

Abstract
Mammalian H3.3 is a variant of the canonical histone H3.1 essential for genome reprogramming in the fertilized eggs and maintenance of chromatin structure in neuronal cells. An H3.3-specific histone chaperone, DAXX, directs the deposition of H3.3 onto pericentric and telomeric heterochromatin. H3.3 differs from H3.1 by only five amino acids, yet DAXX can distinguish the two with high precision. By a combination of structural, biochemical and cell-based targeting analyses, here we show that Ala87 and Gly90 are the principal determinants of H3.3 specificity. DAXX uses a shallow hydrophobic pocket to accommodate the small, hydrophobic Ala87 of H3.3, whereas a polar binding environment in DAXX prefers Gly90 in H3.3 over the hydrophobic Met90 in H3.1. An H3.3-H4 heterodimer is bound by the histone-binding domain of DAXX, which makes extensive contacts with both H3.3 and H4.
Epigenetic information is embedded in covalent modifications of DNA and histones, and in the nature of histones within the nucleosome. In most cases, the nucleosome consists of an octamer of four canonical histones synthesized during the S phase of the cell cycle and incorporated into the nucleosome in a replication-dependent manner. In some genomic loci, cell types or developmental stages, histone variants may replace canonical histones. Expression of histone variants is not restricted to the S phase, and they can be assembled into the nucleosome in replication-independent processes, through alternative histone deposition pathways1–4. Two ubiquitous variants of canonical histones H3.1 in mammals are CENP-A and H3.3. CENP-A is a centromere-specific histone H3 variant assembled onto centromeric chromatin during late M-early G1 phase of the cell cycle1,5. Deposition of CENP-A at the centromere requires histone chaperone HJURP6–8. H3.3 is synthesized throughout the cell cycle and localized in gene bodies as well as in pericentric and telomeric heterochromatin1–4. Two principal H3.3 deposition pathways are mediated by histone chaperones DAXX (death domain associated protein) and HIRA9–13. DAXX cooperates with an SNF2-like chromatin remodeler, ATRX, to direct the deposition of H3.3 onto pericentric and telomeric heterochromatin12–14, whereas HIRA mainly targets H3.3 to gene bodies2–4. A third chaperone, DEK, has been implicated in the assembly of H3.3 nucleosomes and transcriptional activation of a nuclear receptor15. H3.3 is highly enriched during early development and in neuronal cells1–4. It plays important roles in reprogramming the paternal genome in fertilized eggs, and somatic mutations of H3.3 have been implicated in the development of pediatric glioblastomas16–18.
To understand the assembly of H3.3 nucleosomes, it is necessary to know how it is differentially recognized by its chaperones. DAXX and HIRA share no known sequence homology, and their sequences give no clues to how they might bind histones, not to mention the precise knowledge needed to distinguish five amino acid differences between H3.3 and H3.1. Structural studies can provide atomic-resolution details of histone-chaperone interactions, as exemplified by the structures of several H3(variants)-H4 complexes with their respective chaperones19–23. Here we set out to investigate the molecular mechanism of specific recognition of H3.3 by DAXX. We have determined the structure of the histone binding domain of human DAXX (DAXX HBD) in complex with histones H3.3 and H4, and identified and analyzed the determinants for the binding specificity between H3.3 and DAXX, using a combination of structural, biochemical and cellular co-localization approaches.
RESULTS
Overall structure of DAXX HBD-H3.3-H4
A previous study identified that residues 183–417 of DAXX encompasses the domain for binding the H3.3-H4 complex14. By systematic deletion of DAXX and co-purification with the full-length H3.3-H4 complex, we have succeeded in crystallizing and solving a 2.8 Å structure of the histone binding domain of human DAXX (DAXX HBD, a.a. 184–390) in complex with histones H3.3 and H4 (Table 1). The structure shows that DAXX HBD mainly consists of six α-helices, α1–α6, which can be separated into three subdomains based on whether the helices are interacting with each other: subdomain-1 includes α1–α2; subdomain-2 is composed of α3–α5; and subdomain-3 consists of α6 (Fig. 1A). Subdomain-1 interacts with H3.3 regions spanning αN, α1 and its C-terminal loop, and the N-terminal part of α2. Subdomain-2 contacts both H3.3 and H4. α5 packs antiparallelly against α2, the central helix of the histone-fold domain of H3.3, while the N-terminal end of α4 interacts with the loop connecting α2 and α3 of H3.3. The interaction between subdomain-2 of DAXX HBD and H4 occurs in two regions: the loop connecting α3 and α4 of DAXX HBD contacts H4 through its loop between α1 and α2, and the C-terminal end of α1; and the very C-terminal tail of H4 is stabilized by interaction with the C-terminal region of α5 of DAXX HBD. Helix α6 of the DAXX HBD packs antiparallelly against α2 in histone H4 and also interacts with H3.3 via its C-terminal helix, α3 (Fig. 1A). The binding between DAXX HBD and H3.3-H4 involves a mixture of hydrophobic and polar interactions (Fig. 1B). The interaction between DAXX HBD and the H3.3-H4 heterodimer buries a total surface area of 6544 Å2, consistent with a stable heterotrimeric complex that can withstand purification steps at a 1 M salt concentration.
Table 1.
Data collection and refinement statistics (SAD)
| K2PtCl4 derivative | |
|---|---|
| Data collection | |
| Space group | P6122 |
| Cell dimensions | |
| a, b, c (Å) | 72.10, 72.10, 323.55 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution (Å) | 50–2.80 (2.90–2.80) * |
| Rsym or Rmerge | 0.083 (0.521) |
| I / σI | 21.9 (1.9) |
| Completeness (%) | 99.8 (99.1) |
| Redundancy | 7.8 (8.1) |
| Refinement | |
| Resolution (Å) | 50–2.8 |
| No. reflections | |
| Rwork / Rfree | 20.8/27.1 |
| No. atoms | |
| Protein | 3040 |
| Ligand/ion | 13 |
| Water | 6 |
| B-factors (Å2) | 79.7 |
| DAXX (main chain, side chain) | 83.2, 87.9 |
| H3.3 (main chain, side chain) | 69.1, 73.6 |
| H4 (main chain, side chain) | 71.0, 75.2 |
| Water | 65.1 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 0.905 |
Values in parentheses are for highest-resolution shell.
Figure 1.
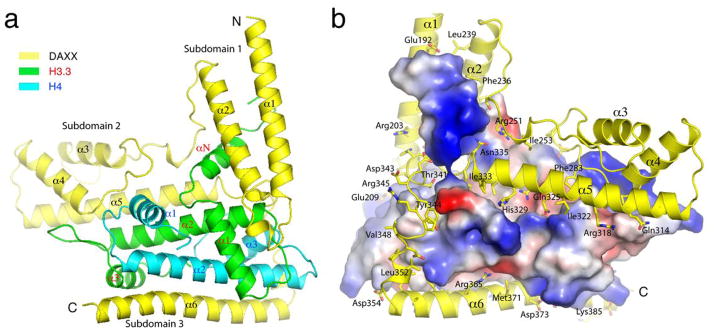
Overall structure of the DAXX HBD-H3.3-H4 complex. (A) A ribbon representation showing DAXX HBD, H3.3 and H4, with secondary structure elements labeled in black, red and blue font, respectively. Helices grouped in subdomains are indicated. (B) DAXX-histone interactions. H3.3 and H4 are shown in a surface representation with electrostatic potential distribution. DAXX residues involved in interactions with DAXX are shown as stick models superimposed on the ribbon diagram of DAXX; certain key residues are labeled.
Comparison of nucleosomal and DAXX-bound H3.3-H4
Comparing the structures of the DAXX HBD-bound H3.3-H4 heterodimer and the H3.3-H4 half tetramer in the H3.3-containing nucleosome core particle (NCP)24 reveals major conformational differences of H3.3 (Figs. 2A & 2B). Most notably, the αN of H3.3, which interacts with helices α1 and α2 of DAXX HBD, is oriented in an opposite direction with respect to the corresponding helix in the nucleosomal complex (Fig. 2A). The αN of H3.3 is normally involved in interacting with DNA near the entry-exit point in the NCP, and the observed orientation in the present structure indicates a major conformational rearrangement of the helix, deviating from the conformation that favors the wrapping the DNA in the nucleosomal complex. Interestingly, helices α1 and α2 of DAXX HBD would block the wrapping of nucleosomal DNA if the DAXX HBD-bound H3.3-H4 heterodimer was superimposed with one of the nucleosomal H3.3-H4 heterodimers (Fig. 2C). In addition, the α3 in H3.3 is tilted away from the α2 of histone H4; the last turn of α2 in H3.3 is unwound, and the loop between α2 and α3 in H3.3 is pulled toward the α4 in DAXX HBD (Figs. 2B & 1A). The conformation of α3 and the C-terminal portion of α2 is critical for the formation of a H3.3-H4 tetramer through H3.3-H3.3 interactions, as observed in the structure of the H3.3-containing NCP24. The present conformation of α2 and α3 prevents the dimerization of H3.3-H4 heterodimers due to potential steric clashes. The middle part of the α5 of DAXX HBD also poses a spatial hindrance on the association of another H3.3-H4 heterodimer (Fig. 2C). It is interesting to note that all structures of histone chaperone-H3(variants)-H4 complexes determined to date show the binding of a chaperone to a heterodimer of H3 (variants) and H4, although inhibition of dimerization via H3 or its variants is achieved differently by different chaperones19–23.
Figure 2.

Conformational differences between the DAXX HBD-bound and nucleosomal H3.3-H4 complexes. (A) An H3.3-H4 dimer from the NCP structure (PDB 3AV2), with H3.3 and H4 colored in wheat and light blue, respectively, is superimposed with the DAXX HBD-H3.3-H4 complex. (B) Superimposed structures viewed from a direction perpendicular to that in Figure 2A (looking up from the bottom). (C) A model replacing one nucleosomal H3.3-H4 heterodimer with that from the DAXX HBD complex (ribbon only). The other nucleosomal H3.3-H4 complex is shown in a semi-transparent surface representation superimposed onto a ribbon model. The coloring scheme is the same as in (A) and (B), except that DAXX is colored yellow here. Regions of spatial hindrance for the formation of an H3.3-H4 tetramer are indicated with red dashed-line circles.
H3.3 residues important for specific recognition by DAXX
H3.3 differs from H3.1 by five amino acids, Ser31(Ala31), Ala87(Ser87), Ile89(Val89), Gly90(Met90), and Ser96(Cys96), where the corresponding H3.1 residues are enclosed in parentheses. Three residues, Ala87(Ser87), Ile89(Val89) and Gly90(Met90) are located on the first turn of the α2 of H3.3 or H3.1 (Fig. 3A). Previous studies have shown that these three residues are critical for specific functions of H3.310. In the cocrystal structure, Ser31 is disordered and Ser96 is involved in interactions with H4, supporting the reported observations that they are not determinants of H3.3 specificity in DAXX binding14. Indeed, the H3.3 region where Ala87, Ile89 and Gly90 are located interacts with DAXX HBD (Fig. 3A). Ala87 in H3.3 is situated in a shallow hydrophobic pocket formed by the Cys338 and Leu340 located on the loop connecting α5 and α6 in DAXX HBD (Fig. 3A). The distance between the Cβ of Ala87 and the nearest atoms of Cys338 and Leu340 is approximately 4 Å, whereas His339, a polar residue, next to Cys338 and Leu340 is more than 6.5 Å away from Ala87. Ile89 in H3.3 is bound in a hydrophobic pocket formed by Leu210, Leu215 and Tyr222, which are located in α2 and its preceding loop in DAXX HBD (Fig. 3A). Finally, Gly90 has no sidechain, and the closest DAXX residue is Tyr222, whose nearest atom is at a distance of approximately 3.4 Å (Fig. 3A). The Cα atom of Gly90 directly faces Glu225 and Lys229 in DAXX, which are approximately 5.3 Å and 6.2 Å away, respectively.
Figure 3.

H3.3 residues responsible for DAXX binding specificity. (A) A stereo view of H3.3-specific interactions with DAXX HBD. The three H3.3-specific residues are highlighted in magenta, and the interacting DAXX residues are shown (DAXX-yellow; H3.3-green or magenta; H4-cyan). Helix α2 of a canonical H3 (gray; PDB 1KX5) is superimposed and the three residues different from H3.3 are shown for comparison. (B) A schematic diagram showing the lac operator-repressor (LacO-LacI) targeting system for assaying DAXX-H3.3 interactions. (C) Co-localization of DAXX with H3.3, H3.1 and indicated mutants. (D) Top panel: co-IP of full-length Flag-tagged DAXX with HA-tagged histones H3.1 and H3.3. IPs were analyzed by western blotting with indicated antibodies. Bottom panel: GST pull-down of wild-type or the C338S L340N double mutant of DAXX HBD with the H3.1-H4, H3.1(S87A)-H4, or H3.3-H4 complexes as indicated. The pull-downs were analyzed by Coomassie-stained SDS-PAGE.
With this structural knowledge in hand, we set out to assess the importance of each of these three residues in H3.3(H3.1) and the contributions of their cognate binding sites in DAXX in the specific recognition of H3.3. A previous study using in vitro GST pull-down and co-IP methods showed that mutating each of the H3.3 residues to their H3.1 counterparts did not abrogate DAXX binding14. Conversely, changing each of the corresponding amino acids in H3.1 did not show markedly increased binding to DAXX either14. We first tested co-localization of DAXX and H3.3 using a LacO-LacI targeting system in the A03_1 cell line25. In these experiments, plasmids carrying full-length DAXX or its derivative fused with mCherry and LacI, and H3.3/H3.1 or their derivatives fused with ECFP, were co-transfected into A03_1 cells (Fig. 3B). Consistent with the reported results14, we observed co-localization of A87S, I89V and G90M mutants of H3.3 with DAXX (Fig. 3C). An A87S G90M double mutant of H3.3 showed reduced co-localization with DAXX, while virtually no co-localization of an A87S I89V G90M triple mutant was detected (Fig. 3C and Supplementary Fig. 1A). In contrast, single amino acid substitution mutants of H3.1, S87A and M90G, gained the ability to co-localize with DAXX. To assess if co-localization of the H3.1 mutants with DAXX implies a physical interaction, an HA-tagged S87A mutant of H3.1, H3.1(S87A), and a Flag-tagged DAXX were co-transfected in HEK293T cells, and their association was analyzed by co-immunoprecipitation (co-IP). Also, a GST pull-down experiment was performed using bacterially expressed GST-DAXX HBD and the H3.1(S87A)-H4 mutant complex. Both the co-IP and GST pull-down results support a direct interaction between H3.1(S87A) and DAXX (Fig. 3D and Supplementary Fig. 2A). The above data suggest that the presence of either an alanine at residue 87 or a glycine at residue 90 is sufficient for DAXX binding.
H3.3-specific binding sites in DAXX HBD
To probe the nature of the binding sites and assess their binding preferences for the three H3.3-specific residues, we mutated DAXX and tested the binding of these mutants to H3.3 and H3.1. We first examined the H3.3’s Ala87 binding site in DAXX, which consists of hydrophobic residues Cys338 and Leu340 (Fig. 3A). Because of the hydrophobic nature of Ala87, we reasoned that changing Cys338 and Leu340 to polar residues would reduce the binding of H3.3 and H3.1(S87A). As expected, GST pull-down using the DAXX C338S L340N mutant showed detectable but subtle reduction of H3.1(S87A) and H3.3 binding (Fig. 3D). Co-localization of H3.1(S87A) with DAXX was affected to a greater extent than that of H3.3 by the C338S L340N mutation (Fig. 4A and Supplementary Fig. 1B). For comparison, the G90M mutant of H3.3, which differs from H3.1(S87A) at the Ile89(Val89) site, displayed a markedly reduced but still detectable co-localization with the C338S L340N mutant of DAXX (Fig. 4A). When the Ser87 of H3.1 is substituted with a similar size, but hydrophobic cysteine, the H3.1(S87C) mutant co-localized with the wild-type DAXX but not the C338S L340N mutant, much like the H3.1(S87A) mutant (Fig. 4A). All together, the above data indicate that the hydrophobic nature of Ala87 and a hydrophobic binding environment in DAXX are important for the preferential binding of H3.3 at this site.
Figure 4.

DAXX residues important for recognition of H3.3. (A) The involvement of Cys338 and Leu340 in the recruitment of H3.3 or H3.1 and their derivatives as indicated (left). The C338S L340N mutant of DAXX interferes with recognition of residue 87 in H3.3 and H3.1 (right). (B) Analyses of DAXX residues important for recognition of Gly90 of H3.3. Top panel: co-IP of DAXX or its E225M K229M double mutant with histones H3.1 and H3.3. Bottom panel: GST pull-down with the wild-type or mutant DAXX HBD (WT, E225M K229M, and Y222E) with H3.1-H4 and H3.3-H4 complexes as indicated. (C) Co-localization of E225M K229M and E225Q K229Q mutants with both H3.3 and H3.1.
Next, we examined H3.3’s G90 binding site in DAXX, which contains a pair of charged residues, Glu225 and Lys229 (Fig. 3A). We first changed them to a pair of hydrophobic residues, two methionines. Co-IP and GST pull-down analyses showed that H3.3 interacted well with the E225M K229M mutant of DAXX (Fig. 4B and Supplementary Fig. 2B). Interestingly, the DAXX mutant also gained the ability to interact with H3.1 (Fig. 4B). Co-localization experiments corroborated the co-IP and GST pull-down results, and showed that a polar but neutral E225Q K229Q mutant of DAXX co-localized with H3.1 (Fig. 4C and Supplementary Fig. 1C). A reasonable explanation of the above results is that Gly90 of H3.3 is not specifically recognized by DAXX, but rather a methionine at this position in H3.1 is incompatible with the binding site in DAXX. The inability of a methionine to bind the Glu225-Lys229 site in DAXX is not due to its size, as superposition of α2 in H3 with that in H3.3 shows that Glu225 and Lys229 allow ample space for a methionine (Fig. 3A). Hence, we conclude that DAXX has a preference against a hydrophobic residue at this position of histone H3.3(H3.1). Lastly, Tyr222 bisects the Gly90 and Ile89 binding sites in DAXX. The latter is hydrophobic; changing Tyr222 to a charged residue, glutamate, disrupted DAXX’s ability to bind H3.3 in a GST pull-down assay (Fig. 4B). Co-localization analyses are consistent with the notion that the H3.3 Ile89 binding site on DAXX needs to be hydrophobic (Supplementary Fig. 3).
Discussion
Here we have focused our study to understand the molecular mechanism by which the specificity for H3.3 is achieved by DAXX. It is remarkable that DAXX can precisely distinguish H3.3 and H3.1 with only three amino acid differences in the histone-fold domain. Our analyses here revealed that two H3.3 residues, Ala87 and Gly90 are principal determinants for specific recognition by DAXX HBD. Mutational studies indicated that any one of the two H3.3 residues could direct the recognition by DAXX. The data presented here suggest a chiefly two-site recognition mechanism: the Ala87 of H3.3 is a small, hydrophobic residue preferred by the shallow hydrophobic pocket formed by Cys338 and Leu340 in DAXX; and DAXX preferentially selects the Gly90 of H3.3 over the hydrophobic residue Met90 of H3.1. Clearly, Ile89 in H3.3 also has an effect in DAXX binding, but most likely through a less specific but stronger hydrophobic interaction between H3.3 and DAXX due to its bulkier side chain, compared that of Val89 in H3.1. It is interesting that an alanine at residue 87 or a glycine at residue 90 can independently direct the recognition by DAXX. This observation suggests that optimal binding at either of these two sites can overcome the presence of a non-optimal amino acid at the other site.
One puzzle from the structural results is that DAXX HBD binds a large surface area, most of which has the same amino acid composition in H3.3-H4 and H3.1-H4 complexes; then, how would the difference of a small number of amino acids in H3.3 and H3.1 results in drastically different DAXX binding properties? In fact, similar phenomena have been seen in other chaperone-histone complexes19–23. A plausible explanation is that DAXX and other histone chaperones may first survey the specific residues in their cognate histones before fully engaged in chaperone-histone interactions. Obviously, additional or alternative possibilities, such as post-translational modifications and participation of other proteins or nucleic acids in an in vivo environment, may contribute to the recognition and regulation of chaperone-histone interactions.
In conclusion, the structural basis of specific interactions between H3.3 and DAXX learned here should be useful for delineating the various pathways of H3.3 deposition, and facilitate our understanding of the molecular mechanisms of the biological and pathological functions of H3.3.
Methods
Protein expression and purification
cDNAs encoding human histones H3.3 and H4 were cloned into a pRSF-Duet vector (Novagen) to generate a bicistronic plasmid, pRSFDuet1-H3.3-H4, for co-expression of both proteins. The cDNA fragment encoding the histone-binding domain (HBD) of human DAXX (a.a. 184–390) was amplified by PCR and cloned into a pGEX-6P-1 vector. The DAXX HBD-H3.3-H4 complex was co-expressed using the two plasmids in E. coli BL21 CodonPlus (DE3) RIL cells. The expression of proteins was induced with 0.3 mM β-D-1-thiogalactopyranoside (IPTG) at 16°C overnight. Cells were then harvested by centrifugation and resuspended in buffer A (50 mM Tris-HCl pH8.0, 1 M NaCl, 10% Glycerol, 1 mM EDTA, 10 mM β-mercaptoethanol, and 1 mM phenylmethylsulphonyl fluoride), and lysed by sonication. Cell debris were removed by centrifugation, and the supernatant was incubated with glutathione (GSH) resins for 2 h at 4°C, followed by washing the resins three times with buffer A. The target protein complex was eluted with buffer B (25 mM Tris-HCl pH8.0, 500 mM NaCl, 5 mM β-mercaptoethanol, and 30 mM GSH). Cleavage of the GST tag was carried out by dialyzing the eluted sample in the presence of PreScission protease (GE Healthcare) against buffer C (25 mM Tris-HCl pH8.0, 300 mM NaCl, 5 mM β-mercaptoethanol) overnight at 4°C. The dialyzed sample was loaded onto an SP column (GE Healthcare) and the DAXX HBD-H3.3-H4 complex was eluted with a linear gradient of 0.3–2.0 M NaCl. The eluted sample was concentrated by ultrafiltration and loaded onto a HiLoad 200 16/60 size-exclusion column (GE Healthcare) in buffer D (10 mM Tris-HCl pH8.0, 1 M NaCl, 1 mM DTT). High purity fractions of the DAXX HBD-H3.3-H4 complex were pooled and concentrated to approximately 30 mg/ml using ultrafiltration and stored at −80°C before use.
Crystallization, data collection, structure determination and refinement
Native crystals of the DAXX HBD-H3.3-H4 complex were grown at 25°C using the sitting-drop vapor diffusion method by mixing 1.0 μl protein solution (12–15 mg/ml) with 1.0 μl reservoir solution containing 0.1 M HEPES-Na, pH 7.6, 23% (v/v) PEG 3350, 0.25 M Ammonium acetate, 1% Tacsimate pH 7.0 and 6% ethanol. After 7–8 days, crystals were transferred to cryo solution containing 10% ethylene glycol and flash-frozen in liquid nitrogen. Platinum derivatives were prepared by soaking the crystals with 5 mM K2PtCl4 for 10 minutes. The soaked crystals were then transferred to cryo solution and flash-frozen for storage. X-ray diffraction data were collected at the Shanghai Synchrotron Radiation Facility (SSRF) on beamline BL17U with a Quantum 315r CCD detector (ADSC) using a wavelength of 0.979 Å for native crystals and 1.070 Å for heavy atom derivatives. Data were indexed, integrated and scaled with the HKL2000 package26.
Four Pt sites were identified with SHELXD27 using the Pt-peak data set, and the Pt positions were imported into PHENIX28 for phase calculation at a 2.8 Å resolution. The initial model was built manually with COOT29, and the model was improved by iterative cycles of refinement with PHENIX. Since the quality of the Pt-derivative data set was higher than that of the native crystal, structure refinement was carried out using the data set from the Pt-derivative. The ordered structure contains residues 184–386 of DAXX HBD, residues 43–135 of histone H3.3, and residues 27–101 of histone H4. The refined model has satisfactory R-values (Rwork/Rfree = 20.8/27.1%) and good Ramachandran statistics (98.1% favored and 1.9% allowed). A section of the SAD-phased electron density map is shown in Supplementary Fig. 4. Detailed statistics from data collection and structure refinement are given in Table 1. Structural figures were prepared using PyMOL (http://www.pymol.org).
GST pull-down and co-immunoprecipitations
GST pull-down assays were performed essentially as described previously14. Briefly, 3 μg of GST or GST-fused DAXX HBD or its mutants was immobilized on 5 μL of Glutathione Sepharose 4 Fast Flow (GE Healthcare) resin, which was then mixed with 5 μg of histone tetramers in 100 μL of the binding buffer (20 mM Hepes-HCl pH7.9, 500 mM NaCl, 5 mM 2-mercaptoethanol, 0.2% NP-40, 1 mM EDTA), and the mixture was rotated overnight at 4 °C. The resins were then washed 5 times with 1 mL of binding buffer containing 1.0 M NaCl. Bound proteins were eluted in SDS sample buffer, separated on a 15% SDS-PAGE gel, and stained with Coomassie Blue. For in vivo interaction studies, 293T cells were transiently transfected with expression vectors encoding Flag-DAXX WT, Flag-DAXX Y222E, Flag-DAXX E225M K229M, HA-H3.1, HA-H3.3 and HA-H3.1(S87A). 24h after transfection, cells were lysed in RIPA buffer (50mM Tris pH7.4, 1% NP-40, 350mM NaCl, 1mM EDTA, 0.25% deoxycholate, 10% glycerol, 1mM phenylmethylsulphonyl fluoride, 1ug/mL aprotinin, 1ug/mL pepstatin, 1ug/mL leupeptin). The cleared lysates were subjected to immunoprecipitation with anti-FLAG M2 affinity gel (Sigma-Aldrich) or monoclonal anti-HA-Agarose (Sigma-Aldrich). The beads were extensively washed, and the bound proteins were eluted by boiling in the SDS sample buffer. The eluted proteins were separated on a 12% SDS-PAGE gel and analyzed by Western blotting, with antibodies specific to HA, FLAG, and α-tubulin (Sigma-Aldrich) used at 3000, 3000, and 5000 fold dilutions, respectively.
Cell lines and plasmids
The A03_1 cell line carrying an array of lacO operators25 was cultured in F-12 Ham’s medium (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% penicillin/streptomycin (Invitrogen). HEK-293T cells were maintained in Dulbecco modified Eagle medium (Hyclone), supplemented with 10% fetal bovine serum (ExCell) and 1% penicillin/streptomycin (Gibco). Plasmids for expressing histones H3.3 or H3.1 were constructed by cloning their respective cDNAs into a pECFP-C1 vector (for LacI-LacO targeting) or pCDNA4.0 vector (co-immunoprecipitation studies), respectively, and the full-length cDNA of human DAXX was cloned into a pmCherry-LacI vector (LacI-LacO targeting) or pFlag-CMV4.0 vector (co-immunoprecipitation), respectively. All site-directed mutagenesis experiments were carried out with Phusion (New England Biolabs) to generate mutant constructs. All of the constructs were confirmed by DNA sequencing.
Transfection and fluorescence microscopy
For transfections, A03_1 cells were seeded onto glass coverslips for 16 h. Plasmids were transfected into cells with LipoD293 (SignaGen) according to the manufacturer’s instructions. At 48 h after transfection, cells were washed with PBS and fixed with 4% paraformaldehyde in PBS for 15 min, then washed with PBS and stained with 4′,6-diamidino-2-phenylindole (DAPI) for 15 min. Confocal laser images were captured with an Olympus Fluview FV500 microscope (Tokyo, Japan) equipped with a 60× oil-immersion lens.
Supplementary Material
Acknowledgments
We thank SSRF beamline scientists for technical support during data collection. The work was supported by grants from the Chinese Ministry of Science and Technology (2009CB825501 to R.M.X. and 2011CB966300 to G.L.), the Natural Science Foundation of China (31210103914, 90919029 & 3098801 to R.M.X., and 31071147 to G.L.), and the Chinese Academy of Sciences (CAS) Strategic Priority Research Program (XDA01010304 to G.L.). R.M.X. is a CAS-Novo Nordisk Great Wall Professor.
Footnotes
Accession Codes
PDB id: 4HGA
Author contributions
R.M.X., G.L. and Z.Z. conceived the project; R.M.X. and G.L. designed the experiments and Z.Z. contributed reagents. C.P.L, C.X., M.W., Z.Y., N.Y. and P.C. performed the experiments, and all authors analyzed the data. C.P.L., G.L. and R.M.X. wrote the paper.
References
- 1.Talbert PB, Henikoff S. Histone variants--ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol. 2010;11:264–75. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 2.Banaszynski LA, Allis CD, Lewis PW. Histone variants in metazoan development. Dev Cell. 2010;19:662–74. doi: 10.1016/j.devcel.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Res. 2011;21:421–34. doi: 10.1038/cr.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campos EI, Reinberg D. New chaps in the histone chaperone arena. Genes Dev. 2010;24:1334–8. doi: 10.1101/gad.1946810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Black BE, Cleveland DW. Epigenetic centromere propagation and the nature of CENP-a nucleosomes. Cell. 2011;144:471–9. doi: 10.1016/j.cell.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunleavy EM, et al. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell. 2009;137:485–97. doi: 10.1016/j.cell.2009.02.040. [DOI] [PubMed] [Google Scholar]
- 7.Foltz DR, et al. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell. 2009;137:472–84. doi: 10.1016/j.cell.2009.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanchez-Pulido L, Pidoux AL, Ponting CP, Allshire RC. Common ancestry of the CENP-A chaperones Scm3 and HJURP. Cell. 2009;137:1173–4. doi: 10.1016/j.cell.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ray-Gallet D, et al. HIRA is critical for a nucleosome assembly pathway independent of DNA synthesis. Mol Cell. 2002;9:1091–100. doi: 10.1016/s1097-2765(02)00526-9. [DOI] [PubMed] [Google Scholar]
- 10.Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell. 2002;9:1191–200. doi: 10.1016/s1097-2765(02)00542-7. [DOI] [PubMed] [Google Scholar]
- 11.Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116:51–61. doi: 10.1016/s0092-8674(03)01064-x. [DOI] [PubMed] [Google Scholar]
- 12.Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010;24:1253–65. doi: 10.1101/gad.566910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldberg AD, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–91. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci USA. 2010;107:14075–80. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawatsubashi S, et al. A histone chaperone, DEK, transcriptionally coactivates a nuclear receptor. Genes Dev. 2010;24:159–70. doi: 10.1101/gad.1857410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khuong-Quang DA, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–47. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu G, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–3. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwartzentruber J, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 19.English CM, Adkins MW, Carson JJ, Churchill ME, Tyler JK. Structural basis for the histone chaperone activity of Asf1. Cell. 2006;127:495–508. doi: 10.1016/j.cell.2006.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Natsume R, et al. Structure and function of the histone chaperone CIA/ASF1 complexed with histones H3 and H4. Nature. 2007;446:338–41. doi: 10.1038/nature05613. [DOI] [PubMed] [Google Scholar]
- 21.Hu H, et al. Structure of a CENP-A-histone H4 heterodimer in complex with chaperone HJURP. Genes Dev. 2011;25:901–6. doi: 10.1101/gad.2045111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou Z, et al. Structural basis for recognition of centromere histone variant CenH3 by the chaperone Scm3. Nature. 2011;472:234–7. doi: 10.1038/nature09854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho US, Harrison SC. Recognition of the centromere-specific histone Cse4 by the chaperone Scm3. Proc Natl Acad Sci USA. 2011;108:9367–71. doi: 10.1073/pnas.1106389108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tachiwana H, et al. Structures of human nucleosomes containing major histone H3 variants. Acta Crystallogr D Biol Crystallogr. 2011;67:578–83. doi: 10.1107/S0907444911014818. [DOI] [PubMed] [Google Scholar]
- 25.Robinett CC, et al. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J Cell Biol. 1996;135:1685–700. doi: 10.1083/jcb.135.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 27.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 28.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.