5.4 (0.22) | ||||
| № improved 13 | 2.49 (0.13) | Effect size 2 | E 4.4 (0.21) | Effect size 2 | ||
| p = 0.004 | p = 0.02 | p = 0.009 | ||||
| VAS (Facco et al. 2011) | B 18.3 (13.0) | Open label | CM subgroup (n = 7) | Problem behaviors | ||
| B 19.3 (7.67) | B 2.2 (0.22) | |||||
| C 15.6 (20.25) | ||||||
| C 22.5 (21.8) | № improved 18 | D 31.84 (14.01–49.67) | E 4.2 (0.32) | |||
| p < 0.001 | ||||||
| Phase IIc | ||||||
| Severity of target behavior 2 (n = 55) | ||||||
| B 2.62 (0.23) | ||||||
| № improved 12 | E 4.91 (0.31) | |||||
| p = 0.06 | ||||||
| Anxiety/mood (n = 26) | p = 0.006 | p = 0.04 | ||||
| B 2.47 (0.25) | ||||||
| E 5.26 (0.46) | ||||||
| p = 0.003 | p = 0.05 | |||||
| Otherd (n = 12) | ||||||
| B 3.49 (0.66) | ||||||
| E 5.84 (0.54) | ||||||
| p = 0.009 | ||||||
| SRS (Constantino et al. 2003) | B 124.5 (26.7) | CM subgroup (n = 7) | ||||
| E 90.1 (31.6) | D −17.91 (−30.4 to −5.77) | |||||
| p < 0.001 | ||||||
| Effect size 1.3 | p = 0.031 | |||||
| RBS-R (Lam and Aman 2007) | D −3.81 (−6.91 to −0.70) | |||||
| p = 0.046 | ||||||
| CM subgroup (n = 7) | ||||||
| D −9.81 (−16.57 to −3.05) | ||||||
| p = 0.038 | ||||||
| VABS-II (Sparrow et al. 1984) | VABS-MAB | SSI subgroup (n = 27) | VABS-C | |||
| B 63.4 (10.1) | ||||||
| E 66.6 (11.2) | ||||||
| p = 0.03 | ||||||
| Effect size 0.32 | ||||||
| VABS-EC | ||||||
| Baseline 22.9 (6.4) | VABS-S | B 69.8 (23.0) | ||||
| Change −4.4 (4.9) | B 80.1 (8.1) | E 78.9 (21.2) | ||||
| № improved 12 | E 99.6 (3.38) | p = 0.003 | ||||
| p = 0.007 | p = 0.03 | Effect size 0.04 | ||||
| Citation | Berry-Kravis et al. (2008) | Leigh et al. (2013); Paribello et al. (2010) | Erickson et al. (2011) | Jacquemont et al. (2011) | Berry-Kravis et al. (2012) | Erickson et al. (2013) |
Data presented as either: mean baseline scores (SD), mean change (SD), mean endpoint score (SD), or mean difference (90 % CI)
B baseline, C change, E endpoint, D difference, CM completely methylated, SSI severe social impairment, CGI Clinical Global Impression, CGI-I CGI of Improvement, CGI-S CGI of Severity, VAS Visual Analogue Scale of Behavior, SRS Social Responsiveness Scale, RBS-R Repetitive Behavior Scale—Revised, VABS-II Vineland Adaptive Behavior Scale—II, VABS-S VABS—Socialization Subscale, VABS-C VABS—Communication subscale, VABS-EC VABS—Expressive Communication Subscale, VABS-MAB VABS—Maladaptive Behavior Subscale
aIf just a subscale was sensitive to change the name is given in parenthesis
bData presented as Least squares mean (SE)
cInvestigators performed an ad hoc analysis of VAS scores by behavior category; they categorized VAS symptoms and defined a combined symptom-specific VAS score as the average of the individual VAS scores for those specific behaviors/symptoms
dOther category included the following behaviors: being organized, potty training, self-calming/self-soothing, verbal initiation of play, chewing objects, overstuffing, scratching stomach, belching, running away, noncompliance/defiance, and self injury
The results of these studies suggest that rating scales can detect therapeutic responses in patients with FXS, and appropriate selection of patients may avoid obscuring a treatment response. They also highlight the importance of using methods such as the ABC-C for FXS scale, which have been validated in this patient population and are therefore more sensitive to changes in specific FXS characteristics. Whether or not the ABC-C for FXS scale can detect changes indicative of disease modification is as yet unknown. Even though the ABC-C may not capture improvements associated with clinical treatment and despite the need to develop and use disease-specific versions, the ABC-C remains the most accepted scale for trials involving patients with developmental delay and behavior issues.
The most appropriate measure for evaluating the extent of disease modification will likely vary according to the age of the patients. It is generally assumed that the younger the patient at the time of treatment onset, the greater the therapeutic benefit may be. This could potentially be assessed by tracking improvements in developmental milestones (e.g., walking, toilet training, and language). Improvements in cognitive function, using study designs investigating literacy and/or numeracy skills after intensive non-pharmacological interventions (delivered with placebo-controlled trials of medications), may also provide an alternative approach to establish disease modification. However, current cognitive outcome measures (in particular standardized IQ tests) have not been validated or standardized for populations with intellectual disability. These measures have inherent problems, such as floor effects and learning effects (test retest effect), and alternative approaches need to be validated within this patient population. One approach, the Test of Attentional Performance for Children (Testbatterie zur Aufmerksamkeitsprüfung für Kinder, KiTAP), uses a computer-based approach to measure the core executive-function deficits of attention and inhibition. In a recent pilot study, the KiTAP showed that it could provide reliable and clinically relevant scores in a population of individuals with FXS over a wide range of age and function (Knox et al. 2012).
The appropriate timing of any intervention aimed at disease modification is also important, as it is not yet clear if there are age limits beyond which disease modification is no longer possible. For example, preclinical data suggest that it is possible to rescue the FXS phenotype in adult Fmr1-knockout mice following chronic treatment with an mGluR5 antagonist (Michalon et al. 2012), and no age-related effects were reported in the recent trial evaluating arbaclofen in patients with FXS aged 6–39 years (Berry-Kravis et al. 2012). Finally, any study designed to quantify disease modification will need to be multidimensional and longitudinal in order to effectively track the rate of any changes that occur.
Multiple treatments targeting different mechanisms involved in FXS
Mavoglurant was developed to therapeutically block mGluR5, targeting the excessive glutamatergic signaling associated with a lack of FMRP (Levenga et al. 2011). Loss of FMRP and the resultant mGluR5-dependent dysregulation of synaptic plasticity are thought to contribute to the pathology and symptomatology of FXS (Darnell et al. 2011; Jacquemont et al. 2007; Levenga et al. 2010). This is referred to as the mGluR theory of FXS, which proposes that inhibition of group I mGluR signaling might be a potential therapeutic target in FXS (Bear et al. 2004).
Following the results of studies that showed treatment with mGluR5 antagonists, including mavoglurant, could rescue several synaptic phenotypes in animal models (Choi et al. 2010; de Vrij et al. 2008; Levenga et al. 2011; McBride et al. 2005; Tucker et al. 2006; Yan et al. 2005), it was hypothesized that mavoglurant had the potential to treat the underlying pathophysiology of FXS, thus differing from current pharmacotherapy for FXS which is symptom-driven.
Other agents targeting specific molecular pathways are in development for FXS (Table 3); these include RG7090 (RO4917523), another mGluR5 antagonist; STX209 (arbaclofen, R-baclofen), a γ-aminobutyric acid type B (GABAB) receptor agonist (Berry-Kravis et al. 2012); and minocycline, a matrix metalloproteinase-9 antagonist (Bilousova et al. 2009; Leigh et al. 2013; Paribello et al. 2010; Utari et al. 2010). All of these agents aim to address the underlying pathology of FXS by targeting specific molecular pathways. However, there are currently no data linking these specific pathways to particular symptoms of FXS. Furthermore, there is a lack of biomarkers related to the mechanism targeted by these new treatments (e.g., synaptic plasticity rescued in animal models) preventing investigators from evaluating how effective these agents are at targeting a particular pathway.
Table 3.
Drugs in clinical development for treating FXS: drug class, action, study phase, and published data
| Class | Drug | Action | Study phase | Published data | |
|---|---|---|---|---|---|
| Completed | Ongoing | ||||
| Glutamatergic | Mavoglurant (AFQ056) | mGluR5 antagonist | II | II; IIb; III | Jacquemont et al. (2011) |
| RG7090 (RO4917523) | mGluR5 antagonist | IIa | II | ||
| STX107 | mGluR5 antagonist | None | |||
| Fenobam (NPL-2009) | mGluR5 antagonist | Open label | None | Berry-Kravis et al. (2009) | |
| Memantinea | NMDA receptor antagonist | Open label | None | Erickson et al. (2009) | |
| GABAergic | Arbaclofen (STX209/R-baclofen) | GABAB receptor agonist | II | III | Berry-Kravis et al. (2012) |
| Ganaxolone | GABAA receptor agonist | None | II | ||
| Acamprosatea,b | GABAA receptor agonist, NMDA receptor antagonist; anti-oxidant | Open label | II/III | Erickson et al. (2010); Erickson et al. (2013) | |
| Atypical antipsychotics | Aripiprazolea | Partial dopamine D2 receptor agonist; serotonin 5-HT1A agonist; SSRI (serotonin 5-HT2A antagonist) | Open label | None | Erickson et al. (2011) |
| Antidepressant/anxiolytic | Sertralinea | SSRI (serotonin 5-HTA antagonist) | Open label | II | Indah Winarni et al. (2012) |
| Mood stabilizer | Lithiuma | Phospholipase C inhibitor; GSK-3 inhibitor | Open label | None | Berry-Kravis et al. (2008) |
| Antibiotic | Minocyclinea | MMP9 inhibitor | Open label II | None | Paribello et al. (2010); Leigh et al. (2013) |
Only open-label studies or phase II and above are reported in the table
mGluR metabotropic glutamate receptor, GABA B γ-aminobutyric acid type B, MMP9 metalloproteinase 9, NMDA N-methyl-d-aspartate, 5-HT 1A 5-hydroxytryptamine 1A, GSK-3 glycogen synthase-3, SSRI selective serotonin uptake inhibitor
aDrug already approved by FDA for other indications
bMode of action still unclear
What insights into ASD can be gained from studying FXS?
ASD encompasses an etiology and clinically heterogeneous population, which has posed challenges in the search to find effective treatments. However, recent research shows promise for the mapping of the multitude of aforementioned genetic variants in ASD onto shared pathways. Given the overwhelming degree of locus heterogeneity, finding convergence in specific molecular pathways will be required in order to perform targeted treatment trials with sufficient sample size. Understanding the molecular overlap between FXS and ASD, combined with lessons learned from planning and running clinical trials for FXS, may provide valuable insight into the most appropriate study designs for investigating potential therapies in ASD.
We have previously highlighted the link between FMRP and autism-candidate genes associated with synaptic plasticity (Ascano et al. 2012; Darnell et al. 2011; Iossifov et al. 2012). Lack of FMRP interferes with synaptic plasticity causing disruption of various pathways, for example, up-regulation of mGluR5 and mTOR signaling (Bear et al. 2004; Sharma et al. 2010) and down regulation of the GABA and dopamine systems (D’Hulst and Kooy 2007; Wang et al. 2008).
The glutamate and GABA pathways have both been implicated in ASD by the use of Fmr1 KO mice as a model system for studying autistic behaviors (Rogers et al. 2013) and the increasing evidence of altered expression of mGluR5, FMRP, and GABA receptors in individuals with autism (Fatemi et al. 2010, 2011; Fatemi and Folsom 2011). Agents targeting these two pathways in FXS (Table 3) may therefore be useful in ASD. Research using models of FXS to elucidate converging molecular pathways behind autistic behaviors might identify novel therapeutic targets for ASD. However, treatments targeting these pathways could have different outcomes for individuals across the ASD spectrum despite similarities in clinical presentation. To illustrate, tuberous sclerosis complex (TSC) and FXS share clinical characteristics (including ASD) and are caused by haploinsufficiency of TSC1 and TSC2 associated with the regulation of protein synthesis at the synapse (for review, see Orlova and Crino 2010). In vivo data indicate that the synaptic dysfunction observed in mouse models of FXS and TSC falls at opposite ends of the physiological spectrum (Auerbach et al. 2011; Bateup et al. 2011) and can be rescued with agents that modulate mGluR5 in opposite directions or by crossing the mouse strains (Auerbach et al. 2011).
Given the wide range of symptoms observed in the ASD patient population, clinical trials in ASD face similar problems to trials in FXS in regard to patient selection and choice of outcome measures. The development of biomarkers stratification and endophenotyping methodologies in FXS trials could be adapted for ASD trials. Likewise, new or modified measures (e.g., ABC-C for FXS) that can effectively track improvements in symptoms may also be transferable to studies in ASD, but their feasibility across the ASD spectrum will need to be tested. Because of the prevalence of autism in FXS and shared neurophysiology with ASD, FXS can be viewed as a model for autism.
Conclusions
Advances in understanding the neurobiology of “monogenic” syndromes such as FXS have revealed heterogeneity at the level of phenotype, manifestations of the causative mutation, and drug response. Complex disorders such as ASD are further complicated by increasing evidence for a heterogeneous etiology and mechanism of disease, unlike FXS which is caused by a mutation within a single genetic locus, the FMR1 gene. Current research is focused on identifying common therapeutic targets among patients with different molecular etiologies. The development of novel treatments for specific molecular targets for these disorders has the potential to rescue specific phenotypes and may result in what can be classified as disease modification.
Developing biomarkers may aid patient stratification for clinical trials and predict response to treatments. In addition to patient stratification methods, future clinical trial designs will also need to consider appropriate endpoints and length and timing of interventions (Fig. 2). Ultimately, it may be possible to stratify patients for genetic risk factors associated with neurodevelopmental disorders such as FXS and ASD, to enable early implementation of customized therapeutic interventions that could normalize brain development and optimize clinical outcomes.
Fig. 2.

Key elements for future clinical trials in FXS
Acknowledgments
Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals. We thank Georgina Collett, Ph.D., and Kerrie O’Rourke, Ph.D., of iMed Comms, who provided medical writing assistance with this review.
Conflict of interest
S Jacquemont has acted as a consultant for Novartis Pharma AG and received honoraria and reimbursement for travel expenses. S Jacquemont has also received grants for the clinical investigation of mavoglurant. R Hagerman is an employee of the University of California and a member of the National Fragile X Foundation Scientific and Clinical Advisory Board Committee. R Hagerman has received compensation from Novartis Pharma AG, F. Hoffman-La Roche, and Seaside Therapeutics for the clinical investigation of new drugs. R Hagerman also received honoraria from Novartis and reimbursement for travel expenses from F. Hoffman-La Roche and Novartis Pharma AG and has acted as a consultant for Novartis Pharma AG and Genentech Inc. R Hagerman has received royalties from Oxford University Press and Johns Hopkins University Press for books published in the field of neurological disorders. E Berry-Kravis has acted as a consultant for Novartis Pharma AG and F. Hoffman-La Roche. E Berry-Kravis received compensation from Novartis Pharma AG, F. Hoffman-La Roche, and Seaside Therapeutics for the clinical investigation of new drugs and an honorarium from Novartis Pharma AG. E Berry-Kravis also received reimbursement for travel expenses from Novartis Pharma AG, Seaside Therapeutics, and F. Hoffman-La Roche. V Des Portes is an employee of the Centre Université de Lyon, France. His institution has received compensation from Novartis Pharma AG for the clinical investigation of new drugs, and he is currently involved in ongoing clinical trials with Novartis Pharma AG and F. Hoffman-La Roche. He also has acted as a consultant for Novartis Pharma AG and received reimbursement for travel expenses. B Gomez-Mancilla, F von Raison, F Gasparini, M Ufer, and G Apostol are employees of Novartis Pharma AG and hold shares with Novartis Pharma AG. F Gasparini and B Gomez-Mancilla have also received reimbursement from Novartis Pharma AG for travel expenses, and the spouse of G Apostol is an employee of Novartis International AG.
References
- Aman MG, Singh NN, Stewart AW, Field CJ. The aberrant behavior checklist: a behavior rating scale for the assessment of treatment effects. Am J Ment Defic. 1985;89:485–491. [PubMed] [Google Scholar]
- Ascano M, Jr, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, Williams Z, Ohler U, Tuschl T. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492:382–386. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480:63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni B, Mandel JL. Advances in understanding of fragile X pathogenesis and FMRP function, and in identification of X linked mental retardation genes. Curr Opin Genet Dev. 2002;12:284–293. doi: 10.1016/S0959-437X(02)00300-3. [DOI] [PubMed] [Google Scholar]
- Bateup HS, Takasaki KT, Saulnier JL, Denefrio CL, Sabatini BL. Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. J Neurosci. 2011;31:8862–8869. doi: 10.1523/JNEUROSCI.1617-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Bell MV, Hirst MC, Nakahori Y, MacKinnon RN, Roche A, Flint TJ, Jacobs PA, Tommerup N, Tranebjaerg L, Froster-Iskenius U. Physical mapping across the fragile X: hypermethylation and clinical expression of the fragile X syndrome. Cell. 1991;64:861–866. doi: 10.1016/0092-8674(91)90514-Y. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Sumis A, Hervey C, Nelson M, Porges SW, Weng N, Weiler IJ, Greenough WT. Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Dev Behav Pediatr. 2008;29:293–302. doi: 10.1097/DBP.0b013e31817dc447. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Hessl D, Coffey S, Hervey C, Schneider A, Yuhas J, Hutchison J, Snape M, Tranfaglia M, Nguyen DV, Hagerman R. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet. 2009;46:266–271. doi: 10.1136/jmg.2008.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E, Hessl D, Rathmell B, Zarevics P, Cherubini M, Walton-Bowen K, Mu Y, Nguyen DV, Gonzalez-Heydrich J, Wang PP, Carpenter RL, Bear MF, Hagerman RJ. Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci Transl Med. 2012;4:152ra127. doi: 10.1126/scitranslmed.3004214. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Hessl D, Abbeduto L, Reiss AL, Beckel-Mitchener A, Urv TK, Outcome measures working groups (2013) Outcome measures for clinical trials in fragile X syndrome. J Dev Behav Pediatr 34:508–522 [DOI] [PMC free article] [PubMed]
- Bilousova TV, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW, Ethell IM. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet. 2009;46:94–102. doi: 10.1136/jmg.2008.061796. [DOI] [PubMed] [Google Scholar]
- Choi CH, McBride SM, Schoenfeld BP, Liebelt DA, Ferreiro D, Ferrick NJ, Hinchey P, Kollaros M, Rudominer RL, Terlizzi AM, Koenigsberg E, Wang Y, Sumida A, Nguyen HT, Bell AJ, McDonald TV, Jongens TA. Age-dependent cognitive impairment in a Drosophila fragile X model and its pharmacological rescue. Biogerontology. 2010;11:347–362. doi: 10.1007/s10522-009-9259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantino JN, Davis SA, Todd RD, Schindler MK, Gross MM, Brophy SL, Metzger LM, Shoushtari CS, Splinter R, Reich W. Validation of a brief quantitative measure of autistic traits: comparison of the social responsiveness scale with the autism diagnostic interview—revised. J Autism Dev Disord. 2003;33:427–433. doi: 10.1023/A:1025014929212. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries BB, Wiegers AM, Smits AP, Mohkamsing S, Duivenvoorden HJ, Fryns JP, Curfs LM, Halley DJ, Oostra BA, van den Ouweland AM, Niermeijer MF. Mental status of females with an FMR1 gene full mutation. Am J Hum Genet. 1996;58:1025–1032. [PMC free article] [PubMed] [Google Scholar]
- de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL, Oostra BA, Willemsen R. Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis. 2008;31:127–132. doi: 10.1016/j.nbd.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Hulst C, Kooy RF. The GABAA receptor: a novel target for treatment of fragile X? Trends Neurosci. 2007;30:425–431. doi: 10.1016/j.tins.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Dyer-Friedman J, Glaser B, Hessl D, Johnston C, Huffman LC, Taylor A, Wisbeck J, Reiss AL. Genetic and environmental influences on the cognitive outcomes of children with fragile X syndrome. J Am Acad Child Adolesc Psychiatry. 2002;41:237–244. doi: 10.1097/00004583-200203000-00002. [DOI] [PubMed] [Google Scholar]
- El Fatimy R, Tremblay S, Dury AY, Solomon S, De Koninck P, Schrader JW, Khandjian EW. Fragile X mental retardation protein interacts with the RNA-binding protein Caprin1 in neuronal RiboNucleoProtein complexes [corrected] PLoS One. 2012;7:e39338. doi: 10.1371/journal.pone.0039338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson CA, Mullett JE, McDougle CJ. Open-label memantine in fragile X syndrome. J Autism Dev Disord. 2009;39:1629–1635. doi: 10.1007/s10803-009-0807-3. [DOI] [PubMed] [Google Scholar]
- Erickson CA, Mullett JE, McDougle CJ. Brief report: acamprosate in fragile X syndrome. J Autism Dev Disord. 2010;40:1412–1416. doi: 10.1007/s10803-010-0988-9. [DOI] [PubMed] [Google Scholar]
- Erickson CA, Stigler KA, Wink LK, Mullett JE, Kohn A, Posey DJ, McDougle CJ. A prospective open-label study of aripiprazole in fragile X syndrome. Psychopharmacology. 2011;216:85–90. doi: 10.1007/s00213-011-2194-7. [DOI] [PubMed] [Google Scholar]
- Erickson CA, Wink LK, Ray B, Early MC, Stiegelmeyer E, Mathieu-Frasier L, Patrick V, Lahiri DK, McDougle CJ. Impact of acamprosate on behavior and brain-derived neurotrophic factor: an open-label study in youth with fragile X syndrome. Psychopharmacology (Berl) 2013;228:75–84. doi: 10.1007/s00213-013-3022-z. [DOI] [PubMed] [Google Scholar]
- Facco E, Zanette G, Favero L, Bacci C, Sivolella S, Cavallin F, Manani G. Toward the validation of visual analogue scale for anxiety. Anesth Prog. 2011;58:8–13. doi: 10.2344/0003-3006-58.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD. Dysregulation of fragile X mental retardation protein and metabotropic glutamate receptor 5 in superior frontal cortex of individuals with autism: a postmortem brain study. Mol Autism. 2011;2:6. doi: 10.1186/2040-2392-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Reutiman TJ, Folsom TD, Rooney RJ, Patel DH, Thuras PD. mRNA and protein levels for GABAAalpha4, alpha5, beta1 and GABABR1 receptors are altered in brains from subjects with autism. J Autism Dev Disord. 2010;40:743–750. doi: 10.1007/s10803-009-0924-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD, Kneeland RE, Liesch SB. Metabotropic glutamate receptor 5 upregulation in children with autism is associated with underexpression of both fragile X mental retardation protein and GABAA receptor beta 3 in adults with autism. Anat Rec (Hoboken) 2011;294:1635–1645. doi: 10.1002/ar.21299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber KB, Visootsak J, Warren ST. Fragile X syndrome. Eur J Hum Genet. 2008;16:666–672. doi: 10.1038/ejhg.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Berry-Kravis EM, Bassell GJ. Therapeutic strategies in fragile X syndrome: dysregulated mGluR signaling and beyond. Neuropsychopharmacology. 2012;37:178–195. doi: 10.1038/npp.2011.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Hagerman PJ. Fragile X syndrome: diagnosis, treatment and research. Baltimore: Johns Hopkins University Press; 2002. [Google Scholar]
- Hagerman RJ, Hull CE, Safanda JF, Carpenter I, Staley LW, O’Connor RA, Seydel C, Mazzocco MM, Snow K, Thibodeau SN. High functioning fragile X males: demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. Am J Med Genet. 1994;51:298–308. doi: 10.1002/ajmg.1320510404. [DOI] [PubMed] [Google Scholar]
- Hagerman R, Hoem G, Hagerman P. Fragile X and autism: intertwined at the molecular level leading to targeted treatments. Mol Autism. 2010;1:12. doi: 10.1186/2040-2392-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall SS, Lightbody AA, Reiss AL. Compulsive, self-injurious, and autistic behavior in children and adolescents with fragile X syndrome. Am J Ment Retard. 2008;113:44–53. doi: 10.1352/0895-8017(2008)113[44:CSAABI]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Harris SW, Hessl D, Goodlin-Jones B, Ferranti J, Bacalman S, Barbato I, Tassone F, Hagerman PJ, Herman H, Hagerman RJ. Autism profiles of males with fragile X syndrome. Am J Ment Retard. 2008;113:427–438. doi: 10.1352/2008.113:427-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessl D, Dyer-Friedman J, Glaser B, Wisbeck J, Barajas RG, Taylor A, Reiss AL. The influence of environmental and genetic factors on behavior problems and autistic symptoms in boys and girls with fragile X syndrome. Pediatrics. 2001;108:E88. doi: 10.1542/peds.108.5.e88. [DOI] [PubMed] [Google Scholar]
- Hessl D, Nguyen DV, Green C, Chavez A, Tassone F, Hagerman RJ, Senturk D, Schneider A, Lightbody A, Reiss AL, Hall S. A solution to limitations of cognitive testing in children with intellectual disabilities: the case of fragile X syndrome. J Neurodev Disord. 2009;1:33–45. doi: 10.1007/s11689-008-9001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indah Winarni T, Chonchaiya W, Adams E, Au J, Mu Y, Rivera SM, Nguyen DV, Hagerman RJ. Sertraline may improve language developmental trajectory in young children with fragile X syndrome: a retrospective chart review. Autism Res Treat. 2012;2012:104317. doi: 10.1155/2012/104317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB, Mardis ER, Wilson RK, Schatz MC, McCombie WR, Wigler M. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahashi C, Tassone F, Hagerman RJ, Yasui D, Parrott G, Nguyen D, Mayeur G, Hagerman PJ. A quantitative ELISA assay for the fragile X mental retardation 1 protein. J Mol Diagn. 2009;11:281–289. doi: 10.2353/jmoldx.2009.080118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Hagerman RJ, Hagerman PJ, Leehey MA. Fragile-X syndrome and fragile X-associated tremor/ataxia syndrome: two faces of FMR1. Lancet Neurol. 2007;6:45–55. doi: 10.1016/S1474-4422(06)70676-7. [DOI] [PubMed] [Google Scholar]
- Jacquemont S, Curie A, Des Portes V, Torrioli MG, Berry-Kravis E, Hagerman RJ, Ramos FJ, Cornish K, He Y, Paulding C, Neri G, Chen F, Hadjikhani N, Martinet D, Meyer J, Beckmann JS, Delange K, Brun A, Bussy G, Gasparini F, Hilse T, Floesser A, Branson J, Bilbe G, Johns D, Gomez-Mancilla B. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med. 2011;3:64ra1. doi: 10.1126/scitranslmed.3001708. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Cortell R, Kau AS, Bukelis I, Tierney E, Gray RM, Cox C, Capone GT, Stanard P. Autism spectrum disorder in fragile X syndrome: communication, social interaction, and specific behaviors. Am J Med Genet A. 2004;129A:225–234. doi: 10.1002/ajmg.a.30229. [DOI] [PubMed] [Google Scholar]
- Knox A, Schneider A, Abucayan F, Hervey C, Tran C, Hessl D, Berry-Kravis E. Feasibility, reliability, and clinical validity of the Test of Attentional Performance for Children (KiTAP) in fragile X syndrome (FXS) J Neurodev Disord. 2012;4:2. doi: 10.1186/1866-1955-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KS, Aman MG. The Repetitive Behavior Scale—Revised: independent validation in individuals with autism spectrum disorders. J Autism Dev Disord. 2007;37:855–866. doi: 10.1007/s10803-006-0213-z. [DOI] [PubMed] [Google Scholar]
- Leigh MJS, Nguyen DV, Mu Y, Winarni TI, Schneider A, Chechi T, Polussa J, Doucet P, Tassone F, Rivera SM, Hessl D, Hagerman RJ (2013) A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile X syndrome. J Dev Behav Pediatr 34:147–155 [DOI] [PMC free article] [PubMed]
- Levenga J, de Vrij FM, Oostra BA, Willemsen R. Potential therapeutic interventions for fragile X syndrome. Trends Mol Med. 2010;16:516–527. doi: 10.1016/j.molmed.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenga J, Hayashi S, de Vrij FM, Koekkoek SK, van der Linde HC, Nieuwenhuizen I, Song C, Buijsen RA, Pop AS, Gomezmancilla B, Nelson DL, Willemsen R, Gasparini F, Oostra BA. AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol Dis. 2011;42:311–317. doi: 10.1016/j.nbd.2011.01.022. [DOI] [PubMed] [Google Scholar]
- Loesch DZ, Huggins RM, Hagerman RJ. Phenotypic variation and FMRP levels in fragile X. Ment Retard Dev Disabil Res Rev. 2004;10:31–41. doi: 10.1002/mrdd.20006. [DOI] [PubMed] [Google Scholar]
- McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, Sehgal A, Siwicki KK, Dockendorff TC, Nguyen HT, McDonald TV, Jongens TA. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005;45:753–764. doi: 10.1016/j.neuron.2005.01.038. [DOI] [PubMed] [Google Scholar]
- McLennan Y, Polussa J, Tassone F, Hagerman R. Fragile X syndrome. Curr Genomics. 2011;12:216–224. doi: 10.2174/138920211795677886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron. 2012;74:49–56. doi: 10.1016/j.neuron.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institute of Mental Health (1970) CGI: clinical global impressions. In: Guy W, Bonato RR (eds) Manual of the ECDEU assessment battery. National Institute of Mental Health, Rockville
- Nowicki ST, Tassone F, Ono MY, Ferranti J, Croquette MF, Goodlin-Jones B, Hagerman RJ. The Prader–Willi phenotype of fragile X syndrome. J Dev Behav Pediatr. 2007;28:133–138. doi: 10.1097/01.DBP.0000267563.18952.c9. [DOI] [PubMed] [Google Scholar]
- Orlova KA, Crino PB. The tuberous sclerosis complex. Ann N Y Acad Sci. 2010;1184:87–105. doi: 10.1111/j.1749-6632.2009.05117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paribello C, Tao L, Folino A, Berry-Kravis E, Tranfaglia M, Ethell IM, Ethell DW (2010) Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol 10:91 [DOI] [PMC free article] [PubMed]
- Reiss AL, Freund LS, Baumgardner TL, Abrams MT, Denckla MB. Contribution of the FMR1 gene mutation to human intellectual dysfunction. Nat Genet. 1995;11:331–334. doi: 10.1038/ng1195-331. [DOI] [PubMed] [Google Scholar]
- Rogers SJ, Wehner DE, Hagerman R. The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J Dev Behav Pediatr. 2001;22:409–417. doi: 10.1097/00004703-200112000-00008. [DOI] [PubMed] [Google Scholar]
- Rogers TD, McKimm E, Dickson PE, Goldowitz D, Blaha CD, Mittleman G. Is autism a disease of the cerebellum? An integration of clinical and pre-clinical research. Front Syst Neurosci. 2013;7:15. doi: 10.3389/fnsys.2013.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansone SM, Widaman KF, Hall SS, Reiss AL, Lightbody A, Kaufmann WE, Berry-Kravis E, Lachiewicz A, Brown EC, Hessl D. Psychometric study of the aberrant behavior checklist in fragile X syndrome and implications for targeted treatment. J Autism Dev Disord. 2012;42:1377–1392. doi: 10.1007/s10803-011-1370-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenck A, Bardoni B, Langmann C, Harden N, Mandel JL, Giangrande A. CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron. 2003;38:887–898. doi: 10.1016/S0896-6273(03)00354-4. [DOI] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow S, Balla D, Cicchetti D. Vineland adaptive behavior scales: interview edition. Circle Pines: American Guidance Service; 1984. [Google Scholar]
- Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, Warren ST. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet. 1992;1:397–400. doi: 10.1093/hmg/1.6.397. [DOI] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, Ikle DN, Dyer PN, Lampe M, Willemsen R, Oostra BA, Taylor AK. FMRP expression as a potential prognostic indicator in fragile X syndrome. Am J Med Genet. 1999;84:250–261. doi: 10.1002/(SICI)1096-8628(19990528)84:3<250::AID-AJMG17>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Tucker B, Richards RI, Lardelli M. Contribution of mGluR and Fmr1 functional pathways to neurite morphogenesis, craniofacial development and fragile X syndrome. Hum Mol Genet. 2006;15:3446–3458. doi: 10.1093/hmg/ddl422. [DOI] [PubMed] [Google Scholar]
- Utari A, Chonchaiya W, Rivera SM, Schneider A, Hagerman RJ, Faradz SM, Ethell IM, Nguyen DV. Side effects of minocycline treatment in patients with fragile X syndrome and exploration of outcome measures. Am J Intellect Dev Disabil. 2010;115:433–443. doi: 10.1352/1944-7558-115.5.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wu LJ, Kim SS, Lee FJ, Gong B, Toyoda H, Ren M, Shang YZ, Xu H, Liu F, Zhao MG, Zhuo M. FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron. 2008;59:634–647. doi: 10.1016/j.neuron.2008.06.027. [DOI] [PubMed] [Google Scholar]
- Willemsen R, Mohkamsing S, de Vries B, Devys D, van den Ouweland A, Mandel JL, Galjaard H, Oostra B. Rapid antibody test for fragile X syndrome. Lancet. 1995;345:1147–1148. doi: 10.1016/S0140-6736(95)90979-6. [DOI] [PubMed] [Google Scholar]
- Willemsen R, Smits A, Mohkamsing S, van Beerendonk H, de Haan A, de Vries B, van den Ouweland A, Sistermans E, Galjaard H, Oostra BA. Rapid antibody test for diagnosing fragile X syndrome: a validation of the technique. Hum Genet. 1997;99:308–311. doi: 10.1007/s004390050363. [DOI] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major fragile X syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49:1053–1066. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Zarnescu DC, Shan G, Warren ST, Jin P. Come FLY with us: toward understanding fragile X syndrome. Genes Brain Behav. 2005;4:385–392. doi: 10.1111/j.1601-183X.2005.00136.x. [DOI] [PubMed] [Google Scholar]